单晶结构分析用SHELXTL程序进行晶体
SHELXTL程序进行晶体结构分析的方法

y
z
占有率 温度因子
C1 1 0.155832 0.182795 0.318988 11.00000 0.00001
温度因子: 原子是否正确 过大: 偏重 过小: 偏轻
B 进一步精修:
主体结构的确定
各向异性: ANIS 针对温度因子
ANIS 温度因子用六个参数表示,原子的热振动为 取向的三轴椭球
进行这步操作,可以大幅降低R1值和goof值
e 点refine菜单,系统会提示有Q 峰没命名,并 选择所有Q 峰,顺序与原来顺序相同
•删除原子或(Q)峰的方法: a 光标指在欲删除位置(原子或键) ,点K 键
b 光标指在欲删除位置,右手键点Bonds and Angles,再在对话框中点“Delete”
c 光标指在欲删除位置,右手键点“Delete” d 先选择,再点Select菜单中的Kill selected e 在ins文件删除
Most Disagreeable Reflections (* if suppressed or used for Rfree)
m 是一或两位数,指定氢的类型: =1 叔-H, =2 仲-H, =3(或13) 伯-H, =4 芳-H, =8(或14) X-O-H, =9 X=CH2或X-NH2, =15 笼状B-H
n 是一位数,指定固定的类型: =1 坐标、占有率、位移因子固定, =2 占有率、位移因子固定, =3(或7) 坐标固定, =4 同3,但允许修正X-H的键长(方向固定)
4.INS文件的建立和更新
结构解析和精修的过程,是ins文件建立和不 断更新的过程,这主要是下列过程实现的:
xprep、xshell—refine、xl、xp、edit、copy
SHELXTL程序进行晶体结构分析的方法报告

XP中做的图形文件
晶体学信息文件
结构因子文件
记录仪器型号、晶体外观等的文件
SHELXTL结构分析的步骤
1、准备反射点文件 .HKL,格式一般为h, k, l, Io, (I) 。 2、使用 XPREP 程序输入晶胞参数,进行数据统计和检查消光规律以确 定空间群,输入分子式等等,程序结束时输出 .INS。该 .INS文件通常设 定了直接法的现行设置。 3、使用 XS 解析结构,读入 .HKL和 .INS文件,结果输出到 .LST和 .RES文件。 4、使用 XP/XSHELL 读入 .RES文件,建立初始结构模型,结果输出到 .INS文件,该 .INS文件通常设定了最小二乘法修正和 Fourier 合成的设 置。 5、使用 XL 读入 .HKL和 .INS文件,进行结构模型的最小二乘法修正和 Fourier合成(通常为差值Fourier合成),结果输出到 .LST和 .RES文件。 6 、重复 4 , 5过程不断扩展完善结构模型和精修,直到结构修正收敛和 偏离因子最低。 7、生成数据CIF表格。
3.其它文件
晶体结构报表文件 4.INS文件的建立和更新 结构解析和精修的过程,是ins文件建立和不 断更新的过程,这主要是下列过程实现的: xprep、xshell—refine、xl、xp、edit、copy
res lst plt cif fcf pcf tex
xs、xl、refine产生的文件
单晶结构分析电子教案
用SHELXTL程序 进行结构分析的方法
H H HO HO HO OH O
H H H
OH
SHELXTL有以下程序: windows 界面
XPREP:为结构解析准备 .INS文件程序。
单晶结构解析

两个必要文件(由XPREP程序产生) name.hkl, name.ins
结构解析和精修的过程,是ins文件建立和不断更新的过程, 这主要是下列过程实现的:XPREP、XS、XL、XP
其它文件
res lst plt cif fcf pcf tex
xs、xl、refine产生的文件 记录xs、xl、refine过程和结果的文件 XP中做的图形文件 晶体学信息文件 结构因子文件 记录仪器型号、晶体外观等的文件
画图
XCIF
打印表格
二、数据处理--XPREP
运行步骤:
1.从name.hkl文件(若存在)或name.raw文件中读入衍射 点;2.从name.p4p或键盘获得单胞参数及误差 3.判断晶格类型 4.寻找最高对称性
5.确定空间群
6.输入分子 7.建立 name.hkl和 name.ins
* XPREP的主要功能和应用
读入、更改、 •单击进入XPREP程序 合并衍射数据 •根据程序的提示输入晶胞参数 •选择可能的晶格 程序则显示以下菜单: 计算显示 Patterson截面 寻找更高 的对称性 确定或输 入已知的 空间群
[D] Read,Modify or Merge DATDSETS [P] Contour PATTERSON Secions [H] Search for HIGHER mertric symmetry [S] Determine or input SPACE GROUP
•XS计算结果的评估
# 直接法,RE越小越好,一般大于0.3,就预示 着不成功,可以尝试用Patterson法来解
N
O
C u (N O )2 3
+
O N
E tO H
用SHELXTL程序进行晶体结构分析的方法 单晶结构分析电子教案

用SHELXTL程序进行晶体结构分析的方法单晶结构分析电子教案第一章:SHELXTL程序简介1.1 背景知识介绍晶体学的基本概念和晶体结构分析的重要性。
解释单晶结构分析与其他结构分析方法的区别。
1.2 SHELXTL程序介绍介绍SHELXTL程序的基本功能和用途。
解释SHELXTL程序在晶体结构分析中的重要性。
1.3 SHELXTL程序的安装和使用说明SHELXTL程序的安装步骤和注意事项。
演示如何运行SHELXTL程序并进行基本操作。
第二章:晶体数据收集2.1 实验设备和技术介绍用于晶体数据收集的实验设备和技术。
解释X射线衍射仪的工作原理和重要性。
2.2 晶体数据的收集过程说明晶体数据收集的步骤和注意事项。
演示如何使用X射线衍射仪进行晶体数据收集。
2.3 数据质量评估介绍评估晶体数据质量的方法和指标。
解释数据质量对晶体结构分析的影响。
第三章:SHELXTL程序的操作3.1 输入数据和参数说明如何将收集到的晶体数据输入到SHELXTL程序中。
介绍如何设置SHELXTL程序的参数和选项。
3.2 解晶体结构解释SHELXTL程序解晶体结构的过程和步骤。
演示如何使用SHELXTL程序解晶体结构并获取结果。
3.3 结构精修和优化介绍结构精修和优化的概念和方法。
解释SHELXTL程序中结构精修和优化的操作步骤。
第四章:结果分析和报告4.1 结构分析结果的查看和解读介绍如何查看和解读SHELXTL程序的结构分析结果。
解释结构分析结果中的关键信息和指标。
4.2 结构分析结果的可视化说明如何使用SHELXTL程序进行结构分析结果的可视化。
演示如何结构分析结果的图像和图表。
第五章:实例分析与讨论5.1 实例分析提供一个具体的晶体结构分析实例。
演示如何使用SHELXTL程序进行实例分析并解释结果。
5.2 结果讨论和解释讨论和解释实例分析的结果和意义。
探讨可能的问题和解决方法。
5.3 练习和习题提供一些相关的练习和习题,帮助学生巩固所学内容。
单晶结构解析XP作图

单晶结构解析XP作图:1、画结构图打开Shelxtl软件→导入res文件→打开XP程序→输入fmol→kill $q→kill $h→envi (中心对称原子)→回车后寻找不同于1555对称操作的代码如2555→sgen 2555→proj(查看结构)→利用操作按钮转动结构找出最佳摆放位置(高度不能大于宽度)→labl 2 500(2 500是默认的大小) →telp 0 -30 0.05 0(后面四个数据分别确定结构的模型和一些参数) →回车回车直到出现Plotfile:(在这里输入名字,这里画结构图,可以统一命名为jiegou) 后回车将会出现命名所有原子的图(根据鼠标位置提醒依次在原子周围左键点击)→draw jiegou→回车后会出现SLPT device[L]:(输入a或者h)再回车输入jiegou,然后回车直到光标不闪位置→出现了xp《图标说明结构图已经画好→quit注:红色字体为结构需要对称操作才能显示一个完整的分子结构所进行的操作。
图片操作解析在这里必须输入命名,否者打不开。
在这里找到res文件的位置2导入res文件后,打开XP操作系统输入fmol后回车3输入kill $h $q4 proj后出现下图所示利用这些按钮旋转得到最佳摆放模式如下图5 按步骤画图二、作堆积图Fmol→matr 1(代表a方向堆积) →pbox 15 15→pack 然后点击按钮sgen/fmol保存(倒数第二个)→proj cell→telp 命名所有原子当出现这个时表明图已经画好cell→命名duiji 后出现一个命名原子的图框,标出O a b c→draw duiji→a或h→duiji→→quit1、此步骤可有可无这个键保存标出Oabc即可后按B键保存退出后面是一样的,quit退出程序三、画弱相互作用:氢键、p···π、π···π、M···M等首先要找到氢键的位置,然后再根据对称操作找到氢键画法基本步骤为fmol→kill $q→uniq 原子1 原子2(为氢键的两个原子如uniq C5 N4) →envi N4 3(通过N4对称操作找到C5的操作代码不能为1555如为2555) →sgen 2555→join 3 H5 N4(3代表虚线、注意氢键的因为H5和N4非C5和N4,如错误,可用undo C5 N4来断开这之间的) →pick(杀掉无关的原子:回车键为杀原子,空白键为跳过,/键为保存,注意杀完后不是按/键退出的将没有保存)→telp后面画图步骤与上面一致(只需要标出C5和N4原子)π···π画法对称出两个需要连接的苯环后sgen后→cent/x C5 C6 C7 C8 C9 C10(定义苯环C5C6C7C8C9C10的中心为X1A) →cent/x C15 C16 C17 C18 C19 C20(定义苯环C15C16C17C18C19C20的中心为X1B) →join 3 X1A X1B →proj摆放最佳→pick→telp(画π···π键不需要命名原子telp后命名后出现的命名原子框可以之间按B键退出)以下面画C15-H15A···N3为例经过对称操作后应该为H15C和N3相连此时若不需要再画其他氢键了,可以将两边没有连键的原子杀掉如下图杀完原子后按/键保存退出将显示所杀掉的原子数依次telp只需要命名两个相关的原子即可enter为跳过原子,然后按B键保存退出,或者一直enter到最后也可以保存退出。
XRD从原理到实践从精修到表达用SHELXTL程序进行晶体结构分析的方法
2. 结构的解析 1) 结构解析的基本原理 XS用直接法或Patterson法解决相角问题,试验 性找出部分原子或重原子的位置(坐标) * 所谓直接法(direct methods),就是运用数 学的方法,利用不同衍射点的关系,从大量强度数据 中,直接找出各个衍射点的相角,从而达到解析晶体 结构的目的。其过程概括如下:
SIZE 0.47 0.43 0.35 HTAB 2 L.S. 8 ACTA 可输入各种让XS和XL执行的命令 BOND FMAP 2 PLAN 20
信 息 区
命 令 区
DFIX 1.43 0.02 o5 c27 WGHT 0.074900 1.631200 FVAR 0.169240 TEMP 25
2) 用XS和XSHELL程序定出结构雏形(初始套) 一般先试直接法,再试Pattson法,用什么方法 是通过改变Edit .ins文件中的指令来实现的
直接法:
TITL 020908b in C2/c CELL 0.710730 30.1927 8.5175 13.9108 90.0000 95.1300 90.0000 ZERR 8.00 0.0146 0.0042 0.0071 0.0000 0.0100 0.0000 LATT 7 SYMM -X, Y, 0.5-Z SFAC C H N O Cr UNIT 144 112 24 56 8 TEMP 25 TREF HKLF 4 END
原子表 命 坐标、占有率、温度因子 令 区
Co1 5 0.639436 0.182296 0.778299 11.000000 0.037950 = 0.043120 0.052330 -0.002120 0.007180 -0.002370 Co2 5 0.132299 0.088237 0.782543 11.000000 0.061270 = 0.080610 0.082660 -0.004140 0.011300 0.005080 N1 3 0.816545 0.181404 0.857527 11.000000 0.048210= 0.044720 0.042580 -0.001460 0.010450 -0.001390 N2 3 0.641818 0.098803 0.864065 11.000000 0.055810 = 0.050870 0.070080 0.000700 0.013560 -0.009990 …………………………………………………………………. HKLF 4 衍射点文件类型 END
SHELXTL程序进行晶体结构分析的方法-1

-1.200000 -1.200000
检查cif若是报错的话 可以通过 命名水分子周围其它方向的
氢键来解决,但是不是绝对能解决
2021/5/27
30
2021/5/27
12
看完整度
空间群越对称,越高,需要点越少 低原因:空间群选低了,找高空间群
晶体数据不好
2021/5/27
13
输入分子式:
输入元素种类,尤其是重原子,但数目不重要(原子要大写) CHONBFeCoCl
2021/5/27
14
Input Name 一般都是输入 1
Input Y 之后点回车即可
m 是一或两位数,指定氢的类型:
=1 叔-H, =2 仲-H, =3(或13) 伯-H,
=4 芳-H, =8(或14) X-O-H,
=9
2021/5/27
X=CH2或X-NH2,
=15 笼状B-H
27
n 是一位数,指定固定的类型: =1 坐标、占有率、位移因子固定, =2 占有率、位移因子固定, =3(或7) 坐标固定, =4 同3,但允许修正X-H的键长(方向固定)
4.INS文件的建立和更新
结构解析和精修的过程,是ins文件建立和不 断更新的过程,这主要是下列过程实现的: 2021/5/x27prep、xshell—refine、xl、xp、edit、copy 4
SHELXTL结构分析的步骤
1、准备反射点文件 .HKL,格式一般为h, k, l, Io, (I) 。 2、使用 XPREP 程序输入晶胞参数,进行数据统计和检查消光规律以确 定空间群,输入分子式等等,程序结束时输出 .INS。该 .INS文件通常设 定了直接法的现行设置。 3、使用 XS 解析结构,读入 .HKL和 .INS文件,结果输出到 .LST和 .RES文件。 4、使用 XP/XSHELL 读入 .RES文件,建立初始结构模型,结果输出到 .INS文件,该 .INS文件通常设定了最小二乘法修正和Fourier 合成的设 置。 5、使用 XL 读入 .HKL和 .INS文件,进行结构模型的最小二乘法修正和 Fourier合成(通常为差值Fourier合成),结果输出到 .LST和 .RES文件。 6、重复4,5过程不断扩展完善结构模型和精修,直到结构修正收敛和 偏离因子最低。 7、生成数据CIF表格。
用SHELXTL程序进行晶体结构分 析的方法共68页文档

31、只有永远躺在泥坑里的人,才不会再掉进坑里。——黑格尔 32、希望的灯一旦熄灭,生活刹那间变成了一片黑暗。——普列姆昌德 33、希望是人生的乳母。——科策布 34、形成天才的决定因素应该是勤奋。——郭沫若 35、学到很多东西的诀窍,就是一下子不要学很多。——洛克
33、如果惧怕前面跌宕的山岩,生命 就永远 只能是 死水一 潭。 34、当你眼泪忍不住要流出来的时候 ,睁大 眼睛, 千万别 眨眼!你会看到 世界由 清晰变 模糊的 全过程 。注定 要融化 的,也 许是用 眼泪的 方式。
35、不要以为自己成功一次就可以了 ,也不 要以为 过去的 光荣可 以被永 远肯定 。
用SHELXTL程序进行晶体 结构分 析的方法
31、别人笑我太疯癫,我笑他人看不 穿。(名 言网) 32、我不想听失意者的哭泣,抱怨者 的牢骚 ,这是 羊群中 的瘟疫 ,我不 能被它 传染。 我要尽 量避免 绝望, 辛勤耕 耘,忍 受苦楚 。我一 试再试 ,争取 每天的 成功, 避免以 失败收 常在别 人停滞 不前时 ,我继 续拼搏 。
单晶结构分析

SHELXTL的主要子程序和文件
XS XPREP XP *.plt *.sav *.ps
*.hkl
*.ins
*
*.pcf XCIF
XL
XSHELL *.cif *.fcf
*.lst
*.tex
二 常用的XS和XL指令
指令 ACTA 产生cif文件 含 义
将原子坐标强制性地固定在指定位臵上, 或在指定位臵上产生原子 ANIS 将各向同性换成各向异性精修 BOND 计算键长、键角(加$H包括H的键长、键角) BIND 计算指定原子对的键长、键角 CONF 计算扭转角 DELU 限制指定原子间键长的标准偏差 DFIX 限定指定原子对间的距离 EADP 给两个或多个原子指定相同的位移参数 AFIX
HFIX mn 需加氢的原子名(产生AFIX固定)
m 是一或两位数,指定氢的类型: =1 叔-H, =2 仲-H, =3(或13) 伯-H, =4 芳-H, =8(或14) X-O-H, =9 X=CH2或X-NH2, =15 笼状B-H
n 是一位数,指定固定的类型: =1 坐标、占有率、位移因子固定, =2 占有率、位移因子固定, =3(或7) 坐标固定, =4 同3,但允许修正X-H的键长(方向固定) 然后进行精修 • 也可在XSHELL中先点Atoms-Hybridize All 再 点Atoms-Calculate Hydrogens 然后检查H加的是否合理,如不合理,可打开 ins 文件修改不合理的部分,再精修 • 还可在XSHELL中先选择同类原子,再用 Select > Atoms > Set HFIX分别进行加氢
原子类型
命 令 区
DFIX 1.43 0.02 o5 c27 WGHT 0.074900 1.631200 FVAR 0.169240 TEMP 25
单晶结构精修__用SHELXTL程序进行晶体结构精修.
* 如果得到的原子或峰的位置能够构成合理的
化学结构,就要通过删除不合理的原子或(Q)峰, 并把Q 峰选择后定为相应的原子。操作方法是: •选择原子或(Q)峰的方法:
a 光标指在欲选择位置,点S 键。可依次选 择所有的要选择的原子或Q 峰
b 光标指在欲选择位置,右手键点“Select”, 可依次选择原子
用这种方法时,首先利用重原子的特征峰,即 Harker峰,求出重原子坐标,再通过Fourier合成 获得其它原子的坐标 Harker峰,或Harker截面,就是同一套等效的 原子组成的Patterson峰,由于平方效应,重原子的 Harker峰会显得十分突出,寻找起来一般比较容易。 因此,可以轻松地从分析重原子的Harker峰,得到 重原子的坐标
主要的三种情况: a 晶胞的错误确定引起的 b 仪器只根据Rint和CFOM(诊断因子)值 来确定,有时会降低晶体的对称性
c 由系统消光规律的错误判断引起 解决方法:在xprep中重新确定空间群;重新做晶胞
* XPREP的主要功能和应用
读入、更改、 •单击进入XPREP程序 合并衍射数据
•根据程序的提示输入晶胞参数 •选择可能的晶格 程序则显示以下菜单: 计算显示 Patterson截面 寻找更高 的对称性 确定或输 入已知的 空间群
设置好ins文件 ,点击XS,就可自动进行计算
•XS计算结果的评估
# 直接法,RE越小越好,一般大于0.3,就预示 着不成功
# 打开Xshell,检查计算出的原子或(Q) 峰位 置是否构成合理的化学结构
# 精修(refint)一次后,R1值是否大小于0.5; 或继续精修R1值是否很快降低
2) 用XS和XSHELL程序定出结构雏形(初始套) 一般先试直接法,再试Pattson法,用什么方法 是通过改变Edit .ins文件中的指令来实现的
[自然科学]单晶结构解析_OK
![[自然科学]单晶结构解析_OK](https://img.taocdn.com/s3/m/bea9818002768e9950e73892.png)
b 建立可以利用正切公式的三相角及四相角关系
c 赋于起始相角
d 利用正切公式精修相角
2021/8/28
12
e 计算诊断指标,判断各套相角的质量
f 采用诊断指标最佳的相角数据计算解析电子密度图,即E图
* Patterson法是其本人1934年提出,通常只用来解 析含有重原子的结构
画图
name.ins
XCIF
打印表格
4
二、数据处理--XPREP
运行步骤:
1.从name.hkl文件(若存在)或name.raw文件中读入衍射点;2. 从name.p4p或键盘获得单胞参数及误差 3.判断晶格类型 4.寻找最高对称性 5.确定空间群 6.输入分子 7.建立 name.hkl和 name.ins
指定原子半径
CELL
加/x同时给中显心示点晶X胞1A参数
CENT/x Atomnames
计算并显示指定原子的中心
DRAW filename指定显示的范打围印结构图或转换图形文件
ENVI n A1
显示指定原子的环境
EXAM
显示该通道中所有的文件
EXIT
退出XP
FILE filenames
存储XP中产生的文件
Option A: FOM = 0.025 deg. ORTHORHOMBIC P-lattice R(int) = 0.022 [ 3032] Cell: 5.965 9.042 18.403 90.00 90.02 90.01 Volume: 992.52 Matrix: 1.0000 0.0000 0.0000 0.0000 1.0000 0.0000 0.0000 0.0000 1.0000
用SHELXTL程序进行晶体结构分析的方法-2

B 刚性基团的固定
苯环的固定(第一种方法)
C5 1 AFIX 66 C6 1 C7 1 C8 1 C9 1 C10 1 C11 1 AFIX 0 C12 1 0.783982 0.389310 0.472723 11.00 0.670845 0.623140 0.132871 0.187310 0.301438 0.350699 0.385330 0.384059 0.107389 0.112700 0.113657 0.112192 0.475554 0.522099 0.616281 0.566092 0.561510 0.515138 0.056310
强制性精修(固定)
A 不同原子在同一位置的固定
……………… TEMP 25 EADP CL1 CL1' EXYZ CL1 CL1' WGHT 0.000000 FVAR 0.09096 0.5 CR1 5 0.113174 0.216284 0.628141 11.00 0.03132 0.04649 = 0.03455 0.00095 0.00183 0.00118 CL1 3 0.118366 0.241830 0.483154 21.00 0.02696 0.03867 = 0.03287 -0.00084 0.00065 0.00346 CL1' 4 0.118366 0.241830 0.483154 -21.00 0.02696 0.03867 = 0.03287 -0.00084 0.00065 0.00346 ………………
高氯酸根无序
WGHT 0.045200 0.000000 FVAR 0.286910 0.5 Cl1 5 …………………… PART 1 O1 4 x y z 21.000 0.05 O2 4 x y z 21.000 0.05 O3 4 x y z 21.000 0.05 O4 4 x y z 21.000 0.05 PART 2 O1' 4 x y z -21.000 0.05 O2' 4 x y z -21.000 0.05 O3' 4 x y z -21.000 0.05 O4' 4 x y z -21.000 0.05 PART 0 高氯酸根键长的固定 DFIX 1.44 0.01 cl1 o1 cl1 o2 cl1 o3 cl1 o4 cl1' o1' cl1' o2' cl1' o3' cl1' o4' DFIX 2.35 0.02 o1 o2 o1 o3 o1 o4 o2 o3 o2 o4 o3 o4 o1' o2' o1' o3' o1' o4' o2' o3' o2' o4' o3' o4' EXYZ CL1 CL1' EADP CL1 CL1'
用SHELXTL程序进行晶体结构分析的方法-2
WGHT 0.045200 0.000000 .in FVAR 0.286910 0.5 s N1 3 0.338342 0.110671 0.424071 11.00 0.072990 0.046510 = 0.040170 0.003320 0.008270 0.021590 part 1 O1 4 0.6986 0.4252 0.6507 21.00 0.05 part 2 O1’ 4 0.6922 0.3560 0.6522 -21.00 0.05 part 0 O2 4 0.529357 0.400370 0.615937 11.00 0.053290 0.087650 = 0.054980 -0.001990 0不 同,则不能用AFIX66或AFIX59进行 固定,而是要用DFIX的键长命令进 行固定 如果只是不共面,可用共平面命令: PLAT 0.01 C1>C6 或者 PLAT 0.01 C1 C2 C3 C4 C5 C6
烷基链无序
C2 C1 C3
C4
无序处理的命令同前 对于无序的烷基链,精修时可以限定相邻原子键长为1.54 0.01 ;间隔一个C原子的两个C原子之间的距离为2.52 0.01 DFIX 1.54 0.01 C1 C2 C2 C3 C3 C4 DFIX 2.52 0.01 C1 C3 C2 C4
化 学 键 (Chemical bond) B —F C —F P —F C—Cl O—Cl C —C C═C C≡C
键 长(Bond length) /(Å) 1.52 1.38 1.58 1.77 1.44 1.54 1.34 1.20
化 学 键 (Chemical bond) C—N C═N C≡N N—N N═N N≡N N—O N═O
用SHELXTL程序进行晶体结构分析的方法-2
B 刚性基团的固定
苯环的固定(第一种方法)
C5 1 AFIX 66 C6 1 C7 1 C8 1 C9 1 C10 1 C11 1 AFIX 0 C12 1 0.783982 0.389310 0.472723 11.00 0.670845 0.623140 0.132871 0.187310 0.301438 0.350699 0.385330 0.384059 0.107389 0.112700 0.113657 0.112192 0.475554 0.522099 0.616281 0.566092 0.561510 0.515138 0.056310
(第二种方法)
DFIX 1.37 0.02 C1 C2 C2 C3 C3 C4 C4 C5 C5 C1 (六条边) DFIX 2.20 0.02 C1 C3 C1 C4 C2 C5 C2 C4 C3 C5 (五条对角线)
常见化学键的键长表
C 限制性精修
a 键长的限 FMAP 2 PLAN 20 dfix 1.50 0.01 C1 C2 C3 C4 TEMP 25 b 同类键键长相近的限制 FMAP PLAN SADI SADI TEMP
化 学 键 (Chemical bond) B —F C —F P —F C—Cl O—Cl C —C C═C C≡C
键 长(Bond length) /(Å) 1.52 1.38 1.58 1.77 1.44 1.54 1.34 1.20
化 学 键 (Chemical bond) C—N C═N C≡N N—N N═N N≡N N—O N═O
茂环的固定
(第一种方法)
C5 1 0.783982 0.389310 0.472723 11.000000 0.056310 AFIX 59 C6 1 0.670845 0.385330 0.475554 11.000000 0.037930 C7 1 0.623140 0.384059 0.522099 11.000000 0.038030 C8 1 0.670845 0.385330 0.475554 11.000000 0.037930 C9 1 0.623140 0.384059 0.522099 11.000000 0.038030 C10 1 0.301438 0.113657 0.561510 11.000000 0.036590 AFIX 0 C11 1 0.350699 0.112192 0.515138 11.000000 0.035930 C12 1 0.284100 0.110708 0.472513 11.000000 0.043900
用xshell进行晶体结构解析和精修
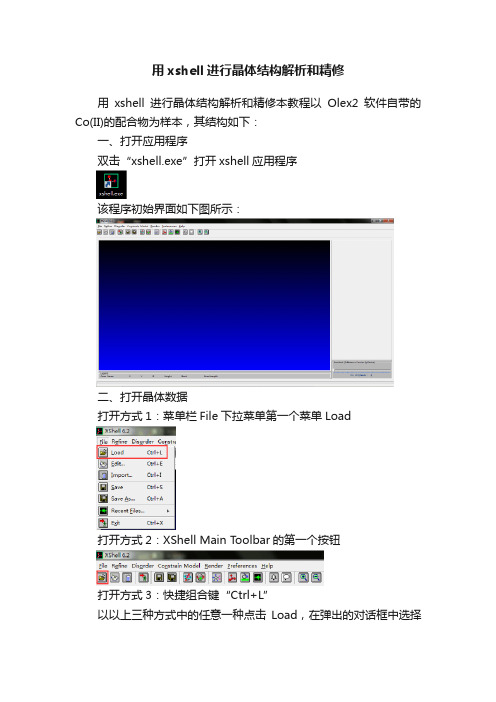
用xshell进行晶体结构解析和精修用xshell进行晶体结构解析和精修本教程以Olex2软件自带的Co(II)的配合物为样本,其结构如下:一、打开应用程序双击“xshell.exe”打开xshell应用程序该程序初始界面如下图所示:二、打开晶体数据打开方式1:菜单栏File下拉菜单第一个菜单Load打开方式2:XShell Main Toolbar的第一个按钮打开方式3:快捷组合键“Ctrl+L”以以上三种方式中的任意一种点击Load,在弹出的对话框中选择要打开的文件,并打开该文件,如下图所示:打开后如下图所示,蓝色区域显示的是化合物的结构,右下角是Q峰条三、结构解析1.Q峰的显示将Q峰条处的“”用鼠标拉至最左端,让左边蓝色界面中的Q峰数目减少至0个,如下图所示:可以看到,系统默认固定下了中心金属Co,并将其命名为Co1,以及三个S原子,并将其命名为S2,S3,S42. 结构的放大与缩小在XShell Main Toolbar中最后有两个按钮,如上图所示,可以将蓝色界面显示的结构放大和缩小以便查看局部结构和全局结构3. 不相连结构的单独显示当结构中有不连在一起的多个单独结构时,可以只显示其中某个单独结构,比如当前状态下Co1是一个单独结构,S3是一个单独结构,S2-S4是一个单独结构。
比如我要显示S2-S4这个单独结构,可以将鼠标放在S2-S4结构中的任意原子或者键上,当鼠标变为白色十字时(如下左图所示),单击鼠标右键,在弹出的菜单(如下右图所示)中点击“Associate Connected Atoms”即可单独显示S2-S4独立结构:单独显示的S2-S4独立结构:在蓝色界面任意处单击鼠标右键,在弹出的对话框中点击“Disassociate Atoms”即可恢复至全局结构显示键长:将鼠标移动至某一个键(此处将鼠标移动至S2-S4键)上,在蓝色界面下方会有键长信息,如下图红色方框所示:原子信息:将鼠标移动至某一个原子(此处将鼠标移动至Co1)上,在蓝色界面下方会显示该原子的名称以及坐标信息5. 选择原子或键原子的选择:将鼠标移动至某个原子上,当鼠标变为白色十字时,单击鼠标左键,该原子及其名称会变成蓝色(如下左图所示),这表示该原子被选中键的选择:将鼠标移动至某个键上,当鼠标变为白色十字时,单击鼠标左键,该键会变成蓝色(如上右图所示),这表示该键被选中但原子或键被选中后,如果点击键盘上的“Delete”按钮,则被选中的原子或键会被删除。
单晶结构分析 用SHELXTL程序进行晶体结构分析的方法

-4 -2 -2 20.459 3.120
…………………………………..
TCIETL*LL.i0n04.17s21晶0文037e格3标件iXn1类1P题光:.528(型1波组7)0/c长成19.上308可0空1分间7.2成1群44五90部.00分0 10晶8.47胞1 参90.0数00
0.044720 0.042580 -0.001460 0.010450 -0.001390
N2 3 0.641818 0.098803 0.864065 11.000000 0.055810 =
0.050870 0.070080 0.000700 0.013560 -0.009990
………………………………………………………………….
DELU 限制指定原子具有相似的位移参数
DFIX 限定指定原子对间的距离
EADP 给两个或多个原子指定相同的位移参数
指令
含
义
END 指令输入结束
EQIV 提供分子内或分子间键合原子的对称操作码
ESEL 限制E值的下、上限
EXTI 对晶体消光效应参数进行精修
EXYZ 让两个或多个原子具有相同的坐标
FLAT 限制指定原子在相同的平面上
HKLF 4
衍射点文件类型
END
文件结束命令
3.其它文件
res xs、xl、refine产生的文件 lst 记录xs、xl、refine过程和结果的文件 plt XP中做的图形文件 cif 晶体学信息文件 fcf 结构因子文件 pcf 记录仪器型号、晶体外观等的文件 tex 晶体结构报表文件
4.INS文件的建立和更新
•删除原子或(Q)峰的方法: a 光标指在欲删除位置(原子或键) ,点K 键
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
•常用 的小技巧:
a 解析和精修结构 的初期,可把C、O、N都定为C
b 如果独立单元中分子较多,可光标指在 一个分 子中的某一原子上,再用右手键菜单中的UNIQ指令把 其分离出来处理
记录xs、xl、refine过程和结果的文件
XP中做的图形文件
晶体学信息文件
结构因子文件
记录仪器型号、晶体外观等的文件
SHELXTL的主要子程序和文件
XS XPREP
XP
*.plt *.sav *.ps
*.hkl
*.ins
*.res
*.pcf XCIF
XL
XSHELL *.cif *.fcf
*.lst
d 先选择,再点Select菜单中的Kill selected
e 在ins文件删除
• 恢复删错的原子或(Q)峰的方法:
点“Edit>Restore Killed Atom”,先选择要恢 复的原子(或Q峰),再点“Restore” • 命名(标记)原子或(Q)峰的方法:
a 先选择,再用Lablels菜单中的Group label
文件结束命令
3.其它文件
晶体结构报表文件 4.INS文件的建立和更新 结构解析和精修的过程,是ins文件建立和不 断更新的过程,这主要是下列过程实现的: xprep、xshell—refine、xl、xp、edit、copy
res lst plt cif fcf pcf tex
xs、xl、refine产生的文件
原子表 命 坐标、占有率、温度因子 令 区
Co1 5 0.639436 0.182296 0.778299 11.000000 0.037950 = 0.043120 0.052330 -0.002120 0.007180 -0.002370 Co2 5 0.132299 0.088237 0.782543 11.000000 0.061270 = 0.080610 0.082660 -0.004140 0.011300 0.005080 N1 3 0.816545 0.181404 0.857527 11.000000 0.048210= 0.044720 0.042580 -0.001460 0.010450 -0.001390 N2 3 0.641818 0.098803 0.864065 11.000000 0.055810 = 0.050870 0.070080 0.000700 0.013560 -0.009990 …………………………………………………………………. HKLF 4 衍射点文件类型 END
指令 END EQIV ESEL EXTI EXYZ FLAT FMAP FREE FVAR HFIX HKLF HTAB
含
义
指令输入结束 提供分子内或分子间键合原子的对称操作码 限制E值的下、上限 对晶体消光效应参数进行精修 让两个或多个原子具有相同的坐标 限制指定原子在相同的平面上 所计算Fourier图的类型
*.tex
二、常用的XS和XL指令 指令 含 义 ACTA 产生cif文件 AFIX 将原子坐标强制性地固定在指定位置上, 或在指定位置上产生原子 ANIS 将各向同性换成各向异性精修 BOND 计算键长、键角(加$H包括H的键长、键角) BIND 计算指定原子对的键长、键角 CONF 计算扭转角 DELU 限制指定原子具有相似的位移参数 DFIX 限定指定原子对间的距离 EADP 给两个或多个原子指定相同的位移参数
2. 结构的解析 1) 结构解析的基本原理 XS用直接法或Patterson法解决相角问题,试验 性找出部分原子或重原子的位置(坐标) * 所谓直接法(direct methods),就是运用数 学的方法,利用不同衍射点的关系,从大量强度数据 中,直接找出各个衍射点的相角,从而达到解析晶体 结构的目的。其过程概括如下:
指令
含
义
SYMM 所属空间群的对称操作 TEMP 衍射数据收集的温度 TITL 样品的编号(或名称)和空间群
UNIT
晶胞中每种原子的总个数
WGHT 指定所用权重
ZERR 晶胞中分子个数和晶胞参数的标准偏差
三、 SHELXTL结构分析的步骤
1.项目的设立 打开SHELXTL程序:点project 输入文件名,查找到hkl 文件并打开 new,
c 用Select菜单(或背景右手键点“Select”)可 选择部分和所有的原子或Q 峰,顺序与原顺序相同 d 按住“Shift”键,左手键拖出一个选择框并同 时轻开,可选择一个或多个原子
e 点refine菜单,系统会提示有Q 峰没命名,并 选择所有Q 峰,顺序与原来顺序相同
•删除原子或(Q)峰的方法: a 光标指在欲删除位置(原子或键) ,点K 键 b 光标指在欲删除位置,右手键点Bonds and Angles,再在对话框中点“Delete” c 光标指在欲删除位置,右手键点“Delete”
设置好ins文件 ,点击XS,就可自动进行计算
•XS计算结果的评估
# 直接法,RE越小越好,一般大于0.3,就预示 着不成功
# 打开Xshell,检查计算出的原子或(Q) 峰位置 是否构成合理的化学结构
# 精修(refint)一次后,R1值是否大小于0.5; 或继续精修R1值是否很快降低
b 仪器只根据Rint和CFOM(诊断因子)值 来确定,有时会降低晶体的对称性
c 由系统消光规律的错误判断引起 解决方法:在xprep中重新确定空间群;重新做晶胞
* XPREP的主要功能和应用
读入、更改、 •单击进入XPREP程序 合并衍射数据
•根据程序的提示输入晶胞参数 •选择可能的晶格 程序则显示以下菜单: 计算显示 Patterson截面 寻找更高 的对称性 确定或输 入已知的 空间群
不计算指定原子对的键长、键角 全比例系数 限制H原子在理想位置上 衍射数据的格式 计算氢键
指令 ISOR L.S. LATT
含 义 限制指定原子的位移参数类似于各向同性 指定XL中用最小二乘法进行精修的轮数 晶格的类型.依次为:P I R F A B C,无心为负值
MOVE 移动或转换坐标 MPLA 计算平面 OMIT PART PLAN SFAC SIMU SIZE 忽略指定的衍射点或限定theta角范围 划分成键原子的范围(用于无序结构) 计算和列出Q峰的数目 晶体中存在的原子的种类 限制指定范围内的原子有相同的位移参数 晶体的大小
-4 4 -4 -4 2 -2 -2 -2 -2 2 -2 -2 21.189 19.539 17.129 20.459 3.050 2.120 2.710 3.120
…………………………………..
标题 晶胞参数 X 光波长 空间群 TITL 041203e in P2(1)/c *.ins 文件:组成上可分成五部分 CELL 0.71073 11.5870 19.3080 17.2144 90.000 108.471 90.000 晶格类型 ZERR 4.00 0.0069 0.0112 0.0102 0.000 0.009 0.000 对称操作码 LATT 1 单胞中 SYMM -X, 0.5+Y, 0.5-Z 单胞中 分子数目 SFAC C H N O Co S 原子数目 晶胞参数的标 准偏差 UNIT 108 104 40 20 8 16 原子类型
化学结构,就要通过删除不合理的原子或(Q)峰, 并把Q 峰选择后定为相应的原子。操作方法是: •选择原子或(Q)峰的方法:
a 光标指在欲选择位置,点S 键。可依次选 择所有的要选择的原子或Q 峰
* 如果得到的原子或峰的位置能够构成合理的
b 光标指在欲选择位置,右手键点“Select”,可 依次选择原子
Patterson法:
TITL 020908b in C2/c CELL 0.710730 30.1927 8.5175 13.9108 90.0000 95.1300 90.0000 ZERR 8.00 0.0146 0.0042 0.0071 0.0000 0.0100 0.0000 LATT 7 SYMM -X, Y, 0.5-Z SFAC C H N O Cr UNIT 144 112 24 56 8 TEMP 25 PATT HKLF 4 END
用这种方法时,首先利用重原子的特征峰,即 Harker峰,求出重原子坐标,再通过Fourier合成 获得其它原子的坐标 Harker峰,或Harker截面,就是同一套等效的 原子组成的Patterson峰,由于平方效应,重原子的 Harker峰会显得十分突出,寻找起来一般比较容易。 因此,可以轻松地从分析重原子的Harker峰,得到 重原子的坐标
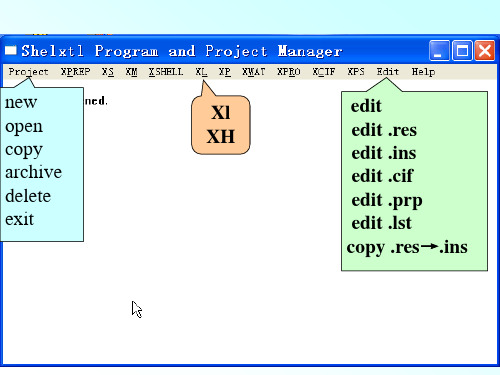
new open copy archive delete exit
Xl XH
edit edit .res edit .ins edit .cif edit .prp edit .lst copy .res→.ins
一、SHELXTL文件 1. 文件名 一般,同一结构,所有文件都用相同的名(不 能超过8个字符),只是扩展名不同 2. 两个必要文件(由XPREP程序产生) *.hkl文件: 所有的衍射点,每一点一行。 格式为:h k l F2 σ (F2)
a 将|Fo|转化为归一化结构因子|Eo|
b 建立可以利用正切公 d 利用正切公式精修相角
e 计算诊断指标,判断各套相角的质量 f 采用诊断指标最佳的相角数据计算解析电子密度图,即E图
* Patterson 法是其本人 1934 年提出,通常只用来 解析含有重原子的结构
SIZE 0.47 0.43 0.35 HTAB 2 L.S. 8 ACTA 可输入各种让XS和XL执行的命令 BOND FMAP 2 PLAN 20