医疗器械认证中TCF文件
CE认证TCF技术文件编写技术服务

CE认证TCF技术文件编写技术服务
keywords:TCF技术文件编写,CE认证TCF文件编写
CE认证TCF技术文件
TCF文件是申请CE认证的制造商向CE认证机构提度交的一份重要文件,它是认证机构审核发证的重要依据。
编制TCF文件必须全部使知用英文。
欧盟法律要求,加贴了CE标签的产品投放到欧洲市场后,其技术文件(TechnicalFiles)必须存放于欧盟境内供监督机构随时检查。
技术文件中所包涵的内容若有变化,技术文件也应及时地更新。
技术文件通常应包括下列内容:
a.制造商(欧盟授权代表(欧盟授权代理)AR)名称,商号,地址
b.产品的型号,编号
c.产品使用说明书
d.安全设计文件(关键结构图,即能反映爬申距离、间隙、绝缘层数和厚度的设计图)
e.产品技术条件(或企业标准)
f.产品电原理图
g.产品线路图
h.关键元部件或原材料清单
i.测试报告((TestingReport)
j.欧盟授权认证机构NB出具的相关证书((2010年后欧盟规定CE认证证书必须是指定机构或者是能力机构出具)
k.产品在欧盟境内的注册证书(对于某些产品比如:ClassI医疗器械,普通IVD体外诊断医疗器械)等等......。
IEC60601-1第三版(中文版)

国际标准 IEC60601-1 医用电气设备—— 第一部分:安全通用要求和基本准则
MEDICAL ELECTRICAL EQUIPMENT – Part 1: General requirements for basic safety and essential pe.......................................................................................................................8 1.1 范围 ........................................................................................................................................8 1.2 目标 ........................................................................................................................................8 1.3 * 并列标准 ................................................................................................................................8 1.4 * 专用标准 ................................................................................................................................8
医疗器械进入欧盟医疗器械市场要求

入欧盟的相关知识一、欧盟国家(共31 家)1、 2004 年 5 月 1 日前:法国、德国、意大利、荷兰、比利时、卢森堡、丹麦、爱尔兰、英国、希腊、西班牙、葡萄牙、奥地利、芬兰、瑞典;2、 2004 年 5 月 1 日加入:波兰、匈牙利、捷克、斯洛伐克、斯洛文尼亚、爱沙尼亚、拉脱维亚、立陶宛、塞浦路斯、马耳他;3、欧洲自由贸易协会EFTA:挪威、列支敦士登、冰岛、瑞士;4、新入欧盟:罗马利亚、保加利亚;5、瑞士不要求产品携带CE标志。
二、医疗器械 CE认证的通用要求1、基本要求(总要求)(1)安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析);(2)风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统);(3)性能符合性(产品的基本要求);(4)器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。
);(5)器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。
2、基本要求的具体包括如下14 条:(1)器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高水平的防护办法。
(2)生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应:首先应尽可能降低甚至避免危险。
其次,对无法避免的危险采取适当的防护措施,包括安装报警装置;最后,告知用户所提供防护措施的弱点及其可能带来的危险。
(3)器械必须取得生产者期望获得的功能。
器械设计制造和包装应有利于第一条(2)(A )D 多规定的各项功能的发挥。
(4)在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3 款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。
(5)器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变。
(完整word版)MDD医疗器械指令

医疗器械指令(MDD)93/42/EEC 简介什么是医疗器械?“医疗器械”是指制造商预定用于人体以下目的的任何仪器、装置、器具、材料或其他物无论它们是单独使用还是组合使用,包括为其正常使用所需的软件:疾病的诊断、预防、监视、治疗或减轻;损伤或残障的诊断、监视、治疗、减轻或修补;解剖学或生理过程的探查,替换或变更;妊娠的控制医疗器械的评估等级:所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的相关指令。
医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。
它根据不同的要求共分为6个等级,供认证机构评估。
设计阶段生产阶段I级自我符合声明自我符合声明I级(测量功能)自我符合声明申报机构I级(灭菌)自我符合声明申报机构IIa级自我符合声明申报机构IIb级申报机构申报机构III级申报机构申报机构认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或EN/ISO 13485评审质量体系。
由于美国、加拿大和欧洲普遍以ISO 9001, EN 46001或ISO 13485作为质量保证体系的要求,故建议质量保证体系的建立均以这些标准为基础。
医疗器械的风险分析:EN1441失效模式及后果分析(FMEA);失效树分析(FTA);上市后的监控(客户投诉情况等);临床经验根据EN1441风险分析的一些例子;器械的预期用途;预期与病人和第三者的接触;有关在器械中所使用的材料/元件的风险;供给患者或来自患者的能量;在无菌条件下生产的器械;用于改变病人环境的器械;说明用器械;用于控制其它器械或药品或与其配合使用的器械;不需要的能量或物质的输出;易受环境影响的器械;带有重要消耗品或附件的器械;必要的日常维护和校正;含有软件的器械;货架寿命有限制的器械;延迟或长期使用可能造成的影响;普通风险;所有的适用项目必须论述包括可能的危险和降低风险的方法。
医疗器械产品怎样获CE认证

医疗器械产品怎样获CE认证CE认证是指符合欧洲市场标准的医疗器械产品的认证。
CE认证是一个强制性的程序,用于确保产品的安全性、卫生性和环境保护。
下面将详细介绍医疗器械产品获得CE认证的过程。
1. 确定适用的指令和标准:首先,根据医疗器械产品的用途和特性,确定适用的指令和标准。
常见的指令包括医疗器械指令(Medical Device Directive,MDD)和体外诊断医疗器械指令(In Vitro Diagnostic Medical Device Directive,IVDD)等。
2.风险评估和分类:根据指令和标准的要求,对医疗器械产品进行风险评估和分类。
风险评估是确定产品的潜在危害和安全性能的过程。
分类是根据产品的危害程度对其进行分级,并确定适用的附录。
3. 设计和开发过程:医疗器械产品的设计和开发过程必须符合相关的标准和技术规范。
其中,关键的要素包括:设计和验证验证(Design Verification and Validation),产品规范质量计划(Quality System Planning),标准操作程序(Standard Operating Procedures,SOP),风险管理计划(Risk Management Plan)等。
4.制造和检验:医疗器械产品的制造过程必须符合相关的标准和要求。
制造过程的文件化、追溯和质量控制措施是获得CE认证的关键。
此外,对产品的检验和测试也是必须的,以确保其符合相关的标准和技术规范。
5.技术文件:医疗器械产品的认证过程中,需要提供相应的技术文件。
技术文件应包括产品的设计、开发、制造和检验等方面的相关信息,以证明产品符合相关的指令和标准。
6.申请认证:在准备好技术文件后,可以向认证机构提交申请。
认证机构会对技术文件进行评估和审查,并对产品进行必要的测试和验证。
如果认证机构确认产品符合相关的指令和标准,会颁发CE认证证书。
7.CE标志:获得CE认证后,在产品上可以使用CE标志,表明产品符合欧洲市场标准。
CE FCC CCC认证标准

CE 认证基本知识1 CE 标记CE-MARKING 即 CE 标签,贴在产品显著位置,以表明产品符合欧共体相关标准.具 有 CE-MARKING 的产品无须进一步检验便可在欧盟成员国销售.产品上的 CE 标记意味着 厂商宣布了该产品已达到相应指令的基本要求, 同时, 也意味着该产品可以在欧盟地区合法 销售. 无 CE 标签的产品,不准进入欧盟市场.这是欧盟关于市场准入的强制性措施.CE 标 签符号如下:2 CE 认证具有市场准入和抗风险两重性CE 认证具有市场准入性: CE 标记认证是产品进入欧盟国家及欧盟自由贸易协会国家市场的通行证. 为各国产品在 欧洲市场进行贸易提供了统一的最低技术标准,简化了贸易程序. 欧盟委员会强制性地规定: 何国家的产品要进入欧洲市场,都必须是已获得 CE 认证的产品.没有进行 CE 认证的 产品是不得进入欧盟市场的. CE 认证具有抗风险两重性: CE 认证是对产品质量最有效的保证,贴有 CE 标记的产品将减除或降低产品在欧洲市 场上的将要出现的风险. 这些风险包括: 1.被市场监督机构查核不符合基本安全与卫生要求的风险; 2.被消费者协会或该类保护团体指控的风险; 3.出于竞争而被同行指控的风险等产品的生产商必须对这些风险承担法律责任和赔偿 责任. 欧洲法律规定: 对于被查证核实的擅自行销者,处罚从重.向由欧盟成员国批准的认证机构申请进行 CE 标记认证,可以最大程度地获取市场监督机构的信任,并能有效地预防那些不负责任的 指控情况的出现,在面临诉讼的情况下,CE 认证标志将成为有力的技术证据.3 CE 标记认证须完善的文件出口商在出口产品之前需准备完整的技术构成档案文件(TCF) ,其中应包含如下文件: · 检验报告 · 符合声明(Declaration of Conformity) · 相关证件复印件 · 描述产品的技术性文件 · 工厂的质量保证文件 · 由指定机构确认声明(COC--Confirmation of Confomity)CE 标记的适用(国家)范围奥地利,比利时,丹麦,英国,芬兰,法国,德国,希腊,意大利,爱尔兰,荷兰,葡 萄牙,西班牙,瑞典,卢森堡,冰岛,列支敦士登,挪威等十八国.4 有关产品 CE 标记认证的新方法指令针对不同种类的产品,CE 目前包括二十五条技术指令.它们分别为: 简单压力容器,玩具,建筑产品,电磁兼容,机械,个人防护设备,非自动称量仪器, 可移植医疗器械,普通医疗器械,燃气炉具,无线电及电信终端设备,锅炉,民用爆破器材, 低电压,通讯卫星地面站,升降设备,使用爆炸性气体的设备,娱乐用船只,非简单压力容 器,载人的索道装置,体外诊断医疗器械,航海设备,由液体或气体燃料燃烧的新型热水锅 炉,跨欧公共高速列车系统,家用电冰箱或电冷柜及组合节能性能.5 欧盟委员会颁布的 CE 标记指令类别基本指令: 它适用于所有产品的制造商,涉及的范围包括贸易,强制性责任和其他许多问题.真正 理解这些指令的含义是十分重要的,特别是对于那些产品已经被强制执行这些指令的制造 商.例如:CE 认证,符合性评估,通用产品安全和产品的义务,这些指令的适用范围非常 广,有些会涉及大部分的产品和供应商,如通用产品安全指令和符合性评估程序及 CE 认证 准则等等. 通用指令: 涉及的则是某一专业范围内的产品,如针对低电压产品的 LVD 指令及针对会产生电磁 干扰的产品的 EMC 指令.专业产品指令:专门适用于一些特定的产品,如电信设备和医疗 设备等危险性较高的产品. CE 证书如下:CCC 认证CCC 认证的概念 产品质量认证— 是由经认可的产品认证机构,依据规定的认证程序和特定产品的认证 标准(产品技术标准和质量体系标准)对认证产品进行检测,并对其生产企业质量体系进行 审核,确认合格后发给认证证书和认证标志的过程. 强制性产品认证— 国家对关系到人类健康和安全,环境保护和公共安全的产品实施强 制认证制度,国家定期发布《强制认证产品目录》 ,在"目录"中的产品只有通过强制性认证, 才可生产,进口,销售和使用. 我国原有强制性认证包括国家质量技术监督局,中国电工委员会实施的"长城"标志认证 和国家出入境检验检疫局实施的"CCIB"标志认证.随着我国加入 WTO,各项与贸易有关的 活动(包括产品认证)必须在 WTO 的规则下进行,以适应 WTO 的要求.2001 年经国务院 决定把原国家质量技术监督局与国家出入境检验检疫局合并, 组建国家质量技术监督检验检 疫总局.并成立国家认证认可委员会统一负责国家强制性产品认证制度的管理和实施工作, 对于国家实施强制性认证的产品, 由国家认监委公布统一的产品目录, 确定统一的产品技术 标准和认证实施程序,制定统一的认证标志,规定统一的收费标准.中国强制认证 CCC 标志— 根据我国加入 WTO 的承诺和体现国民待遇的原则,国家对强制性产品认 证使用统一标志.新的国家强制性认证称为"中国强制认证",英文名称为"China Compulsory Certification" 英文缩写"CCC".中国强制认证实施后,取代原来的"长城 CCEE"和商检的 "CCIB"标志.目前的"CCC"认证标志分为四类,分别为: 1,CCC+S 2,CCC+EMC 3,CCC+S&E 4,CCC+F 安全认证标志 电磁兼容类认证标志 安全与电磁兼容认证标志 消防认证标志上述四类标志每类都有大小五种规格. 认证规则和程序 1 适用范围 1)CNCA-01C-014:电动工具; 2)CNCA-01C-016:家用和类似用途设备; 3)CNCA-01C-017:音视频设备; 4)CNCA-01C-018:声音和电视信号的电缆分配系统设备与部件(EMC) ; 5)CNCA-01C-019:卫星电视广播接收机(EMC) ; 6)CNCA-01C-020:信息技术设备; 7)CNCA-01C-021:金融及贸易结算电子设备(EMC) ; 8)CNCA-01C-022:照明电器; 9)CNCA-07C-031:电信终端设备2 引用文件 1) 《强制性产品认证管理规定》 ; 2) 《强制性产品认证标志管理办法》 ; 3) 《第一批实施强制性产品认证的产品目录》 ; 4) 《电气电子产品强制性认证实施规则》-电动工具 (CNCA-01C-014:2001); 5) 《电气电子产品强制性认证实施规则》-家用和类似用途设备(CNCA-01C-016:2001); 6) 《电气电子产品强制性认证实施规则》-音视频设备 (CNCA-01C-017:2001); 7) 《电气电子产品强制性认证实施规则》-音视频设备-声音和电视信号的电缆分配系统 设备与部件(电磁兼容) (CNCA-01C-018:2001); 8) 《电气电子产品强制性认证实施规则》-音视频设备-卫星电视广播接收机 (电磁兼容)(CNCA-01C-019:2001); 9) 《电气电子产品强制性认证实施规则》-信息技术设备(CNCA-01C-020:2001); 10) 《电气电子产品强制性认证实施规则》-信息技术设备-金融及贸易结算电子设备 (电磁兼容) (CNCA-01C-021:2001); 11) 《电气电子产品强制性认证实施规则》-照明电器 (CNCA-01C-022:2001); 12) 《电信设备强制性认证实施规则》-电信终端设备 (CNCA-07C-031:2001).3 认证的基本环节 3.1 认证申请和受理 3.2 型式试验 3.3 初始工厂审查 3.4 认证结果评价和批准 3.5 获证后的监督FCC 认证介绍FCC(Federal Communications Commission,美国联邦通信委员会) 1934 年由 COMMUNICATIONACT 建立是美国政府的一个独立机构, 于 直接对国会负责.FCC 通过控制无线电广播,电视,电信,卫星和电缆来协调 国内和国际的通信.涉及美国 50 多个州,哥伦比亚以及美国所属地区,为确保 与生命财产有关的无线电和电线通信产品的安全性,FCC 的工程技术部 (Office of Engineering and Technology)负责委员会的技术 支持,同时负责设备认可方面的事务.许多无线电应用产品,通讯产品和数字产 品要进入美国市场,都要求 FCC 的认可.FCC 委员会调查和研究产品安全性的 各个阶段以找出解决问题的最好方法,同时 FCC 也包括无线电装置,航空器的 检测等等. 根据美国联邦通讯法规相关部分(CFR 47 部分)中规定,凡进入美国的电子 类产品都需要进行电磁兼容认证(一些有关条款特别规定的产品除外),其中比 较常见的认证方式有三种:Certification,DoC,Verification. 这三种产品的认证方式和程序有较大的差异,不同的产品可选择的认证方式在FCC 中有相关的规定.其认证的严格程度递减.针对这三种认证,FCC 委员会对各试验室也有相关的要求. 目前, 美国已连续几年成为我国第二大贸易伙伴, 中美贸易额呈逐年上升趋势, 因此对美出口不容小觑.美国的产品技术标准,进口法规的严谨堪称世界第一, 了解美国市场准入规则将会帮助我国产品进一步打开美国市场. 联邦通讯委员会(FCC)----管理进口和使用无线电频率装置,包括电脑, 传真机,电子装置,无线电接收和传输设备,无线电遥控玩具,电话,个人电脑 以及其他可能伤害人身安全的产品.这些产品如果想出口到美国,必须通过由政府授权的实验室根据 FCC 技术标准来进行的检测和批准.进口商和海关代理人 要申报每个无线电频率装置符合 FCC 标准,即 FCC 许可证.FCC 认证流程1.符合性声明:产品负责方(制造商或进口商)将产品在 FCC 指定的合格检测机构对产品进行检测,做出检测报告,若产品符合 FCC 标准,则在产品上加贴 相应标签,在用户使用手册中声明有关符合 FCC 标准规定,并保留检测报告以 备 FCC 索要.2.申请 ID,先申请一个 FRN,用来填写其它的表格.如果申请人是第一次申请 FCC ID,就需要申请一个永久性的 Grantee Code.在等待 FCC 批准分发给申请人 Grantee Code 的同时,申请人应抓紧时间将设备进行检测.待准 备好所有 FCC 要求提交的材料并且检测报告已经完成时, FCC 应该已经批准了Grantee Code.申请人用这个 Code,检测报告和要求的材料在网上完成 FCC Form 731 和 Form 159.FCC 收到 Form 159 和汇款后,就开始受理认证的申请.FCC 受理 ID 申请的平均时间为 60 天.受理结束时,FCC 会将 FCC ID 的 Original Grant 寄给申请人.申请人拿到证书后就可以 出售或出口相应产品了.FCC 认证样机1.每个申请认证型号至少提供一台合格样机.(推荐二台或二台以上) 2.提供的样机必须保证是正式合格样机, 其内部电器结构和外观必须和 以后出口的批量样机一致 3.样机上的商标型号必须清晰可靠办理 FCC 认证需提交的资料申请认证产品的生产厂商和申请认证方的全称和详细的联系 通信地址.将提供给用户的认证产品的安装和使用手册的副本. (如该产 品还没有用户手册,则可提供相关内容的草稿副本)产品电气原理图及工作原理说明. (如产品有接地或天线,应 加以描述)有关产品的工作振荡频率表,表中应列出信号的传播路径和 相应振荡频率.其它一些需要说明的产品特点.备注说明 1.相关的文件资料需为中英文两种. 2.为缩短认证周期,提供的资料最好为电子文档形式. 3.在认证过程中,针对一些特殊情况,可能需要企业补交其它额 外相关资料FCC 认证收费标准1. Certification(认可验证) 大多数用于一般无线电产品申请方面. 必须由 FCC 委员会人员审查 实验报告,经核准后发给认可证书. 2. Declaration of Conformity (DoC) 这类申请产品主要针对于 IT 产品和周边辅助设备. 不需 FCC 委员会 人员审查测试报告,厂商可使用自我认证的方式. 自我认证测试报告 必须由经 NVLAP 标准认可实验室发出. FCC Class A 的产品可延用 自我认证方法,申请程序不需经由 FCC 委员会审核.认证方式 Certification Doc 约 25,000 约 17,000 四周 三周备注 该报价含资料 审查, 测试, 报 告和证书费用FCCCFR47 Part15 Part18。
医疗器械技术文档(TCF)内容

医疗器械技术文档(TCF)内容什么是医疗器械·医疗器械是指任何仪器、设备、器具、材料或者其它物品, 不论是单独使用还是组合使用的,包括使用所需软件在内, 由制造者为下列用于人类的预期用途(目的) ,这些目的是:1.疾病的诊断、预防、监护、治疗或者缓解;2.损伤或残疾的诊断、监护、治疗、缓解或者补偿;3.解剖或生理过程的研究、替代或者补偿; · 妊辰控制;4.其作用于人体体表及体内的主要设计作用不是用药理学、免疫学或代谢的手段获得,但可能有些手段参与并起一定辅助作用。
注:西方医学认可的理论,不可以是中医的理论5.所有的预期用途都要有理论及临床证据(奥咨达医疗器械咨询)医疗器械不良事件这个概念对市民来说可能还比较陌生,病床出现问题导致病人受伤了、植入性医疗器械发生问题、使用药贴过敏之外,治疗仪时皮肤灼伤、打针时针头断裂、骨折手术装的接骨板断在体内,这都属于医疗器械不良事件。
这些现象有多方面的原因,如产品的设计缺陷、医护人员过失、病患自身使用不当等都会引起不良事件。
(只专注于医疗器械领域)CE认证是欧盟的产品安全认证,所有进入欧盟市场的医疗器械都必须进行医疗器械CE认证,医疗器械需要满足的CE指令有《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、《医疗器械指令》(MDD,93/42/EEC)和体外诊断器械指令(IVDD, 98/79/EC)。
医疗器械技术文档(TCF)内容技术档案(TCF文档)是欧盟医疗器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和证明,供主管机关抽查,或发生诉讼纠纷时使用。
医疗器械指令93/42/EEC要求"技术档案"可能包含下列项目:(奥咨达医疗器械咨询)A、企业的质量手册和程序文件;B、企业简介及欧洲代理名称、联系方式;C、CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的证明材料);1、产品名称、分类及引用标准条款的简要描述2、产品概述(包括类型和预期用途)a) 产品的历史沿革b) 技术性能参数c) 产品配合使用的附件、配合件和其它设备清单d) 产品的图示与样品e) 产品所用原材料及供应商3、使用该产品的调和标准/或其它标准4、风险分析评估结论和预防措施(EN1441 产品服务危险分析报告)5、生产质量控制a) 产品资料和控制文档(包括产品生产工艺流程图)b) 产品的灭菌方法和确认的描述c) 灭菌验证d) 产品质量控制措施e) 产品稳定性和效期的描述6、包装和标识a) 包装材料说明b) 标签c) 使用说明书7、技术评价a) 产品检验报告及相关文献b) 技术概要及权威观点8、潜在风险评价a) 产品潜在风险测试报告及相关文献b) 潜在风险的概要及权威观点9、临床评价a) 产品临床测试报告及相关文献b) 临床使用概述及权威观点。
医疗产品TCF技术文件编写技术服务

医疗产品TCF技术文件编写技术服务
TCF文件是医疗器械向欧盟市场做CE认证,需要提供的一套技术文件。
是欧盟医疗器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和证明,供主管机关抽查,或发生诉讼纠纷时使用。
各欧盟指令对于" 技术文件" 的要求有所差别,在这里谨以中国出口企业最常用的“医疗器械”的要求为例,加以说明。
主要内容如下:
1 、产品名称、分类及引用标准条款的简要描述
2 、产品概述(包括类型和预期用途)
3 、使用该产品的调和标准/ 或其它标准
4 、风险分析评估结论和预防措施(EN1441 产品服务危险分析报告)
5 、生产质量控制
产品资料和控制文档(包括产品生产工艺流程图)
产品的灭菌方法和确认的描述
灭菌验证
产品质量控制措施
产品稳定性和效期的描述
6 、包装和标识
7 、技术评价
8 、潜在风险评价
产品潜在风险测试报告及相关文献
潜在风险的概要及权威观点
9 、临床评价
产品临床测试报告及相关文献
临床使用概述及权威观点
附1 、产品出厂检测报告
附2 、产品型式检测报告
附3 、基本要求检查表。
医用吊塔如何取得CE认证?

医用吊塔如何取得欧盟CE认证文章来源:/cechangjianwenti/xinwenzixun/16.html产品要顺利通过CE认证,需要做好三方面的工作。
其一,收集与认证产品有关的欧盟技术法规和欧盟(EN)标准,通过消化、吸收、纳入企业产品标准。
其二,企业严格按照以上产品标准组织生产,也就是把上述技术法规和EN标准的要求,贯彻到企业产品的设计开发和生产制造的全过程。
第三,企业必须按照IS O9000+ISO13485标准建和维护质量体系,并取得ISO9000+ISO13485认证。
医用吊塔CE认证应遵循的欧盟技术法规和EN标准对于目前欧盟已发布的18类工业产品指令,从这些指令的结构看,它们可分为垂直指令和水平指令。
垂直指令是以具体产品为对象,如医疗器械指令;水平指令适用于各种产品系列,如电磁兼容性指令,它适用于全部电器及电子零部件产品。
对于医用吊塔,适用的指令有第十四项、第一项和第五项,即:93/42/EEC医疗器械指令、2006/95EEC低电压(LVD)指令和2004/108/EEC电磁兼容性(EMC)指令。
支持这些指令的欧盟标准是:(1)EN60601-1医用电气设备第一部分:安全通用要求;(2)EN60601-1-1医用电气设备第一部分:安全通用要求及第一号修正;(3)EN60601-2-11医用电气设备第二部分:(4)EN60601-1-2医用电气设备第一部分:安全通用要求1.2节并行标准电磁兼容性——要求和测试。
其中第(1)、(2)、(3)项标准是医用吊塔低电压(LVD)测试的依据:第(4)项标准是医用吊塔电磁兼容性(EMC)测试的依据。
医用吊塔CE认证程序、内容欧盟把医疗器械产品分为四类,即:第Ⅰ类、第Ⅱa类、第Ⅱb类、第Ⅲ类。
第Ⅰ类产品要加贴CE标志,可采取自行宣告的方式。
即厂商编制产品的技术文件档案,同时自行按有关EN标准对产品进行测试或委托有能力的试验室进行测试合格。
第Ⅱa类、第Ⅱb类、第Ⅲ类产品要加贴CE标志,则必须由欧盟指定的验证机构验证。
MDD指令测验(范卷)

欧盟医疗器械指令MDD试卷姓名:岗位:得分:一、是非题:(对“√”错“×”)每题3分共30分1.2007/47/EC 是对93/42/EEC 的第5 次修订。
(√)2.含有致癌、导致基因突变及毒性的可能性物质,必须遵循67/548/EC。
(√)3.说明书应有版本及发行日期。
(√)4.对孕妇、哺乳期女性、婴幼儿等产品应明确使用产品的风险。
(√)5.2007/47/EC软件被明确定义为有源医疗器械,必须证明其有效性。
(√)6.注册责任人的信息,需要保密。
(×)7.医疗产品分类,将根据主要作用方式来确定,而不是预期用途。
(√)8.所有器械都将需要临床数据,包括Ⅰ类产品。
(√)9.临床评价可以做对比的方法,不一定做临床。
(√)10.一个设备停用或移走,被另一个相同的设备所替代,将影响分类。
(√)二、选择题:每题5分,共20分。
1..在英国,违反CE标记的惩罚为:(G )A.扣留,罚没 B.5000英镑的罚款C.3个月的监禁 D.通报欧盟-产品消失E.撤出市场或回收所有在用产品 F.追究刑事责任 G.以上的一种和数种并罚2.2007年9月欧盟正式发布对93/42/EEC更新指令2007/47/EC (D )A.这份新指令是自1993年起,对医疗器械指令的一个重大的修改B.2010年3月21日为最终执行期C.所有已经申请CE或即将申请认证的客户所提供的技术文件必须完全符合新指令要求。
D.以上全部3.2007/47/ec医疗器械指令( D )A.2007/47/ec 是新的医疗器械指令。
B.2007/47/ec中仅仅包含3个指令中被修订部分的局部内容,而不是3个指令的完整内容。
C.2007/47/ec 并非新的医疗器械指令。
D. B+C4.2007/47/EC中明确规定记录保存时间不得小于产品使用周期( D )A.自生产之日起五年以备检查B.对植入设备产品生产之日起15年C.拿到证书后,必须到销售国注册,包括Ⅰ类产品D.以上全部三、填充:每空格3分,共30分1.MDD 93/42/EEC共包括23个条款和12附件。
欧洲不同分类医疗器械认证要求

欧洲不同分类医疗器械认证要求欧洲分为不同的类别来认证医疗器械,根据欧洲医疗器械法规(Medical Device Regulation, MDR)的规定,医疗器械的分类主要根据其风险级别和使用目的来确定。
根据MDR的分类规定,医疗器械被分为四个不同的类别:1. 一类医疗器械(Class I Medical Device): 这类器械具有最低的风险级别,包括非主动性医疗器械、不侵入性医疗器械和低风险侵入性医疗器械。
这类器械通常不需要进行独立的认证,但需要符合一定的欧盟标准和规定。
2. 二类医疗器械(Class IIa/IIb Medical Device): 这类器械具有中等风险级别,包括一些活动性的医疗器械、侵入性医疗器械和医疗诊断装置等。
这类器械需要进行CE认证,以证明其符合欧盟的安全和性能要求。
3. 三类医疗器械(Class III Medical Device): 这类器械具有高风险级别,包括一些植入性医疗器械和高风险诊断装置等。
这类器械需要进行CE认证,以证明其符合欧盟的安全和性能要求,并且可能需要进行更严格的审查和评估。
4. 特殊类医疗器械(Special Class Medical Device): 这类器械通常用于管理或治疗罕见的疾病,具有特殊的技术特征和高度的风险。
这类器械需要进行CE 认证,以证明其符合欧盟的安全和性能要求,同时还需要获得欧盟委员会的特别许可。
除了以上的分类认证要求外,欧洲还对医疗器械的生产过程和质量管理系统有着严格的要求,生产商需要遵守ISO 13485质量管理系统的规定,并且需要依据欧洲的要求进行产品的临床评估和市场监测。
需要注意的是,以上的分类和认证要求仅为一般性说明,具体的认证要求可能根据不同的医疗器械种类和规格有所不同。
生产商在进行医疗器械认证前应仔细研究和了解相关的欧洲法规,并根据具体情况进行相应的操作。
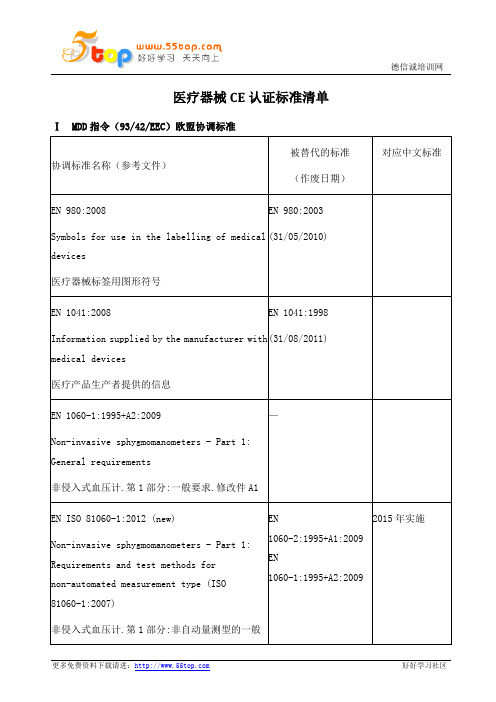
医疗器械CE认证标准清单
EN ISO 14971:2009
YY/T0316- 2008, IDT
EN ISO23747:2009
Anaesthetic andrespiratoryequipment — Peakexpiratory flow metersfor the assessment ofpulmonary function inSpontaneouslybreathing humans
实验于人体的医疗器械临床研究---有效的临床实践
EN ISO 14155-1:2009&EN ISO 14155-2:2009
(03/04/2012)
EN ISO 14971:2012(new)
Medical devices - Application of risk management to medical devices (ISO 14971:2007, Corrected version 2007-10-1)
医疗电气设备.第1-8部分:对基本安全和主要性能的一般要求.附属标准:对医疗电器设备及医疗电器系统报警系统的测试和指导的一般要求
EN 60601-1-8:2004
and its amendment
(01/06/2012)
EN 60601-1-11:2010
Medical electrical equipment -- Part 1-11: General requirements for basic safety and essential performance - Collateral standard: Requirements for medical electrical equipment and medical electrical systems used in the home healthcare environment
UL,GS认证

CE\EMC\GS\UL认证简介CE"标志,进入欧盟的护照在欧盟市场,"CE"标志属强制性认证标志,不论是欧盟内部企业生产的产品,还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴"CE"标志,以表明产品符合欧盟《技术协调与标准化新方法》指令的基本要求。
这是欧盟法律对产品提出的一种强制性要求。
对于欧盟通过的技术标准接轨指令的产品必须佩带欧盟CE标志.至今,欧盟市场已执行以下技术标准接轨指令:(1)简单加压容器(欧盟404/87指令);(2)玩具安全(欧盟378/88指令);(3)建筑产品(欧盟106/89指令);(4)兼容家用电子产品(欧盟336/89指令);(5)机械安全(欧盟392/89指令);(6)个人保护设备(欧盟686/89指令);(7)非自动衡器(欧盟384/90指令);(8)煤气用具(欧盟396/90指令);(9)拆卸式医疗器械(对欧盟385/90指令的补充);(10)活动式机械和升降设备(对欧盟392/89指令的修改);(11)通讯终端设备(欧盟263/91指令);(12)节能锅炉(欧盟42/92指令)加贴"CE"标志的相关要求"CE"标志无处不在,由新方法指令所涉及的所有产品在投放市场前都必须加贴"CE"标志。
所有从其他国家进口的使用过的产品及所有经过重大修改的产品(可视为新产品)上市前都要求加贴"CE"标志;"CE"标志必须加贴在显要位置上;"CE"标志最低高度不得少于5mm,如果缩小或扩大应按比例进行;"CE"标志取代各成员国的符合标志,它表明产品符合欧洲指令(取代所有国家法规)的唯一标志。
如何获得"CE"标志1989年欧盟理事会批准的关于《全球合格评定方法》的决定(以下简称《全球方法》)是对1985年《新方法》决议作的补充,旨在表明可用更多的方法证明产品符合指令的基本要求,即如果制造商选择了其他生产准则,可通过合格评定形式证明产品符合指令的基本要求,但必须由第三方进行测试或认证。
医疗器械各国认证要求

医疗器械各国认证要求医疗器械标准这些医疗器械标准已被许多国家广泛采用。
医疗器械指令列表欧盟指令中涵括许多基本保健及安全规定,以及评估产品符合规定程度的订定程序.每项指令在区域性标准制订机构所订定的调和欧洲标准中均会具体说明详细的基本规定。
因此,相关产品的制造商、进口商及配销商都必须明显标示,产品完全符合每项指令在「基本规定」所列出的保健及安全规定。
形成「新方法」基础的指令,包含了广泛的产品类别(横向指令)或某种特定的产品类别(纵向指令)。
指令标题欧盟所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的相关指令。
医疗器械需要满足的指令有:《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、《医疗器械指令》(MDD,93/42/EEC)体外诊断器械指令(IVDD, 98/79/EC)。
医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备.它根据不同的要求共分为6个等级,供认证机构评估.认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或EN/ISO 13485评审质量体系.由于美国、加拿大和欧洲普遍以ISO 9001,EN 46001或ISO 13485作为质量保证体系的要求,故建议质量保证体系的建立均以这些标准为基础。
体外诊断医疗器械指令(IVDD),IVDD的要求与MDD相似,可按以下分类申请:—北美在美国,食品和药物管理局(FDA)是监督和管理获准向消费者进行销售的食物,药物,化妆品和医疗器械的法定机构.器械及放射线健康中心(CDRH),作为FDA的一个分支,专管医疗器械。
其对医疗器械按不同等级进行不同程度的监管(医疗器械分为I级,II级或III级,I 级作为低风险范畴,而III级属高风险范畴):在加拿大方面,加拿大医疗器械认证认可机构(CMDCAS)要求医疗器械厂商提前获得经CMDCAS认可的第三方机构,如UL的质量体系审查,证明其质量系统符合CMDCAS的ISO13485/ISO13488标准.对CMDCAS认证的了解对于完成FDA 的质量系统注册(QSR)非常有帮助,因为如上所述的QSR是以ISO 9001和ISO 13485标准为基础的。
医疗器械认证流程

医疗器械认证流程医疗器械认证包括.1、产品安全认证.2、质量管理体系认证医疗器械怎样取得“安全认证标志”.以下以取得CE认证为例说明:.产品要顺利通过产品要顺利通过产品要顺利通过产品要顺利通过CE认证认证认证认证,需要做好三方面的工作需要做好三方面的工作需要做好三方面的工作需要做好三方面的工作。
其一,收集与认证产品有关的欧盟技术法规和欧盟(EN)标准,通过消化、吸收、纳入企业产品标准。
其二,企业严格按照以上产品标准组织生产,也就是把上述技术法规和EN标准的要求,贯彻到企业产品的设计开发和生产制造的全过程。
第三,企业必须按照ISO9000+ISO13485标准建和维护质量体系,并取得ISO9000+ISO13485认证。
CE认证应遵循的欧盟技术法规和认证应遵循的欧盟技术法规和认证应遵循的欧盟技术法规和认证应遵循的欧盟技术法规和EN标准标准标准标准。
对于目前欧盟已发布的18类工业产品指令,从这些指令的结构看,它们可分为垂直指令和水平指令。
垂直指令是以具体产品为对象,如医疗器械指令;水平指令适用于各种产品系列,如电磁兼容性指令,它适用于全部电器及电子零部件产品。
对于医疗器械,适用的指令有第十四项、第一项和第五项,即:93/42/EEC医疗器械指令、73/23/EEC低电压(LVD)指令和89/336/EEC电磁兼容性(EMC)指令。
支持这些指令支持这些指令支持这些指令支持这些指令的欧盟标准是的欧盟标准是的欧盟标准是的欧盟标准是:.(1)EN60601-1医用电气设备第一部分:安全通用要求;.(2)EN60601-1-1医用电气设备第一部分:安全通用要求及第一号修正;.(3)EN60601-2-11医用电气设备第二部分:γ射束治疗设备安全专用要求;.(4)EN60601-1-2医用电气设备第一部分:安全通用要求1.2节并行标准电磁兼容性——要求和测试。
其中第(1)、(2)、(3)项标准是医疗器械低电压(LVD)测试的依据:第(4)项标准是医疗器械电磁兼容性(EMC)测试的依据。
医疗器械欧盟认证质量手册mdr法规要求

医疗器械欧盟认证质量手册mdr法规要求在医疗器械行业中,欧盟认证质量手册是一份非常重要的文件,它是根据欧洲联盟医疗器械监管法规(Medical Device Regulation, MDR)而编制的。
该手册是用来确保医疗器械制造商在设计、生产和销售医疗器械时符合欧盟的质量要求和法规。
根据MDR法规要求,医疗器械制造商需要遵循以下几个方面的要求来编制质量手册:1. 组织和管理体系:制造商需要建立一个有效的组织和管理体系,以确保医疗器械质量的持续改进和符合法规要求。
这包括确定质量目标、管理风险、建立合适的流程和程序等。
2. 设计控制:医疗器械的设计必须符合欧盟的法规要求,包括安全性、性能和可用性等方面。
制造商需要制定适当的设计控制程序,包括设计验证、设计验证和可行性研究等。
3. 生产控制:医疗器械的生产必须符合欧盟的法规要求,包括工艺控制、材料控制、设备校准等。
制造商需要建立适当的生产控制程序,确保生产的医疗器械符合质量标准。
4. 产品标识和追溯:制造商需要确保医疗器械能够进行有效的标识和追溯。
这包括正确标注产品信息,如产品批次、序列号等,并建立适当的追溯系统,以便在需要时能够追踪产品的来源和使用情况。
5. 风险管理:制造商需要对医疗器械的风险进行评估和管理。
这包括确定和评估可能存在的风险,并采取措施来降低风险。
制造商还需要建立适当的风险管理计划和记录。
6. 校准和验证:医疗器械的校准和验证非常重要,以确保其性能和准确性。
制造商需要制定适当的校准和验证程序,并记录校准和验证的结果。
医疗器械欧盟认证质量手册mdr法规要求着眼于保证医疗器械在欧盟市场的质量和安全性,制造商需要严格遵守这些要求,以获得认证。
这不仅有助于提高医疗器械的质量水平,还能够增加消费者的信任度,并进一步扩大市场份额。
因此,制造商应该充分了解这些要求,并在生产过程中严格执行它们,以确保其产品符合欧盟质量标准。
医疗器械认证中TCF文件

医疗器械认证中TCF文件包括那些内容?TCF文件包括七个方面的内容:①简介;②产品的规格叙述;③设计之主要档案内容;④风险分析及评估;⑤测试报告及临床诊断资料;⑥文件设计的管制;⑦产品申请的声明宣言。
技術文檔(TCF)內容「技術文檔」是歐盟醫療器械指令中很重要的一個事項,它的目的是要求企業準備充份的技術資料和證明,供主管機關抽查,或發生訴訟糾紛時使用。
各歐盟指令對於"技術檔案"的要求有所差別,在這裡謹以中國出口企業最常用的「醫療器械」的要求為例,加以說明。
1、企業的質量手冊和程序文件;(奥咨达医疗器械咨询)2、企業簡介及歐洲代理名稱、聯繫方式;3、CE符合性聲明(或稱自我保證聲明,若該產品是和其它設備聯合運用,則應有整體符合基本要求的證明材料);4、產品名稱、分類及引用標準條款的簡要描述5、產品概述(包括類型和預期用途)a) 產品的歷史沿革b) 技術性能參數c) 產品配合使用的附件、配合件和其它設備清單d) 產品的圖標與樣品e) 產品所用原材料及供貨商6、使用該產品的調和標準/或其它標準(只专注于医疗器械领域)7、風險分析評估結論和預防措施(ISO14971/2007 產品服務危險分析報告)8、生產質量控制a) 產品數據和控制文文件(包括產品生產工藝流程圖)b) 產品的滅菌方法和確認的描述c) 滅菌驗證d) 產品質量控制措施e) 產品穩定性和效期的描述9、包裝和標識a) 包裝材料說明b) 標籤c) 使用說明書10、技術評價a) 產品檢驗報告及相關文獻(奥咨达医疗器械咨询)b) 技術概要及權威觀點11、潛在風險評價a) 產品潛在風險測試報告及相關文獻b) 潛在風險的概要及權威觀點12、臨床評價a) 產品臨床測試報告及相關文獻b) 臨床使用概述及權威觀點。
不同风险等级医疗器械CE认证MDR法规合规路径区别

不同风险等级医疗器械CE认证MDR法规合规路径区别经常遇到这种情况,客户报一个产品名称,立刻就期望能够知道他的产品的咨询费用和认证费用。
而几乎所有的咨询机构和认证机构,在没有看到客户详细的产品说明书之前,是不会给出报价的。
因为市面上有几千种医疗器械,不同的医疗器械、相同的器械不同的交付状态其对TD文件的要求、对应的认证模式都是相去甚远的,只有在获取准确的产品信息,进行准确的产品风险类别定义后,我们才可能给出一个大致的报价区间。
那MDR对不同风险等级的医疗器械的认证路径是如何区别的呢?1:Class Is/Im/Ir devices:要注意,Class Is/Im/Ir devices虽然是一类的器械,但是控制的方式是参考IIa的等级来进行控制的,需要提供完整的TD技术文件供认证机构审核,也需要对生产现场进行审核,Is 现场主要审核灭菌的控制,Im 现场审核主要是关于计量管理这一块。
2:Class IIa devices:Class IIa devices需要提供完整的TD技术文件供认证机构审核,也需要对生产现场进行审核。
但是IIa的TD的文件审核公告机构可以进行抽样,同一个Basic UDI 下的产品只需抽一个的TD进行审核。
3:Class IIb Annex VIII Rule 12 devicesRule 12 - Active devices intended to administer and/or remove medicinal products, body liquids or other substances to or from the body旨在将药品、体液或其他物质输送到体内或从体内取出的有源装置,IIb: 是以一种危险的方式实现上述目的的有源装置,比如说:输液泵呼吸机麻醉机麻醉蒸发器透析设备用于心肺机的血泵高压氧舱医用气体压力调节器医用气体混合器如果使用呼吸回路中的水分交换器用于无意识或非自主呼吸患者用于输送氧气的氧气浓缩器(将富氧空气直接送至患者)对于以上情况,有两种路径,一是全面质量体系审核+TD文件审核。
GScertification

产品认证说明:
• 贵公司产品按照欧盟MDD 93/42 EEC指令中医疗器械分类属于Class IIa产品,该产品在CE认证过程中,可以按照附件中Class IIa产 品的认证形式来进行。认证要求见说明中有关Class IIa产品认证要求。 • 产品检测需要按照报价表中所列标准来进行检测,检测必须由已经通过ISO17025认可的实验室进行。检测报告出具英文检测报告。 同时要出具灭菌有效性的说明,以及生物生物相容性的检测。企业目前如有检测报告,可先向ITC机构提供检测报告内容,以便确认检 测项目是否符合EN要求,以尽量减少企业产品检测费用。 • 企业如具备制作CE认证TCF(英文或欧盟通用语言编写)文件能力,报价单中的技术指导费可以不发生,在企业具备自己进行TCF文 件制作中,ITC亚洲会给企业相关技术指导,尽可能减少企业认证费用。所需技术文件内容见获得CE/MDD认证书必备技术文件列表。 • 企业如已经具备ISO13485全质量体系认证,ITC机构仅对已有体系进行审查,无需另行ISO13485体系认证,如企业为方便年审同 时进行体系和产品的年度工厂审查,可更换ITC认证机构颁发的ISO13485证书,费用如报价表中所列。 • 特别要指出的是贵公司产品在认证过程中,欧盟要求必需要首先具备ISO13485体系,同时在认证当中需要进行事后年度审查,以确 保产品在证书有效期内产品质量能够得到保证。 • 以上费用包括: 申请费、技术文件评审费,以及认证书费用和现场审查费。不包含认证机构审核员的差旅费用以及产品邮寄实验室 的寄送费用。 • 审核时间可根据客户要求而定,在企业认为达到可以进行工厂审查条件情况下,请客户提前两周通知ITC 。 • 在该项目开始前企业需向ITC ASIA支付20% 定金及30%合同款,另50%付款在相应的证书签发前在向ITC ASIA付讫。 • 认证时间取决于企业产品基本情况和TCF制作质量,ITC认证机构会在文件审核合格后15-20天左右发给企业有效认证证书正本。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
医疗器械认证中TCF文件包括那些内容?
TCF文件包括七个方面的内容:
①简介;
②产品的规格叙述;
③设计之主要档案内容;
④风险分析及评估;
⑤测试报告及临床诊断资料;
⑥文件设计的管制;
⑦产品申请的声明宣言。
技術文檔(TCF)內容
「技術文檔」是歐盟醫療器械指令中很重要的一個事項,它的目的是要求企業準備充份的技術資料和證明,供主管機關抽查,或發生訴訟糾紛時使用。
各歐盟指令對於"技術檔案"的要求有所差別,在這裡謹以中國出口企業最常用的「醫療器械」的要求為例,加以說明。
1、企業的質量手冊和程序文件;(奥咨达医疗器械咨询)
2、企業簡介及歐洲代理名稱、聯繫方式;
3、CE符合性聲明(或稱自我保證聲明,若該產品是和其它設備聯合運用,則應有整體符合基本要求的證明材料);
4、產品名稱、分類及引用標準條款的簡要描述
5、產品概述(包括類型和預期用途)
a) 產品的歷史沿革
b) 技術性能參數
c) 產品配合使用的附件、配合件和其它設備清單
d) 產品的圖標與樣品
e) 產品所用原材料及供貨商
6、使用該產品的調和標準/或其它標準(只专注于医疗器械领域)
7、風險分析評估結論和預防措施(ISO14971/2007 產品服務危險分析報告)
8、生產質量控制
a) 產品數據和控制文文件(包括產品生產工藝流程圖)
b) 產品的滅菌方法和確認的描述
c) 滅菌驗證
d) 產品質量控制措施
e) 產品穩定性和效期的描述
9、包裝和標識
a) 包裝材料說明
b) 標籤
c) 使用說明書
10、技術評價
a) 產品檢驗報告及相關文獻(奥咨达医疗器械咨询)
b) 技術概要及權威觀點
11、潛在風險評價
a) 產品潛在風險測試報告及相關文獻
b) 潛在風險的概要及權威觀點
12、臨床評價
a) 產品臨床測試報告及相關文獻
b) 臨床使用概述及權威觀點。