Guidance105sample
(完整版)高考英语完形填空里的419个高频词

周末学生课堂之: 高考英语完形填里 419 个高频词,必须记住!(请打印留存,考生背熟)1. occasion 场合2. situation 情况,处境3. take over 接管4. exchange 交换5. command 命令6. confirm 证实7. cultivate 培养8. prosperously 繁荣9. suspect 怀疑10. relatively的相对,比较11. acknowledge 承认,鸣谢12. ambition 抱负,野心13. quality 质量,品质14. protection 保护15. equally 平等地16. promise 承诺17. clearly 清楚地18. grateful 感激19. remove 移开20. force 强迫21. apologize 道歉22. terrible 可怕,糟糕23. stubborn 固执24. actively 积极地,主动地25. spiritual的精神,心灵26. magical 魔力27. willingly 愿意地28. strengthen 加强29. image 形象30. complexity 复杂31. cautious 小心32. manage 管理,成功做成33. prejudice 偏见34. economic 经济,合算35. academic 学术36. control 控制37. adopt 收养,采取38. consume 消费,消耗39. unique 独一无二40. benefici有益41. varied 多变,各种各样的42. demanding 要求高43. appropriate 合理44. entertainment 娱乐45. deliberately 故意地46. purchase 购买47. tough 艰难48. bright 明亮49. remain 留下,保持50. terrify 使害怕51. disappointing 令人失望的52. formal 正式53. desire 愿望54. share 分享,共有55. fulfill 履行(诺言),执行(命令)56. admit 承认57. evident 明显58. consequently 因此,所以59. accustomed 习惯60. accumulate 积累61. participate参加62. absence 缺席63. presence 出席64. bravery 勇敢65. horror 恐惧66. spotless 无暇67. fundamental 基础68. employment 就业,雇用69. involve 包含,使参与70. actually 事实上71. harmony 和谐72. basically基本73. inspire 激发,鼓舞74. imitate 模仿75. awful 糟糕76. generous 慷慨,大方77. wealthy 富有78. function 功能79. stressful有压力80. persiste坚持不懈81. reluctant 勉强的,不愿意的82. diligent 勤奋的83. attentive 注意的,周到的84. unbearable 不能忍受的85. accommodation 住所86. attractive 有吸引力的87. constant 连续的88. brilliant 杰出的,才华横溢的89. clumsy 笨拙的90. declare 宣布,声明91. obtain 获得92. interactive 相互的,互动的93. incident 事件94. adventure 冒险95. in particular 尤其96. in reality 事实上97. emphasize 强调98. overlook 忽视99. deny 否认100. ensure 确保101. financial 金融102. budget 预算103. on the whole 整体上104. potential 潜在的,潜能105. on the contrary 相反106. loyalty 忠实107. assume 假设108. establish 建设109. flexible 灵活的110. sensitive 敏感的111. essential 必不可少的112. unfair 不公平的113. expectation 期待114. impression 印象115. examination 考试,检116. contribution 贡献117. certainty 肯定118. confuse 使迷惑,使混淆119. trap 陷阱,困住120. secondary 次要的121. turn up 出现122. show off 炫耀123. break in 闯进124. settle down 定居,安定下来125. relief 安慰,减轻126. justice 公正127. previous 先前的,早先的128. instantly 立即地129. regularly 规则地,规律地130. occasionally 偶尔地131. independence 独立132. keep up with 跟上133. catch up with 追赶上134. come up with 想出135. put up with 忍受136. guarantee 保证137. convince 使确信,说服138. atmosphere气氛139. sympathy 同情140. punish 惩罚141. puzzled 感到迷惑的142. scared 害怕的143. embarrassed 尴尬的144. reaction 反应145. forgiveness原谅146. imaginary 想象力丰富的147. be filled with 充满148. be pleased with 对感到满意峭防思149. be crowded with 挤满150. be equipped with 配备有 .151. possession财产152. precious 珍贵的153. appreciat欣赏,感激154. admire 钦佩155. wander 闲逛156. get rid of 消除,摆脱157. particularly尤其,特别158. purpose 目的,意图显示159. courage 勇气197. realize 意识到160. determination 决心198. meaningful 有意义的161. 162. roll 滚drop 掉下199. faithful 忠实的,忠诚的163. undoubtedly 毫无疑问地200. grasp 抓住,理解164. temporarily 暂时地201. decorate 装饰165. thankfully 幸运地202. pressure 压力166. lean against 靠着203. obvious 明显的167. challenge 挑战204. predict 预测168. fierce 激烈的205. audience 观众169. practical 实际的,实用206. contain 包含的207. gesture 姿势170. straight 直接的208. pioneer 先锋171. delighted 高兴的209. afford 支付得起172. congratulate 祝贺210. affair 事情173. faint 头晕的211. dependent 依赖的174. consult 咨询212. properly 合理地175. messy 乱的213. sincerely 真诚地176. review 复习,评论214. severely 严厉地177. curious 好奇的215. guidance 指导178. graduation 毕业216. appearance 出现,外貌179. honor 荣耀217. astonishment 惊讶180. comfortable 舒服的,舒218. sharpen 使锋利适的219. comment 评论181. tiresome 令人生厌的,220. privately 私人地无聊的221. frequently 频繁地182. 183. set up 建立hold up 举起,支撑222. physical 身体的,物理的184. pick up 拾起,学会,223. donate 捐赠接185. possess 拥有224. manufacture 制造,制造186. 187. crowded 拥挤的march 行军,前进225. original 原始的,起初的188. apartment 公寓226. effective 有效的189. frightened 害怕的227. object 反对190. turn out 结果,证明228. typical 典型的是229. possibility 可能性191. 192. figure out 算出,想出anxious 焦虑的230. accidentally 意外地,偶然地193. hunt 寻找,打猎231. immediately 立刻194. amused 愉快的,顽皮的232. concern 关心,担心195. optimistic 乐观的233. concentrate 集中196. demonstrate 证明,演示,234. tragedy 悲剧235. neglect 忽视271. countless 数不尽的236. approach 接近,途径,272. thoroughly 彻底地方法273. standard 标准237. disturbanc打扰274. tolerate 忍受238. arrest 逮捕275. memorable 值得纪念的239. restore 恢复276. leave for 离开去到240. available 可得到277. adapt 适应,改编的,可利用的278. somehow 不知怎么地241. accompany 陪伴279. somewhat 稍微,有点242. characteristic 特征,280. acquire 获得特性281. occupy 占据243. automatic 自动的282. accompany 陪伴244. approve 批准,同意283. urgent 紧急的245. roughly 粗略地284. technology 技术246. indicate □立285. giant 三大的247. hesitation 犹豫286. invisible 看不见的248. attach 系着,附着287. evaluation 评估249. plain 平的,朴素的,简288. magnificent 壮丽的单的289. inefficient 效率低的250. mercy 怜悯290. nonsense 废话,荒谬的251. sample 样本291. preserve 保护,保存252. considerable 相当大292. career 事业的,值得考虑的293. profession 职业,专业253. enthusiasm 热情294. exhausted 筋疲力尽的254. confirm 证实295. crash 撞碎,坠毁255. phenomenon 现象296. suspicious 怀疑的256. modify 修改297. requirement 要求257. annual 每年的298. athletic 运动的,运动258. isolation 隔离,孤立员的259. ceremony 典礼,仪式299. prohibit 禁止,阻止260. acceptable 可接受的300. depressed 沮丧的,萧条261. eventually 最后的262. target 目标301. instruction教导,说明263. expose 暴露302. slightly 轻微地264. absolutely 绝对地303. meaningfu有意义的265. negative 消极的,否定304. principle 原则的305. undeserve不应得的266. steady 稳定的306. betray 背叛267. consistent一贯地,307. moderate 中等的,适度一致地的268. permanent 永久的308. evaluate 评价,估价269. professiona 职业的,309. honorable 值得尊敬的专业的310. artificial人造的270. dismiss 解雇311. concept 概念312. symboliz象征353certainly 肯定地,当313. promising 有前途354.national 国家,民族314. conflict 冲突的315. display 显示,陈列355. cultural 文化316. definite明确地,肯356entrance 入口定地317. interrupt 打断357.358annoying 令人恼怒polite 有礼貌318. recall 回想起359unconscious 无意识319. recite 背诵360. patient 有耐心320. resist 抵制361. theory 理论321. desperate 绝望362.classification 分类322. elegant 优雅363smart 聪明323. attempt 试图364. mature 成熟324. random 随机365average 平均325. profitab有利可图366creative 有创造力326. treasure 珍惜,宝藏367. graduate 毕业,毕业生327. poisonous 有毒368.program 节目328. dilemma 窘境369intelligent 智能,聪329. correctly 正确地明330. system 系统370. competent 有能力331. complicated 复杂371. independent 独立332. applicat申请,应用372separate 单独,分开程序333. simple 简单的373voluntarily 自愿地334. recogniz认出,认可374doubt 怀疑335. cruel 残忍375reflection 反射,沉336. comfortable 舒服,舒思,映象映象适337. feared 害怕376.377conclusion 结论adult 成年人338. replace 代替378. talented 有天赋339. method 方法379. outstanding 出色340. seize 抓住380expression 表达,表情341. misunderstand 误解381check 检查342. mistake 错误,弄错382. attend 参加,照料343. imply □立暗示383.be aware of 意识到344. disease 疾病384be fond of 喜欢345. repay 偿还,报答385. be worthy of 值得346. describe 描述386expect 期待,预计347. contract 合同387. decision 决定348. promote 促进,提升388. style 风格349. sacrific牺牲389pretend 假装350. generally 一般来说390.explain 解释351. trouble 麻烦391cause 原因,事业352. pleased 满意392perform 表演,执行393.probably 394.struggle pletely 396.wonder397.experience 可能地挣扎,奋斗完全地失败发展有责任的有价值的,贵能力影响毁坏自信的要求传统的现代的关系合作区398.fail399.develop 400.responsibl e401.valuable 重的402.ability 403.affect 404.destroy 405.confident 406.request 407.traditiona l408.modern 409.relationsh ip410.cooperatio n411.distinguis h412.identify 413.thought 414.discourage 沮丧415.prepare 416.observe 417.energy 418.nervous419. proud 自豪的。
OECD 106

OECD GUIDELINE FOR THE TESTING OF CHEMICALS
Adsorption - Desorption Using a Batch Equilibrium Method
INTRODUCTION 1. This Guideline is based on the proposal submitted by the European Commission in 1993 and takes into account the comments on that proposal submitted by the Member countries. Several activities undertaken in the European Union bore on the development of an adsorption test. These included an extensive investigation due to the collaboration of the German Umweltbundesamt (UBA), the University of Kiel, and the European Commission and its Joint Research Centre at Ispra, Italy (1)(2). In 1988 UBA organised a ring-test in which 27 laboratories in EU Member States participated (3). An OECD Workshop on Soil Selection was held at Belgirate, Italy, in 1995 (4). The Workshop agreed on the main elements of the updated Test Guideline 106 for an adsorption/desorption test and, in particular, on the characterisation and selection of the soil types for use in the test. 2. Other guidelines concerning adsorption/desorption exist only at national level and are mainly focused on pesticides testing (5)(6)(7)(8)(9)(10)(11). These documents as well as an extensive relevant literature were considered when developing this Guideline. SIGNIFICANCE AND USE 3. Adsorption/desorption studies are useful for generating essential information on the mobility of chemicals and their distribution in the soil, water and air compartments of our biosphere (12)(13)(14)(15)(16)(17)(18)(19)(20)(21). They can be used in the prediction or estimation, for example, of the availability of a chemical for degradation (22)(23), transformation and uptake by organisms (24); leaching through the soil profile (16)(18)(19)(21)(25)(26)(27)(28); volatility from soil (21)(29)(30); and run-off from land surfaces into natural waters (18)(31)(32). Adsorption data can also be used for comparative and modelling purposes (19)(33)(34)(35). 4. The distribution of a chemical between soil and aqueous phases is a complex process depending on a number of different factors: the chemical nature of the substance (12)(36)(37)(38)(39)(40), the characteristics of the soil (4)(12)(13)(14)(41)(42)(43)(44)(45)(46)(47)(48)(49), and climatic factors such as rainfall, temperature, sunlight and wind. Thus, the numerous phenomena and mechanisms involved in the process of adsorption of a chemical by soil cannot be completely defined by a simplified laboratory model such as the present Guideline. However, even if this attempt cannot cover all the environmentally possible cases, it provides valuable information on the environmental relevance of the adsorption of a chemical. SCOPE 5. This Guideline is aimed at estimating the adsorption/desorption behaviour of a substance on soils. The goal is to obtain a sorption value which can be used to predict partitioning under a variety of environmental conditions; to this end, equilibrium adsorption coefficients for a chemical on various soils are determined as a function of soil characteristics (e.g. organic carbon content, clay content, soil texture, and
Guidance document on pesticide residue

EUROPEAN COMMISSION12Directorate General Health and Consumer Protection345SANCO/825/00 rev. 8.1 616/11/2010 78Guidance document on pesticide residue 9analytical methods10111213141516[Revision 8 is the version of this guidance document that is currently valid. It is, however, under1718continuous review and will be updated when necessary. The document is aimed at 19manufacturers seeking pesticides authorisations and parties applying for setting or modification 20of an MRL. It gives requirements for methods that would be used in post-registration 21monitoring and control by the competent authorities in Member States in the event that 22authorisations are granted. For authorities involved in post-registration control and monitoring, the document may be considered as being complementary to the documents: Method Validation2324and Quality Control Procedures for Pesticide Residues Analysis in Food and Feed (for the valid revision visit http://ec.europa.eu/food/plant/protection/resources/publications_en.htm) and the2526OECD document “Guidance Document on pesticide residue analytical methods”, 2007.27(ENV/JM/ ENV/JM/MONO(2007)17).1Preamble (4)28292General (5)302.1Good Laboratory Practice (5)312.2Selection of analytes for which methods are required (5)322.3Description of an analytical method and its validation results (5)332.4Hazardous reagents (6)342.5Acceptable analytical techniques considered commonly available (6)352.6Multi-residue methods (7)362.7Single methods and common moiety methods (7)372.8Single methods using derivatisation (7)382.9Method validation (8)392.9.1Calibration (8)2.9.2Recovery and Repeatability (9)40412.9.3Selectivity (11)422.10Confirmation (11)432.10.1Confirmation simultaneous to primary detection (11)442.10.2Confirmation by an independent analytical technique (12)452.11Independent laboratory validation (ILV) (12)2.12Availability of standards (13)46472.13Extraction Efficiency (13)483Analytical methods for residues in plants, plant products, foodstuff (of plant origin),feedingstuff (of plant origin) (Annex IIA Point 4.2.1 of Directive 91/414/EEC; Annex Point IIA,4950Point 4.3 of OECD) (14)513.1Purpose (14)523.2Selection of analytes (14)533.3Commodities and Matrix Groups (14)543.4Limit of quantification (15)553.5Independent laboratory validation (ILV) (15)564Analytical methods for residues in foodstuff (of animal origin) (Annex IIA Point 4.2.1 of 57Directive 91/414/EEC; Annex Point IIA, Point 4.3 of OECD) (16)584.1Purpose (16)594.2Selection of analytes (16)604.3Commodities (16)614.4Limit of quantification (16)4.5Independent laboratory validation (ILV) (16)62635Analytical methods for residues in soil (Annex IIA, Point 4.2.2 of Directive 91/414/EEC;64Annex Point IIA, Point 4.4 of OECD) (17)655.1Purpose (17)665.2Selection of analytes (17)675.3Samples (17)685.4Limit of quantification (17)696Analytical methods for residues in water (Annex IIA, Point 4.2.3 of Directive 91/414/EEC;70Annex Point IIA; Point 4.5 of OECD) (19)716.1Purpose (19)726.2Selection of analytes (19)736.3Samples (19)746.4Limit of quantification (19)756.5Direct injection (20)767Analytical methods for residues in air (Annex IIA, Point 4.2.4 of Directive 91/414/EEC; 77Annex Point IIA; Point 4.7 of OECD) (21)7.1Purpose (21)78797.2Selection of analytes (21)807.3Samples (21)7.4Limit of quantification (21)81827.5Sorbent characteristics (22)837.6Further validation data (22)7.7Confirmatory methods (22)84858Analytical methods for residues in body fluids and tissues (Annex IIA, Point 4.2.5 of86Directive 91/414/EEC; Annex Point IIA Point 4.8 of OECD) (23)8.1Purpose (23)87888.2Selection of analytes (23)898.3Samples (23)908.4Sample set (23)918.5Limit of quantification (23)929Summary - List of methods required (24)10Abbreviations (25)939411References (27)951Preamble96This document provides guidance to applicants, Member States and EFSA on the data 97requirements and assessment for residue analytical methods for post-registration control and 98monitoring purposes. It is not intended for biological agents such as bacteria or viruses. It 99recommends possible interpretations of the provisions of section 3.5.2 of Annex II of 100Regulation (EC) No 1107/2009 [1] and of the provisions of section 4, part A of Annex II and 101section 5, part A of Annex III of Council Directive 91/414/EEC [2]. It also applies to 102applications for setting or modification of an MRL within the scope of Regulation (EC) No 103396/2005 [3]. It has been elaborated in consideration of the ‘Guidance Document on pesticide 104residue analytical methods’ of the OECD [4] and SANCO/10684/2009 “Method validation 105and quality control procedures for pesticide residue analysis in food and feed” [5].106This document has been conceived as an opinion of the Commission Services and elaborated 107in co-operation with the Member States. It does not, however, intend to produce legally 108binding effects and by its nature does not prejudice any measure taken by a Member State nor 109any case law developed with regard to this provision. This document also does not preclude 110the possibility that the European Court of Justice may give one or another provision direct 111effect in Member States.112This guidance document must be amended at the latest if new data requirements as referred to 113in Article 8 (1)(b) and 8 (1)(c) of Regulation (EC) No 1107/2009 will have been established 114in accordance with the regulatory procedure with scrutiny referred to in Article 79 (4).1152General1162.1Good Laboratory Practice117According to Guidance Document 7109/VI/94-Rev. 6.c1 (Applicability of Good Laboratory 118Practice to Data Requirements according to Annexes II, Part A, and III, Part A, of Council 119Directive 91/414/EEC) [6] the development and validation of an analytical method for 120monitoring purposes and post-registration control is not subject to GLP. However, where the 121method is used to generate data for registration purposes, for example residue data, these 122studies must be conducted to GLP.1232.2Selection of analytes for which methods are required124The definition of the residues relevant for monitoring in feed and food as well as in 125environmental matrices and air is not the subject matter of this document. Criteria for the 126selection of analytes in case that no legally binding definition is available are given in the 127respective sections 3 - 8. In addition, sections 5.2, 6.2, 7.2 and 8.2 clarify under which 128circumstances analytical methods for residues may not be necessary.1292.3Description of an analytical method and its validation results130Full descriptions of validated methods shall be provided. The submitted studies must include 131the following points:132•Itemisation of the fortified compounds and the analytes, which are quantified133•Description of the analytical method134•Validation data as described in more detail below135•Description of calibration including calibration data136•Recovery and Repeatability137•Data proving the selectivity of the method138•Confirmatory data, if not presented in a separate study139•References (if needed)140141The following information should be offered in the description of the analytical method:142•An introduction, including the scope of the method143•Outline/summary of method, including validated matrices, limit of quantification (LOQ), 144range of recoveries, fortification levels and number of fortifications per level145•Apparatus and reagents146•instrument parameters used as example if appropriate147•Description of the analytical method, including extraction, clean-up, derivatisation (if148appropriate), chromatographic conditions (if appropriate) and quantification technique149•Hazards or precautions required150•Time required for one sample set151•Schematic diagram of the analytical method152•Stages where an interruption of the method is possible153•Result tables (if results are not presented in separate studies)154•Procedure for the calculation of results from raw data155•Extraction efficiency of solvents used156•Important points and special remarks (e.g. volatility of analyte or its stability with regard 157to pH)158•Information on stability of fortified/incurred samples, extracts and standard solutions (If 159the recoveries in the fortified samples are within the acceptable range of 70-120 %,160stability is sufficiently proven.)161Sometimes it may be necessary for other information to be presented, particularly where 162special methods are considered.1632.4Hazardous reagents164Hazardous reagents (carcinogens category I and II [7]) shall not be used. Among these 165compounds are diazomethane, chromium (VI) salts, chloroform and benzene.1662.5Acceptable analytical techniques considered commonly available167Analytical methods shall use instrumentation regarded as "commonly available":168•GC detectors: FPD, NPD, ECD, FID, MS, MS n (incl. Ion Traps and MS/MS), HRMS169•GC columns: capillary columns170•HPLC detectors: MS, MS/MS, HRMS, FLD, UV, DAD171•HPLC columns: reversed phase, ion-exchange, normal phase172•AAS, ICP-MS, ICP-OES173Other techniques can be powerful tools in residue analysis, therefore the acceptance of 174additional techniques as part of enforcement methods should be discussed at appropriate 175intervals. Whilst it is recognised that analytical methodology is constantly developing, some 176time elapses before new techniques become generally accepted and available.1772.6Multi-residue methods178Multi-residue methods that cover a large number of analytes and that are based on GC-MS 179and/or HPLC-MS/MS are routinely used in enforcement laboratories for the analysis of plant 180matrices. Therefore, validated residue methods submitted for food of plants, plant products 181and foodstuff of plant origin (Section 3) should be multi-residue methods published by an 182international official standardisation body such as the European Committee for 183Standardisation (CEN) (e.g. [8 - 12]) or the AOAC International (e.g. [13]). Single residue 184methods should only be provided if data show and are reported that multi-residue methods 185involving GC as well as HPLC techniques cannot be used.186If validation data for the residue analytical method of an analyte in at least one of the 187commodities of the respective matrix group have been provided by an international official 188standardisation body and if these data have been generated in more than one laboratory with 189the required LOQ and acceptable recovery and RSD data (see Section 2.9.2), no additional 190validation by an independent laboratory is required.1912.7Single methods and common moiety methods192Where a pesticide residue cannot be determined using a multi-residue method, one or where 193appropriate more alternative method(s) must be proposed. The method(s) should be suitable 194for the determination of all compounds included in the residue definition. If this is not 195possible and an excessive number of methods for individual compounds would be needed, a 196common moiety method may be acceptable, provided that it is in compliance with the residue 197definition. However, common moiety methods shall be avoided whenever possible.1982.8Single methods using derivatisation199For the analysis of some compounds by GC, such as those of high polarity or with poor 200chromatographic properties, or for the detection of some compounds in HPLC, derivatisation 201may be required. These derivatives may be prepared prior to chromatographic analysis or as 202part of the chromatographic procedure, either pre- or post-column. Where a derivatisation 203method is used, this must be justified.204If the derivatisation is not part of the chromatographic procedure, the derivative must be 205sufficiently stable and should be formed with high reproducibility and without influence of 206matrix components on yield. The efficiency and precision of the derivatisation step should be 207demonstrated with analyte in sample matrix against pure derivative. The storage stability of 208the derivative should be checked and reported. For details concerning calibration refer to 209Section 2.9.1.210The analytical method is considered to remain specific to the analyte of interest if the 211derivatised species is specific to that analyte. However, where – in case of pre-column 212derivatisation – the derivative formed is a common derivative of two or more active 213substances or their metabolites or is classed as another active substance, the method should be 214considered non-specific and may be deemed unacceptable.2152.9Method validation216Validation data must be submitted for all analytes included in the residue definition for all 217representative sample matrices to be analysed at adequate concentration levels.218Basic validation data are:219•Calibration data220•Concentration of analyte(s) found in blank samples221•Concentration level(s) of fortification experiments222•Concentration and recovery of analyte(s) found in fortified samples223•Number of fortification experiments for each matrix/level combination224•Mean recovery for each matrix/level combination225•Relative standard deviation (RSD) of recovery, separate for each matrix/level combination 226•Limit of quantification (LOQ), corresponding to the lowest validated level227•Representative clearly labelled chromatograms228•Data on matrix effects, e.g. on the response of the analyte in matrix as compared to pure 229standards230.Further data may be required in certain cases, depending on the analytical method used, and 231the residue definition to be covered.2322.9.1Calibration233The calibration of the detection system shall be adequately demonstrated at a minimum of 3 234concentration levels in duplicate or (preferably) 5 concentration levels with single 235determination. Calibration should be generated using standards prepared in blank matrix 236extracts (matrix matched standards) for all sample materials included in the corresponding 237validation study (Sections 3 - 8). Only, if experiments clearly demonstrate that matrix effects 238are not significant (i.e. < 20 %), calibration with standards in solvent may be used. Calibration 239with standards in solvent is also acceptable for methods to detect residues in air (Section 7). 240In case that aqueous samples are analysed by direct injection HPLC-MS/MS calibration shall 241be performed with standards in aqueous solution.242The analytical calibration must extend to at least the range which is suitable for the 243determination of recoveries and for assessment of the level of interferences in control 244samples. For that purpose a concentration range shall be covered from 30 % of the LOQ to 24520 % above the highest level (Section 2.9.2).246All individual calibration data shall be presented together with the equation of the calibration. 247Concentration data should refer to both, the mass fraction in the original sample (e.g. mg/kg) 248and to the concentration in the extract (e.g. µg/L). A calibration plot should be submitted, in 249which the calibration points are clearly visible. A plot showing the response factor1 versus the 250concentration for all calibration points is preferred over a plot of the signal versus the 251concentration.252Linear calibrations are preferred if shown to be acceptable over an appropriate concentration 253range. Other continuous, monotonically increasing functions (e.g. exponential/power, 254logarithmic) may be applied where this can be fully justified based on the detection system 255used.256When quantification is based on the determination of a derivative, the calibration shall be 257conducted using standard solutions of the pure derivative generated by weighing, unless the 258derivatisation step is an integral part of the detection system. If the derivative is not available 259as a reference standard, it should be generated within the analytical set by using the same 260derivatisation procedure as that applied for the samples. Under these circumstances, a full 261justification should be given.2622.9.2Recovery and Repeatability263Recovery and precision data must be reported for the following fortification levels, except for 264body fluids and body tissues (Section 8):265•LOQ 5 samples266•10 times LOQ, or MRL (set or proposed) or other relevant level (≥ 5 x LOQ)2675 samples268Additionally, for unfortified samples residue levels must be reported:269samples•blankmatrix 2270According to the residue definition the LOQ of chiral analytes usually applies to the sum of 271the two enantiomers. In this case it is not necessary to determine the enantiomers separately. 2721 The response factor is calculated by dividing the signal area by the respective analyte concentration.Enantioselective methods would only be required if a single enantiomer is included in the 273residue definition.274In cases of complex residue definitions (e.g. a residue definition which contains more than 275one compound) the validation results shall be reported for the single parts of the full residue 276definition, unless the single elements cannot be analysed separately.277The mean recovery at each fortification level and for each sample matrix should be in the 278range of 70 % - 120 %. In certain justified cases mean recoveries outside of this range will be 279accepted.280For plants, plant products, foodstuff (of plant and animal origin) and in feeding stuff recovery 281may deviate from this rule as specified in Table 1.2282Table 1: Mean recovery and precision criteria for plant matrices and animal matrices [4]283Concentration level Range of mean recovery(%)Precision, RSD(%)> 1 µg/kg ≤ 0.01 mg/kg 60 - 120 30> 0.01 mg/kg ≤ 0.1 mg/kg 70 - 120 20> 0.1 mg/kg ≤ 1.0 mg/kg 70 - 110 15> 1 mg/kg 70 - 110 10284If blank values are unavoidable, recoveries shall be corrected and reported together with the 285uncorrected recoveries.286The precision of a method shall be reported as the relative standard deviation (RSD) of 287recovery at each fortification level. For plants, plant products, foodstuff (of plant and animal 288origin) and feeding stuff the RSD should comply with the values specified in Table 1. In other 289cases the RSD should be ≤ 20 % per level. In certain justified cases, e.g. determination of 290residues in soil lower than 0.01 mg/kg, higher variability may be accepted.291When outliers have been identified using appropriate statistical methods (e.g. Grubbs or 292Dixons test), they may be excluded. Their number must not exceed 1/5 of the results at each 293fortification level. The exclusion should be justified and the statistical significance must be 2942 According to Annex IIA 4.2 of Directive 91/414/EEC the mean recovery should normally be 70 % - 110 % andthe RSD should preferably be ≤ 20 %.clearly indicated. In that case all individual recovery data (including those excluded) shall be 295reported.2962.9.3Selectivity297Representative clearly labelled chromatograms of standard(s) at the lowest calibrated level, 298matrix blanks and samples fortified at the lowest fortification level for each analyte/matrix 299combination must be provided to prove selectivity of the method. Labelling should include 300sample description, chromatographic scale and identification of all relevant components in the 301chromatogram.302When mass spectrometry is used for detection, a mass spectrum (in case of MS/MS: product 303ion spectrum) should be provided to justify the selection of ions used for determination.304Blank values (non-fortified samples) must be determined from the matrices used in 305fortification experiments and should not be higher than 30 % of the LOQ. If this is exceeded, 306detailed justification should be provided.3072.10Confirmation308Confirmatory methods are required to demonstrate the selectivity of the primary method for 309all representative sample matrices (Sections 3 – 8). It has to be confirmed that the primary 310method detects the right analyte (analyte identity) and that the analyte signal of the primary 311method is quantitatively correct and not affected by any other compound.3122.10.1Confirmation simultaneous to primary detection313A confirmation simultaneous to the primary detection using one fragment ion in GC-MS and 314HPLC-MS or one transition in HPLC-MS/MS may be accomplished by one of the following 315approaches:316•In GC-MS, HPLC-MS, by monitoring at least 2 additional fragment ions (preferably317m/z > 100)for low resolution system and at least 1 additional fragment ion for high318resolution/accurate mass system319•In GC-MS n (incl. Ion Traps and MS/MS), HPLC-MS/MS, by monitoring at least 1320additional SRM transition321The following validation data are required for the additional fragment ions (MS and HRMS) 322or the additional SRM transition (MS n and MS/MS): calibration data (Section 2.9.1), recovery 323and precision data according to Section 2.9.2 for samples fortified at the respective LOQ (n = 3245) and for 2 blank samples.325For all mass spectrometric techniques a mass spectrum (in case of single MS) or a product ion 326spectrum (in case of MS n) should be provided to justify the selection of the additional ions. 3272.10.2Confirmation by an independent analytical technique328Confirmation can also be achieved by an independent analytical method. The following are 329considered sufficiently independent confirmatory techniques:330•chromatographic principle different from the original method331• e.g. HPLC instead of GC332•different stationary phase and/or mobile phase with significantly different selectivity333•the following are not considered significantly different:334•in GC: stationary phases of 100 % dimethylsiloxane and of 95 % dimethylsiloxane 335+ 5 % phenylpolysiloxane336•in HPLC: C18- and C8-phases337•alternative detector338• e.g. GC-MS vs. GC-ECD, HPLC-MS vs. HPLC-UV/DAD339•derivatisation, if it was not the first choice method340•high resolution/accurate mass MS341•in mass spectrometry an ionisation technique that leads to primary ions with different m/z 342ratio than the primary method (e.g. ESI negative ions vs. positive ions)343It is preferred that confirmation data are generated with the same samples and extracts used 344for validation of the primary method.345The following validation data are required: calibration data (Section 2.9.1), recovery and 346precision data (Section 2.9.2) for samples fortified at the respective LOQ (n ≥ 3) and of a 347blank sample and proof of selectivity (Section 2.9.3).3482.11Independent laboratory validation (ILV)349A validation of the primary method in an independent laboratory (ILV) must be submitted for 350methods used for the determination of residues in plants, plant products, foodstuff (of plant 351and animal origin) and in feeding stuff. The ILV shall confirm the LOQ of the primary 352method, but at least the lowest action level (MRL).353The extent of independent validation required is given in detail in sections 3 and 4.354In order to ensure independence, the laboratory chosen to conduct the ILV trials must not 355have been involved in the method development and in its subsequent use. In case of multi-356residue methods it would be accepted if the ILV is performed in a laboratory that has already 357experience with the respective method.358The laboratory may be in the applicant’s organisation, but should not be in the same location. 359In the exceptional case that the lab chosen to conduct the ILV is in the same location, 360evidence must be provided that different personnel, as well as different instrumentation and 361stocks of chemicals etc have been used.362Any additions or modifications to the original method must be reported and justified. If the 363chosen laboratory requires communication with the developers of the method to carry out the 364analysis, this should be reported.3652.12Availability of standards366All analytical standard materials used in an analytical method must be commonly available. 367This applies to metabolites, derivatives (if preparation of derivatives is not a part of the 368method description), stable isotope labelled compounds or other internal standards.369If a standard is not commercially available the standard should be made generally available by 370the applicant and contact details be provided.3712.13Extraction Efficiency372The extraction procedures used in residue analytical methods for the determination of residues 373in plants, plant products, foodstuff (of plant and animal origin) and in feeding stuff should be 374verified for all matrix groups for which residues ≥ LOQ are expected, using samples with 375incurred residues from radio-labelled analytes.376Data or suitable samples may be available from pre-registration metabolism studies or 377rotational crop studies or from feeding studies. In cases where such samples are no longer 378available to validate an extraction procedure, it is possible to "bridge" between two solvent 379systems (details in [4]). The same applies if new matrices are to be included.3803Analytical methods for residues in plants, plant products, foodstuff (of 381plant origin), feedingstuff (of plant origin)382(Annex IIA Point 4.2.1 of Directive 91/414/EEC; Annex Point IIA, Point 3834.3 of OECD)3843.1Purpose385•Analysis of plants and plant products, and of foodstuff and feeding stuff of plant origin for 386compliance with MRL [3].3873.2Selection of analytes388The selection of analytes for which methods for food and feed are required depends upon the 389definition of the residue for which a maximum residue level (MRL) is set or is applied for 390according to Regulation (EC) No 396/2005.3913.3Commodities and Matrix Groups392Methods validated according to Section 2.9 and 2.10 must be submitted for representative 393commodities (also called “matrices” by analytical chemists) of all four matrix groups in 394Table 2.395396Table 2: Matrix groups and typical commoditiesMatrix group Examples for commoditiesbarley, rice, rye, wheat, dry legume vegetables dry commodities (high protein/highstarch content)commodities with high water content apples, bananas, cabbage, cherries, lettuce, peaches,peppers, tomatoescommodities with high oil content avocados, linseed, nuts, olives, rape seedcommodities with high acid content grapefruits, grapes, lemons, oranges397Important Note: This list of commodities is not a comprehensive list of commodities/matrices.398Applicants may consult regulatory authorities for advice on the use of other commodities.If samples with high water content are extracted at a controlled pH a particular method or 399validation for commodities with high acid content is not required.400Where a previously validated method has been adopted to a new matrix group, validation data 401must be submitted for representative matrices of this group.402。
USP-1092-溶出度试验的开发和验证(中英文对照版)之欧阳地创编

(1092)溶出度试验的开发和验证【中英文对照版】INTRODUCTION前言Purpose目的The Dissolution Procedure: Developmentand Validation <1092> provides a comprehensive approach covering items to considerfor developing and validating dissolution procedures and the accompanyinganalytical procedures. It addresses the use of automation throughout the testand provides guidance and criteria for validation. It also addresses thetreatment of the data generated and the interpretation of acceptance criteriafor immediate and modifiedrelease solid oral dosage forms.溶出实验:开发和验证(1092)指导原则提供了在溶出度方法开发和验证过程中以及采用相应分析方法时需要考虑的因素。
本指导原则贯穿溶出度实验的全部过程,并对方法提供了指导和验证标准。
同时它还涉及对普通制剂和缓释制剂所生成的数据和接受标准进行说明。
Scope范围Chapter <1092> addresses the development andvalidation of dissolution procedures, with a focus on solid oral dosage forms.Many of the concepts presented, however, may be applicable to other dosageforms and routes of administration. General recommendations are given with theunderstanding that modifications of the apparatus and procedures as given in USPgeneral chapters need to be justified.<1092>章节讨论了溶出度实验的开发和验证,重点是口服固体制剂。
ICRP报告名称(至112号)

ICRP 01号出版物—— Recommendations of the International Commission on Radiological Protection (Superseded by ICRP 26).ICRP 02号出版物—— Report of Committee II on Permissible Dose for Internal Radiation (Superseded by ICRP 30).ICRP 03号出版物—— Report of Committee III on Protection Against X-rays up to Energies of 3 MeV and Beta- and Gamma-rays from Sealed Sources ICRP 04号出版物—— Report of Committee IV on Protection Against X-rays Electromagnetic Radiation Above 3 MeV and Electrons, Neutrons and Protons ICRP 05号出版物—— Report on Committee V in the Handling and Disposal of Radioactive Materials in Hospitals and Medical Research Establishments (Superseded by ICRP 25).ICRP 06号出版物—— Recommendations of the ICRP (Revision to ICRP 1) (Superseded by ICRP 26).ICRP 07号出版物—— Principals of Environmental Monitoring Related to the Handling of Radioactive MaterialICRP 08号出版物—— The Evaluation of Risks from RadiationICRP 09号出版物—— Recommendations of the International Commission on Radiological Protection (Revision of ICRP 6) (Superseded by ICRP 26) ICRP 10A号出版物—— The Assessment of Internal Contamination Resulting from Recurring of Prolonged Uptakes (Superseded by ICRP 54)ICRP 10号出版物—— Evaluation of Radiation Doses to Body Tissues from Internal Contamination due to Occupational Exposure (Superseded by ICRP 54)ICRP 11号出版物—— A Review of the Radiosensitivity of the Tissues in BoneICRP 12号出版物—— General Principles of Monitoring for Radiation Protection of Workers (Superseded by ICRP 35)ICRP 13号出版物—— Radiation Protection in Schools for Pupils up to the Age of 18 Years (Superseded by ICRP 36)ICRP 14号出版物—— Radiosensitivity and Spatial Distribution of DoseICRP 15号出版物—— Protection Against Ionizing Radiation from External Sources(Superseded by ICRP 33).ICRP 16号出版物—— Protection of the Patient in X-ray Diagnosis (Superseded by ICRP 34).ICRP 17号出版物—— Protection of the Patient in Radionuclide Investigations (Superseded by ICRP 52).ICRP 18号出版物—— The RBE for High-LET Radiations with Respect to MutagenesisICRP 19号出版物—— The Metabolism of Compounds of Plutonium and Other Actinides (See also ICRP 48).ICRP 20号出版物—— Alkaline Earth Metabolism in Adult ManICRP 21号出版物—— Data for Protection Against Ionizing Radiation from External Sources - Supplement to ICRP Publication 15 (Superseded by ICRP 33 & 51).ICRP 22号出版物—— Implication of Commission Recommendations that Doses be Kept as Low as Readily AchievableICRP 23号出版物—— Reference Man: Anatomical, Physiological and Metabolic CharacteristicsICRP 24号出版物—— Radiation Protection in Uranium and Other MinesICRP 25号出版物—— The Handling, Storage, Use and Disposal of Unsealed Radionuclides in Hospitals and Medical Research Establishments (Supersedes ICRP 5).ICRP 26号出版物—— Recommendations of the International Commission on Radiological Protection (Superseded by ICRP 60)(Supersedes ICRP 1, 6 & 9) ICRP 27号出版物—— Problems Involved in Developing an Index of HarmICRP 28号出版物—— The Principles and General Procedures for Handling Emergency and Accidental Exposure of WorkersICRP 29号出版物—— Radionuclide Release into the Environment - Assessment of Doses to ManICRP 30号出版物—— Limits for Intakes of Radionuclides by WorkersICRP 31号出版物—— Biological Effects of Inhaled RadionuclidesICRP 32号出版物—— Limits for Inhaled Radionuclides by WorkersICRP 33号出版物—— Protection Against Ionizing Radiation from External Sources Used in Medicine (Supersedes ICRP 15 & 21)ICRP 34号出版物—— Protection of the Patient in Diagnostic RadiologyICRP 35号出版物—— General Principles of Monitoring for Radiation Protection of Workers (Supersedes ICRP 12)ICRP 36号出版物—— Protection Against Ionizing Radiation in the Teaching of ScienceICRP 37号出版物—— Cost-Benefit Analysis in the Optimization of Radiation ProtectionICRP 38号出版物—— Radionuclide Transformations: Energy and Intensity of EmissionsICRP 39号出版物—— Principles of Limiting Exposure of the Public to Natural Sources of RadiationICRP 40号出版物—— Protection of the Public in the Event of Major Radiation Accidents:Principles for Planning (Superseded by ICRP 63)ICRP 41号出版物—— Nonstochastic Effects of Ionizing RadiationICRP 42号出版物—— A Compilation of the Major Concepts and Quantities in Use by the ICRPICRP 43号出版物—— Principles of Monitoring for the Radiation Protection of the PublicICRP 44号出版物—— Protection of the Patient in Radiation TherapyICRP 45号出版物—— Quantitative Bases for Developing a Unified Index of HarmICRP 46号出版物—— Radiation Protection Principles for the Disposal of Solid Radioactive WasteICRP 47号出版物—— Radiation Protection of Workers in MinesICRP 48号出版物—— The Metabolism of Plutonium and Related ElementsICRP 49号出版物—— Developmental Effects of Irradiation on the Brain of the Embryo and FetusICRP 50号出版物—— Lung Cancer Risk from Indoor Exposures to Radon DaughtersICRP 51号出版物—— Data for Use in Protection Against External Radiation (Supersedes ICRP 21)ICRP 52号出版物—— Protection of the Patient in Nuclear Medicine (Supersedes ICRP 17).ICRP 53号出版物—— Radiation Dose to Patients from RadiopharmaceuticalsICRP 54号出版物—— Individual Monitoring for Intakes of Radionuclides by Workers: Design and InterpretationICRP 55号出版物—— Optimization and Decision-Making in Radiological ProtectionICRP 56号出版物—— Age-dependent Doses to Members of the Public from Intake of RadionuclidesICRP 57号出版物—— Radiological Protection of the Worker in Medicine and DentistryICRP 58号出版物—— RBE for Deterministic EffectsICRP 59号出版物—— The Biological Basis for Dose Limitation in the SkinICRP 60号出版物—— 1990 Recommendations of the International Commission on Radiological ProtectionICRP 61号出版物—— Annual Limits on Intake of Radionuclides by Workers Based on the 1990 RecommendationsICRP 62号出版物—— Radiological Protection in Biomedical ResearchICRP 63号出版物—— Principles for Intervention for Protection of the Public in a Radiological EmergencyICRP 64号出版物—— Protection from Potential Exposure: A Conceptual FrameworkICRP 65号出版物—— Protection Against Radon-222 at Home and at WorkICRP 66号出版物—— Human Respiratory Tract Model for Radiological ProtectionICRP 67号出版物—— Age-dependent Doses to Members of the Public from Intake of RadionuclidesICRP 68号出版物—— Dose Coefficients for Intakes of Radionuclides by WorkersICRP 69号出版物—— Age-dependent Doses to Members of the Public from Intake of Radionuclides: Part 3 Ingestion Dose CoefficientsICRP 70号出版物—— Basic Anatomical & Physiological Data for use in Radiological Protection: The SkeletonICRP 71号出版物—— Age-dependent Doses to Members of the Public from Intake of Radionuclides: Part 4 Inhalation Dose CoefficientsICRP 72号出版物—— Age-dependent Doses to the Members of the Public from Intake of Radionuclides Part 5, Compilation of Ingestion and Inhalation CoefficientsICRP 73号出版物—— Radiological Protection and Safety in MedicineICRP 74号出版物—— Conversion Coefficients for use in Radiological Protection against External RadiationICRP 75号出版物—— General Principles for the Radiation Protection of WorkersICRP 76号出版物—— Protection from Potential Exposures: Application to Selected Radiation SourcesICRP 77号出版物—— Radiological Protection Policy for the Disposal of Radioactive WasteICRP 78号出版物—— Individual Monitoring for Internal Exposure of WorkersICRP 79号出版物—— Genetic Susceptibility to CancerICRP 80号出版物—— Radiation dose to patients from radiopharmaceuticalsICRP 81号出版物—— Radiation Protection Recommendations as Applied to the Disposal of Long-lived Solid Radioactive WasteICRP 82号出版物—— Protection of the Public in Situations of Prolonged Radiation ExposureICRP 83号出版物—— Risk Estimation for Multifactorial DiseasesICRP 84号出版物—— Pregnancy and Medical RadiationICRP 85号出版物—— Avoidance of Radiation Injuries from Medical Interventional ProceduresICRP 86号出版物—— Prevention of Accidents to Patients Undergoing Radiation TherapyICRP 87号出版物—— Managing Patient Dose in Computed TomographyICRP 88号出版物—— Doses to the Embryo and Fetus from Intakes of Radionuclides by the MotherICRP 89号出版物—— Basic Anatomical and Physiological Data for Use in Radiological Protection: Reference ValuesICRP 90号出版物—— Biological Effects after Prenatal Irradiation (Embryo and Fetus)ICRP 91号出版物—— A Framework for Assessing the Impact of Ionising Radioation on Non-Human SpeciesICRP 92号出版物—— Relative Biological Effectiveness (RBE), Quality Factor (Q), and Radiation Weighting Factor (wR)ICRP 93号出版物—— Managing Patient Dose in Digital RadiologyICRP 94号出版物—— Release of patients after therapy with unsealed radionuclidesICRP 95号出版物—— Doses to Infants from Ingestion of Radionuclides in Mother's MilkICRP 96号出版物—— Protecting People Against Radiation Exposure in the Event of a Radiological AttackICRP 97号出版物—— Prevention of High-dose-rate Brachytherapy AccidentsICRP 98号出版物—— Radiation Aspects of Brachytherapy for Prostate CancerICRP 99号出版物—— Low-Dose Extrapolation of Radiation Related Cancer RiskICRP 100号出版物—— Human Alimentary Tract Model for Radiological ProtectionICRP 101号出版物—— Assessing Dose of the Representative Person for the Purpose of Radiation Protection of the Public and the Optimisation of Radiological ProtectionICRP 102号出版物—— Managing Patient Dose in Multi-Detector Computed Tomography(MDCT)ICRP 103号出版物—— The 2007 Recommendations of the International Commission on Radiological ProtectionICRP 104号出版物—— Scope of Radiological Protection Control MeasuresICRP 105号出版物—— Radiological Protection in MedicineICRP 106号出版物—— Radiation Dose to Patients from Radiopharmaceuticals - A third amendment to ICRP Publication 53ICRP 107号出版物—— Nuclear Decay Data for Dosimetric CalculationsICRP 108号出版物—— Environmental Protection: the Concept and Use of Reference Animals and PlantsICRP 109号出版物—— Application of the Commission's Recommendations for the Protection of People in Emergency Exposure SituationsICRP 110号出版物—— Adult Reference Computational PhantomsICRP 111号出版物——Application of the Commission’s Recommendations to the Protection of People Living in Long-term Contaminated Areas After a Nuclear Accident or a Radiation EmergencyICRP 112号出版物—— Preventing Accidental Exposures from New External Beam Radiation TherapyICRP Supporting Guidance 2—— Radiation and Your Patient: A Guide for Medical PractitionersICRP Supporting Guidance 3—— Guide for the Practical Application of the ICRP Human Respiratory Tract ModelICRP Supporting Guidance 4—— Development of the Draft 2005 Recommendations of the ICRP——A Collection of PapersICRP Supporting Guidance 5——Analysis of the Criteria Used by the International Commission on Radiological ProtectionICRP 01号出版物——Recommendations of the International Commission on Radiological Protection (Superseded by ICRP 26).国际放射防护委员会01号出版物 - 关于国际放射防护委员会建议(取代了国际放射防护委员会26)。
现行的土壤质量ISO标准
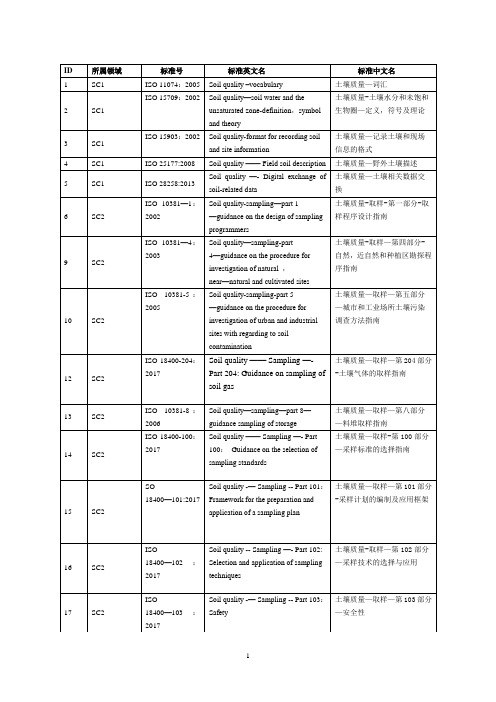
Soil quality-determination of organochlorine pesticide and polychlorinated biphenyls -GM with electronic capture detection
土壤质量-电子俘获探测气相色谱法测定有机氯农药和多氯联苯
16
SC2
ISO 18400—102:2017
Soil quality -- Sampling —- Part 102: Selection and application of sampling techniques
土壤质量-取样—第102部分—采样技术的选择与应用
17
SC2
ISO 18400—103:2017
土壤质量—用于理化分析样品的预处理
37
SC3
ISO 11465:1993
Soil quality—- determination of dry matter and water content on a mass basis –gravimetric method
土壤质量—土壤生物的干物质和水含量的测定—重量法
土壤质量—土壤王水萃取物中镉,铬,钴,铜,鉛,,锰,镍和锌含量的测定—火焰和电热原子吸收光谱测定法
28
SC3
ISO11048:1995
Soil quality-determination of water –soluble and acid-soluble sulfate
土壤质量—水溶性和酸溶性硫酸盐的测定
土壤质量—取样-第105部分-样品的包装、运输、储存和保存
19
SC2
ISO 18400—106:2017
FDA指南 混合均匀性接受标准

Powder Blends and Finished Dosage Units — Stratified In-Process Dosage Unit Sampling and AssessmentDRAFT GUIDANCEThis guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should be submitted within 60 days of publication in the Federal Register of the notice announcing the availability of the draft guidance. Submit comments to Dockets Management Branch (HFA-305), Food and Drug Administration, 5630 Fishers Lane, rm. 1061, Rockville, MD 20852. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register.For questions regarding this draft document contact Jon E. Clark, 301-594-5613 or Mike Gavini, 301-827-9053.U.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)October 2003Pharmaceutical CGMPsPowder Blends and Finished Dosage Units — Stratified In-Process Dosage Unit Sampling and AssessmentAdditional copies are available from:Office of Training and CommunicationDivision of Drug Information, HFD-240Center for Drug Evaluation and ResearchFood and Drug Administration5600 Fishers LaneRockville, MD 20857(Tel) 301-827-4573/cder/guidance/index.htmU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Office of Pharmaceutical Science (OPS)Office of Compliance (OC)October 2003Pharmaceutical CGMPsDraft — Not for ImplementationTABLE OF CONTENTSI.INTRODUCTION (1)II.BACKGROUND (1)III.SCOPE (2)IV.CORRELATION OF IN-PROCESS STRATIFIED SAMPLING WITH POWDER MIX AND FINISHED PRODUCT (4)A.Assessment of Powder Mix Uniformity (4)B.Correlation of Powder Mix Uniformity with Stratified In-Process Dosage Unit Data (5)C.Correlation of Stratified In-Process Samples with the Finished Product (6)V.EXHIBIT/VALIDATATION BATCH POWDER MIX HOMOGENEITY (6)VI.VERIFICATION OF MANUFACTURING CRITERIA (7)A.In-Process Dosage Unit Sampling and Analysis (7)B.Criteria to Meet the Readily Pass Classification (8)C.Criteria to Meet the Marginally Pass Classification (8)D.Sample Locations for Routine Manufacturing (9)VII. ROUTINE MANUFACTURING BATCH TESTING METHODS (9)A.Standard Criteria Method (SCM) (9)1.Stage 1 Test (10)2.Stage 2 Test (10)B.Marginal Criteria Method (MCM) (10)C.Switching to Standard Test Method from Marginal Test Method (11)VIII.REPORTING THE USE OF STRATIFIED SAMPLING (11)A.Applications Not Yet Approved (11)B.Postapproval Change (12)GLOSSARY (13)ATTACHMENT 1: VERIFICATION OF MANUFACTURING CRITERIA (14)ATTACHMENT 2: ROUTINE MANUFACTURING BATCH TESTING (15)Guidance for Industry112Powder Blends and Finished Dosage Units — Stratified In-Process 3Dosage Unit Sampling and Assessment4567This draft guidance, when finalized, will represent the Food and Drug Administration's (FDA's) current 8thinking on this topic. It does not create or confer any rights for or on any person and does not operate to 9bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of 10the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA 11staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call 12the appropriate number listed on the title page of this guidance.13.141516I.INTRODUCTION1718This guidance is intended to assist manufacturers of human drug products in meeting the19requirements of 21 CFR 211.110 for demonstrating the adequacy of mixing to ensure uniformity of in-process powder blends and finished dosage units. This guidance describes the procedures 2021for assessing powder mix adequacy, correlating in-process dosage unit test results with powder 22mix test results, and establishing the initial criteria for control procedures used in routine23manufacturing.2425FDA's guidance documents, including this guidance, do not establish legally enforceable26responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should 27be viewed only as recommendations, unless specific regulatory or statutory requirements are28cited. The use of the word should in Agency guidances means that something is suggested or 29recommended, but not required.3031II.BACKGROUND323334This guidance is the result of an Agency effort to achieve a science-based policy and regulatory enforcement. Experts from industry, academia, and the FDA developed the principles3536underlying this guidance after extensive public discussion. A brief history of the evolution of 37this guidance is provided in the following paragraphs.1 This guidance has been prepared by the Office of Pharmaceutical Science and the Office of Compliance in theCenter for Drug Evaluation and Research (CDER) at the Food and Drug Administration in cooperation with the Product Quality Research Institute (PQRI) (see footnote 3). This guidance document represents the Agency'scurrent thinking on assessment of the uniformity of powder blends and finished dosage units in the absence of new technology development or implementation.3839In response to industry concerns regarding regulations for demonstrating the adequacy of in-40process powder mixing, the FDA published a draft guidance for industry on blend uniformity 41analysis in August 1999.2 Comments submitted to the docket resulted in the formation of the 42Blend Uniformity Working Group (BUWG) by the Product Quality Research Institute (PQRI).3 43The PQRI BUWG conducted a public meeting, PQRI Workshop on Blend Uniformity, on44September 7 and 8, 2000.4546Using the consensus reached by participants in this workshop, the BUWG developed a draft47recommendation, The Use of Stratified Sampling of Blend and Dosage Units to DemonstrateAdequacy of Mix for Powder Blends. The draft recommendation received examination and peer 4849review in multiple scientific and public venues. In addition, the Advisory Committee for50Pharmaceutical Science (ACPS) reviewed the draft recommendation and received public51comment during scheduled meetings of the committee.4 The draft recommendation was revised 52to incorporate the results of peer review and public comment and was presented to CDER's53Center Director in final form on December 30, 2002. The recommendation was subsequently 54published in the PDA Journal of Pharmaceutical Science and Technology.5 This draft guidance 55reflects CDER's effort to incorporate the draft recommendation into regulatory policy.565758III.SCOPE5960Stratified sampling is the process of sampling dosage units at predefined intervals and collecting representative samples from specifically targeted locations in the compression/filling operation 6162that have the greatest potential to yield extreme highs and lows in test results. These test results 63are used to monitor the manufacturing process output that is most responsible for causing64finished product variability. The test results can be used to develop a single control procedure to 65ensure adequate powder mix and uniform content in finished products.6667The methods described in this guidance are not intended to be the only methods for meeting68Agency requirements to demonstrate the adequacy of powder mix. Traditional powder blend 69sampling and testing, in conjunction with testing for uniformity of content in the finishedproduct, can be used to comply with current good manufacturing practice requirements702 The FDA withdrew the guidance for industry ANDAs: Blend Uniformity Analysis o n May 17, 2002.3 PQRI is a collaborative body involving FDA's Center for Drug Evaluation and Research (CDER), industry, andacademia. Since its inception in January 1996, the mission of PQRI has been to generate scientific information in support of regulatory policies through research. Additional information about PQRI is available at .4 The PQRI BUWG recommendation appeared on the public ACPS agenda on November 28, 2001 (introduction),May 8, 2002 (distribution and comment), and October 22, 2002 (final comment).5 G Boehm, J Clark, J Dietrick, L Foust, T Garcia, M Gavini, L Gelber, J Geoffry, J Hoblitzell, P Jimenez, GMergen, F Muzzio, J Planchard, J Prescott, J Timmermens, and N Takiar, "The Use of Stratefied Sampling of Blend and Dosage Units to Demonstrate Adequacy of Mix for Powder Blends, PDA J. Pharm. Sci Technol,. 57:59-74, 2003.71(CGMPs). Use of at-, in-, or on-line measurement systems can also be appropriate and are72described in other guidance documents.67374This guidance provides recommendations on how to:7576•Conduct powder blend sampling and analyses.77•Establish initial criteria for stratified sampling of in-process dosage units7 and evaluation 78of test results.79•Analyze the stratified samples and evaluate data.•Correlate the stratified sample data with the powder blend data.8081•Assess powder mix uniformity.•Correlate the stratified sample data with the finished dosage unit data and assess8283uniformity of content.•Test exhibit and validation batches for adequacy of powder mix.8485•Test and evaluate routine manufacturing batches.86•Report the use of stratified sampling in the application.8788The methods described in this guidance can be used to monitor active ingredient homogeneity of 89powder blends and to ensure uniform content of the finished product for solid oral drug products. 90These methods are only one way to satisfy the CGMP and application review requirements for 91in-process testing to demonstrate adequacy of powder mix and uniform content of the finished 92product. The method assumes appropriate monitoring of all manufacturing steps as required by 93the regulations or application commitments. This guidance does not discuss the assessment of the potency and other attributes that can affect the finished dosage units, or the homogeneity of 9495inactive ingredients. Formulations with extremely low dose and/or high potency may call for96more rigorous sampling than that described in this guidance to assess the uniformity of powder 97blends or the uniformity of content of the finished dosage units.9899When using the methods described in this guidance, certain data or trends may be observed. We 100recommend that manufacturers scientifically evaluate these types of research data to determine if they affect the quality of a product and, if so, how. The FDA does not intend to inspect research 101102data collected on an existing product for the purpose of evaluating the suitability of proposedmethods. Any FDA decision to inspect research data would be based on exceptional situations 1036 In August 2003, the Agency issued the draft guidance for industry PAT – A Framework for InnovativePharmaceutical Manufacturing and Quality Assurance. Once finalized, it will represent the Agency's perspective on this issue.7 The in-process dosage unit is a capsule or tablet as it is formed in the manufacturing process before it is coated orpackaged.similar to those outlined in Compliance Policy Guide Sec. 130.300.8 Those data used to support 104105validation or regulatory submissions will be subject to inspection in the usual manner.106107108IV.CORRELATION OF IN-PROCESS STRATIFIED SAMPLING WITH POWDER 109MIX AND FINISHED PRODUCT110111If you plan to follow the procedures described in this guidance document, we recommend that 112you first complete the process development procedures described in this section before using the methods described in sections V, VI, VII. The subsections below describe how to assess the 113114adequacy of powder mix, uniformity of content of the in-process and finished dosage unitsthrough correlation and assessment of data from development, validation and manufacturing 115116batches. These procedures can reveal deficiencies in the blending operation that may not have 117been previously detected. We recommend that manufacturers correct deficiencies in the118blending operation before implementing the routine manufacturing control methods described in 119this guidance.120121A.Assessment of Powder Mix Uniformity122123We recommend the assessment of powder mix uniformity using the following procedures:124•Conduct blend analysis on batches by extensively sampling the mix in the blender and/or 125126intermediate bulk containers (IBCs).127•Identify appropriate blending time and speed ranges, dead spots in blenders, and locations 128of segregation in IBCs. Determine sampling errors.129•Define the effects of sample size (e.g., 1-10X dosage unit range) while developing a130technique capable of measuring the true uniformity of the blend. Sample quantities larger 131than 3X can be used with adequate scientific justification. Appropriate blend sampling 132techniques and procedures should be developed for each product with consideration to 133various designs of blend powder sampling and the physical and chemical properties of 134the blend components.135•Design blend-sampling plans and evaluate them using appropriate statistical analyses. 136•Quantitatively measure any variability that is present among the samples. Attribute the 137sample variability to either lack of uniformity of the blend or sampling error. Significant 138within-location variance in the blend data can be an indication of one factor or acombination of factors such as inadequacy of blend mix, sampling error9 or1398 FDA/ORA Compliance Policy Guide, Sec. 130.300, FDA Access to Results of Quality Assurance Program Auditsand Inspections (CPG7151.02)9 If blend sampling error is detected, more sophisticated, statistical analyses should be applied to assess the situation,such as the use of methods described in J Berman, DE Elinski, CR Gonzales, JD Hofer, PJ Jimenez, JA Planchard, RJ Tlachac, PF Vogel, “Blend Uniformity Analysis: Validation and In-Process Testing.” Technical Report No. 25, PDA J Pharm. Sci. Technol. 51(Suppl 3i-iii), S1-99, 1997.140agglomeration.10, 11 Significant between-location variance in the blend data can indicate141that the blending operation is inadequate.142143B.Correlation of Powder Mix Uniformity with Stratified In-Process Dosage 144Unit Data145146We recommend the following steps for correlation:147•Conduct periodic sampling and testing of the in-process dosage units by sampling them at 148149defined intervals and locations throughout the compression or filling process. Use aminimum of 20 appropriately spaced in-process dosage unit sampling points. There150151should be at least 7 samples taken from each of these locations for a total minimum of at152least 140 samples.153•Take 7 samples from each additional location to further assess each significant event,12 154such as filling or emptying of hoppers and IBCs, start and end of the compression or155filling process and equipment shutdown. This may be accomplished by using processdevelopment batches, validation batches, or by using routine manufacturing batches for 156157approved products.•Significant events may also include observations or changes from one batch to another 158159(e.g., batch scale-up and observations of undesirable trends in previous batch data).160• Prepare a summary of the data and analysis used to correlate the stratified sampling161locations with significant events in the blending process. We recommend you submit this162summary with the application as described in section VIII of this guidance.163•Compare the powder mix uniformity with the in-process dosage-unit data described above.164165•Investigate any discrepancies observed between powder mix and dosage-unit data and establish root causes. At least one trouble-shooting guide is available that may be helpful 166167with this task.13 Possible corrections may range from going back to formulation168development to improve powder characteristics to process optimization. Sampling10OS Sudah, PE Arratia, D. Coffin-Beach, FJ Muzzio, "Mixing of Cohesive Pharmaceutical Formulations in Tote (Bin)-Blenders,” Drug Dev. Ind. Pharm, 28(8): 905-918, 2002.11V Swaminathan, DO Kildsig, “Polydisperse powder mixtures: effect of particle size and shape on mixturestability,” Drug Dev. Ind. Pharm., 28(1):41-48, 2002.12 A s ignificant event is any operation during the solid dosage production process that can affect the integrity of thein-process materials – see section IX Glossary.13 JK Prescott, TJ Garcia, "A Solid Dosage and Blend Content Uniformity Troubleshooting Diagram," Pharm.Technol., 25 (3):68-88, 2001.problems may also be negated by use of alternate state-of-the-art methods of in situ real-169170time sampling and analysis.171C.Correlation of Stratified In-Process Samples with the Finished Product172173174We recommend the following steps:175176•Conduct testing for uniform content of the finished product using an appropriate177procedure or as specified in the Abbreviated New Drug Application (ANDA) or the NewDrug Application (NDA) for approved products.178179•Compare the results of stratified in-process dosage unit analysis with uniform content of the finished dosage units from the previous step. This analysis should be done without 180181weight correction.14182•Prepare a summary of the data and analysis used to conclude that the stratified in-process 183sampling provides assurance of uniform content of the finished product. We recommend184you submit this summary with the application as described in section VIII of thisguidance.185186187188V.EXHIBIT/VALIDATATION BATCH POWDER MIX HOMOGENEITY189This section describes sampling and testing the powder mix of exhibit and process validation 190191batches used to support implementing the stratified sampling method described in this guidance.192193194We recommend that during the manufacture of exhibit and process validation batches, you assess195the uniformity of the powder blend, the in-process dosage units, and the finished product196independently. We recommend you use the following steps to identify sampling locations and197acceptance criteria prior to the manufacture of the exhibit and/or validation batches.151981991.Carefully identify at least 10 sampling locations in the blender to represent potential areas200of poor blending. For example, in tumbling blenders (such as V-blenders, double cones,or drum mixers), samples should be selected from at least two depths along the axis of 201202the blender. For convective blenders (such as a ribbon blender), a special effort should203be made to implement uniform volumetric sampling to include the corners and discharge204area (at least 20 locations are recommended to adequately validate convective blenders).2052062.Collect at least 3 replicate samples from each location. Samples should meet the207following criteria:14 Weight correction is a mathematical correction to eliminate the effect of potentially variable tablet weight onmeasurement of mix adequacy—see Glossary, Section IX.15 This is described in Section IV of this guidance.208209•Assay one sample per location (number of samples (n) ≥ 10)210(n = 20 for ribbon blender).211212•RSD (relative standard deviation) of all individual results ≤ 5.0 percent.213214•All individual results are within 10.0 percent (absolute) of the mean of the results. 215216If samples do not meet these criteria, we recommend that you investigate the failure according to 217the flow chart in Attachment 1. We also recommend that you not proceed any further with 218implementation of the methods described in this guidance until the criteria are met.219220Sampling errors may occur in some powder blends, sampling devices, and techniques that make 221it impractical to evaluate adequacy of mix using only the blend data. In such cases, we222recommend that you use in-process dosage unit data in conjunction with blend sample data to 223evaluate blend uniformity.224225Some powder blends may present unacceptable safety risk when directly sampled. The safety 226risk, once described, may justify an alternate procedure. In such cases, process knowledge and 227data from indirect sampling combined with additional in-process dosage unit data may be228adequate to demonstrate the adequacy of the powder mix. Data analysis used to justify using 229these alternate procedures should be described in a summary report that is maintained at the 230manufacturing facility.231232As an alternative, you can substitute the procedures described in the PDA Technical Report No. 23325, (see reference in footnote 8) to ensure that the blend is uniform and that the method meets or 234exceeds the criteria described above.235236237VI.VERIFICATION OF MANUFACTURING CRITERIA238239You should complete the assessment of powder mix uniformity and correlation of stratified in-240process dosage unit sampling development procedures before establishing the criteria andcontrols for routine manufacturing. We also recommend that you assess the normality and241242determine RSD from the results of stratified in-process dosage unit sampling and testing that 243were developed. The RSD value should be used to classify the testing results as either readily 244pass (RSD ≤ 4.0%), marginally pass (RSD ≤ 6.0%) or inappropriate for demonstration of batch 245homogeneity (RSD > 6.0%). The procedures are discussed in the following sections:246247A.In-Process Dosage Unit Sampling and Analysis248249We recommend the following steps:250•Carefully identify locations throughout the compression or filling operation to sample in-251252process dosage units. The sampling locations should also include significant processevents such as hopper changeover, filling or machine shutdown and the beginning and 253254end of the compression or filling operation.16 There should be at least 20 locations with 7 255samples each for a minimum total of 140 samples. These include periodic sampling256locations and significant event locations.257•Sample at least 7 in-process dosage units from each sampling location.258•Assay at least 3 of the 7 and weight correct each result. (The number of samples should 259be specified and justified for a given product and process.)•Conduct an analysis of the dosage unit stratified sampling data to demonstrate that the 260261batch has a normal distribution of active ingredient. Indications of trends, bimodal262distributions, or other forms of a distribution other than normal should be investigated. If 263these occurrences significantly affect your ability to ensure batch homogeneity, they264should be corrected.•Prepare a summary of this analysis. Potential investigation results along with a265description of batch normality should be included in the summary. Submit this summary 266267with the application as described in section VIII of this guidance.268In addition to this analysis of batch normality, we recommend that you classify the test results as 269270readily pass or marginally pass according to the following procedure:271B.Criteria to Meet the Readily Pass Classification272273274For each separate batch, compare the test results to the following criteria:275276•For all individual results (for each batch n ≥ 60) the RSD ≤ 4.0 percent.277•Each location mean is within 90.0 percent to110.0 percent of target strength.278279280•All individual results are within the range of 75.0 percent to 125.0 percent of target strength.281282283If your test results meet these criteria, they are classified as readily pass and you can start routine 284batch testing using the Standard Verification Method (SVM) described in section VII. If your 285test results fail to meet these criteria, we recommend that you compare the results with themarginally pass criteria described below.286287288C.Criteria to Meet the Marginally Pass Classification289290If your dosage unit test results fail to meet the criteria for the r eadily pass classification, you 291should assay the remaining dosage units (all 7 units per location) and compare the test results to 292the following criteria:16 The beginning and end samples are taken from dosage units that would normally be included in the batch.293294•For all individual results (for one batch n ≥ 140) the RSD ≤ 6.0 percent.295•Each location mean is within 90.0 percent to 110.0 percent of target strength.296297298•All individual results are within the range of 75.0 percent to 125.0 percent of target299strength.300301If your test results meet these criteria, results can be classified as marginally pass. If yoursamples do not meet these criteria, we recommend that you investigate the failure, find justified 302303and assignable cause(s), correct the deficiencies, and repeat the powder mix homogeneity304assessment, in-process dosage unit sampling correlation, and initial criteria establishment305procedures. The disposition of batches that have failed the m arginally pass criteria is outside the 306scope of this guidance.307308D.Sample Locations for Routine Manufacturing309310We recommend that you prepare a summary of the data analysis from the powder mixassessment and stratified sample testing. From the data analysis, you should establish the311312stratified sample locations for routine manufacturing, taking into account significant process 313events and their effect on in-process dosage unit and finished dosage unit quality attributes. You should identify at least 10 sampling locations during capsule filling or tablet compression to 314315represent the entire routine manufacturing batch.316317318VII. ROUTINE MANUFACTURING BATCH TESTING METHODS319We recommend that you evaluate the routine manufacturing batches against the following320321criteria after completing the procedures described above to assess the adequacy of the powder 322mix and uniform content in finished dosage form.323324These routine manufacturing batch-testing methods include the Standard Criteria Method (SCM) 325and the Marginal Criteria Method (MCM). The SCM consists of two stages, each with the same accept/reject criteria. The second of the two stages recommends using a larger sample size to 326327meet these criteria. The MCM uses accept/reject criteria that are different from the SCM.328329If the batch data fail to conform to the SCM criteria, we recommend continued sampling and 330testing to intensified criteria (MCM). Both verification methods and the procedures for331switching from one to the other are detailed below and in the flow chart in Attachment 2.332333A.Standard Criteria Method (SCM)334335We recommend using the SCM verification method when either of the following conditions is 336met:•Results of establishing initial criteria are classified as readily pass.337。
Life Sciences Reporting Summary说明书

June 2017Corresponding Author:Johanna A. JoyceDate:Jun 13, 2017Life Sciences Reporting SummaryNature Research wishes to improve the reproducibility of the work we publish. This form is published with all life science papers and is intended to promote consistency and transparency in reporting. All life sciences submissions use this form; while some list items might not apply to an individual manuscript, all fields must be completed for clarity.For further information on the points included in this form, see Reporting Life Sciences Research . For further information on Nature Research policies, including our data availability policy , see Authors & Referees and the Editorial Policy Checklist.` Experimental design 1. Sample sizeDescribe how sample size was determined.Sample sizes were chosen based on power of 0.9 to detect a difference of>1.5 standard deviations between LF vs. HF means with 95% confidence.However, in most cases, previous data was sufficient to inform sample sizefor subsequent experiments. 2. Data exclusionsDescribe any data exclusions.Exclusion criteria were not needed. 3. ReplicationDescribe whether the experimental findings were reliably reproduced.All data included in the study are reproducible: all flow cytometryexperiments were repeated 3 or more times with similar results; all animaltrials were repeated with at least n=4 in at least 2 independent cohortswith similar results; all in vitro assays were repeated in at least 3independent experiments with similar results.4. RandomizationDescribe how samples/organisms/participants were allocated into experimental groups.For most experiments, a method for randomization was not needed since experimental groups were pre-determined by diet/weight. In cases whererandomization was required (e.g. Figure 7), average weight was used toensure balanced representation between experimental groups.5. BlindingDescribe whether the investigators were blinded to group allocation during data collection and/or analysis.For all experiments, automated quantitative methods were used. We chose to analyze data in this manner to avoid investigator bias.Note: all studies involving animals and/or human research participants must disclose whether blinding and randomization were used.6. Statistical parametersFor all figures and tables that use statistical methods, confirm that the following items are present in relevant figure legends (or the Methods section if additional space is needed).n/aConfirmedThe exact sample size (n) for each experimental group/condition, given as a discrete number and unit of measurement (animals, litters, cultures, etc.)A description of how samples were collected, noting whether measurements were taken from distinct samples or whether the same samplewas measured repeatedly.A statement indicating how many times each experiment was replicatedThe statistical test(s) used and whether they are one- or two-sided (note: only common tests should be described solely by name; morecomplex techniques should be described in the Methods section)A description of any assumptions or corrections, such as an adjustment for multiple comparisonsThe test results (e.g. pvalues) given as exact values whenever possible and with confidence intervals notedA summary of the descriptive statistics, including central tendency (e.g. median, mean) and variation (e.g. standard deviation, interquartile range)Clearly defined error barsSee the web collection on statistics for biologists for further resources and guidance.`SoftwarePolicy information about availability of computer code7. SoftwareDescribe the software used to analyze the data in this study. GraphPad Prism Pro5 was used for all data analysis.For all studies, we encourage code deposition in a community repository (e.g. GitHub). Authors must make computer code available to editors and reviewers upon request. The Nature Methods guidance for providing algorithms and software for publication may be useful for any submission.`Materials and reagentsPolicy information about availability of materials8. Materials availabilityIndicate whether there are restrictions on availability of uniquematerials or if these materials are only available for distribution by afor-profit company.All materials used in this study are available from commercial sources.9. AntibodiesDescribe the antibodies used and how they were validated for use inthe system under study (i.e. assay and species).Detailed information on antibody vendors, catalog and clone numbers anddilutions used can be found in Supplementary Table 3. All antibodies usedin this study were titrated for each lot, and an optimal dilution wasselected (see also Supplementary Table 4 for each type of assay). Mouseantibodies included: CD45 A700 and APC, Rat anti-mouse; Gr1 FITC andPerCP-Cy5.5, Rat anti-mouse; CD11b PE-Cy7 and PE, Rat anti-mouse/human; Ly6G PE, Rat anti-mouse; Ly6C BV421 and APC-Cy7, Rat anti-mouse; CD3 PE-Cy7, Rat anti-mouse; CD4 BV605, Rat anti-mouse; CD8AFITC, Rat anti-mouse; NK1.1 PerCP-Cy5.5, Rat anti-mouse; CD107a APC,Rat anti-mouse; SiglecF APC, Rat anti-mouseIL5ra A488, Rat anti-mouse. Human antibodies included: CD45 BV605, Ratanti-mouse/human; CD11B BUV395, Mouse anti-human; CD14 FITC,Mouse anti-human; CD16 A700, Mouse anti-human; CD66B PE-Cy7,Mouse anti-human; IL5RA PE, Mouse anti-human.June 201710. Eukaryotic cell linesa. State the source of each eukaryotic cell line used.Breast tumor cell lines were isolated from the MMTV-PyMT mouse model,backcrossed into the BL6 background for >10 generations. These cell lineswere subsequently selected for their capacity to grow in the mammary fatpad (7x105 cells injected/mouse) of WT BL6 animals within a reasonabletime period (<2 months). Three cell lines were selected for subsequentexperiments, including 99LN, 86R2, and 91R2. All breast tumor cell lines inculture were maintained in DMEM supplemented with 10% FBS, and werevalidated to be mycoplasma-free. For assays involving immune cell culture,primary cells were isolated from peripheral sources (i.e. blood or bonemarrow) by FACS and used immediately for functional assays, for exampleNK cell co-culture or T cell CFSE.b. Describe the method of cell line authentication used.Cell line authentication was not needed - all cell lines from this study weregenerated in-house for immediate downstream application.c. Report whether the cell lines were tested for mycoplasmacontamination.All cell lines were confirmed to be mycoplasma negative.d. If any of the cell lines used in the paper are listed in the databaseof commonly misidentified cell lines maintained by ICLAC,provide a scientific rationale for their use.No cell lines used in this study were found in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI Biosample.June 2017`Animals and human research participantsPolicy information about studies involving animals; when reporting animal research, follow the ARRIVE guidelines 11. Description of research animalsProvide details on animals and/or animal-derived materials used in the study.Diet-induced obesity (DIO) model: To model obesity with diet, 5w-oldfemale BL6 mice (Jackson Laboratory) were enrolled on either high fat (HF;60% kcal, Research Diets D12492) or low fat (LF; 10% kcal, Research DietsD12450) irradiated rodent diet for 15w. After 15w, animals were either sacrificed for flow cytometry, or injected with tumor cells. For the diet-switch model (HF-LF), 5w-old female BL6 mice were fed for 15w with HFdiet, and then switched to LF diet for 7w prior to sacrifice.Ob/ob model: To control for the effects of adipose tissue content in mice,4w-old female B6.Cg-Lep-ob (ob/ob; Jackson Laboratory) mice werepurchased and maintained on normal rodent diet. These mice gain weightdue to a homozygous mutation in the leptin (Lep) gene that causesexcessive eating and rapid weight gain. Weight was monitored over time beginning at 5w-old, and mice were euthanized when they reached >40g.This time period was significantly shorter (6w) than that of the DIO model(15w). After 6w, animals were sacrificed for flow cytometry analysis ofmyeloid cell populations in the lung, or injected with tumor cells for 48h metastasis assays.Balb/c obesity-resistant model: To control for the effects of nutrientcontent in diet, 5w-old WT female Balb/c mice (Jackson Laboratory) were enrolled on either HF or LF diet (Research Diets, see ‘DIO model’) for 15w.Balb/c animals do not gain weight in response to HF feeding. After 15w,animals were sacrificed for flow cytometry analysis of myeloid cellpopulations in the lung.Immune compromised mouse models: Two immune compromised mouse models were used in this study, athymic nude (lack T cells) and NOD-scidIL2r-gamma null (NSG; lack mature T, B and NK cells). In both cases, 5w-old female mice (Jackson Laboratory) were used. Mice were treated for 5dwith recombinant antibodies, and then sacrificed for flow cytometryanalysis of myeloid populations in blood and lung.Preparation of mouse samples for flow cytometry or FACS: Mice were anesthetized with avertin, blood was collected by submandibular bleeding,and cardiac perfusion with PBS was performed. All tissues weremechanically dissociated and filtered through a 40 um mesh to generate asingle cell suspension, and red blood cells were lysed (Pharm Lyse; BD Biosciences).Preparation of mouse serum: To collect serum from mice, blood wascollected by submandibular bleeding into eppendorf tubes and allowed toclot at room temperature for ~20 minutes. Samples were centrifuged at2000 x g, 4°C, 10 min. Supernatant was transferred to a polypropylenetube either individually or pooled. For pooled serum, three individualmouse samples were combined and stored at -80°C for downstream applications.June 2017Policy information about studies involving human research participants 12. Description of human research participantsDescribe the covariate-relevant population characteristics of the human research participants.Human blood samples: Blood collection from human donors was approvedby the Institutional Review Board of Rockefeller University, fully compliantwith all relevant ethical regulations regarding research involving human participants, and obtained with informed consent. Fresh whole-bloodsamples were obtained from healthy female donors (including 5 leandonors (BMI=18-25), and 2 obese donors with BMI equal to or more than35; donors were all postmenopausal) at Rockefeller University.Human serum samples: Human serum samples were obtained fromconsenting healthy female donors, and banked as individual or pooled.Serum samples were pooled from 9 lean (BMI=18-25) or 10 obese(BMI>35) postmenopausal women (median age=56 years old; age range45-66) and stored at -80°C for downstream application. For collection of matched human weight loss serum samples, the clinical trial wasconducted at Rockefeller University under identifierNCT01699906. Sample collection was approved by the Institutional ReviewBoard of Rockefeller University (New York, NY). Mean BMI before weightloss= 38.8 +/-3.4 sd; mean BMI after weight loss= 35.1 +/-3.0 sd; meanage= 60.6 years +/- 3.6 sd.June 2017June 2017Corresponding Author:Johanna A. Joyce Date:Jun 13, 2017Flow Cytometry Reporting Summary Form fields will expand as needed. Please do not leave fields blank.` Data presentationFor all flow cytometry data, confirm that:1. The axis labels state the marker and fluorochrome used (e.g. CD4-FITC).2. The axis scales are clearly visible. Include numbers along axes only for bottom left plot of group (a 'group' is an analysis of identical markers).3. All plots are contour plots with outliers or pseudocolor plots.4. A numerical value for number of cells or percentage (with statistics) is provided.` Methodological details5. Describe the sample preparation.For flow cytometry of mouse samples, mice were anesthetizedwith avertin, blood was collected by submandibular bleeding, andcardiac perfusion with PBS was performed. All tissues weremechanically dissociated and filtered through a 40 um mesh togenerate a single cell suspension and red blood cells were lysed(Pharm Lyse; BD Biosciences). Cells were counted, incubated withFc block (1h; BD Biosciences; 1:100/10^6 cells), incubated withfixable live/dead stain (30 min; Invitrogen), and then incubatedwith conjugated antibodies (1h). Alternatively, DAPI was used fordead cell exclusion instead of fixable live/dead stain. Mouseneutrophils were defined as CD45+CD11b+Gr1+/hi or CD45+CD11b+Ly6CloLy6G+. CD45+CD11b+Gr1lo cells were determined to beCD45+CD11b+Ly6Chi monocytes, and were therefore excludedfrom all neutrophil gating. OneComp eBeads (eBioscience) or ArC™Amine Reactive Compensation Beads (Invitrogen) were used for compensation.6. Identify the instrument used for data collection. A BD LSRFortessa was used for flow cytometry, and a BD FACSAria III was used for FACS.7. Describe the software used to collect and analyze the flow cytometry data.FlowJo was used for all flow cytometry and FACS data analysis, and for generating representative flow plots.8. Describe the abundance of the relevant cell populations within post-sort fractions.In all cases, post-sort purity was confirmed to be greater than>90% for downstream applications, including qRT-PCR and cytospin.9. Describe the gating strategy used.Mouse gating strategy: In all cases, dead cells and debris wereexcluded from analyses using FSC x SSC, a live/dead stain and/orDAPI. CD45+ was used as a marker for total leukocytes, CD11b+was used as a marker for myeloid cells. Neutrophils were furtherdefined as CD45+CD11b+Gr1+/hi or CD45+CD11b+Ly6CloLy6G+.CD45+CD11b+Gr1lo cells were determined to be CD45+CD11b+Ly6Chi monocytes, and were therefore excluded from allneutrophil gating. Eosinophils were defined as CD45+CD11b+myeloid cells with high side scatter, and Siglec-f+. In some cases,further gating on IL5ra+ or Ki67+ populations was performed foreosinophils, monocytes, and neutrophils as defined here. For CFSEassays, bulk T cells were gated as CD45+CD3+ and then furthergating according to positive CD4 or CD8 status for helper andcytotoxic T cells, respectively. For NK cell cytotoxicity assays, NKcells were defined as CD45+ with low side scatter, NK1.1+. Gatingon CD107a+ populations was used to further define NK cells withcytotoxic function.Human gating strategy: In all cases, dead cells and debris wereexcluded from analyses using FSC x SSC and DAPI. CD45+ was usedas a marker for total leukocytes, CD11b+ was used as a marker formyeloid cells. Gating strategy using cell surface markers were asfollows: Peripheral blood neutrophils (CD45+CD11b+CD66b+CD16+CD14lo), eosinophils (CD45+CD11b+CD66b+CD16-CD14lo), non-classical monocytes (CD45+CD11b+CD66b-CD16+CD14lo),intermediate monocytes (CD45+CD11b+CD66b-CD16+CD14hi),and classical monocytes (CD45+CD11b+CD66b-CD16-CD14+).Gating on IL5R+ cells was also included in analysis for eachpopulation. Eosinophils were used as a positive gating control forIL5R positivity as this is a canonical marker/signaling pathway forthis cell type.Tick this box to confirm that a figure exemplifying the gating strategy is provided in the Supplementary Information.June 2017。
美国DMF目录及要求

Drug SubstanceChemistry, Manufacturing, and Controls InformationDRAFT GUIDANCEThis guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should be submitted within 180 days of publication in the Federal Register of the notice announcing the availability of the draft guidance. Submit comments to Dockets Management Branch (HFA-305), Food and Drug Administration, 5630 Fishers Lane, rm. 1061, Rockville, MD 20852. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register.For questions regarding this draft document contact (CDER) Stephen Miller (301) 827-2392, (CBER) Chris Joneckis (301) 435-5681, or (CVM) Dennis Bensley (301) 827-6956.U.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Biologics Evaluation and Review (CBER)Center for Veterinary Medicine (CVM)January 2004CMCDrug Substance Chemistry, Manufacturing, and Controls InformationAdditional copies are available from:Office of Training and CommunicationDivision of Drug Information, HFD-240Center for Drug Evaluation and ResearchFood and Drug Administration5600 Fishers LaneRockville, MD 20857(Tel) 301-827-4573/cder/guidance/index.htmorOffice of Communication, Training andManufacturers Assistance, HFM-40Center for Biologics Evaluation and ResearchFood and Drug Administration1401 Rockville Pike, Rockville, MD 20852-1448/cber/guidelines.htm.(Tel) Voice Information System at 800-835-4709 or 301-827-1800orCommunications Staff, HFV-12Center for Veterinary MedicineFood and Drug Administration7519 Standish PlaceRockville, MD 20855(Tel) 301-827-3800/cvm/guidanc/published.htmU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Biologics Evaluation and Review (CBER)Center for Veterinary Medicine (CVM)January 2004CMCTABLE OF CONTENTS1I. INTRODUCTION (1)II. BACKGROUND (3)A. The Common Technical Document — Quality (CTD-Q) Format (3)B. Content of an Application (4)C. Additional Guidance (4)D. References to Other Applications or Master Files (MFs) (5)1. Other Applications (5)2. Master Files (MFs) (6)III. GENERAL INFORMATION (S.1) (8)A. Nomenclature (S.1.1) (8)B. Structure (S.1.2) (8)C. General Properties (S.1.3) (9)IV. MANUFACTURE (S.2) (10)A. Manufacturers (S.2.1) (10)B. Description of Manufacturing Process and Process Controls (S.2.2) (10)1. Flow Diagram (11)2. Description of the Manufacturing Process and Process Controls (12)3. Reprocessing, Reworking, Recycling, Regeneration, and Other Operations (15)C. Control of Materials (S.2.3) (18)1. Starting Materials (18)2. Reagents, Solvents, and Auxiliary Materials (19)3. Diluents (20)D. Controls of Critical Steps and Intermediates (S.2.4) (20)E. Process Validation and/or Evaluation (S.2.5) (23)F. Manufacturing Process Development (S.2.6) (23)V. CHARACTERIZATION (S.3) (24)A. Elucidation of Structure and Other Characteristics (S.3.1) (24)1. Elucidation of Structure (24)2. Physicochemical Characterization (25)3. Biological and Other Relevant Characteristics (26)B. Impurities (S.3.2) (27)VI. CONTROL OF DRUG SUBSTANCE (S.4) (29)1 Alphanumeric designations in parentheses that follow headings show where information should be placed in applications that are submitted in Common Technical Document (CTD) format.A. Specification (S.4.1) (29)B. Analytical Procedures (S.4.2) (34)C. Validation of Analytical Procedures (S.4.3) (35)D. Batch Analyses (S.4.4) (35)1. Batch Analysis Reports (36)2. Collated Batch Analyses Data (36)E. Justification of Specification (S.4.5) (37)VII. REFERENCE STANDARDS OR MATERIALS (S.5) (40)VIII. CONTAINER CLOSURE SYSTEM (S.6) (40)IX. STABILITY (S.7) (41)A. Stability Summary and Conclusions (S.7.1) (41)B. Postapproval Stability Protocol and Stability Commitment (S.7.2) (41)C. Stability Data (S.7.3) (41)1. Primary Stability Studies (41)2. Supporting Stability Studies (42)3. Stress Studies (42)X. APPENDICES (A) (43)A. Facilities and Equipment (A.1) (43)B. Adventitious Agents Safety Evaluation (A.2) (44)1. Nonviral Adventitious Agents (45)2. Viral Adventitious Agents (45)XI. REGIONAL INFORMATION (R) (46)A. Executed Production Records (R.1.S) (46)B. Comparability Protocols (R.2.S) (46)C. Methods Validation Package (R.3.S) (46)XII. LITERATURE REFERENCES (3.3) (47)ATTACHMENT 1: (48)STARTING MATERIALS FOR SYNTHETIC DRUG SUBSTANCES (48)ATTACHMENT 2: (56)STARTING MATERIALS OF PLANT OR ANIMAL ORIGIN (56)GLOSSARY (59)GUIDANCE FOR INDUSTRY2Drug SubstanceChemistry, Manufacturing, and Controls Information12345678910111213If you plan to submit comments on this draft guidance, to expedite FDA review of your14comments, please:15∙Clearly explain each issue/concern and, when appropriate, include a proposed revision and the rationale and/or justification for the proposed revision.1617∙Identify specific comments by line numbers; use the pdf version of the document whenever 18possible.19∙If possible, e-mail an electronic copy (Word) of the comments you have submitted to the20docket to cummingsd@.212223I. INTRODUCTION2425Information on the chemistry, manufacturing, and controls (CMC) for the drug substance must 26be submitted to support the approval of original new drug applications (NDAs), abbreviated new 27drug applications (ANDAs), new animal drug applications (NADAs), and abbreviated new28animal drug applications (ANADAs).3 This guidance provides recommendations on the CMC 29information for drug substances that should be submitted to support these applications. The30guidance is structured to facilitate the preparation of applications submitted in Common31Technical Document (CTD) format.3233This guidance addresses the information to be submitted for drug substances to ensure continued 34drug substance and drug product quality (i.e., the identity, strength, quality, purity, and potency).2 This guidance has been prepared by Drug Substance Technical Subcommittee of the Chemistry Manufacturing andControls Coordinating Committee (CMC CC) in the Center for Drug Evaluation and Research (CDER), the Center for Biologics Evaluations and Research (CBER) and the Center for Veterinary Medicine (CVM) at the FDA.3 See 21 CFR 314.50(d)(1) and 514.1(b)This guidance provides recommendations on the information that should be included for the3536following topics:37∙Nomenclature, structure, and general drug substance properties3839∙Manufacture40∙Characterization41∙Control of drug substance42∙Reference standards or materials43∙Container closure system44∙Stability45The recommendations provided in this guidance apply to the following types of drug substances:464748∙Drug substances manufactured by chemical synthesis49∙Highly purified and well characterized drug substances derived from plants or animals 4 50∙Semisynthetic drug substances manufactured by the chemical modification of a highly 51purified and well characterized intermediate derived from plants or animals52∙The synthetic portion of the manufacturing process for semisynthetic drug substances53manufactured by the chemical modification of an intermediate produced by conventional 54fermentation.5556The guidance does not provide specific recommendations relating to the following:5758∙Monoclonal antibodies59∙Peptides60∙Oligonucleotides61∙Radiopharmaceuticals62∙Medical gases63∙Drug substances that are not well characterized (e.g., botanicals, some proteins) derived 64from plants or animals65∙Drug substances derived using transgenic technology66∙Drug substances derived directly from or manufacturing operations involving67fermentation (conventional fermentation or using rDNA technology) or tissue or cell68culture.6970More detailed guidance on the content of an application may be available in separate guidance 71documents for specific types of drug substances (see section II.C). Applicants with drug72substances not specifically covered by this (Drug Substance guidance) or another guidance can 73apply the content recommendations in this guidance, as scientifically appropriate, and/or can74contact the appropriate chemistry review teams for guidance.754 For purposes of this guidance, d rug substances derived from plants or animals does not include materials producedby plant cell fermentation, animal cell or tissue culture, or through use of transgenic technology (e.g.,biotechnology-derived protein drug products).FDA’s guidance docume nts, including this guidance, do not establish legally enforceable7677responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should 78be viewed only as recommendations, unless specific regulatory or statutory requirements arecited. The use of the word should in Agency guidances means that something is suggested or7980recommended, but not required.8182This guidance, when finalized, will replace the guidance entitled Submitting Supporting83Documentation in Drug Applications for the Manufacture of Drug Substances (February 1987).848586II. BACKGROUND8788A. The Common Technical Document — Quality (CTD-Q) Format89In November 2000, the International Conference on Harmonisation of Technical9091Requirements for Registration of Pharmaceuticals for Human Use (ICH) issued92harmonized guidance for the format of drug product applications (i.e., Common93Technical Document (CTD)). The CTD describes a format for applications that94(supplemented with regional information) can be used for submission to the regulatory 95authorities in the United States, European Union, and Japan. One focus of this effort was 96harmonizing the format for quality information (i.e., chemistry, manufacturing, and97controls) that will be submitted in an application. FDA’s guidance on M4Q: The CTD —98Quality describes the format for the quality information submitted in Module 3 of an99application and provides additional information on formatting aspects of an application. 100Applicants can submit NDAs, ANDAs, NADAs, and ANADAs using the CTD-Q101format.5Applicants should review FDA’s guidance on M4Q: The CTD — Quality and 102other related CTD guidance documents for detailed formatting recommendations on103preparing an application in CTD format.104105Module 3 of each NDA and ANDA should include the specified CTD sections: Drug 106Substance (3.2.S), Drug Product (3.2.P), Appendices (3.2.A), Regional Information107(3.2.R), and Literature References (3.3). In some cases, the majority of information to 108address the drug substance sections will be incorporated by reference from a master file 109(see section II.D.2). However, an applicant should still provide information to address 110some of the drug substance subsections. Recommendations on the content of the drug 111product section (3.2.P) of Module 3 will be the provided in the guidance Drug Product —112Chemistry, Manufacturing, and Controls Information (Drug Product guidance), when 113finalized.6 The Appendices, Regional Information, and Literature References sections 114include information for both drug substance and drug product, as appropriate.1155 The information in animal drug applications is commonly presented in the order of the required CMC informationspecified under section § 514.1(b)(4) and (5). Although the CTD-Q format was developed for human drugs, thedrug substance information to support NADAs and ANADAs can be formatted according to the CTD-Q format or any alternative format that provides the appropriate information to support the application.6 A draft version of this guidance published on January 28, 2003 (68 FR 4219).116This Drug Substance guidance has been organized in a format conforming to Module 3 of 117the CTD, and it provides CMC content recommendations specific to drug substance,118including recommendations for the Appendices, Regional Information, and Literature 119References sections. Alphanumeric designations in parentheses corresponding to the 120CTD format follow relevant headings and text to show where information is to be placed 121in the CTD.7 Recommendations specific to drug product, including recommendations for 122the Appendices, Regional Information and Literature References sections, will be123provided in the Drug Product guidance.124125Multiple Drug Substances in an Application126127When an application is submitted for a drug product involving two or more drug128substances (e.g., combination drug product, copackaged drug products), information for 129each drug substance should be presented separately in the application. Information130presented separately means one complete S section for one drug substance followed by 131other complete S sections for additional drug substances. All of the information pertinent 132to each one of the drug substances (general information, manufacture, characterization, 133control, standards, container closure system, and stability) should be provided in a single 134section.135136B. Content of an Application137The application should include information in every S subsection for each of the drug 138139substances and manufacturing schemes (e.g., alternative processes, manufacturing site) 140intended for approval under the application. Information should be provided in theAppendices, Regional Information, and Literature References sections for each of the 141142drug substances and manufacturing schemes, as appropriate. If an Appendices or143Regional Information subsection or the Literature References section is not applicable, 144this should be stated in the application.145146C. Additional Guidance147148This Drug Substance guidance and the Drug Product guidance, when finalized, will be 149the primary content guidances for NDA and ANDA applicants. For quality, the general 150format guidance is M4Q: The CTD — Quality. These are the first guidances an applicant 151should consider when preparing the quality section (i.e., chemistry, manufacturing, and 152controls) of an NDA or ANDA (Module 3).153This guidance references ICH guidance documents cited in the CTD-Q and FDA’s154155guidances on general technical topics (i.e., stability, container closure systems, analytical 156procedures and methods validation, sterilization process validation, drug master files, and7 Arabic numbers have been assigned to specific sections of the CTD. For example, the designation 3.2 before S, P,A, and R indicates Module 3, Body of Data section 2. Where this guidance discusses Module 3, Body of Datasection 2, for brevity, the initial designation 3.2 is not repeated throughout the rest of the guidance (e.g., 3.2.S.1.3reads S.1.3).157environmental assessments) rather than incorporating this detailed information. These 158guidances are referenced in the text and/or listed at the end of a section. An applicant159should refer to these guidances for recommendations on the detailed information that160should be included in the application to address the general technical topic.161162Finally, an applicant should consider guidances that are available for specific technical 163issues or type (e.g., synthetic peptides) of drug substance when preparing its application. 164These guidances provide additional recommendations on unique scientific and technical 165aspects of the topic. Some references to these types of guidances are included in this166guidance. However, the references are given only as examples, and the list is not meant 167to be all-inclusive. Some examples of these types of guidance include the following:168∙Submission of Chemistry, Manufacturing, and Controls Information for Synthetic 169170Peptide Substances171∙Submission of Chemistry, Manufacturing and Controls Information for a172Therapeutic Recombinant DNA-Derived Product or a Monoclonal Antibody173Product for In Vivo Use, CBER/CDER (under development)174∙Botanical Drug Products (under development)∙Fermentation Derived Drug Substances and Intermediates and Associated Drug 175176Products (under development)177∙Synthetic Oligonucleotides; Submission of Chemistry, Manufacturing, and178Controls Information (under development)179∙Radiopharmaceutical Drug Products: Chemistry, Manufacturing and ControlsInformation (under development)180181182FDA continues to update existing and publish new guidance documents. An applicant 183should use current guidance when preparing an NDA, ANDA, NADA or ANADA184submission.8185186D. References to Other Applications or Master Files (MFs)1871881. Other Applications189190In some cases, chemistry, manufacturing, and controls information about drug substances 191is provided in one application by reference to pertinent information in another application. 192This situation is less common than inclusion of information by reference to a MF and193usually occurs when the same firm submits both applications.194An applicant must identify in the application all other referenced applications, and each 195reference to information submitted in another application must identify where the196information can be found in the referenced application (21 CFR 314.50(a)(1) and197514.1(a)). If the referenced application was submitted by a firm other than the applicant,the referencing application must contain a written statement that authorizes the reference, 1988Current guidance documents are available on the Internet at /cder/guidance/index.htm,/cber/guidelines.htm, and /cvm/guidance/published.htm.199signed by the holder of the referenced application (21 CFR 314.50(g)(1), 314.420(b). and 200514.1(a)).9 Copies of letters of authorization (LOAs) should be submitted in Module 1 of 201the NDA or ANDA or in the appropriate section of an NADA or ANADA.2022032. Master Files (MFs)204205This guidance describes chemistry, manufacturing, and controls information for drug206substances that should be submitted to the Agency as part of the process of seeking theapproval of an NDA, ANDA, NADA, or ANADA. When a drug substance is207208manufactured by a firm other than the applicant, much of this information is frequently 209provided by reference to one or more Type II MFs rather than directly in an application. 210The CMC information in a Type II MF can be organized in CTD-Q format. Under FDA's 211regulations, an application can incorporate by reference all or part of the contents of any 212MF to address particular drug substance issues if the MF holder provides written213authorization (i.e., LOA) to the applicant and the authorization is included in the214application (Module 1 for an NDA or ANDA or in the appropriate section of an NADAor ANADA). The authorization must specifically identify the material being215216incorporated by reference (21 CFR 314.420 and 514.1(a)). The incorporated material217should be identified by name, reference number, volume and page number of the MF, anddate of submission. See 21 CFR 314.420, CDER’s guidance on Drug Master Files, and 218219CVM’s guidance on Preparation and Submission of Veterinary Master Files for moreinformation.220221222Both the applicant and the drug substance manufacturer (MF holder) contribute to223establishing and maintaining the identity, strength, quality, purity, and potency of the224applicant's drug products by manufacturing and controlling the drug substance in225accordance with the information submitted in the application and, by reference, in the MF. 226The following recommendations pertain to location of information in the MF and/or227application when an applicant and Type II MF holder are different firms.228229∙General Information (S.110): Both the MF and the application should include this 230information. These sections should contain similar, though not necessarily identical, 231information. For example, if an applicant performed screening studies and232established the existence of multiple polymorphs, information concerning these233polymorphs might be present in the application but not in the MF.234235∙Manufacture (S.2): The application should identify in S.2.1 the manufacturers of 236each drug substance with appropriate administrative information (see section IV.A). 237The MF should include this information for its manufacturing operations and any9 CVM discourages the reference of NDAs or ANDAs for drug substance information. In these instances, CVMrecommends that the drug substance information be included in a master file or incorporated in the applicant’sNADA or ANADA.10 Alphanumeric designations in parentheses that follow headings show where information should be placed inapplications that are submitted in Common Technical Document (CTD) format.238contract facilities that are used (e.g., intermediate manufacturers, laboratories). In239general, a MF can be referenced for the information recommended in S.2.2 through240S.2.6. However, the information should be augmented by the applicant, as241appropriate. For example, if the applicant micronizes drug substance purchased from242a MF holder the information on the micronization process should be included in the243application.244245∙Characterization (S.3): In general, a MF can be referenced for this information.However, the information should be augmented by the applicant, as appropriate. For 246247example, characterization information on physical properties critical to the applicant’s248product, such as solid state form or particle size distribution, should be included in249S.3.1 by the applicant under certain circumstances (e.g., applicant manipulates the250physical property (micronizes), the MF holder has not characterized the physical251property). Furthermore, information on an applicant’s studies to characterizeimpurities (S.3.2) can be warranted to support the applicant’s drug substance controls. 252253254∙Control of Drug Substance (S.4): In general, information recommended in S.4 should be provided in both the MF and the application. However, reference to an MF 255256can be appropriate for some of the information in S.4.2 through S.4.5 if the MF257holder and applicant are working together to develop the drug substance controls.Both the MF and the application should include a drug substance specification (S.4.1). 258259The MF could include more than one drug substance specification if the holder sellsdifferent technical grades of the drug substance (e.g., micronized and nonmicronized). 260261262∙Reference Standards (S.5): In general, information should be provided in both the 263MF and the application. However, reference to a MF can be appropriate for some of264the information if the MF holder and applicant are working together to develop the265reference standard.266267∙Container Closure System (S.6): In general, MFs can be referenced for this268information. However, the information should be augmented by the applicant, as269appropriate.270271∙Stability (S.7): In general, MFs can be referenced for this information. However, the information should be augmented by the applicant, as appropriate. For example, 272273an applicant might perform stress studies to support the analytical procedures it used274to control the drug substance.275276∙Appendices (A): In general, MFs can be referenced for this information. However, 277the information should be augmented by the applicant, as appropriate.278279∙Regional Information (R): Comparability protocols can be included in both the MF 280and application (R.2.S). A methods validation package should be included in theapplication (R.3.S).281282∙Literature References (3.3): Both the MF and the application should include283284literature references as warranted.285Type II MFs for drug substance intermediates can also be submitted in the CTD-Q format. 286287However, not all sections of the CTD-Q format would apply (e.g., S.4). The CMC288information provided to support an intermediate should be appropriate for the particularsituation (e.g., process, complexity of the molecule).289290291III. GENERAL INFORMATION (S.1)292293294General information on the nomenclature, structure, and general properties of the drug substance, should be provided in S.1.295296297A. Nomenclature (S.1.1)298299All appropriate names or designations for the drug substance should be provided in S.1.1. 300Any codes, abbreviations, or nicknames used in the application to identify the drug301substance should also be listed, including the following, if they exist or have been302proposed. A name that has not yet been finalized should be identified as proposed in the 303list.304305∙United States Adopted Name (USAN)∙Compendial name11306307∙Chemical names (e.g., Chemical Abstracts Service (CAS), International Union of 308Pure and Applied Chemistry (IUPAC))∙Company names or laboratory codes309310∙Other nonproprietary names (e.g., International Nonproprietary Name (INN),311British Approved Name (BAN), Japanese Accepted Name (JAN))312∙Chemical Abstracts Service (CAS) Registry Number313314B. Structure (S.1.2)315316Information on the chemical structure of the drug substance should be provided in S.1.2. 317This information should include:318319∙one or more drawings to show the overall chemical structure of the drug substance, 320including stereochemistry321∙molecular formula322∙molecular weight323324For a naturally derived protein drug substance, the information should include:11 A compendial name is a name that appears in an official compendium as defined in the Federal Food, Drug, andCosmetic Act (e.g., United States Pharmacopeia (USP)) (§ 201(j) (21 U.S.C. 32(i)).325326∙the schematic amino acid sequence indicating glycosylation sites or other327posttranslational modifications∙ a general description of the molecule (e.g., shape, disulfide bonds, subunit328329composition)330∙number of amino acid residues331∙molecular weight332333C. General Properties (S.1.3)334A list should be provided of the general physicochemical properties of the drug substance. 335336Other relevant properties of the drug substance should also be listed. Relevant properties 337are those physical, chemical, biological and microbiological attributes relating to theidentity, strength, quality, purity, and/or potency of the drug substance and, as338339appropriate, drug product. The information should include, as appropriate:340341∙ A general description (e.g., appearance, color, physical state)342∙Melting or boiling points343∙Optical rotation344∙Solubility profile (aqueous and nonaqueous, as applicable)345∙Solution pH346∙Partition coefficients347∙Dissociation constants348∙Identification of the physical form (e.g., polymorph, solvate, or hydrate) that will 349be used in the manufacture of the drug product350∙Biological activities351352For a naturally derived protein drug substance, additional information should be included, 353such as:354355∙Isoelectric point356∙Extinction coefficient357∙Any unique spectral characteristics358359If the drug substance can exist in more than one physical form, the information included 360in S.1.3 should be for the form (or forms) of the drug substance that will be used in the 361manufacture of the drug product. Detailed information on the characterization (e.g., X-362ray powder diffraction data, thermal analysis curves) of these and other physical forms 363and conditions required to produce one form or another should be provided in S.3.1.364。
Guidance120sample

To Pharmaceutical ManufacturingIntroductionThis document discusses the basic principles of water activity and the importance it has in the manufacture of pharmaceuticals. It also provides direction on when and where testing for water activity can be most beneficial.Water activity (a w) is defined as the ratio of product vapour pressure to pure water vapour pressure. It is a measure of the water available for chemical or microbiological activity within a product. It is not a measure of the total water content of an item, as water can be chemically bound and not be available for use. Its values are typically expressed as a decimal value and can range from 0.0 (completely dry) up to 1.0 (pure water).Though the principles of water activity have been used for centuries (e.g. salting, drying, mummification), its use by the FDA occurred in the 1980¶s when water activity testing was added to existing strategies for microbiological control in food products. Water activity is significant to the pharmaceutical industry in that it affects the quality of ingredients and finished product through their chemical stability, a reduced need for chemical preservatives, and a potential reduction in the need for microbial limits testing.Microbial growth requires water. Water dissolves solutes within a viable cell and is required for metabolic function. When an (a w) value is associated with a micro-organism it serves as an indication of potential metabolic activity. While organism proliferation ceases below certain water activity levels, some species have the ability to adapt slightly and continue to grow at levels below their optimum range. Organisms grown in an environment outside of their optimal a w range will most likely be more resistant to thermal means of destruction.There is a comprehensive understanding of microorganisms and their associated a w values. The optimal a w range for microorganism growth lies between 0.995 and 0.980. Most bacteria cannot grow below levels of 0.90. Yeasts typically survive down to levels of 0.87. Molds can tolerate a w levels down to about 0.80. Microbiological growth at any level ceases at a w values of about 0.60. Thus, by controlling the water1。
GuidanceforIndustry-FoodandDrugAdministration

E15 Definitions for
Genomic Biomarkers,
Pharmacogenomics,
Pharmacogenetics, Genomic
Data and Sample Coding
Categories
Additional copies are available from:
2. Additional Information (2.1.2)......................................................................................................... 2
B. Pharmacogenomics and Pharmacogenetics (2.2) ........................................................................ 3
5. Additional Information (2.3.5)......................................................................................................... 6
Table 1: Summary of Genomic Data and Sample Coding Categories.............................................. 7
/cber/guidelines.htm.
U.S. Department of Health and Human Services
关于CIA报考科目

关于报考科目。
CIA考试一共有四门课程,分别是《内部审计在治理、风险和控制中的作用》、《实施内部审计业务》、《经营分析和信息技术》和《经营管理技术》。
第一、二门交叉的内容比较多,模拟题中经常有第一部分的题目出现在第二部分,反之亦然,有时都分不清这是第一部分还是第二部分的题;而且考试时也会经常碰到串题的现象。
因此新考生报考时最好第一、二门一起报,这样看书时能全面复习就不怕考试串题了。
但如果有一门未通过,下次再考时就只能两门一起看了。
如果有比较充裕时间看书的话,最好前三门一起报,因为第一、二门的考试范围里也涉及第三门中信息技术审计的内容。
关于平时看书做题。
只要掌握了适合你的学习方法,逐步培养出比较准确的题感,通过CIA考试并不难。
我的总体感觉是CIA还是一门需要花费大量时间去努力准备的考试,如果想花很少时间就能通过几乎不可能,除非你平时就是干审计这一职业的。
知识在于平时的积累,对考CIA的大多数人来说都是在职学习,平时工作很忙还要兼顾家庭,不能保证有大段连续的学习时间用于CIA备考复习。
但时间是可以一点点挤出来的,只要做个有心人,每天挤出个把小时来看书应该能做得到,就算有个十分钟空闲做个三五道习题也行,积少成多可以达到同样的效果,题感也是靠经常不断的学习思考、反复练习方能培养出来的。
所以考CIA还贵在每天坚持,不能看三天停个十天半月的,那样效果适得其反。
只有持之以恒的决心和坚持不懈的努力,方能体会到成功的喜悦。
关于考试心态。
既然准备考这个试,就应该充分重视它,但也不必压力过大、看得过重。
心态放平和不必把目标定的过高(诸如一定要一次通过三门或四门),平时只要做到认真对待,保证看书时间和适量的做题就可以了。
在考场上,一开始的紧张情绪绝大多数考生都会有,做了几题后这种紧张情绪就应该慢慢化解,取而代之的是全身心的投入和思考,尽快地把自己调整到最佳的考场状态,正常发挥平时的积累。
对一些没遇到过或不能肯定选哪个答案的题目,则在脑子里飞快的思索和联想,还不能确定的就再读一遍题目和答案,划出题干中的关键句子和词语,同时进行扩散性思维,运用排除法、联想法等方法进行综合判断、选择。
guidance-technical-documentation-and-design-dossiers

Whereas the term “Technical Documentation” or “Technical File“ is used for medical devices of class I, class IIa, andclass IIb, the term “Design Dossier“ is used for class III products.术语“技术文档”或“技术文件”用于医疗设备的I类,IIA类和IIB类,术语“设计档案”用于第三类产品。
Technical Documentation is retained in the premises of the manufacturer or the Authorized Representative for potential review of Competent Authorities and Notified Bodies.技术文件保留在制造商或授权代表处,以备主管部门和公告机构审查。
Part B of the Technical File may be available at the manufacturer only, whereas Design Dossiers have to be submitted to the Notified Body for review prior to CE marking of the product技术文件的B部分可能仅在制造商提供,然而在产品标识"CE"之前,设计档案材料必须提交给认证机构(use form MDD Application for CE Conformity Assessment MED_F_03.15; http://www.tuev-sued.de/industry-and-consumer- products/download-center/applications).(使用MDD应用程序MED_F_03.15进行CE合格评估)We will assign a project coordinator who will entrust one or more further experts with the review of particular modules.我们将指派一个项目协调员,他将委托一个或多个进一步的专家对特定的模块进行评审。
高考英语选词填空高频词汇

高考英语选词填空高频词汇1. occasion 场合2. situation 情况,处境3. take over 接管4. exchange 交换5. command 命令6. confirm 证实7. cultivate 培养8. prosperously 繁荣的9. suspect 怀疑10. relatively 相对的,比较的11. acknowledge 承认,鸣谢12. ambition 抱负,野心13. quality 质量,品质14. protection 保护15. equally 平等地16. promise 承诺17. clearly 清楚地18. grateful 感激的19. remove 移开20. force 强迫21. apologize 道歉22. terrible 可怕的,糟糕的23. stubborn 固执的24. actively 积极地,主动地25. spiritual 精神的,心灵的26. magical 魔力的27. willingly 愿意地28. strengthen 加强29. image 形象30. complexity 复杂31. cautious 小心的32. manage 管理,成功做成33. prejudice 偏见34. economic 经济的,合算的35. academic 学术的36. control 控制37. adopt 收养,采取38. consume 消费,消耗39. unique 独一无二的40. beneficial 有益的41. varied 多变的,各种各样的42. demanding 要求高的43. appropriate 合理的44. entertainment 娱乐45. deliberately 故意地46. purchase 购买47. tough 艰难的48. bright 明亮的49. remain 留下,保持50. terrify 使害怕51. disappointing 令人失望的52. formal 正式的53. desire 愿望54. share 分享,共有55. fulfill 履行(诺言),执行(命令)56. admit 承认57. evident 明显的58. consequently 因此,所以59. accustomed 习惯的60. accumulate 积累61. participate 参加62. absence 缺席63. presence 出席64. bravery 勇敢65. horror 恐惧66. spotless 无暇的67. fundamental 基础的68. employment 就业,雇用69. involve 包含,使参与70. actually 事实上71. harmony 和谐72. basically 基本的73. inspire 激发,鼓舞74. imitate 模仿75. awful 糟糕的76. generous 慷慨的,大方的77. wealthy 富有的78. function 功能79. stressful 有压力的80. persistent 坚持不懈的81. reluctant 勉强的,不愿意的82. diligent 勤奋的83. attentive 注意的,周到的84. unbearable 不能忍受的85. accommodation 住所86. attractive 有吸引力的87. constant 连续的88. brilliant 杰出的,才华横溢的89. clumsy 笨拙的90. declare 宣布,声明91. obtain 获得92. interactive 相互的,互动的93. incident 事件94. adventure 冒险95. in particular 尤其96. in reality 事实上97. emphasize 强调98. overlook 忽视99. deny 否认100. ensure 确保101. financial 金融102. budget 预算103. on the whole 整体上104. potential 潜在的,潜能105. on the contrary 相反106. loyalty 忠实107. assume 假设108. establish 建设109. flexible 灵活的110. sensitive 敏感的111. essential 必不可少的112. unfair 不公平的113. expectation 期待114. impression 印象115. examination 考试,检查116. contribution 贡献117. certainty 肯定118. confuse 使迷惑,使混淆119. trap 陷阱,困住120. secondary 次要的121. turn up 出现122. show off 炫耀123. break in 闯进124. settle down 定居,安定下来125. relief 安慰,减轻126. justice 公正127. previous 先前的,早先的128. instantly 立即地129. regularly 规则地,规律地130. occasionally 偶尔地131. independence 独立132. keep up with 跟上133. catch up with 追赶上134. come up with 想出135. put up with 忍受136. guarantee 保证137. convince 使确信,说服138. atmosphere 气氛139. sympathy 同情140. punish 惩罚141. puzzled 感到迷惑的142. scared 害怕的143. embarrassed 尴尬的144. reaction 反应145. forgiveness 原谅146. imaginary 想象力丰富的147. be filled with 充满148. be pleased with 对......感到满意149. be crowded with 挤满150. be equipped with 配备有...... 151. possession 财产152. precious 珍贵的153. appreciate 欣赏,感激154. admire 钦佩155. wander 闲逛156. get rid of 消除,摆脱157. particularly 尤其,特别158. purpose 目的,意图159. courage 勇气160. determination 决心161. roll 滚162. drop 掉下163. undoubtedly 毫无疑问地164. temporarily 暂时地165. thankfully 幸运地166. lean against 靠着167. challenge 挑战168. fierce 激烈的169. practical 实际的,实用的170. straight 直接的171. delighted 高兴的172. congratulate 祝贺173. faint 头晕的174. consult 咨询175. messy 乱的176. review 复习,评论177. curious 好奇的178. graduation 毕业179. honor 荣耀180. comfortable 舒服的,舒适的181. tiresome 令人生厌的,无聊的182. set up 建立183. hold up 举起,支撑184. pick up 拾起,学会,接185. possess 拥有186. crowded 拥挤的187. march 行军,前进188. apartment 公寓189. frightened 害怕的190. turn out 结果是,证明是191. figure out 算出,想出192. anxious 焦虑的193. hunt 寻找,打猎194. amused 愉快的,顽皮的195. optimistic 乐观的196. demonstrate 证明,演示,显示197. realize 意识到198. meaningful 有意义的199. faithful 忠实的,忠诚的200. grasp 抓住,理解201. decorate 装饰202. pressure 压力203. obvious 明显的204. predict 预测205. audience 观众206. contain 包含207. gesture 姿势208. pioneer 先锋209. afford 支付得起210. affair 事情211. dependent 依赖的212. properly 合理地213. sincerely 真诚地214. severely 严厉地215. guidance 指导216. appearance 出现,外貌217. astonishment 惊讶218. sharpen 使锋利219. comment 评论220. privately 私人地221. frequently 频繁地222. physical 身体的,物理的223. donate 捐赠224. manufacture 制造,制造业225. original 原始的,起初的226. effective 有效的227. object 反对228. typical 典型的229. possibility 可能性230. accidentally 意外地,偶然地231. immediately 立刻232. concern 关心,担心233. concentrate 集中234. tragedy 悲剧235. neglect 忽视236. approach 接近,途径,方法237. disturbance 打扰238. arrest 逮捕239. restore 恢复240. available 可得到的,可利用的241. accompany 陪伴242. characteristic 特征,特性243. automatic 自动的244. approve 批准,同意245. roughly 粗略地246. indicate 暗示247. hesitation 犹豫248. attach 系着,附着249. plain 平的,朴素的,简单的250. mercy 怜悯251. sample 样本252. considerable 相当大的,值得考虑的253. enthusiasm 热情254. confirm 证实255. phenomenon 现象256. modify 修改257. annual 每年的258. isolation 隔离,孤立259. ceremony 典礼,仪式260. acceptable 可接受的261. eventually 最后262. target 目标263. expose 暴露264. absolutely 绝对地265. negative 消极的,否定的266. steady 稳定的267. consistently 一贯地,一致地268. permanent 永久的269. professional 职业的,专业的270. dismiss 解雇271. countless 数不尽的272. thoroughly 彻底地273. standard 标准274. tolerate 忍受275. memorable 值得纪念的276. leave for 离开去到277. adapt 适应,改编278. somehow 不知怎么地279. somewhat 稍微,有点280. acquire 获得281. occupy 占据282. accompany 陪伴283. urgent 紧急的284. technology 技术285. giant 巨大的286. invisible 看不见的287. evaluation 评估288. magnificent 壮丽的289. inefficient 效率低的290. nonsense 废话,荒谬的291. preserve 保护,保存292. career 事业293. profession 职业,专业294. exhausted 筋疲力尽的295. crash 撞碎,坠毁296. suspicious 怀疑的297. requirement 要求298. athletic 运动的,运动员的299. prohibit 禁止,阻止300. depressed 沮丧的,萧条的301. instruction 教导,说明302. slightly 轻微地303. meaningful 有意义的304. principle 原则305. undeserved 不应得的306. betray 背叛307. moderate 中等的,适度的308. evaluate 评价,估价309. honorable 值得尊敬的310. artificial 人造的311. concept 概念312. symbolize 象征313. promising 有前途的314. conflict 冲突315. display 显示,陈列316. definitely 明确地,肯定地317. interrupt 打断318. recall 回想起319. recite 背诵320. resist 抵制321. desperate 绝望的322. elegant 优雅的323. attempt 试图324. random 随机的325. profitable 有利可图的326. treasure 珍惜,宝藏327. poisonous 有毒的328. dilemma 窘境329. correctly 正确地330. system 系统331. complicated 复杂的332. application 申请,应用程序333. simple 简单的334. recognize 认出,认可335. cruel 残忍的336. comfortable 舒服的,舒适的337. feared 害怕的338. replace 代替339. method 方法340. seize 抓住341. misunderstand 误解342. mistake 错误,弄错343. imply 暗示344. disease 疾病345. repay 偿还,报答346. describe 描述347. contract 合同348. promote 促进,提升349. sacrifice 牺牲350. generally 一般来说351. trouble 麻烦352. pleased 满意的353. certainly 肯定地,当然354. national 国家的,民族的355. cultural 文化的356. entrance 入口357. annoying 令人恼怒的358. polite 有礼貌的359. unconscious 无意识的360. patient 有耐心的361. theory 理论362. classification 分类363. smart 聪明的364. mature 成熟的365. average 平均366. creative 有创造力的367. graduate 毕业,毕业生368. program 节目369. intelligent 智能的,聪明的370. competent 有能力的371. independent 独立的372. separate 单独的,分开的373. voluntarily 自愿地374. doubt 怀疑375. reflection 反射,沉思,映象376. conclusion 结论377. adult 成年人378. talented 有天赋的379. outstanding 出色的380. expression 表达,表情381. check 检查382. attend 参加,照料383. be aware of 意识到384. be fond of 喜欢385. be worthy of 值得386. expect 期待,预计387. decision 决定388. style 风格389. pretend 假装390. explain 解释391. cause 原因,事业392. perform 表演,执行393. probably 可能地394. struggle 挣扎,奋斗395. completely 完全地396. wonder 想知道397. experience 经验,经历398. fail 失败399. develop 发展400. responsible 有责任的401. valuable 有价值的,贵重的402. ability 能力403. affect 影响404. destroy 毁坏405. confident 自信的406. request 要求407. traditional 传统的408. modern 现代的409. relationship 关系410. cooperation 合作411. distinguish 区分412. identify 鉴定,识别413. thought 想法,思想414.discourage 使气馁,使沮丧415. prepare 准备416. observe 观察,遵守417. energy 精力418. nervous 紧张的419. proud 自豪的。
高中英语完形填空800个高频词(全)

高中英语完形填空800个高频词(全)名词(114)1.business生意,事情2.occasion场合3.situation情况,处境4.power权利5.memory记忆6.challenge挑战7.doubt疑惑8.image形象9.ambition野心quality质量,品质h.protection保护12.promise承诺4prejudice偏见14.entertainment娱乐15.absence缺席圮.presence出席17.bravery勇敢】8.horror恐惧】9.function功能2。
.accommodation住所21.incident事件之2.adventure冒险23.expectation期待24.impression印象25.contribution贡献plexity复杂27.desire愿望28.relief松了一口气9.courage勇气3。
.determination决心31.graduation毕业32.honor荣耀33.atmosphere气氛34.sympathy同情35.justice公正‘6.budget预算‘7.kindness善良38.reaction反应39.forgiveness原谅40.guidance指导41.astonishment惊讶43.gesture姿势‘4.pioneer先锋‘5.audience观众46.system系统47.application申请,应用程序48.isolation孤立49.ceremony仪式50.pressure压力51.target目标^2.standard标准53.technology技术54.evaluation评估55.nonsense无意义的,荒谬的56.requirement要求57.instruction教诲58.career事业S9.profession职业60.method方法61.disease疾病62.contract合同63.sacrifice牺牲64.affair事情.时间65.possibility可能性66.tragedy悲剧67.disturbance打扰68.mercy怜悯69.sample样本70.enthusiasm热情71.phenomenon现象72.conflict冲突73.dilemma窘境74.reflection反映,倒影75.conclusion结论76.program节目77.intelligence智力78.relationship关系79.cooperation合作80.decision决定81.style风格82.expression表达,表情83.theory理论84.classification分类85.entrance入口87.thought想法88.employment就业,雇用89.harmony和谐90.possession财产91.energy精力。
MDCG 2020-7_guidance_pmcf_plan_template_en

The aim of the PMCF plan is: confirming the safety1 and performance, including the clinical benefit if applicable, of the device throughout its expected lifetime; identifying previously unknown side-effects and monitor the identified side-effects and contraindications; identifying and analysing emergent risks on the basis of factual evidence; ensuring the continued acceptability of the benefit-risk ratio, referred to in Section 1 and 9 of Annex I in the MDR; identifying possible systematic misuse or off-label use of the device, with a view to verifying that the intended purpose is correct.
甲醛溶液浸泡的人类组织样本基因组DNA的提取方法与质量鉴定

激光生物学报ACTA LASER BIOLOGY SINICAVol. 31 No. 2Apr. 2022第31卷第2期2022年4月收稿日期:2022-02-07;修回日期:2022-03-25。
基金项目:国家自然科学基金面上项目(81970324);湖南省自然科学基金资助项目(2018JJ 2666,2018JJ 2611,2019JJ 50394);湖南省科技厅普惠项目(2020ZK 4028);动物多肽药物创制国家地方联合工程实验室开放基金课题项目(2018KF 002);省部共建淡水鱼类发育生物学国家重点实验室开放课题项目(2017KF 005);湖南省教育厅优秀青年项目(19B 342)。
作者简介:杨博宇,硕士研究生。
* 通信作者:江志钢,高级实验师,主要从事心脏发育方面的研究。
E-mail: 270212729@ 。
甲醛溶液浸泡的人类组织样本基因组DNA 的提取方法与质量鉴定杨博宇1,蔡 毅1,孙鲁宁1,栾泽东2,庞文艳2,吴秀山1,范雄伟1,江志钢1*(1. 湖南师范大学心脏发育研究中心,长沙 410081;2. 莱州市妇幼保健院,烟台 261400)摘 要:甲醛溶液浸泡的人类组织样本很多情况下是可遇不可求的珍贵资源,而其 DNA 的提取是一大难题。
本文分别使用改进的甲醛溶液浸泡样本基因组DNA 提取方法以及通用型基因组DNA 提取试剂盒,对四份不同时间固定的福尔马林浸泡的人类胚胎皮肤和肌肉的样本进行基因组DNA 提取。
使用紫外分光光度计鉴定所提取DNA 的纯度和质量浓度,DNA 溶液点样进行琼脂糖凝胶电泳及成像,结果显示,改进的提取方法所得基因组DNA 的质量浓度和纯度均优于通用型基因组DNA 提取方法。
以提取所得的基因组DNA 模板进行PCR 扩增,电泳结果显示,改进的提取方法所提取的基因组DNA 有更好的PCR 扩增效果。
本研究为甲醛溶液浸泡的珍贵样本的基因组DNA 的提取提供了方法指导和改进方向的参考。
Protocol105

Protocol105PROTOCOL # 105-4Raised & Non Raised Silk / Silk BlendsKnit & WovenPERFORMANCE TEST TEST METHOD REQUIREMENT INITIAL PACKAGEPhysical PropertiesFiber Content AATCC 20/20A16 CFR 303Wool Product Labeling Act –16 CFR 300Fur Products Labeling Act –16 CFR 301Fur Products IdentificationAct – 15 U.S.A 69 One Fiber Only: No ToleranceTwo or More Fibers: +/- 3% max deviation from contracted fiber contentFunctional Fibers i.e. Spandex: +/- 2% max deviation from contracted fiber contentYarn Size ASTM D 1059 Report Actual DataCount ASTM D 3887,ASTM D 3775,AATCC/ASTM TS-002Report Actual DataFabric Weight ASTM D 3776 Deviation from contracted weight (g/m2 OR oz/yd2): +/- 5%maximumFlammability(Refer to Kohl’s Safety Standards)16 CFR 161016 CFR 1615/1616 Class 1Non-exempt fabrics must be tested in every colorPass (Acceptance Criteria)The supplier must keep all children's sleepwear test records and samples for a period of 7 yearsAnalyticalFormaldehyde Kohl’s TM-5JIS L 1041:2000 –Sec. 6.3.1 (b) Method A or B Negative, If Spot Test is Positive, proceed with step 2 Children over 4 & Adults = 75 ppm max.Children’s 0-4T = 0.05 Absorbency max.Dimensional ChangeDimensional Change to Home Laundering, Dry Cleaning or Hand Wash AATCC 135 (3 washes)/AATCC 158 (1 cycle)AATCC/ASTM TS-006(Length X Width)Ultra Tide ? PowderDetergentWovens and Knits Dry Clean Items-5% X -5% max -2% X -2% maxGrowth +1% X +1% maxAppearance Retention AATCC/ASTM TS-008ModifiedUltra Tide ? PowderDetergent Must meet all applicable Kohl's Appearance Evaluation RequirementsStrength & DurabilityTensile Strength - Wovens Only ASTM D 5034 ≤2.0 oz/sq. yd.– 20 lbs.>2.0 oz/sq. yd.- 25 lbs.Tear Strength - Wovens Only ASTM D 1424(Elmendorf) ≤2.0 oz/sq. yd. – 1.5 lbs. >2.0 oz/sq. yd.- 2 lbs.Bursting Strength – Knits Only ASTM D 3786 30 lbs./sq.in min Pilling Resistance ICI – ISO 12945-1(ICI Box)As Received: Grade 3.5 min for 2 hours ColorfastnessColorfastness to ChlorineBleach AND Non-ChlorineBleachAATCC/ASTM TS-001 Shade Change: Grade 4.0 minRaised & Non Raised Silk / Silk BlendsKnit & WovenPERFORMANCE TEST TEST METHOD REQUIREMENTColorfastness to Crocking AATCC 8/116Original StateIf results fail in original state,perform after 1 Wash/DryCycle All fabrics (except listed below): Dry - Grade 3.5 min Wet - Grade 2.5 minPigment Dyed/Printed, Lightly Brushed, Piled, Sulphur, Indigo:Dry - Grade 3 minWet - Grade 2 minADDITIONAL COLORWAY TESTINGFlammability* Required where applicable at additional cost(Refer to Kohl’s Safety Standards)16 CFR 161016 CFR 1615/1616Class 1Non-exempt fabrics must be tested in every colorPass (Acceptance Criteria)The supplier must keep all children's sleepwear test records and samples for a period of 7 yearsFormaldehyde Kohl’s TM-5JIS L 1041:2000 –Sec. 6.3.1 (b) Method A or B Negative, If Spot Test is Positive, proceed with step 2 Children over 4 & Adults = 75 ppm max.Children’s 0-4T = 0.05 Absorbency max.Appearance Retention AATCC/ASTM TS-008ModifiedUltra Tide ? PowderDetergent Must meet all applicable Kohl's Appearance Evaluation RequirementsColorfastness to ChlorineBleach AND Non-ChlorineBleachAATCC/ASTM TS-001 Shade Change: Grade 4 minColorfastness to Crocking AATCC 8/116Original StateIf results fail in original state,perform after 1 Wash/DryCycle All fabrics (except listed below): Dry - Grade 3.5 min Wet - Grade 2.5 minPigment Dyed/Printed, Lightly Brushed, Piled, Sulphur, Indigo:Dry - Grade 3 minWet - Grade 2 minREQUIRED WHERE APPLICABLE AT ADDITIONAL COST ColorfastnessColorfastness to Perspiration - Acid Only*(Activewear/Intimates/Linings/Dk. Garment Dye) AATCC 15 Shade Change: Grade 4 minSelf Staining: Grade 4.5 minStaining: Grade 3.0 minColorfastness to Water* (Color block fabrics & contrast trims) AATCC 107 Shade Change: Grade 4 minSelf Staining: Grade 4 minStaining: Grade 3.5 minRaised & Non Raised Silk / Silk BlendsKnit & WovenPERFORMANCE TEST TEST METHOD REQUIREMENT Strength & DurabilityStretch and Recovery(For Knits containing spandex) ASTM D 2594 Mod.Use 4 lbs. load1-5% Spandex: Measure Time Dependent Growth andRecovery using 60 minutes. Report both growth andrecovery. Pass or fail based on 7% max. growth.> 5% but <10% Spandex: Measure Time Dependent Growth and Recovery using 30 minutes. Report both growth andrecovery. Pass or fail based on 5% max. growth.> 10% Spandex: Measure Time Dependent Growth andRecovery using 60 seconds. Report both growth andrecovery. Pass or fail based on 85% min. recoveryStretch and Recovery(For Wovens containing Spandex – only in the direction of stretch) ASTM D 3107 ModifiedUse 4 lbs. loadStretch: Calculate available stretch after 10 seconds(sec. 10.3.6.1) and test at 85% of average fabric stretch1-5% Spandex: Measure Time Dependent Growth andRecovery using 30 minutes. Report both growth andrecovery. Pass or fail based on 7% max. growth.> 5% but <10% Spandex: Measure Time Dependent Growth and Recovery using 30 minutes. Report both growth andrecovery. Pass or fail based on 5% max. growth.> 10% Spandex: Measure Time Dependent Growth andRecovery using 30 seconds. Report both growth andrecovery. Pass or fail based on 85% min. recovery.(*) asterisk = ALL COLORWAYS MUST BE TESTEDMinimum Sample Size Necessary for Testing Initial Package = 3 yards X full width of darkestcolorwayAdditional Colorway = 1 yard X full width percolorwayAdditional samples may be needed for items that contain two care instructions, i.e. “For Best Results, Dry Clean”. Please consult with INTERTEK for the number of samples. Package Price (USD)USA ROWInitial Package $225 $165 Additional Colorways $100 $55 Optional Testing Section – Refer to Price ListIf Formaldehyde Spot Test is found positive, additional charges will incur for additional testing. Turn Around Time = 4 working daysCare Instructions with “line dry or flat dry” may require an additional dayRaised & Non Raised Silk / Silk BlendsKnit & WovenPROTOCOL REVISION DESCRIPTION OF CHANGE DATE REVISION/APPROVED BYApril 2007/ Ro Jain 105-1 1. Fabric Count = Added test method AATCC/ASTM TS-0022. Addition Colorway Package = Colorfastness to CrockingRequirements:Dry - Grade 3.5 minWet - Grade 2.5 min105-2 1. Appearance Retention (KAR) –Method has been changed toNov. 2008/Ro JainAATCC/ASTM TS-008 Modified.2.Yarn Size & Fabric Count – Changed requirement +/- 5% to ReportActual Data.3. Colorfastness to Accelerated Laundering/Dry Cleaning –Test hasbeen removed. Have been added to Appearance Retention.4. Colorfastness to Crocking - Test specimen to be tested originalstate and after 1 wash/dry cycle.5. Formaldehyde Content – Test to be used on all products.Changed requirement to:Children over 36 months & Adults = 75 ppm max.Infants under 36 months = 0.05 Absorbency max.6. Stretch Properties – Changed requirements. Refer to page 2 and 3of this protocol for requirements.105-3 1. Removed - No tolerance allowed for wool/wool blend products inNov.2009/Ro Jainthe fiber content test2. Combined Formaldehyde spot test and full test into one line &updated test method3. Pilling Resistance updated requirement to Grade 3.5 min/2 hrs4. Stretch Properties- modified load (4 lbs) / updated requirements105-4 1. Fiber ContentNov. 2011/Ro Jain Added the Fur labeling and Identification acts and wool #16 cfr3002. AnalyticalFormaldehyde-added Kohls TM-53. Updated Tide to “Ultra”。
FIRA 6250 2005
FIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .FIRA STANDARD 6250: 2005 SPECIFICATION: FURNITURE MATERIALS (INTERIOR)1.SCOPEFIRA Standard 6250: 2005 Specification - Furniture Materials (Interior) specifies requirements and guidance information for the performance of materials used in interior furniture, including domestic and contract furniture applications.The specification is sub-divided into the following sub sectionsClassification: Guidance on service useSECTION 2 TABLE 1Specification: Surface finishes.(e.g. paint, lacquer plastic foil, paper foil, plastics laminate)SECTION 3 TABLE 2Specification: Adhesive bonds.(e.g. edgings, surface materials).SECTION 3 TABLE 3Specification: Environmental response.(e.g. cabinet doors, worktops, panels).SECTION 3TABLE 4 TABLE 5 TABLE 6Specification: Workmanship.SECTION 3 TABLE 7Specification: Exposure to xenon arc lightSECTION 3 TABLE 8Specification: Gloss.SECTION 3 TABLE 9Specification: Timber Quality Guidance (Non-structural). SECTION 3 TABLE 10Marking SECTION 4 TABLE 11FIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .2. CLASSIFICATIONPerformance requirements for furniture materials are divided into three main classification ratings with the appropriate rating selected according to service use.Table 1 provides general guidance on the main service use applications and recommended classification rating. Classification ratings for different properties may be mixed according to requirements. For example an office desk will typically require a ‘severe’ rating for finish performance and ‘general’ rating for adhesion. (FIRA can advise on the appropriate test specification for a product).TABLE 1. GUIDANCE ON SERVICE USECLASSIFICATION RATINGCAREFULGENERALSEVEREDomestic e.g. delicate, occasional furniture.z z Domestic e.g. tables and cabinets: kitchen, office, bedroom, lounges, dining.z Office e.g. workstations, desks, filing cabinets, conference tables z z Educational e.g. school desks general furniture (not laboratory furniture)z z Institutional –retirement homesz z Hotel e.g. bedroom and lounge furniturez z Hospitals general office and patient furniture (not specialised) z z Military z Bar z Church z z Police station z Recreation room z Common room z Public hallz Student accommodation z 24hrs AirportszFIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .3.SPECIFICATION: FURNITURE MATERIALS (INTERIOR)TABLE 2: FIRA STANDARD 6250: SPECIFICATION- FINISH PERFORMANCEPROPERTYMINIMUM PERFORMANCE RATINGWORKING SURFACES (KITCHEN WORKTOPS)HORIZONTAL SURFACES (EXCLUDING KITCHEN WORKTOPS)OTHER SURFACESCarefulGeneralSevereCarefulGeneralSevereCarefulGeneralSevereBS 3962: Part 6 Resistance to mechanical damage crosscut 3 3 4 3 3 4 scrape, surface penetration 2 5 5 2 2 3 2 3 scrape, substrate penetration 3 5 5 3 3 4 3 4impact - - - 2 3 4 (1)2 3 3impact large ball-BS6222 1999 Part 3 450mm drop,10mm max,no cracking - - - - - - BS EN 12721: Resistance to wet heat (5)55°C4 5 5 3 3 4 2 3 3 70°C3 4 5 2 2 3 2 2 85°C3 4 5 2 2 2 2 BS EN 12722: Resistance to dry heat (5)85°C3 34 3 100°C2 23 3 120°C3 5 5 140°C2 4 4 160°C2 3 4 BS EN 12720: Resistance to marking by liquids (1 hour covered) acetone 3 5 5 3 3 ethanol 96% 4 5 5 3 3 3 2 3 ethanol 48% 5 5 5 3 4 4 3 4 tea 5 5 5 4 5 5 4 5 5 coffee 5 5 5 4 5 5 4 5 5disinfectant (phenol) 5 5 5 3 (2) 3 (2)disinfectant (chloro) 5 5 5 3 (2) 3 (2)paraffin oil 5 5 5 3 (2) 3 (2)blackcurrant juice 5 5 5 3 (2) 3 (2)ammonia solution (3) 5 5 5 3(2) 3 (2)acetic acid (4.4 % solution) 5 5 5 3 (2) 3 (2)olive oil 5 5 5 5 (2) 5 (2)BS EN 12720 / FIRA Procedure (4): Resistance to oils and fats (24 hours uncovered)Oils (solid vegetable oil) 5 5 5 4 5 4 (2) 4 (2)Fats (butter) 5 5 5 4 5 4 (2) 4 (2)BS EN 438 Resistance to wear ( revs) 150 350 350 Notes : (1)Rating 3 acceptable on solid wood substrate (2)Levels apply only to surfaces in kitchens. (3)This requirement may not be met when using ‘waterborne’ finishes. (4)Hard oil and fat placed and spread directly onto surface of panel. (5)Deviation: block aluminium content - minimum 95%, base roughness ra 0.8 – 1.6 μm. Flexible rating allowanceFIRA Standard 6250: 2007 - contains the following flexible allowance – “A maximum of two results may fall by one rating below the ratings specified, provided the result is not less than a rating of 2”.FIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .3.SPECIFICATION: FURNITURE MATERIALS (INTERIOR)TABLE 3: FIRA STANDARD 6250: SPECIFICATION- ADHESION PERFORMANCEMINIMUM PERFORMANCE FOR ADHESION OF SURFACE AND EDGING BONDSTEST CONDITIONSSURFACE BONDSEDGING BONDSRIGIDEDGING BONDS FLEXIBLEFIRA STANDARD 51CAREFUL GENERALSEVERE CAREFUL GENERALSEVERE CAREFUL GENERALSEVERE40°C 44444450°C 44444460°C 44443370°C4 (1)4 (1)4 (1)3 (1)FIRA STANDARD 48CAREFUL GENERALSEVERECAREFUL GENERALSEVERECAREFUL GENERALSEVERE85%RH 25°C44 3(1) Applicable only to worktop surface bonds in kitchens and edging bonds in high temperature environments.TABLE 4: FIRA STANDARD 6250: SPECIFICATION - ENVIRONMENTAL PERFORMANCECABINET DOORS (and other frame jointed components)TEST CONDITIONSFIRA STANDARD 48MAXIMUM PERMITTED FRAME JOINT MOVEMENT 1 (mm)CAREFULGENERAL SEVERE35%RH 25°C Dry0.5 0.5 65%RH 23°C Medium0.5 0.5 85%RH 25°CDamp 1.0 1.The frame joint movements specified are permitted, provided joint integrity is not adversely affected e.g. frame joints do not open, become loose or distorted.CONSTRUCTION:There shall be no obvious joint opening when the sample is viewed with normal or corrected vision at a distance of 400 ± 200mm. Minor hairline thickness cracks are permitted provided they do detract from the visual appearance of the sample.3.SPECIFICATION: FURNITURE MATERIALS (INTERIOR)TABLE 6: FIRA STANDARD 6250: SPECIFICATION - RATINGTEST CONDITIONSFIRA STANDARD 48 BOW PERFORMANCE REQUIREMENTCAREFULGENERAL SEVERE 35%RH 25°C Dry z z65%RH 23°C Medium z z85%RH 25°C Damp zBow measurements can be made using a standard 600 mm span bow meter*.(* A suitable instrument for carrying out bow measurements is described in BS 4965 1999. The instrument can also be purchased direct from FIRA).FIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .FIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .3.SPECIFICATION: FURNITURE MATERIALS (INTERIOR)TABLE 7: FIRA STANDARD 6250: SPECIFICATION – WORKMANSHIP. FIRA TEST METHOD 41.FREEDOM FROM SHARP EDGESFinished components shall not have any sharp edges or splinters that are likely to cause discomfort or injury to persons during normal use of the furniture.FREEDOM FROM VISUAL DEFECTS:When components are viewed with normal or corrected vision at 400 ± 200 mm there shall be no obvious defects that would detract from the appearance of the furniture.There shall be no obvious joint opening when the sample is viewed with normal or corrected vision at a distance of 400 ± 200mm. Minor hairline thickness cracks are permitted provided they do detract from the visual appearance of the sample.TABLE 8: FIRA STANDARD 6250: SPECIFICATION – COLOUR FASTNESS TO XENON ARC LIGHT.(Based on BS EN ISO 105-B02: 1999 – Textiles -Tests for colour fastness – Part B02 Colour fastness to artificial light: Xenon arc fading lamp test).The degree of colour change of exposed and non-exposed areas is compared with Standard Grey Scales (ISO 105 - AO2) where 1 = maximum colour change and 5 = no colour change.Maximum permitted colour change Natural materials e.g. solid wood, veneers.Synthetic materials e.g. PVC paper foilGrey Scale 3Grey Scale 4TABLE 9: FIRA STANDARD 6250: SPECIFICATION – GLOSS (BS EN ISO 9241 (Ergonomics of VDT’s) Part 5 (Postural requirements) 1999).TEST METHODGLOSSBSEN 13722:2004 Furniture Assessment of the surface gloss.(60° geometry method)Gloss levels of desk finishes shall be less than 45 gloss units at an angle of incidence of 60 degrees.Based on BS EN ISO 9241 Part 5:1999 Clause 5.4.4 Finish of the Work Surface specifies: ‘The finish of the work surfaces should not exceed silky matt (corresponding to 45 gloss units) to minimise specular reflections’.FIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .3.SPECIFICATION: FURNITURE MATERIALS (INTERIOR)TABLE 10: FIRA STANDARD 6250: SPECIFICATION – TIMBER QUALITY GUIDANCE*(NON-STRUCTURAL)Timber Source Preferably from a 'sustained yield' source. E.g. FSC managed source. Not a species listed under CITES. Moisture Content 9% ± 2 (at point of manufacture)Growth defectsTimber other than that intended for appearance effects e.g. ‘rustic’ or ‘hidden’ surfaces shall be free of major growth defects such as dead knots, knotholes, edge knots, ingrown bark, ring shake, tension wood, wane, juvenile wood pith, brittle heart, cross grain cracks.Live knots permitted but not to exceed 2 live knots (maximum diameter 6mm) in any visible area of the product.Sapwood Maximum of 10% sapwood, provided that it is free from fungal stain.GrainThe slope of the grain shall not exceed 1 in 8 when measured from the outside edge of components.Rot, mould and fungal stainNot permissible on visible surfaces.Wood boring insects ‘worm holes’No live wood boring insects are permissibleNo wormholes are permitted on exposed surfacesUp to 10 wormholes, less than 2mm, are permissible on hidden surfaces provided that they are filled.Termite damage not permissibleKiln Drying Defects Defects due to drying such as shakes, splits, cracks, honey combing and case hardening notpermissible.Minor non-structural surface splits and checks permissible on hidden surfaces.Distortion ≤ 0.5mm variation from the straight per 100mm on any width.≤ 3mm variation from the straight on any length*Note: The quality of timber varies significantly according to wood species, price and other processing and grading factors. It is therefore recommended timber specifications should fully discussed and agreed in detail with the timber supplier.4. MARKINGTABLE 11: FIRA STANDARD 6250: – MARKINGEach item for which conformity to this FIRA Standard is claimed the following information shall be made available to the purchaser, distributor or retailer.a) the name, trademark or other means of identification of either the manufacturer, distributor or retailerb) the number and date of this FIRA Standard, i.e. FIRA Standard 6250 :2005, together with the appropriate performancetest rating /classificationNote: Marking FIRA Standard 6250: 2005 on or in relation to a product represents a manufacturer's declaration of conformity that the product meets the relevant requirements of the standard. The accuracy of the claim is therefore solely the responsibility of the person making the claim. For such a declaration third party certification is desirableAmendments01/03/2005 Issue 2Revision of FIRA 6250 1999 Standard FIRA TM 01319/11/2007 Issue 3Flexible rating allowance text added - previously omitted in error from specification table.Colour fastness to light test changed from a ‘FIRA a test procedure’ to that based on – ‘BS EN ISO 105-B02: 1999 – Textiles -Tests for colour fastness – Part B02 Colour fastness to artificial light: Xenon arc fading lamp test’. Similar method but with improved temperature and humidity control.FIRA STANDARD 6250: 2005Authorised by: V Taylor Date: 19/11/2007 Issue 3 .。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。