新药Tipifarnib(替吡法尼)合成检索总结报告
新药Masitinib(马赛替尼)合成检索总结报告

新药Masitinib(马赛替尼)合成检索总结报告
一、Masitinib(马赛替尼)简介
Masitinib(马赛替尼)是一种特异性的Ckit抑制剂,主要用于治疗因为Ckit突变(gain of function)引起的胃肠道间质瘤。
但是因为其可以抑制肥大细胞的活性,现在也开始用于治疗哮喘,多发硬化症。
Masitinib(马赛替尼)分子结构式如下:
英文名称:Masitinib
中文名称:马赛替尼
本文主要对Masitinib(马赛替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Masitinib(马赛替尼)合成路线
三、Masitinib(马赛替尼)合成检索总结报告(一) Masitinib(马赛替尼)中间体3的合成
(二) Masitinib(马赛替尼)中间体5的合成方法一
(三) Masitinib(马赛替尼)中间体5的合成方法二
①Masitinib(马赛替尼)中间体6的合成
②Masitinib(马赛替尼)中间体5的合成
(四) Masitinib(马赛替尼)中间体8的合成。
上市新药厄达替尼(Erdafitinib)合成检索总结报告
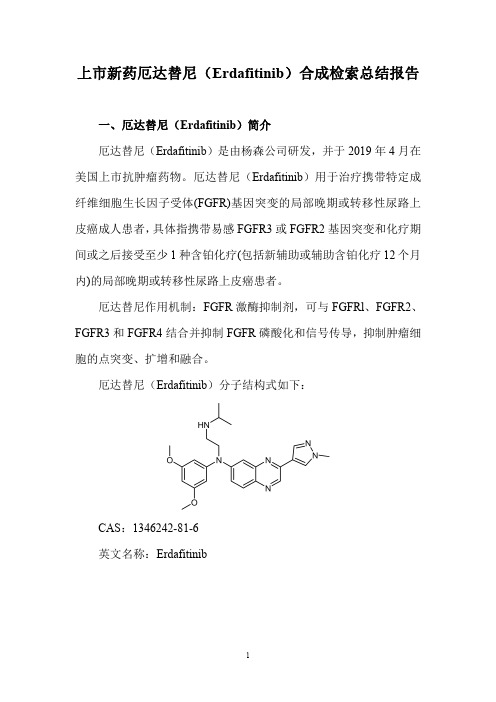
上市新药厄达替尼(Erdafitinib)合成检索总结报告一、厄达替尼(Erdafitinib)简介厄达替尼(Erdafitinib)是由杨森公司研发,并于2019年4月在美国上市抗肿瘤药物。
厄达替尼(Erdafitinib)用于治疗携带特定成纤维细胞生长因子受体(FGFR)基因突变的局部晚期或转移性尿路上皮癌成人患者,具体指携带易感FGFR3或FGFR2基因突变和化疗期间或之后接受至少1种含铂化疗(包括新辅助或辅助含铂化疗12个月内)的局部晚期或转移性尿路上皮癌患者。
厄达替尼作用机制:FGFR激酶抑制剂,可与FGFRl、FGFR2、FGFR3和FGFR4结合并抑制FGFR磷酸化和信号传导,抑制肿瘤细胞的点突变、扩增和融合。
厄达替尼(Erdafitinib)分子结构式如下:CAS:1346242-81-6英文名称:Erdafitinib二、厄达替尼(Erdafitinib)合成路线三、厄达替尼(Erdafitinib )合成检索总结报告(一)厄达替尼中间体2的合成序号实验步骤参考文献1To a suspension of 1,2-diaminobenzene 1(1equiv.)in ethanol (1mol/L)was added ethyl 2-oxoacetate (1.1equiv.).The mixture was stirred at reflux for 1h,then at room temperature overnight.The precipitated solid was filtered and washed with ethanol,then dried to give quinoxalinone 2.Carrer,Amandine;Brion,Jean-Daniel;Messaoudi,Samir;Alami,Mouad;Organic Letters ;vol.15;nb.21;(2013);p.5606-56092To a suspension of o-arylenediamine 1(4.0mmol,1.0equiv)and potassium carbonate (2.0equiv.)in ethanol (1mol/L)was added ethyl 2-oxoacetate (1.1equiv).The reaction mixture was stirred and heated at reflux in an oil bath for 12h,then at room temperature for 12h.Upon completion,the suspension was washed with ethanol,then filtered and dried to give quinoxalinone 2.Noikham,Medena;Kittikool,Tanakorn;Yotphan,Sirilata;Synthesis ;vol.50;nb.12;(2018);p.2337-2346Ethyl 2-oxoacetate (1.1equiv.)was added to a suspension of o -arylenediamine 1(4mmol,1equiv.)in ethanol (1mol/L).The reaction mixture was stirred andSumunnee,Ladawan;Pimpasri,Chaleena;Noikham,Medena;3heated at reflux in anoil bath for 1h,then at room temperature for 16h.Upon completion (as monitored byTLC),the precipitate was filtered and washed with ethanol,then dried to give quinoxalinone 2.Yotphan,Sirilata;Organic and Biomolecular Chemistry ;vol.16;nb.15;(2018);p.2697-27044To a stirred suspension of o-phenylenediamine (50g,462.9mmol)in ethanol(200ml),at rt was added a solution of ethyl glyoxalate in toluene (50;113ml_,555.48mmol)over a period of 45min.After heating to 45°C for 10h,the mixture was left at rt under stirring.The precipitate was filtered and the residue was washed with water and dried to give 1H-quinoxalin-2-one as an off-white powder (63g,93%).WO2011/26579;(2011);(A1)English(二)厄达替尼中间体3的合成序号实验步骤参考文献1To a solution of quinoxalin-2(lH)-one 2(54.64g,374mmol,1.0eq.)in HOAc (1000mL)was added a solution of Br 2(19.18mL,374mmol,1.0eq.)in HOAc (200mL)dropwise.The resulting mixture was stirred at rt for 12h,then poured into ice-water.The precipitate was collected by filtration and dried to afford 7-bromoquinoxalin-2(lH)-one 3as an off-white solid (74g,88%).NEUPHARMA,INC.;QIAN,Xiangping;ZHU,Yong-liang;WO2013/40515;(2013);(A1)2013/53384;(2013);(A1)English 2Quinoxalone 2(250g,1.7mol)is dissolved in acetic acid (4500mL).A mixture of acetic acid (988mL)and bromine (108mL,2.1mol)is added dropwise,and the mixture stirred at room temperature for 12hours,then heated to 60°C for 12hours.After cooling to room temperature,the reaction is filtered and the solid washed with water.The wet cake (500g)is then dissolved in 1500mL of methanol and heated to 60°C,then filtered and dried at 60°C to give 3in 85%yield CLAVIUS PHARMACEUTICALS,LLC;SAWYER,J.,Scott;(109pag.);WO2019/5241;(2019);(A1)English 3To a cooled 0°C solution of quinoxalinone 2(50g,342.2mmol)in acetic acid (800ml)was added in a dropwise manner a solution of bromine (32ml)in acetic acid (200ml_)over a period of 30min.Solids formed within the reaction upon addition of bromine,and the reaction was allowed to stir slowly for a further 90min.WO2016/97918;(2016);(A1)English。
新药Itacitinib(伊他替尼)合成检索总结报告

一、Itacitinib(伊他替尼)简介 Incyte 宣布一项重要的 3 期 GRAVITAS-301 研究结果,该研究评 估了 Itacitinib(伊他替尼)联合皮质类固醇在未接受过治疗的急性移 植物抗宿主病(GVHD)患者中的疗效。与安慰剂联合皮质类固醇相 比,Itacitinib(伊他替尼)联合皮质类固醇在第 28 天未达到改善总缓 解率(ORR)的主要终点(分别为 74.0%和 66.4%,p = 0.08)。在未 接受过治疗的急性 GVHD 患者中,将 Itacitinib(伊他替尼)联合皮 质类固醇中可改善总体缓解率。 Itacitinib(伊他替尼)分子结构式如下:
五、Itacitinib(伊他替尼)合成路线一检索总结报告 (一) Itacitinib(伊他替尼)中间体 2 的合成(路线一)
2
合成方法 操作方法
一
操作方法 二
操作方法 三
操作方法 olution of 3-hydroxyazetidine hydrochloride 1 (2.20 g) and triethylamine (4.0 mL) in MeOH (20 mL) at 0°C, di-tert-butyl dicarbonate (3.12 g) was added. After stirring at room temperature for 6 h, the solvent was evaporated. The residue was diluted with CH2Cl2, washed with water and the organic phase was evaporated to dryness to give tert-butyl 3-hydroxy-l-azetidinecarboxylate 2 (3.22 g, 93%) which was used without purification in the next step. To a solution of azetidin-3-ol hydrochloride 1 (45 g, 410.75 mmol, 1 eq) in MeOH (1.2 L) was added TEA (83.13 g, 821.51 mmol, 2 eq) and di-tert-butyl dicarbonate (89.65 g, 410.75 mmol, 1 eq). The mixture was stirred at 25°C for 16 hours. Then the reaction mixture was concentrated in vacuo. The residue was re-dissolved in EtOAc (1 L). The mixture was washed with H2O (3 ×500 mL) and brine (3× 500 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give the title compound 2 (65 g, 91 %) as a yellow oil, which was used directly in the next step. To a solution of azetidin-3-ol hydrochloride 1 (2.00 g, 18.3 mmcl) in CH2Cl2 (20 mL) was added TEA (5 mL) and (Boc)2O (4.80 g, 22.0 mmcl). The mixture was stirred at rt overnight. The reaction mixture was concentrated. The residue was dissolved in EtOAc (20 mL). The mixture was washed with water (20 mL×2) and brine (20 mL), dried over Na2SO4 and concentrated to give the title compound 2 (2.80 g, yield 88%) as yellow oil. To a stirred cold (0°C) solution of 3-hydroxyazetidine hydrochloride 1 (75 g, 0.68 mol) in ethanol (1300 mL) was added triethylamine (208g/280mL, 2.05mol) followed by Boc2O (164 g, 0.75 mol). The resultant solution was stirred at ambient temperature for 16 hours. GC/MS analysis of the reaction mixture revealed complete reaction. Volatiles were removed in vacuo and the residue was diluted with EtOAc (1300 mL) and washed with 10% citric acid (700 mL), water (700 mL) and brine (700 mL). The organics were dried over sodium sulfate filtered, and concentrated to give the desired product 2 (100.8 g, 85% yield). A mixture of 3-azetidinol hydrochloride 1 (10 g, 91 mmol), di-tert-butyl dicarbonate (18.8 g, 86.3 mmol) and sodium bicarbonate (15.3 g, 182 mmol) in dioxane:water (400 mL, 1:1) was stirre'd at room temperature for 15 hours. The organic portion was removed in vacuo and the aqueous portion was extracted with ethyl acetate three times. The
新药Fisogatinib(非索替尼)合成检索总结报告

新药Fisogatinib(非索替尼)合成检索总结报告一、Fisogatinib(非索替尼)简介Fisogatinib(非索替尼)是由Blueprint Medicines开发的一款在研的强效、高选择性成纤维细胞生长因子受体-4(FGFR4)抑制剂,用于治疗FGFR4驱动的晚期或转移性肝细胞癌HCC。
Fisogatinib(非索替尼)具有成为首个分子水平生物标记物驱动的HCC靶向药物的潜力。
Fisogatinib(非索替尼)分子结构式如下:英文名称:Fisogatinib中文名称:非索替尼本文主要对Fisogatinib(非索替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Fisogatinib(非索替尼)合成路线三、Fisogatinib (非索替尼)合成检索总结报告(一)Fisogatinib (非索替尼)中间体3的合成合成方法实验步骤参考文献操作方法一1(5.00g,20.5mmol)and 2(3.73g,20.5mmol)were dissolved in tetrahydrofuran (30ml),added with a solution of cesium carbonate (20.00g,61.5mmol)in water (30ml),and added with a catalytic amount of Pd(PPh 3)Cl 2.Theresulting mixture was heated to reflux for 4h under nitrogen atmosphere.The reaction solution was concentrated to dryness and extracted with ethyl acetate.The organic phase was washed once with saturated sodium chloride,dried over anhydrous sodium sulfate,and concentrated under reduced pressure.The resulting crude product was subjected tocolumn chromatography (200-300mesh silica gel,petroleum ether/ethyl acetate=10/1)to obtain intermediate 3(3.80g,yield of 62%)as a pale yellow solid.US2019/209564;(2019);(A1)English;CN110386921;(2019);(A).操作方法二A mixture of 6-bromo-2-chloroquinazoline (1)(5.0g,20.5mmol),3,5-dimethoxyphenylboronic acid (2)(3.7g,20.5mmol),Cs 2CO 3(20.0g,61.5mmol)and Pd(PPh 3)2Cl 2(1.4g,2.1mmol)in THF (50mL),dioxane (50mL)and water (10mL)was degassed with N 2three times,and stirred at 80°C for 3hours.An aliquot of the reaction mixture was analyzed by both TLC and LCMS,which indicated that the reaction had proceeded to completion.The mixture was cooled to room temperature,and extracted with EtOAc (3×200mL).The combined organic layers were washed with water and brine,dried over sodium sulfate,filtered and concentrated.The residue was purified by silica gel chromatography (petroleum ether/EtOAc =8:1)to obtain 2-chloro-6-(3,5-dimethoxyphenyl)quinazoline (3)as a light yellow solid (2.4g,38%).WO2014/11900;(2014);(A2)English;US2015/197519;(2015);(A1)English;US2017/9695165;(2017);(B2)English(二)Fisogatinib(非索替尼)中间体4的合成合成方法实验步骤参考文献操作方法一To a solution of2-chloro-6-(3,5-dimethoxyphenyl)quinazoline(3)(2.7g,8.9mmol)indry THF(80mL)wasadded dropwise sulfuryl chloride(3.0g,22.3mmol)at-20°C,and the reactionmixture was stirred for an additionalhour.An aliquot of the reaction mixture was analyzed byboth TLC and LCMS,which indicated that the reaction hadproceeded to completion.The reaction mixture wasquenched with water(1mL),and the solvents were removedunder reduced pressure.The precipitate was washed withCH3CN and dried to obtain2-chloro-6-(2,6-dichloro-3,5-dimethoxyphenyl)quinazoline(4)(2.6g,79%)as a whitesolid.WO2014/11900;(2014);(A2)English;US2015/197519;(2015);(A1)English;US2017/9695165;(2017);(B2)English操作方法二3(3.80g,12.6mmol)was dissolved in tetrahydrofuran(100ml),under nitrogen cooled to-20~30o C,was addeddropwise sulfuryl chloride(5.11g,37.9mmol),the resultingmixture at the same temperature reaction for2hours.Thereaction mixture was slowly raised to ambient temperature,was added acetonitrile(100ml),stirred for10minutes,theresulting solid was collected by filtration.Drying,to giveintermediate4(2.80g,60%yield)as a pale yellow solid.US2019/209564;(2019);(A1)English;CN110386921;(2019);(A).(三)Fisogatinib(非索替尼)中间体6的合成合成方法实验步骤参考文献操作方法一(3R,4R)-3-(((S)-1-phenylethyl)amino)tetrahydro-2H-pyran-4-ol5(2.0g,9.04mmol)was taken up in methanol(10ml)followed by addition of Et3N(1.260ml,9.04mmol)andBOC-anhydride(2.308ml,9.94mmol).The reaction mixturewas stirred at room temperature overnight.The solvents werethen removed in vacuo and the residue was taken up in DCM(10ml)and hexane(20ml)and heated to80°C.until thesolvent level was reduced by half.The reaction mixture wasremoved from heat and cooled to room temperature whilestirring.5ml of ether was then added and the reaction wasstirred at room temperature for2hours.The reaction mixtureUS2015/119405;(2015);(A1)English。
药物阿法替尼(Afatinib)合成检索总结报告

药物阿法替尼(Afatinib)合成检索总结报告
一、阿法替尼(Afatinib)简介
阿法替尼(Afatinib)是表皮生长因子受体(EGFR)和人表皮生长因子受体2(HER2)酪氨酸激酶的强效、不可逆的双重抑制剂。
阿法替尼(Afatinib)适应于具有表皮生长因子受体(EGFR)基因敏感突变的局部晚期或转移性非小细胞肺癌(NSCLC),既往未接受过EGFR酪氨酸激酶抑制剂(TKI)治疗;也适应于含铂化疗期间或化疗后疾病进展的局部晚期或转移性鳞状组织学类型的非小细胞肺癌。
阿法替尼(Afatinib)分子结构式如下:
英文名称:Afatinib
中文名称:阿法替尼
本文主要对阿法替尼(Afatinib)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、阿法替尼(Afatinib)合成路线
中间体7合成路线一:
中间体7合成路线二:
中间体7合成路线三:
中间体7合成路线四:
阿法替尼(Afatinib)16合成路线一:阿法替尼(Afatinib)16合成路线二:
阿法替尼(Afatinib)16合成路线三:
三、阿法替尼(Afatinib)合成检索总结报告(一) 阿法替尼(Afatinib)中间体2的合成
(二) 阿法替尼(Afatinib)中间体3的合成
(三) 阿法替尼(Afatinib)中间体5的合成。
新药Tepotinib(特泊替尼)合成检索总结报告

新药Tepotinib(特泊替尼)合成检索总结报告一、Tepotinib(特泊替尼)简介2019年09月12日,美国食品和药物管理局已授予其靶向抗癌药MET抑制剂Tepotinib(特泊替尼)突破性药物资格,用于治疗接受含铂化疗后病情进展、携带MET基因第14号外显子跳跃突变的转移性非小细胞肺癌患者。
2018年3月,Tepotinib(特泊替尼)被日本卫生劳动福利部授予了治疗携带MET基因第14号外显子跳跃突变的晚期NSCLC患者的SAKIGAKE资格(创新药物)。
Tepotinib(特泊替尼)分子结构式如下:英文名称:Tepotinib中文名称:特泊替尼本文主要对Tepotinib(特泊替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Tepotinib(特泊替尼)合成路线三、Tepotinib (特泊替尼)合成检索总结报告(一)Tepotinib (特泊替尼)中间体3的合成合成方法实验步骤参考文献操作方法一2.2L of a freshly prepared 1.5M sodium methoxide solution are added dropwise with stirring to a suspension of 259g (1.09mol)of 3-methoxycarbonylbenzamidinium acetate 1and 528g (1.08mol)of ({2-dimethylamino-1-[dimethyli-mmoniomethyl]vinylamino}methylene)dimethyl-ammonium dihexafluorophosphate 2(“a minoreductone precursor”,prepared in accordance with C.B.Dousson et al.,Synthesis 2005,1817)in 1L of methanol.The reaction mixture is then warmed to 60°C.over the course of 40min and held at this temperature for 30min.The reaction mixture is then cooled to room temperature,diluted with 10L of dichloromethane and washed three times with 5L of water each time.The organic phase is dried over sodium sulfate and evaporated.The residue is recrystallised from ethyl acetate:methyl 3-[5-(dimethylaminomethyleneamino)pyrimidin-2-yl]-benzo ate 3as beige crystals;m.p.146°2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1)English;US2011/269765;(2011);(A1)English;US2011/269756;(2011);(A1)English 操作方法二100g of 3-hydroxymethylbenzamidinium acetate 1(419.75mmol)and 204.93g of a minoreductone precursor 2(419.74mmol)are suspended in 1000ml of dried MeOH in an N 2-flushed 2L three-necked flask,and a freshly prepared solution of 28.99g of sodium in 300ml of MeOH is added dropwise with stirring,and the mixture is subsequently stirred at 60°C.for 30min,giving a clear solution.For work-up,the reaction batch is cooled,diluted with dichloromethane,washed 2×with water,dried over sodium sulfate and evaporated to dryness in a rotary evaporator.The residue 3is crystallised from a little methanol and diethyl 2010/311733;(2010);(A1)English(二)Tepotinib (特泊替尼)中间体4的合成合成方法实验步骤参考文献操作方法一160ml(2.88mol)of concentrated sulfuric acid are added to a suspension of 103.5g (364mmol)of methyl 3-[5-(di-methylaminomethyleneamino)-pyrimidin-2-yl]benzoate 3in 1.3L of water,and the mixture is heated at the boil for 4hours.The reaction mixture is cooled to room temperature,diluted with water and filtered with suction.The residue is washed with water and dried in vacuo:3-(5-hydroxypyri-midin-2-yl)benzoic acid as brownish crystals;m.p.293-295°2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1);US2011/269765;(2011);(A1);US2011/269756;(2011);(A1).操作方法二103.5g of methyl 3-[5-(dimethylaminomethylenamino)-pyrimidin-2-yl]benzoate 3(364.04mmol)are suspended in 1300ml of water in a 2l single-necked flask,and 160ml of conc.sulfuric acid (95-97%)(2.88mol)are subsequently added,and the reaction batch is warmed at 130°C.(oil-bath temperature)for 4h.For work-up,the reaction batch is cooled,and the precipitate formed is filtered off,washed with water and dried at 50°C.in a vacuum drying cabinet.Yield:78.9g (364.5mmol)of 3-(5-hydroxypyrimidin-2-yl)-benzoic acid 2010/311733;(2010);(A1)English(三)Tepotinib (特泊替尼)中间体5的合成合成方法实验步骤参考文献操作方法一32.7ml (445mmol)of thionyl chloride are added to a suspension of 88.0g (366mmol)of 3-(5-hydroxypyrimidin -2-yl)benzoic acid 4in 1.4l of methanol,and the mixture is heated at 80°C.for 2hours.20ml (276mmol)of thionyl chloride and,after 2hours,a further 10ml (138mmol)of thionyl chloride are then added.After each addition,the reaction mixture is stirred at 80°C.for 2hours.The reaction mixture is concentrated to a volume of about 300ml in vacuo.The resultant precipitate is filtered off and dried in vacuo:methyl 3-(5-hydroxypyrimidin-2-yl)benzoate 5as brownish crystals;m.p.219-223°C.US2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1)English;US2011/269765;(2011);(A1)English;US2011/269756;(2011);(A1).78.8g of 3-(5-hydroxypyrimidin-2-yl)benzoic acid 4are suspended in 1.4l of absolute methanol,and 32.7ml of。
靶向药物总结

靶向药物总结1、Axitinib,中文称号:阿西替尼;商品名:Inlyta;中文明学称号: N-甲基-2-( ( 3-( ( 1E) -2-( 吡啶-2-基) 乙烯) -1H-吲唑-6-基) 硫) 苯甲酰胺。
特点及其作用靶点:阿西替尼是由美国Pfizer 公司研发的一种多靶点酪氨酸激酶抑制剂,是一种口服的第2 代血管内皮生长因子受体抑制剂,选择性作用于VEGFR1,VEGFR2 和VEGFR3,经过抑制VEGF 介导的内皮细胞增殖和存活,起到抑制肿块生长和癌症停顿的作用。
于2021年1月27 日获美国FDA 同意上市。
常用剂量与用法:该药为片剂,引荐的剂量为每天 5 mg ,每天2 次,给药距离约12 h,如患者呕吐或漏失 1 次给药,不应添加服用,应按通常时间服用下一次剂量。
顺应症:适用于既往全身治疗失败后早期肾细胞癌的治疗。
主要反作用:最罕见的不良反响是腹泻、高血压、疲惫、食欲减低、恶心、发音阻碍、手掌-足底( 手-足) 综合征、体重减轻、呕吐、乏力和便秘等。
2、Regorafenib,中文称号:瑞格非尼。
特点及其作用靶点:Regorafenib 是一种触及正常细胞功用和病理进程中多种膜结合和细胞内激酶的小分子抑制剂,例如肿瘤发作,肿瘤血管生成和肿瘤微环境维持。
在体外生化或细胞剖析regorafenib或其主要的活性代谢物M-2和M-5抑制RET,VEGFR1,VEGFR2,VEGFR3,KIT,PDGFR-α,PDGFR- β,FGFR1,FGFR2,TIE2,DDR2,Trk2A,Eph2A,RAF-1,BRAF,BRAFV600E,SAPK2,PTK5,和Abl 的活性。
在体内大鼠肿瘤模型中regorafenib显示抗-血管生成活性,以及在一些小鼠移植瘤模型中对人类大肠癌有抑制肿瘤生长以及抗转移活性作用。
常用剂量与用法:1〕引荐剂量:160 mg口服,每天1次,2〕与食物服用。
顺应症:2021年9月27日,FDA同意了口服药物Regorafenib(瑞格非尼)〔Stivarga,拜耳〕治疗转移性结直肠癌,Regorafenib被同意时同时带有黑框正告,指出能够有严重或致命性的肝毒性。
新药Tucatinib(图卡替尼)合成检索总结报告

本文主要对 Tucatinib(图卡替尼)的合成路线、关键中间体的合 成方法及实验操作方法进行了文献检索并作出了总结。
二、Tucatinib(图卡替尼)合成路线
三、Tucatinib(图卡替尼)合成检索总结报告 (一) Tucatinib(图卡替尼)中间体 3 的合成
参文献
US2010/9958; (2010); (A1) English
Bioorganic and Medicinal Chemistry; vol. 19; nb. 6; (2011); p. 1987- 1998.
European Journal of Medicinal Chemistry; vol. 61; (2013); p. 132 -145. Bioorganic and Medicinal Chemistry Letters; vol. 25; nb. 22; (2015); p. 5147-5154.
新药 Tucatinib(图卡替尼)合成检索总结报告
一、Tucatinib(图卡替尼)简介 2020 年 4 月 17 日,美国食品药品监督管理局(FDA)批准 Tucatinib (图卡替尼)片剂与曲妥珠单抗和卡培他滨合用,用于晚期无法切除 (不能手术切除)或转移性 HER2 阳性乳腺癌治疗。 Tucatinib(图卡替尼)分子结构式如下:
(二) Tucatinib(图卡替尼)中间体 4 的合成
合成方法
操作方法 一
实验步骤
A solution of 6.0 g (27.5 mmol) of N'-(2-cyano-4-nitrophenyl)-N,N-dimethylformamidine 3, 33.9 g (41.8 mL, 412.4 mmol) of cyclohexene, and 0.6 g of 10% Pd/C in 360 mL of methanol was refluxed for 4 hrs. The hot mixture was filtered through Celite. Solvent was removed and the residue was recrystallized from chloroform-carbon tetrachloride giving 4.9 g (95%) of the title compound 4 as a light gray crystalline solid.
新药Ripretinib(瑞普替尼)合成检索总结报告

新药Ripretinib(瑞普替尼)合成检索总结报告
一、Ripretinib(瑞普替尼)简介
2019年8月13日,Deciphera宣布,其用于治疗四线及四线以上的胃肠间质瘤(GIST)的基因突变激酶抑制剂Ripretinib,在关键性的3期临床研究中获得了阳性结果。
这项名为INVICTUS的3期临床研究是一项随机、双盲和安慰剂对照的国际多中心研究,旨在评估ripretinib在晚期GIST患者中的安全性,耐受性和疗效。
对于药物的不良事件,Ripretinib的总体耐受性表现良好,发生贫血、腹痛和高血压事件的概率和安慰剂类似,分别为49%和44%。
Ripretinib(瑞普替尼)分子结构式如下:
英文名称:Ripretinib
中文名称:瑞普替尼
本文主要对Ripretinib(瑞普替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Ripretinib(瑞普替尼)合成路线
三、Ripretinib(瑞普替尼)合成检索总结报告(一) Ripretinib(瑞普替尼)中间体2的合成
(二) Ripretinib(瑞普替尼)中间体3的合成
(三) Ripretinib(瑞普替尼)中间体4的合成
(四) Ripretinib(瑞普替尼)中间体7的合成方法一
(五) Ripretinib(瑞普替尼)中间体7的合成方法二。
上市新药恩曲替尼(entrectinib)合成检索总结报告
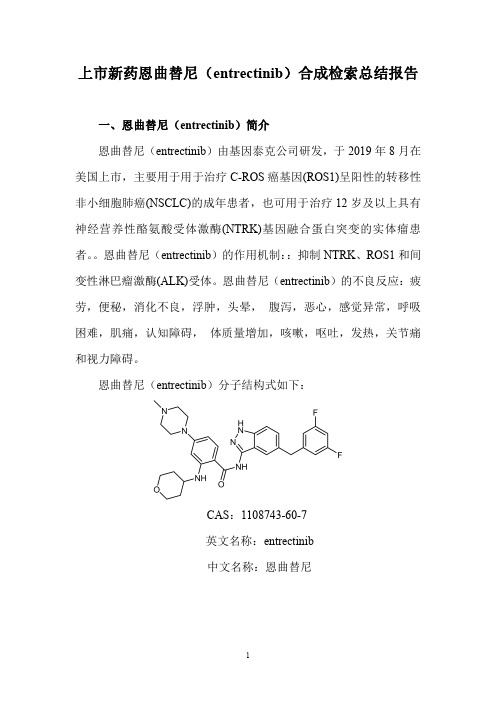
上市新药恩曲替尼(entrectinib)合成检索总结报告一、恩曲替尼(entrectinib)简介恩曲替尼(entrectinib)由基因泰克公司研发,于2019年8月在美国上市,主要用于用于治疗C-ROS癌基因(ROS1)呈阳性的转移性非小细胞肺癌(NSCLC)的成年患者,也可用于治疗12岁及以上具有神经营养性酪氨酸受体激酶(NTRK)基因融合蛋白突变的实体瘤患者。
恩曲替尼(entrectinib)的作用机制::抑制NTRK、ROS1和间变性淋巴瘤激酶(ALK)受体。
恩曲替尼(entrectinib)的不良反应:疲劳,便秘,消化不良,浮肿,头晕,腹泻,恶心,感觉异常,呼吸困难,肌痛,认知障碍,体质量增加,咳嗽,呕吐,发热,关节痛和视力障碍。
恩曲替尼(entrectinib)分子结构式如下:CAS:1108743-60-7英文名称:entrectinib中文名称:恩曲替尼二、恩曲替尼(entrectinib)合成路线三、恩曲替尼(entrectinib )合成检索总结报告(一)恩曲替尼(entrectinib )中间体2的合成序号实验步骤参考文献1With magnetic stirring,Toluene (50mL)was added to the 250mL three-necked flask of the pound 1(2.5g,15.15mmol),boronic acid (3.9g,16.65mmol)and K 3PO 4(6.4g,30.30mmol).Pd(PPh 3)4(385mg,0.33mmol)was added,vacuumed again and replaced with nitrogen three times.The temperature was raised to 110°C,and the reaction was stirred for 5hours with heat.After cooling to room temperature,the solid formed by adding ethyl acetate (100mL)was filtered.The filtrate is concentrated,The residue was passed through a silica gel column to give 2.7g of a white solidCN108623576;(2018);(A)Chinese。
药物Enasidenib(恩西地平)合成检索总结报告

药物Enasidenib(恩西地平)合成检索总结报告一、Enasidenib(恩西地平)简介Enasidenib(恩西地平)于2017年8月在美国上市,主要用于治疗急性白血病的成人患者。
Enasidenib(恩西地平)常见的不良反应有恶心、呕吐、腹泻、胆红素升高、食欲下降。
Enasidenib(恩西地平)分子结构式如下:CAS:1446502-11-9CAS:1650550-25-6英文名称:Enasidenib英文名称:Enasidenib mesylate中文名称:恩西地平中文名称:恩西地平甲磺酸盐本文主要对Enasidenib(恩西地平)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Enasidenib(恩西地平)合成路线三、Enasidenib (恩西地平)合成检索总结报告(一)Enasidenib (恩西地平)中间体2的合成方法一合成方法实验步骤参考文献操作方法一Add palladium(II)acetate (200mg,2w/w%)and 1,1'-bis(diphenylphosphino)ferrocene (dppf)(400mg,4w/w%)to a solution of 2-chloro-6-trifluoromethyl-pyridine 1(10.0g,55.1mmol)in methanol (30mL).To the orange solution add triethylamine (8.45mL,60.6mmol).Purge the mixture with nitrogen and then maintain under an atmosphere of carbon monoxide (40psig)at 60o C for 17h.Cool to ambient temperature and concentrate under a reduced pressure.Dissolve the solid in tert-butylmethyl ether (70mL).Filter the resulting slurry over silica gel (10g)and diatomaceous earth (10g).Concentrate the filtrate to afford the title compound 2as a light orange solid (10.8g,96%).WO2009/131814;(2009);(A2)English操作方法二To a solution of 2-chloro-6-trifluoromethyl-pyridine 1(2g,11.1mmol,1.0eq)in MeOH (20mL)was add Pd(OAc)2(124mg,0.05eq)and dppf (600mg,0.1eq)under an atmosphere of nitrogen.Et 3N (2.3mL,1.5eq)was then added to the resulting orange solution.The reaction solution was then stirred under an atmosphere of carbon monoxide (40psi)at 60°C.for 22hr.Once the reaction completed,the mixture was filtered and the filtrate was concentrated in high vacuum.The residue was purified by column chromato-graphy to afford the desired product 2.US2015/18328;(2015);(A1)English;WO2015/3640;(2015);(A1)English操作方法三Under the protection of nitrogen gas,to a solution of2-bromo-6-trifluoromethylpyridine 1(1.48g,6.55mmol)in methanol (50.0mL)were successively added palladium acetate (74.0mg,0.33mmol),1,1'-bis(diphenylphosphino)-ferrocene (363.0mg,0.655mmol)and triethylamine (0.92g,9.8mmol),and reacted at a temperature of 60°C for 18hours under carbon monoxide atmosphere (2atm).After the reaction was complete,the reaction solution was cooled to room temperature,and filtered.The filtrate was concentrated in vacuo,and the resulting residue was purified by column chromatography on silica gel to afford methyl6-trifluoromethyl-pyridine-2-carboxylate 2(0.9g,yield 67.0%).EP3330258;(2018);(A1)English操作方法四To a solution of 2-bromo-6-trifluoromethylpyridine 1(1.48g,6.55mmol)in methanol (50.0mL)were added in sequence palladium acetate (74.0mg,0.33mmol),1,1'-bis(diphenylphosphino)ferrocene (363.0mg,0.655mmol)and triethylamine (0.92g,9.1mmol)under the protection of nitrogen gas.The reaction solution reacted at 60°C.under a carbon monoxide atmosphere with 2atmospheric pressures for 18hours.After the reaction was completed,the reaction solution was cooled to roomtemperature and filtered.The filtrate was concentrated under vacuum and reduced pressure,and the resulting residue was purified by silica gel column chromatography to afford the title compound 2(0.9g,yield:67.0%).US2019/352281;(2019);(A1)English(二)Enasidenib (恩西地平)中间体2的合成方法二合成方法实验步骤参考文献操作方法一To a solution of 6-trifluoromethyl-pyridine-2-carboxylic acid 3(300g,1.57mol)in methanol (2.25L)is added SOCl 2(225g,1.88mol)dropwise at room temperature,whilemaintaining room temperature.After addition,the mixture is heated to reflux and stirred for two hours then concentrated to remove the solvent.The crude product is diluted with ethyl acetate and washed with saturated NaHCO 3solution.The organic layer is dried over anhydrous Na 2SO 4and concentrated to give 6-trifluoromethyl-pyridine-2-carboxylic acid methyl ester 2.WO2015/18060;(2015);(A1)English操作方法二Methanol is added to the reaction vessel under nitrogen atmosphere.6-trifluoromethyl-pyridine-2-carboxylic acid 3(150g,0.785mol)is added and dissolved at ambienttemperature.Acetyl chloride (67.78g,0.863mol)is added dropwise at a temperature below 45°C.The reaction mixture is maintained at 65-70°C for about 2-2.5h,and then concentrated at 35-45°C under vacuum and cooled to25-35°C.The mixture is diluted with ethyl acetate and rinsed with saturated NaHCO 3solution then rinsed with brinesolution.The mixture is concentrated at temp 35-45°C under vacuum and cooled to 25-35°C,then rinsed with n-heptane and concentrated at temp 35-45°C under vacuum,then degassed to obtain brown solid,which is rinsed with n-heptane and stirred for 10-15minute at 25-35°C.TheWO2016/53850;(2016);(A1)English;WO2017/66611;(2017);(A1)English。
药物Neratinib(来那替尼)合成检索总结报告

药物Neratinib(来那替尼)合成检索总结报告一、Neratinib(来那替尼)简介Neratinib(来那替尼)于2017年7月在美国上市,主要用于阳性乳腺癌成人患者的延长辅助治疗。
Neratinib(来那替尼)常见的不良反应有腹泻、恶心、腹痛、乏力、呕吐、皮疹等等。
Neratinib(来那替尼)分子结构式如下:CAS:698387-09-6CAS:915942-22-2英文名称:Neratinib英文名称:Neratinib Maleate中文名称:来那替尼中文名称:来那替尼马来酸盐本文主要对Neratinib(来那替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Neratinib(来那替尼)合成路线(中间体其他合成路线请参考总结报告)三、Neratinib (来那替尼)合成检索总结报告(一)Neratinib (来那替尼)中间体3的合成合成方法实验步骤参考文献操作方法一50mmolof 3-aminoacrylonitrile 2,50mmol solid base catalyst ZrO 2-Cr 2O 3and 50mL of tetrahydrofuran was added to the reaction flask,Stir at room temperature,a mixture of 55mmol of methyl 4-ethoxy-2-chloro-5-nitrobenzoate 1and 15mL of tetrahydrofuran was added dropwise to the above reaction flask,after the reaction was stirred at room temperature for 2h,Then reflux 2h,After cooling the catalyst was filtered,The catalyst can be reused after drying.The solvent was distilled off under reduced pressure,To the residue was added 400mL of dichloromethane,Wash with 50mL of distilled water three times,The combined organic layers,After drying over anhydrous sodium sulfate,the solvent was distilled off under reduced pressure,The compound of formula (3).Yield 87%.CN106905234;(2017);(A)Chinese (二)Neratinib (来那替尼)中间体4的合成合成方法实验步骤参考文献40mmol of compound 3,40mmol anhydrous potassium carbonate and 40mL DMF was added to the reaction flask,CN106905234;操作方法一The reaction was stirred at50~60o C for 4h,cool to room temperature,Add 60mL of water,Stirring 0.5h,The precipitated solid was filtered,The crude solid was recrystallized from ethanol,Drying under reduced pressure to obtain compound of formula (4).Yield 90%.(2017);(A)Chinese(三)Neratinib (来那替尼)中间体5的合成合成方法实验步骤参考文献操作方法一A mixtureof 20g (77mmol)of 4-hydroxy-7-ethoxy-6-nitroquinoline-3-carbonitrile 4was refluxed with 120mL of POCl 3under N 2for 2hours.The mixture was cooled to room temperature and excess POCl 3was removed in vacuo.The residue was then cooled in an ice bath and 500mL of CH 2Cl 2was slowly added to dissolve the residue.The resulting solution was added to a flask containing 250mL of ice-cold saturated K 2CO 3and stirred for 30minutes.The organic layer was extracted,washed,dried over MgSO 4and evaporated to give 18g (86%)of the title compound 5as an off-white solid mp 200-202°2018/208584;(2018);(A1)English 操作方法二A mixture of 3.45g (13mmol)of 7-Ethoxy-4-hydroxy-6-nitro-quinoline-3-carbonitrile 4,5.55g (26mmol)of phosphorous pentachloride,and 10ml of phosphorous oxychloride was refluxed for 3hours.The mixture was diluted with hexane and the solid was collected.The solid was dissolved in 500ml of ethyl acetate and washed with cold diluted sodium hydroxide solution.The solution was dried over magnesium sulfate and filtered through a pad of silica gel.The solvent was removed giving 2.1g of beige solid 5.EP1950201;(2008);(A1)English;EP1117659;(2003);(B1)English;EP973746;(2003);(B1)English(四)Neratinib (来那替尼)中间体6的合成合成方法实验步骤参考文献50mmol of compound of formula (5),100mL ethanol and 100mL distilled water was added to the reaction flask,The system temperature was raised to 50o C,Followed by adding。
新药Tucatinib(图卡替尼)合成检索总结报告

新药Tucatinib(图卡替尼)合成检索总结报告新药Tucatinib(图卡替尼)合成检索总结报告⼀、Tucatinib(图卡替尼)简介2020年4⽉17⽇,美国⾷品药品监督管理局(FDA)批准Tucatinib (图卡替尼)⽚剂与曲妥珠单抗和卡培他滨合⽤,⽤于晚期⽆法切除(不能⼿术切除)或转移性HER2阳性乳腺癌治疗。
Tucatinib(图卡替尼)分⼦结构式如下:英⽂名称:Tucatinib中⽂名称:图卡替尼本⽂主要对Tucatinib(图卡替尼)的合成路线、关键中间体的合成⽅法及实验操作⽅法进⾏了⽂献检索并作出了总结。
⼆、Tucatinib(图卡替尼)合成路线三、Tucatinib(图卡替尼)合成检索总结报告(⼀)Tucatinib(图卡替尼)中间体3的合成合成⽅法实验步骤参考⽂献操作⽅法⼀To a solution of5-nitroanthranilonitrile1(1.00g,6.13mmol)in dioxane(25mL)was added dimethylformamide dimethylacetal2(0.88g,7.36mmol).After stirred at100°C. for2h,the reaction mixture was cooled to room temperatureand refrigerated.The precipitate was filtered out,washedwith cold ether several times,and dried in vacuo to give1.30g(97%)of product(E)-N'-(2-cyano-4-nitrophenyl)-N,N-dimethylformamidine3as a yellow solid.US2010/9958;(2010);(A1)English操作⽅法⼆5-Nitroanthranilonitrile1(20.0g,122.5mmol)wassuspended in dimethylformamide dimethylacetal2(43mL,360.0mmol).The mixture was heated up to refluxtemperature for1.5h.The resulting mixture was cooled toroom temperature and refrigerated overnight.The yellowprecipitated was filtered,washed with ethyl ether to give3,25.0g(96%);mp153-154°C.Bioorganic andMedicinalChemistry;vol.19;nb.6;(2011);p.1987-1998.操作⽅法三5-Nitroanthranilonitrile1(5.00g,30.7mmol)was refluxedin20ml dimethylformamide dimethyl acetal2.After90min,the mixture was allowed to cool at room temperature.Thesolid was filtered off,washed with several portions of diethylether and dried to yield3as yellow crystals(6.50g,96.9%),mp153-155°C.EuropeanJournal ofMedicinalChemistry;vol.61;(2013);p.132-145.操作⽅法四A mixture of5-nitroanthranilonitrile(1)(1.90g,11.8mmol) and dimethylformamidedimethyl acetal2(8.0mL)was stirred at reflux for1.5h.The mixture was cooled to room temperature and refrigerated.The solid was filtered,washed with several portions of ether,and dried to yield90%(2.30g)of the titled compound3as yellow solid,mp:143-145°C.Bioorganic andMedicinalChemistryLetters;vol.25;nb.22;(2015);p.5147-5154.(⼆)Tucatinib(图卡替尼)中间体4的合成合成⽅法实验步骤参考⽂献操作⽅法⼀A solution of6.0g(27.5mmol)of N'-(2-cyano-4-nitro-phenyl)-N,N-dimethylformamidine3,33.9g(41.8mL,412.4mmol)of cyclohexene,and0.6g of10%Pd/C in360mL of methanol was refluxed for4hrs.The hot mixture was filtered through Celite.Solvent was removed and the residue was recrystallized from chloroform-carbon tetrachloride giving4.9g(95%)of the title compound4as a light gray crystalline solid.EP2014/1000039;(2004);(B1)English。
新药Sitravatinib(司曲替尼)合成检索总结报告

新药Sitravatinib(司曲替尼)合成检索总结报告
一、Sitravatinib(司曲替尼)简介
新药Sitravatinib(司曲替尼)可以靶向消除免疫抑制性的M2型巨噬细胞(M2 TAM)、调节性T细胞(Treg)、骨髓来源的抑制性细胞(MDSC),增加树突状细胞(DC)的抗原呈递能力,从而增强免疫系统的抗肿瘤效应,因此Sitravatinib(司曲替尼)联合PD-1单抗有可能克服免疫治疗耐药。
Sitravatinib(司曲替尼)分子结构式如下:
英文名称:Sitravatinib
中文名称:司曲替尼
本文主要对Sitravatinib(司曲替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Sitravatinib(司曲替尼)合成路线
三、Sitravatinib(司曲替尼)合成检索总结报告(一) Sitravatinib(司曲替尼)中间体3的合成
(二) Sitravatinib(司曲替尼)中间体4的合成
(三) Sitravatinib(司曲替尼)中间体6的合成。
上市新药Fedratinib(菲达替尼)合成检索总结报告

上市新药Fedratinib(菲达替尼)合成检索总结报告一、Fedratinib(菲达替尼)简介Fedratinib(菲达替尼)是由Impact公司研发,化学名为于2019年8月在美国上市,主要用于治疗患有中危-2或高危原发性或继发性(真性红细胞增多症或实质性血小板增多症)骨髓纤维化的成人患者。
Fedratinib(菲达替尼)作用机制是抑制野生型及突变激活的两面神激酶-2(JAK2)和FLT3活性。
Fedratinib(菲达替尼)不良反应是腹泻,恶心,贫血和呕吐。
Fedratinib(菲达替尼)分子结构式如下:CAS:936091-26-8英文名称:Fedratinib中文名称:菲达替尼化学名:N-叔丁基-3-[(5-甲基-2-[[4-(2-吡咯烷-1-基乙氧基)苯基]氨基]嘧啶-4-基)氨基]-苯磺酰胺二盐酸盐一水合物二、Fedratinib(菲达替尼)合成路线三、Fedratinib(菲达替尼)合成检索总结报告(一)Fedratinib(菲达替尼)中间体3的合成方法一序号实验步骤参考文献A mixture of2-chloro-5-methyl-pyrimidin-4-ylamine1(0.4g,2.8mmol),3-bromo-iV-te/t-butyl-benzenesul1fonamide 2(1.0g,3.4mmol),Pd2(dba)3(0.17g,0.19mmol),Xantphos (0.2g,3.5mmol)and cesium caibonate (2.0g,6.1mmol)was suspended in dioxane (25mL)and heated at reflux under the argon atmosphere for 3h.The reaction mixture was cooled to room temperature and diluted with DCM (30mL).The mixture was filtered and the filtrate concentrated in vacuo.The residue was dissolved in EtOAc and hexanes added until solid precipitated.After filtration,the title compound 3(1.2g,98%)was obtained as a light brown solid.It was used in the next step without purification.WO2007/53452;(2007);(A1)English WO2012/60847;(2012);(A1)English(二)Fedratinib (菲达替尼)中间体3的合成方法二序号实验步骤参考文献1Magnetic stirring at room temperature Compound 4(1.0g,6.13mmol)was added to a 50mL single-mouth bottle.And methanol/water mixture (15mL,1/1),Stir and dissolve,Compound 5(1.26g,5.52mmol)was added.The reaction mixture was warmed to 45°C under nitrogen and stirred to react overnight.Cool to room temperature,A large amount of white solid precipitated,filter,The filter cake was washed with methanol/water (3.4mL /4.0mL).Drain,Drying at 50°C under vacuum gave 1.31g of a white solid.The yield was 60.17%.CN109232440;(2019);(A)Chinese 2To a solution of 3-amino-N-(tert-butyl)benzene sulfonamide 5(3.4g,14.9mmol)and 2,4-dichloro-5-methyl-pyrimidine 4(608.0mg,3.7mmol)in dioxane (20mL)was added DIEA (955.0mg,7.4mmol).The reaction mixture was stirred at 120°C for 72hrs,cooled to rt,concentrated,and the residue was purified on silica gel with 20%-50%EA/petroleum ether)to afford N-(tert-butyl)-3-((2-chloro-5-methylpyrimidin-4-yl)amino)benzenesulfonamide 3(544.0mg,41%yield)as yellow solid.WO2017/201069;(2017);(A1)English。
新药Pemigatinib(培米加替尼)合成检索总结报告

新药Pemigatinib(培米加替尼)合成检索总结报告
一、Pemigatinib(培米加替尼)简介
2020年3月,Pemigatinib(培米加替尼)的2期关键性注册临床研究完成中国首例患者给药。
该项研究的目的是评估Pemigatinib(培米加替尼)在既往至少接受过一线系统治疗、成纤维细胞生长因子受体2(FGFR2)基因融合或重排的中国晚期胆管癌患者中的有效性和安全性。
2019年11月,美国食品药品监督管理局(FDA)正式受理Incyte递交的Pemigatinib(培米加替尼)用于治疗复发的FGFR2基因融合或重排的局部晚期胆管癌的NDA,并授予其优先审评资格。
根据美国处方药使用者费用法案,预计Pemigatinib在美国获批的日期为2020年5月30日。
Pemigatinib(培米加替尼)分子结构式如下:
英文名称:Pemigatinib
中文名称:培米加替尼
本文主要对Pemigatinib(培米加替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Pemigatinib(培米加替尼)合成路线
Pemigatinib(培米加替尼)中间体13其他合成方法
三、Pemigatinib(培米加替尼)合成检索总结报告(一) Pemigatinib(培米加替尼)中间体3的合成
(二) Pemigatinib(培米加替尼)中间体4的合成
(三) Pemigatinib(培米加替尼)中间体7的合成。
替吡法尼——精选推荐

替吡法尼详细信息
本文详细介绍了替吡法尼的产品信息,包括中英文名称、别名、cas号、分子结构等基本信息,以及产品的物化性质、产品用途、产品上下游产品等综合信息,为广大化学品研究、化工产品生产制造从业者提供专业的产品信息。
本文所有信息来源化工字典。
替吡法尼
/detail-替吡法尼.html
产品介绍:
中文名称:替吡法尼
中文别名: (R)-6-(氨基(4-氯苯基)(1-甲基-1H-咪唑-5-基)甲基)-4-(3-氯苯基)-1-甲基-2(1H)-喹啉酮
英文名称:Tipifarnib
英文别名:
CAS:192185-72-1
分子结构式:
EINECS:
分子式:C27H22Cl2N4O
分子量:489.3958
风险术语:
安全术语:
物化性质:
熔点:
相对密度:1.33g/cm3 溶解性:
用途:
上游原料:
下游产品:。
FDA授予替吡法尼用于治疗HRAS突变型HNSCC的突破性疗法认定

FDA授予替吡法尼用于治疗HRAS突变型HNSCC的突破性疗法认定FDA已授予试验性药物替吡法尼(tipifarnib)突破性疗法认定,用于治疗复发性或转移性HRAS突变型头颈部鳞状细胞癌(HNSCC)患者,这些患者在铂类化疗后出现疾病进展,之后其变异等位基因频率至少为20%,该药的研发商Kura Oncology宣布。
试验设计该认定是基于2期RUN-HN临床试验(NCT02383927)中报告的初步活性,该试验评估了替吡法尼对复发或转移性HRAS突变型HNSCC患者的疗效。
2020年6月,在美国临床肿瘤学会(ASCO)虚拟科学项目上公布了该试验的数据。
在这项试验中,所有患者在试验开始时都有按RECIST v1.1标准可测量的疾病。
受试者在28天治疗周期的第1天到第7天和第15天到第21天口服替吡法尼,起始剂量为600mg或900mg,每天两次,直到疾病进展(PD)或不可接受的毒性。
试验的主要终点是客观缓解率(ORR)。
关键次要终点包括安全性和耐受性。
此外,探索性终点为无进展生存期(PFS)和缓解持续时间(DOR)。
试验结果截至2019年9月30日,替吡法尼在18名可评估患者中显示的ORR为50%。
中位DOR为14.7个月。
替吡法尼的中位PFS为5.9个月,而上次疗法的中位PFS为2.8个月。
中位总生存期为15.4个月。
总结及其他目前,正在进行的国际性、多中心、开放标签、2队列、非比较性、关键2期AIM-HN 试验(NCT03719690)正在研究替吡法尼用于该适应症。
在这项试验中,研究人员将评估替吡法尼在HRAS突变型HNSCC患者中的ORR。
Kura Oncology公司总裁兼首席执行官Troy Wilson博士说:“我们非常高兴FDA能授予替吡法尼突破性疗法认定,我们很感激FDA对其治疗这一毁灭性疾病的潜力的肯定。
我们会继续专注于开展AIM-HN注册导向试验,并期待与FDA密切合作,尽快将这种疗法应用于患者。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
新药Tipifarnib(替吡法尼)合成检索总结报告
一、Tipifarnib(替吡法尼)简介
Tipifarnib(替吡法尼)适应症有:T细胞淋巴瘤,头部和颈部鳞状细胞癌,急性髓性白血病,非小细胞肺癌,骨髓增生异常综合征,子宫内膜间肉瘤,肾上腺复发性嗜铬细胞瘤等。
2020年3月3日FDA 已将Tipifarnib(替吡法尼)授予快速通道称号,用于治疗患有复发性或难治性血管免疫母细胞性T细胞淋巴瘤(AITL),滤泡性T细胞淋巴瘤(FTCL)和淋巴结转移的成年患者外周血T细胞淋巴瘤。
Tipifarnib(替吡法尼)分子结构式如下:
英文名称:Tipifarnib
中文名称:替吡法尼
本文主要对Tipifarnib(替吡法尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Tipifarnib(替吡法尼)合成路线一
三、Tipifarnib(替吡法尼)合成路线二
四、Tipifarnib(替吡法尼)合成路线一检索总结报告(一) Tipifarnib(替吡法尼)中间体2的合成(路线一)
(二) Tipifarnib(替吡法尼)中间体4的合成(路线一)
(三) Tipifarnib(替吡法尼)中间体5的合成(路线一)。