Activation of the mammalian immune system by siRNAs
基于“蓄毒致毒”探讨痛风性关节炎的态靶治毒思路

2006 环球中医药2023年10月第16卷第10期 Global Traditional Chinese Medicine,October 2023,Vol.16,No.10㊃理论探讨㊃基金项目:国家自然科学基金面上项目(82274544);广州中医药大学 双一流”与高水平大学学科协同创新团队重大项目(2021XK05);广州中医药大学 双一流”与高水平大学学科后备人才培育项目作者单位:510378 广州中医药大学第三附属医院[何宪顺(硕士研究生)㊁田佳庆(硕士研究生)㊁魏腾飞(博士研究生)㊁韦雨柔(硕士研究生)㊁詹芝玮(硕士研究生)],关节中心关节科(魏秋实);广东省中医骨伤研究院(魏秋实㊁何敏聪㊁林天烨㊁何晓铭)作者简介:何宪顺(1993-),2021级在读硕士研究生㊂研究方向:中医药防治骨与关节疾病㊂E⁃mail:hexianshun123@通信作者:魏秋实(1982-),博士,副主任中医师,副教授,博士生导师㊂研究方向:中医药防治骨与关节疾病㊂E⁃mail:weiqshi@基于 蓄毒致毒”探讨痛风性关节炎的态靶治毒思路何宪顺 田佳庆 何晓铭 何敏聪 魏腾飞 韦雨柔 詹芝玮 林天烨 魏秋实【摘要】 痛风性关节炎是与高尿酸血症密切相关的一种反复炎症性疾病,专家团队在长期临证学习及总结古人经验的基础上,认为痛风性关节炎因湿㊁热相搏,郁而不解,热极成毒,蓄毒于内,阳动生风,助热毒攻注骨节而发病,疾病发展演变历经 蓄毒而发,毒损骨络 湿滞难去,毒伏骨脉”过程㊂近年研究发现尿酸钠晶体刺激体内炎性体的激活和肠道菌群失调是痛风性关节发生的重要机制,其中炎性体激活可促进机体分泌白介素1β㊁肿瘤坏死因子⁃α等加重炎症过程,并且肠菌失调还会导致尿酸的分解㊁排泄减少㊂团队认为治疗痛风性关节炎疾病可借鉴仝小林院士的 态靶结合”辨治方略,对痛风辨毒识态定其靶,热毒为痛风急性期所处之 态”,湿毒为痛风缓解期所处之 态”,炎性体㊁肠菌失调为痛风治疗的重要靶点;治疗上遵循 解毒 化毒”原则,急性期以清热解毒定风为法,缓解期以除湿化毒通络为法㊂结合态靶辨治方略,分期辨态治毒打靶,减低疾病发病率和复发率,为治疗痛风性关节炎提供理论依据及方法㊂【关键词】 痛风性关节炎; 蓄毒致毒; 态靶结合; 清热解毒定风; 除湿化毒通络【中图分类号】 R259 【文献标识码】 A doi:10.3969/j.issn.1674⁃1749.2023.10.011Discussion on syndrome differentiation and stage treatment of gouty arthritis by Status⁃Target ’theory based on Toxic Accumulation and Pathogenesis ’HE Xianshun ,TIAN Jiaqing ,HE Xiaoming ,HE Mincong ,WEI Tengfei ,WEI Yurou ,ZHAN Zhiwei ,LIN Tianye ,WEI QiushiThe Third Affiliated Hospital of Guangzhou University of Chinese Medicine ,Guangzhou 510378,China Corresponding author :WEI Qiushi ,E⁃mail :weiqshi@【Abstract 】 Gouty arthritis is a recurrent inflammatory disease closely related to hyperuricemia.On the basis of long⁃term clinical learning and summarizing the experience of the ancient people,the expert inteam thinks that gouty arthritis is caused by dampness and heat fighting,depression and confusion,then heat becomes toxic and accumulates poison inside,Yang moves to generate wind,and helps heat andpoison attack the bone joints,so the patient will have pain attacks.The development and evolution of the disease goes through poison accumulates,poison damage bone and arthritis⁃wet stagnation difficult to go,poison latent bone veins”process.Recent studies have found that sodium urate crystals stimulate theactivation of inflammasome in the body and the imbalance of intestinal flora,which is an important mechanism for the occurrence of gout joints.Inflammasome activation can promote the secretion of interleukin1β,tumor necrosis factor⁃αand other aggravating inflammatory processes,and the imbalance of环球中医药2023年10月第16卷第10期 Global Traditional Chinese Medicine,October2023,Vol.16,No.102007 intestinal flora can also lead to the decomposition and reduced excretion of uric acid.The expert in teambelieves that academician Tong Xiaolin’s Combination of State and Target”strategy can be used for references in the treatment of gout arthritis.The target of gout can be identified by heat poison as thestate”in the acute stage of gout,wet poison as the state”in the remission stage of gout,and the imbalance of inflammasome and intestinal bacteria as an important target for the treatment of gout.The treatment follows the principle of detoxification⁃detoxification”.In the acute phase,the method of clearingheat,detoxification and eliminating wind is treated for used,and in period of remission,the method of de⁃humidification and detoxification and dredging collateral is treated for bined with the treatmentstrategy of state and target differentiation,the treatment of poison and target differentiation by stages candecrease the incidence and recurrence rate,and provide theoretical basis and methods for the treatment ofgouty arthritis.【Key words】 gouty arthritis; accumulation of toxic pathogenic; combination of state and target; clearing away heat and toxic; eliminating dampness and dredging channels 痛风性关节炎(Gout Arthritis,GA)是与高尿酸血症密切相关的一种反复炎症性疾病㊂随着生活水平的提高,GA的患病率呈逐年上升趋势,需引起重视[1]㊂GA属中医 痹症” 历节”范畴,历代医家对其病因总结为先天禀赋不足㊁饮食不节㊁脏腑虚损㊁脾运失司㊁湿热内生㊁久蕴不解㊁发为浊毒,攻注于关节而作痛[2]㊂‘中藏经㊃论痈疽疮肿第四十一“曾言 夫痈疽疮肿之所作也,皆五脏六腑蓄毒不流则生矣,非独因荣卫壅塞而发者也”,首次提出蓄毒致病观点㊂团队专家融合古今医家观点,结合临床实践经验,认为GA发病为湿㊁热相搏成毒,引动风扰,三邪交织体内,蓄而成毒发病㊂GA急性期因湿热搏结,热极成毒,阳动生风,风循经流窜,攻注骨节而遍身作痛,此阶段表现为毒损骨络,痛如虎噬;缓解期为风湿盘踞骨节脉络,久留不去,痹阻气血,聚而成毒,当再次与热搏结,便可发病㊂此阶段表现为毒伏筋脉,遇诱再发㊂中医学角度总结为GA发病历经 蓄毒而发,毒损骨络 湿滞难去,毒伏骨脉”演变过程㊂近年研究证实GA的发病机制与炎症微环境失衡密切相关,包括了炎性体激活㊁肠道菌群失调,其中肠菌失调能加重炎症反应和减少尿酸的分解㊁排泄[3⁃5]㊂因此在诊治中提高对GA炎症微环境失衡的靶向性和精准度,以期减低发病率和复发率㊂团队基于GA蓄毒致病观点,结合仝小林院士的态靶辨治思维,构建了GA的态靶辨治体系,对GA辨以 解毒 化毒”的分期治毒思路,从而提高中医药对GA治疗的靶向性和精准度,为治疗GA提供理论依据及方法㊂1 毒邪在痛风性关节炎的理论溯源与悟萃1.1 湿热风三邪交织,蓄而成毒毒”一词源于在‘辞源“,解释为 恶也㊁害也㊁痛也,及物之能害人者,皆曰毒”;其为医所用肇始于‘内经“,以示病害之意,后世渐拓其义至涵盖病因病机㊁病名病证㊁病理因素㊁药性治法等,尤以示病性之骤烈重笃;‘华氏中藏经“中首见 毒邪”之名,首次提出 蓄毒致病”理论学说;到‘金匮要略心典“所曰 毒,邪气蕴结不解之谓”㊂中医认为毒为邪之渐,当病邪亢盛到一定程度,引起机体阴阳失衡,则称之为 毒”㊂对GA而言,先后天失调均可导致脏腑功能紊乱,津液运化失司,湿困于内,聚而成毒,成为GA发病的成毒之源,为疾病 蓄毒”初段㊂‘素问㊃生气通天论篇“曾记载 膏粱之变,足生大丁”,喻嘉言‘寓意草“曰 醇酒厚味之热毒也,郁怒横决之火毒也”㊂后天嗜食膏粱厚味㊁酒酪之属,易在初期助湿生痰化热;另一方面,GA毒的生成与先天禀赋不足,脾胃㊁肝㊁肾脏功能失调密切相关㊂脾胃亏虚,运化失司,水反为湿,谷反为滞,湿滞久则化为毒,而且脾亦不能分解运化已成之毒;肝主疏泄,若肝失调达,气郁而不畅,则毒滞不去,毒一旦产生,则积毒入内,形成复杂的病证,恶性循环; 肾者,胃之关也,关门不利,故聚水而从其类也”,肾脏升清降浊失常,导致解毒失司,蓄毒体内㊂因此,当湿邪聚而不化,则生痰化热,湿与热相互搏结进一步发展时,则亢而成毒,燔灼于内,热极阳动生风,此时三邪交织成毒,风助毒循经走脉,攻注骨节,遍身疼痛,故见GA发病㊂2008 环球中医药2023年10月第16卷第10期 Global Traditional Chinese Medicine,October2023,Vol.16,No.101.2 谨守病机,分期辨毒1.2.1 急性期 蓄毒而发,毒损骨络 GA急性期时,因湿热二邪亢盛化毒,引动风邪助毒流窜于全身骨节筋脉,遍身而疼㊂‘诸病源候论“曰: 今毒气从脏腑而出,循于经络,攻于手足,故手足指皆肿赤焮痛也㊂”毒积蕴结,阻滞气血,凝滞于经脉之中,遏而不得行,故热;大热不止,肿痛不休,由此脏腑蕴热㊁燔灼津液㊁湿热搏结㊁热极化毒攻注骨节经络,故作痛㊂所以GA急性期因热毒蓄内而发,毒留骨络损其气血,正如‘千金翼方“载: 热毒流于四肢,历节肿痛㊂”1.2.2 缓解期 湿滞难去,毒伏骨脉 缓解期时,因热退毒解风消,但湿邪缠绵,易留滞于骨节经络,加之湿性重浊,易聚而成毒,遇诱再发㊂若既成之毒日久不化,阻滞脉络,则会 热气淳盛,下陷肌肤,筋髓枯”,引起骨质的侵蚀与破坏㊂临床上可见GA 反复发作,肢体困重酸软,患侧皮肤色暗㊁尿黄㊁口干等㊂故GA缓解期治疗以除湿化毒通络为法,喻除湿化GA蓄毒之源,通络疏骨节经脉气血,则毒无自留之意㊂2 痛风性关节炎的态靶辨治体系构建2.1 分期辨毒识态根据态靶辨治理论,结合痛风蓄毒发病两阶段,可分为热毒态㊁湿毒态㊂2.1.1 急性期热毒态 发病初因湿毒不化,郁久化热,湿热搏结,燔灼津液,热极化毒,阳动生风,助毒循经流注,攻于手足而作痛的状态㊂辨识要点有: (1)平素饮食上嗜食酒肉等肥甘厚腻㊁辛辣炙煿之品;(2)患侧红肿㊁灼热,发作时有痛如虎噬之状㊁活动疼痛受限㊁恶热喜冷㊁口渴喜饮㊁小便短赤㊁大便干结等症,舌红㊁苔黄燥,脉数;(3)检验指标上白细胞㊁血沉㊁C反应蛋白等炎症指标可见上升,而血尿酸此时可不伴升高㊂2.1.2 缓解期湿毒态 缓解期因湿滞留恋难去,加之中焦脾胃亏虚㊁受损,津液运化障碍,毒从湿化的状态㊂辨识要点有:(1)有神疲乏力,肢体困重㊁酸软,患侧皮肤暗淡㊁局部有压痛等症,舌胖大,苔厚腻,脉沉㊁滑;(2)血尿酸指标升高(男性>420μmol/ L,女性>357μmol/L)㊂反复发作则见肾功能损害(血肌酐:男性>106μmol/L,女性>97μmol/L;尿素氮>7.1mmol/L)为毒损及肾;影像学双能CT可见关节有尿酸盐沉积㊂2.2 分期论毒定靶态靶结合另一核心是强调打靶, 靶”是参照现代医学疾病框架,结合传统中医学 态”去寻找疾病的靶方㊁靶药,属于微观范畴[6⁃7]㊂目前,各国指南对于GA急性期的治疗都明确首要快速消炎止痛击打症靶,初期和缓解期则主以降尿酸为主,但居高不下的复发率重新审视单一击打症靶㊁标靶是不完备的,最终要精准靶向疾病发生机制,因此,对GA 的病靶认识必不可少㊂近年研究已表明炎症微环境失衡是GA重要发生机制,其中包括炎性体激活和肠道菌群失调,两者已被证明和GA的发生发展密切相关,其中炎性体激活和肠菌失调导致炎症因子大量的释放导致GA急性期发作[8⁃9];并且肠菌失调,能导致肠道对尿酸的排泄及肠菌对尿酸的分解减少,增加体内尿酸蓄积,当菌群平衡被打破,有害菌丰度上调,其分泌的致炎因子白细胞介素⁃1β(in⁃terleukin⁃1β,IL⁃1β)㊁白细胞介素⁃6(interleukin⁃6, IL⁃6)㊁肿瘤坏死因子⁃α(tumor necrosis factor⁃α, TNF⁃α)等炎症因子会再次诱发GA的急性发作,因此缓解期要注重肠菌失调靶点[10]㊂借鉴态靶理论,打靶需先定靶,专家团队认为抑制炎性体激活是GA急性期的治疗靶点,调节肠菌失调是GA缓解期的治疗靶点㊂2.2.1 急性期抑制炎性体激活为靶点 GA急性期中患者最明显症状是关节痛如虎噬,现代医学上因关节中㊁滑膜中大量炎症因子浸润,使痛觉传入神经超敏化[11]㊂临床评价炎症反应最直观是临床表现和血液炎症指标,当前大量研究证明,尿酸钠晶体激活的核苷酸结合寡聚化结构域样受体蛋白3 (Nucleotide⁃binding oligomeric domain⁃like receptor protein3,NLRP3),是痛风急性发作的重要促炎机制[12]㊂其通过下游凋亡相关斑点样蛋白(Apoptosis⁃associated speck⁃like protein,ASC),将半胱天冬酶⁃1前体(precursor cysteine aspartate specific protease⁃1,pro⁃caspase⁃1)切割成为具有活性的半胱天冬酶⁃1(cysteine aspartate specific protease⁃1, caspase⁃1),促使IL⁃1β成熟进而上调促炎介质的表达[13⁃14]㊂故GA急性期产生强烈的炎症级联放大,临床表现和血液炎症指标可佐证㊂其中炎性体活化,上调因子浸润关节与热极成毒,毒损骨络有着共同点,所以急性期以解毒为治则,清热解毒定风为治法,抑制炎性体激活为急性期治疗的靶点㊂2.2.2 缓解期调节肠道菌群失调为靶点 肠道菌环球中医药2023年10月第16卷第10期 Global Traditional Chinese Medicine,October2023,Vol.16,No.102009群作为肠道另一 器官”,在尿酸排泄中也起着重要作用,约25%的尿酸排泄到肠道并被肠道细菌进一步代谢[15]㊂中医学角度上肠道菌群作用与小肠秘别清浊之功相合,如‘医原“曰: 人纳水谷,脾化精微之气以上升,小肠化糟粕传于大肠而下降㊂”若肠菌发生失衡,无法将尿酸等糟粕向下转导,导致尿酸的蓄积㊂GA急性炎症反应被抑制后,首要治疗的目标的控制患者的血尿酸指标,现代降尿酸机制中包括抑制尿酸生成的黄嘌呤氧化酶抑制剂:别嘌醇和非布司他,促进尿酸排泄的尿酸盐重吸收转运子⁃1(Urate Transporter⁃1,URAT1)抑制剂:苯溴马隆㊂这两种降酸机制的常用药物都存在者明显的肝肾副作用,而肠道菌群作为降尿酸的新靶点之一,通过调节自身肠道微生态从而减少尿酸的蓄积,是治疗GA缓解期和(或)高尿酸血症的新方法㊂3 痛风性关节炎态靶体系分期治毒应用3.1 急性期主以解毒:清热解毒定风为法,靶向炎性体GA急性期因湿热相搏,热极成毒,阳动生风,热毒随风循经走脉,毒损骨脉,临床最常见为热毒态,故此时以解毒为原则,以清热解毒定风为法,纠热毒之偏态㊂其中解毒包含治热以寒,清热盛苗头;将一切有形㊁无形之邪从身体玄府㊁孔窍㊁二焦分利逐邪而出,通则不痛,解毒不仅要清,更要通㊂现代痛风发病机制上已明确与NLRP3炎症相关,从中医学角度出发,当体内湿热积聚之毒氤氲凝结之间,相当于上文所述炎性体激活组装过程,因此GA 分期治毒的核心在急性期主以解毒阻断NLRP3的激活㊁组装㊂现代药理学证实,清热解毒中药活性成分能有效提高选择性活化型巨噬细胞(Alternative activated Macrophage2type,M2型)阻断炎性体NLRP3的活化,从而起到抗炎作用[16]㊂中药可选择黄柏㊁土茯苓㊁生大黄㊁连翘㊁蒲公英等清热解毒类药物,热毒重㊁炎症反应剧烈的可加用白花蛇舌草㊁苦参㊁紫花地丁加强靶向对炎性体的生成抑制,最终宏观反应在血液炎症指标得到有效改善,热毒得解,疼痛减轻㊁骨络可愈㊂但要注意清热解毒药应中病即止,不可过用,以防过服伤及阳气㊂临床上痛风发病好发于足大趾,但不唯足大趾,全身大小关节均可发生,为风循经走脉推动热毒流注,成游走性疼痛,故GA急性发作还需 定风 打次靶,可应用独活㊁羌活㊁防风等定风止痛㊂现代研究发现,中药治风之药,能降低血管通透性[17],GA急性期热毒亢盛,血管壁通透性增加,炎性体NLRP3加剧炎症因子通过全身毛细血管流注全身,定风治法能截断炎症因子的全身趋附作用㊂3.2 缓解期主以化毒:除湿化毒通络为法,靶向肠道菌群GA缓解期因热毒以解,但湿邪缠绵留恋㊁弥漫难化,盘踞骨络,内伏骨节经脉,引诱再发,临床上最常见为湿毒态㊂此时湿毒伏于骨脉是外在表现,内在原因为湿毒无以化解,包括脾脏运化失司,湿困肠道,无以驱邪㊂‘伤寒论“云: 阳明居中,主土也,万物所归,无所复传㊂”脾土健运则可以纳毒㊁降毒㊁分解化毒,不仅可以运化水湿去毒之依附,又可降低毒的烈性,促进毒的分解代谢,防其传变而损害它脏㊂小肠主津,大肠主液,当脾运化津液障碍,下焦水湿无以开利,进一步湿聚成毒㊂此阶段以化毒为原则,主以除湿化毒通络为法㊂运用健脾化湿,使津液运行得以舒布,肠道湿困得解,肠菌失衡得调㊂恢复肠菌稳态后,肠道益生菌乳酸杆菌和假单胞菌丰度上调能促进合成尿酸代谢酶,如尿酸酶㊁尿囊素酶和尿囊素酶的能力,这些酶可依次将尿酸降解为5⁃羟基异戊酸㊁尿囊素㊁尿囊酸,最终降解为尿素,减少嘌呤蓄积[18]㊂临床上可用陈皮㊁枳壳㊁山药㊁党参等健脾化湿类中药,若湿毒重,伴纳差㊁腹泻㊁尿酸偏高者,可加用苍术㊁厚朴㊁半夏㊁茯苓㊁藿香除湿之品,加强除湿化毒靶向肠道菌群的调节㊂缓解期时关节可因湿毒久伏,局部气血运行不畅,导致关节屈伸不利,现代医学中表现为尿酸盐沉积于关节引起关节的无菌粘连㊁僵硬㊂因此,同时辅以通络药物,如桑枝㊁威灵仙㊁桑寄生等中药,联用通络法靶向关节不利次靶㊂4 小结与展望综上,GA疾病发生发展离不开 毒”的生成流注和潜藏伏骨,因此治疗上可遵循 解毒 化毒”原则㊂GA急性期需知热极成毒蓄于内,热极阳动生风,助毒循经走脉损于骨络,应清热解毒定风治其病;缓解期需知热虽退但湿毒留滞难去,伏于骨络,遇诱再发,应除湿化毒通络防其变㊂以此为原则,依据态㊁靶逐层辨毒识态,定靶治毒,瞄准炎性体激活和肠菌失调靶点,治以态靶结合,全程辨毒治毒(见图1)㊂用 炎性体 肠道菌群”失衡关系深入探讨既往中医学者从毒辨治GA的认识㊂随着科2010 环球中医药2023年10月第16卷第10期 Global Traditional Chinese Medicine,October2023,Vol.16,No.10图1 痛风性关节炎态靶治毒思路学技术㊁医疗水平发展进步与疾病谱的改变,对GA 构建态靶辨治体系可进一步丰富传统中医对GA的认识,提供了一种新的治疗思维模式㊂今后随着态靶辨治体系的进一步深入研究,不断提炼有效的靶方靶药,对GA病变全程治疗更加精准化,除本文介绍的靶点外,未来还可针对GA急性期巨噬细胞M2极化机制㊁细胞自噬机制,缓解期降酸机制中抑制肝脏黄嘌呤氧化酶酶㊁抑制尿酸盐重吸收转运子⁃1㊁以及促进肾脏三磷酸腺苷结合转运蛋白G超家族成员2蛋白进行多维度㊁多靶点的中药治疗,以期进一步提高中医药对GA的疗效㊂参考文献[1] 冯少华,张影,刘颖新.高尿酸血症与痛风的现代中医认识及展望[J].世界最新医学信息文摘,2018,18(84):158⁃173.[2] 杨良山,钟琴.痛风性关节炎中医病因病机研究综述[J].风湿病与关节炎,2014,3(8):53⁃56.[3] Bodofsky S,Merriman T R,Thomas T J,et al.Advances in ourunderstanding of gout as an auto⁃inflammatory disease[J].SeminArthritis Rheum,2020,50(5):1089⁃1100.[4] Clavijo⁃Cornejo D,Hernandez⁃Gonzalez O,Gutierrez M.Thecurrent role of NLRP3inflammasome polymorphism in gout sus⁃ceptibility[J].Int J Rheum Dis,2021,24(10):1257⁃1265.[5] 张奎,邓向亮,傅南琳.肠道菌群与痛风性关节炎的研究进展[J].广东药科大学学报,2020,36(6):903⁃906. [6] 仝小林.态靶医学 中医未来发展之路[J].中国中西医结合杂志,2021,41(1):16⁃18.[7] 仝小林,何莉莎,赵林华.论 态靶因果”中医临床辨治方略[J].中医杂志,2015,56(17):1441⁃1444.[8] 任鸿雁,邢爱萍,茹晋丽.尿酸的肠道代谢及调节肠道菌群防治高尿酸血症的研究进展[J].中华风湿病学杂志,2021,25(10):708⁃711.[9] 苏友新,滕方舟,蔡唐彦,等.痛风宁含药血清对尿酸盐诱导THP⁃1中NALP3炎性体相关蛋白及炎症因子表达的影响[J].中国中西医结合杂志,2019,39(3):323⁃329. [10] 金钗,徐明智.肠道菌群与高尿酸血症及痛风的相关性研究[J].中国微生态学杂志,2019,31(8):980⁃984. [11] 杨静,李鑫楠,金华.白介素在慢性疼痛的产生和维持中的作用[J].生命科学,2022,34(6):732⁃741. [12] Szekanecz Z,Szamosi S,Kovacs G E,et al.The NLRP3inflam⁃masome⁃interleukin1pathway as a therapeutic target in gout[J].Arch Biochem Biophys,2019,670:82⁃93. [13] So A K,Martinon F.Inflammation in gout:mechanisms andtherapeutic targets[J].Nat Rev Rheumatol,2017,13(11):639⁃647.[14] WANG L,Hauenstein A V.The NLRP3inflammasome:Mechanism of action,role in disease and therapies[J].MolAspects Med,2020,76:100889.[15] GUO Z,ZHANG J,WANG Z,et al.Intestinal MicrobiotaDistinguish Gout Patients from Healthy Humans[J].Sci Rep,2016,6:20602.[16] 田维毅.基于M1/M2型Mφ极化及炎症调控效应探索黄连解毒汤干预AS的作用与机制[D].长沙:湖南中医药大学,2015.[17] 于柳,王哲,熊瑞,等.荆防散抗炎抗过敏作用有效部位初步筛选的实验研究[J].时珍国医国药,2013,24(2):271⁃273.[18] CHU Y,SUN S,HUANG Y,et al.Metagenomic analysisrevealed the potential role of gut microbiome in gout[J].NPJBiofilms Microbiomes,2021,7(1):66.(收稿日期:2022⁃09⁃18)(本文编辑:王馨瑶)。
植物-病原菌互作的分子机制

M. grisea
Plant disease
C. fulvum
B. cinerea
P. infestans
I 植物病原菌的侵染机理
侵染途径 特征 寄主范围
植物病原菌寄生方式
腐生 (necrotroph)
活体寄生 (biotroph)
半活体寄生 (semibiotroph)
分泌胞壁降解酶、毒 菌体进入寄主细胞内 先活体寄生,
有菌系均有抗性。是植物防御潜在病原菌的主要机制,也是 植物最基础最普遍的抗病类型
• 抗病(resistance):植株能限制病原菌在侵染点附近、病斑不 扩展或只产生小斑点: 非亲和性反应(incompatibility))
• 感病(susceptibility)病斑扩大形成典型病斑: 亲和性反应 (compatibility)
素
后腐生
寄主组织死亡、病原 寄主细胞一般保持成 侵染早期寄主
菌定殖、大面积组织 活状态
组织仍成活,
软化
而后死亡
广
窄,一般侵染个别植 两者之间
物
病原真菌在植物表皮细胞内形成吸器(Haustorium)从寄主内吸收营养
病原细菌定殖于寄主细胞间隙
植物病毒可在寄主细胞内大量增殖并通过 胞间连丝进行“cell to cell”扩散
抗病反应。
I. PAMP-Triggered Immunity (PTI) PAMP的作用:病原菌的适应性与生存
已知的主要PAMP
Bacterial flagellin (flg22) Bacterial PAMPs EF-Tu Xoo Ax21 Fungal xylanase Fungal chitin Oomycete glucans
上海交大外科学外科学与免疫学

Phagocytes
上海交大外科学外科学与免疫学
Mononuclear phagocytes
Polymorphs
Neutrophils
Eosinophils
Basophils and mast cells
Platelets
Natural killer (NK) cells
Cells of the Adaptive Immune System
Surface Molecules Expressed on DC
抗原递呈细胞的表面标志
上海交大外科学外科学与免疫学
T B 细胞表面标志的主要差别
上海交大外科学外科学与免疫学
Molecules of immune sys
● immunoglobulin / BCR 上海交大外科学外科学与免疫学
效应相
● Response: Lymphocytes coordinate an immune response that eliminates the source of the Ag
免疫系统 Immune System
●Organs of th上e海交Im大外m科u学n外e科学S与y免s疫te学m 免疫器官
● Transferable
上海交大外科phases of immune response
上海交大外科学外科学与免疫学
识别相
● Recognition: Ag recognized by lymphocytes
激活相
● Activation: lymphocytes activation
● Cells of the Immune System 免疫细胞
● Molecule of the Immune System 免疫分子
细胞自噬机制--2016年诺贝尔生理或医学奖

Scientific Background Discoveries of Mechanisms for AutophagyThe 2016 Nobel Prize in Physiology or Medicine is awarded to Yoshinori Ohsumi for his discoveries of mechanisms for autophagy. Macroautophagy (“self-eating”, hereafter referred to as autophagy) isan evolutionarily conserved process whereby the eukaryotic cell can recycle part of its own contentby sequestering a portion of the cytoplasm in a double-membrane vesicle that is delivered to the lysosome for digestion. Unlike other cellular degradation machineries, autophagy removes long-lived proteins, large macro-molecular complexes and organelles that have become obsolete or damaged. Autophagy mediates the digestion and recycling of non-essential parts of the cell during starvation and participates in a varietyof physiological processes where cellular components must be removed to leave space for new ones. In addition, autophagy is a key cellular process capable of clearing invading microorganisms and toxic protein aggregates, and therefore plays an important role during infection,in ageing and in the pathogenesis of many human diseases. Although autophagy was recognized already in the 1960’s, the mechanism and physiological relevance remained poorly understood for decades. The work of Yoshinori Ohsumi dramatically transformed the understanding of this vital cellular process. In 1993, Ohsumi published his seminal discovery of 15 genes of key importance for autophagy in budding yeast. In a series of elegant subsequent studies, he cloned several of these genes in yeast and mammalian cells and elucidated the function of the encoded proteins. Based on Yoshinori Ohsumi’s seminal discoveries, the importance of autophagyin human physiology and disease is now appreciated.The mystery of autophagyIn the early 1950’s, Christian de Duve was interested in the action of insulin and studied the intracellular localization of glucose-6-phosphatase using cell fractionation methods developed by Albert Claude. In a control experiment, he also followed the distribution of acid phosphatase, but failed to detect any enzymatic activity in freshly isolated liver fractions. Remarkably, the enzymatic activity reappeared if the fractions were stored for five days in a refrigerator1. It soon became clear that proteolytic enzymes were sequestered within a previously unknown membrane structure that de Duve named the lysosome1,2. Comparative electron microscopy of purified lysosome-rich liver fractions and sectioned liver identified the lysosome as a distinct cellular organelle3. Christian de Duve and Albert Claude, together with George Palade, were awarded the 1974 Nobel Prize in Physiology or Medicine for their discoveries concerning the structure and functional organization of the cell.Soon after the discovery of the lysosome, researchers found that portions of the cytoplasm are sequestered into membranous structures during normal kidney development in the mouse4. Similar structures containing a small amount of cytoplasm and mitochondria were observed in the proximal tubule cells of rat kidney during hydronephrosis5. The vacuoles were found to co-localize with acid-phosphatase-containing granules during the early stages of degeneration and the structures were shown to increase as degeneration progressed5. Membrane structures containing degenerating cytoplasm were also present in normal rat liver cells and their abundance increased dramatically following glucagon perfusion6 or exposure to toxic agents7. Recognizing that the structures had the capacity to digest parts of the intracellular content, Christian de Duve coined the term autophagy in 1963, and extensively discussed this concept in a review article published a few years later8. At that time, a compelling case for the existence of autophagy in mammalian cells was made based on results from electron microscopy studies8. Autophagy was known to occur at a low basal level, and to increase during differentiation and remodeling in a variety of tissues, including brain, intestine, kidney, lung, liver, prostate, skin and thyroid gland4,7-13. It was speculated that autophagy might be a mechanism for coping with metabolic stress in response to starvation6and that it might have roles in the pathogenesis of disease5. Furthermore, autophagy was shown to occur in a wide range of single cell eukaryotes and metazoa, e.g. amoeba, Euglena gracilis, Tetrahymena, insects and frogs8,14, pointing to a function conserved throughout evolution.During the following decades, advances in the field were limited. Nutrients and hormones were reported to influence autophagy; amino acid deprivation induced15, and insulin-stimulation suppressed16 autophagy in mammalian tissues. A small molecule, 3-methyladenine, was shown to inhibit autophagy17. One study using a combination of cell fractionation, autoradiography and electron microscopy provided evidence that the early stage of autophagy included the formation of a double-membrane structure, the phagophore,that extended around a portion of the cytoplasm and closed into a vesicle lacking hydrolytic enzymes, the autophagosome18 (Figure 1).Despite many indications that autophagy could be an important cellular process, its mechanism and regulation were not understood. Only a handful of laboratories were working on the problem, mainly using correlative or descriptive approaches and focusing on the late stages of autophagy, i.e. the steps just before or after fusion with the lysosome. We now know that the autophagosome is transient and only exists for ~10-20 minutes before fusing with the lysosome, making morphological and biochemical studies very difficult.Figure 1. Formation of the autophagosome. The phagophore extends to form a double-membrane autophagosome that engulfs cytoplasmic material. The autophagosome fuses with the lysosome, where the content is degraded.In the early 1990’s, almost 30 years after de Duve coined the term autophagy, the process remained a biological enigma. Molecular markers were not available and components of the autophagy machinery were elusive. Many fundamental questions remained unanswered: How was the autophagy process initiated? How was the autophagosome formed? How important was autophagy for cellular and organismal survival? Did autophagy have any role in human disease? Discovery of the autophagy machineryIn the early 1990’s Yoshinori Ohsumi, then an Assistant Professor at Tokyo University, decided to study autophagy using the budding yeast Saccharomyces cerevisae as a model system. The first question he addressed was whether autophagy exists in this unicellular organism. The yeast vacuole is the functional equivalent of the mammalian lysosome. Ohsumi reasoned that, if autophagy existed in yeast, inhibition of vacuolar enzymes would result in the accumulation of engulfed cytoplasmic components in the vacuole. To test this hypothesis, he developed yeast strains that lacked the vacuolar proteases proteinase A, proteinase B and carboxy-peptidase19. He found that autophagic bodies accumulated in the vacuole when the engineered yeast were grown in nutrient-deprived medium19, producing an abnormal vacuole that was visible under a light microscope. He had now identified a unique phenotype that could be used to discover genes that control the induction of autophagy. By inducing random mutations in yeast cells lacking vacuolar proteases, Ohsumi identified the first mutant that could not accumulate autophagic bodies in the vacuole20; he named this gene autophagy 1 (APG1). He then found that the APG1 mutant lost viability much quicker than wild-type yeast cells in nitrogen-deprived medium. As a second screen he used this more convenient phenotype and additional characterization to identify 75 recessive mutants that could be categorized into different complementation groups. In an article published in FEBS Letters in 1993, Ohsumi reported his discovery of as many as 15 genes that are essential for the activation of autophagy in eukaryotic cells20. He named the genes APG1-15. As new autophagy genes were identified in yeast and other species, a unified system of gene nomenclature using the ATG abbreviation was adopted21. This nomenclature will be used henceforth in the text.During the following years, Ohsumi cloned several ATG genes22-24and characterized the function of their protein products. Cloning of the ATG1gene revealed that it encodes a serine/threonine kinase, demonstrating a role for protein phosphorylation in autophagy24. Additional studies showed that Atg1 forms a complex with the product of the ATG13 gene, and that this interaction is regulated by the target of rapamycin (TOR) kinase23,25. TOR is active in cells grown under nutrient-rich conditions and hyper-phosphorylates Atg13, which prevents the formation of the Atg13:Atg1 complex. Conversely, when TOR is inactivated by starvation, dephosphorylated Atg13 binds Atg1 and autophagy is activated25. Subsequently, the active kinase was shown to be a pentameric complex26 that includes, in addition to Atg1 and Atg13, Atg17, Atg29 and Atg31. The assembly of this complex is a first step in a cascade of events needed for formation of the autophagosome.Figure 2. Regulation of autophagosome formation. Ohsumi studied the function of the proteins encoded by key autophagy genes. He delineated how stress signals initiate autophagy and the mechanism by which protein complexes promote distinct stages of autophagosome formation.The formation of the autophagosome involves the integral membrane protein Atg9, as well as a phosphatidylinositol-3 kinase (PI3K) complex26 composed of vacuolar protein sorting-associated protein 34 (Vps34), Vps15, Atg6, and Atg14. This complex generates phosphatidylinositol-3 phosphate and additional Atg proteins are recruitedto the membrane of the phagophore. Extension of the phagophore to form the mature autophagosome involves two ubiquitin-like protein conjugation cascades (Figure 2).Studies on the localization of Atg8 showed that, while the protein was evenly distributed throughout the cytoplasm of growing yeast cells, in starved cells, Atg8 formed large aggregates that co-localized with autophagosomes and autophagic bodies27. Ohsumi made the surprising discovery that the membrane localization of Atg8 is dependent on two ubiquitin-like conjugation systems that act sequentially to promote the covalent binding of Atg8 to the membrane lipid phosphatidylethanolamine. The two systems share the same activating enzyme, Atg7. In the first conjugation event, Atg12 is activated by forming a thioester bond with a cysteine residue of Atg7, and then transferred to the conjugating enzyme Atg10 that catalyzes its covalent binding to the Atg5 protein26,28,29. Further work showed that the Atg12:Atg5 conjugate recruits Atg16 to form a tri-molecular complex that plays an essential role in autophagy by acting as the ligase of the second ubiquitin-like conjugation system30. In this second unique reaction, the C-terminal arginine of Atg8 is removed by Atg4, and mature Atg8 is subsequently activated by Atg7 for transfer to the Atg3 conjugating enzyme31. Finally, the two conjugation systems converge as the Atg12:Atg5:Atg16 ligase promotes the conjugation of Atg8 to phosphatidylethanolamine26,32.Lipidated Atg8 is a key driver of autophagosome elongation and fusion33,34. The two conjugation systems are highly conserved between yeast and mammals. A fluorescently tagged version of the mammalian homologue of yeast Atg8, called light chain 3 (LC3), is extensively used as a marker of autophagosome formation in mammalian systems35, 36.Ohsumi and colleagues were the first to identify mammalian homologues of the yeast ATG genes, which allowed studies on the function of autophagyin higher eukaryotes. Soon after, genetic studies revealed that mice lacking the Atg5gene are apparently normal at birth, but die during the first day of life due to inability to cope with the starvation that precedes feeding37. Studies of knockout mouse models lacking different components of the autophagy machinery have confirmed the importance of the process in a variety of mammalian tissues26,38.The pioneering studies by Ohsumi generated an enormous interest in autophagy. The field has become one of the most intensely studied areas of biomedical research, with a remarkable increase in the number of publications since the early 2000’s.Different types of autophagyFollowing the seminal discoveries of Ohsumi, different subtypes of autophagy can now be distinguished depending on the cargo that is degraded. The most extensively studied form of autophagy, macroautophagy, degrades large portions of the cytoplasm and cellular organelles. Non-selective autophagy occurs continuously, andis efficiently induced in response to stress, e.g.starvation. In addition, the selective autophagy of specific classes of substrates - protein aggregates, cytoplasmic organelles or invading viruses and bacteria - involves specific adaptors that recognize the cargo and targets it to Atg8/LC3 on the autophagosomal membrane39. Other forms of autophagy include microautophagy40, which involves the direct engulfment of cytoplasmic material via inward folding of the lysosomal membrane, and chaperone-mediated autophagy (CMA). In CMA, proteins with specific recognition signals are directly translocated into the lysosome via binding to a chaperone complex41.Autophagy in health and diseaseInsights provided by the molecular characterizationof autophagy have been instrumental in advancing the understanding of this process and its involvement in cell physiology and a variety of pathological states (Figure 3). Autophagy was initially recognized as a cellular response to stress, but we now know that the system operates continuously at basal levels. Unlike the ubiquitin-proteasome system that preferentially degrades short-lived proteins, autophagy removes long-lived proteins and is the only process capable of destroying whole organelles, such as mitochondria, peroxisomes and the endoplasmic reticulum. Thus, autophagy plays an essential rolein the maintenance of cellular homeostasis. Moreover, autophagy participates in a variety of physiological processes, such as cell differentiation and embryogenesis that require the disposal of large portions of the cytoplasm. The rapid inductionof autophagy in response to different types of stress underlies its cytoprotective function and the capacity to counteract cell injury and many diseases associated with ageing.Because the deregulation of the autophagic flux is directly or indirectly involved in a broad spectrum of human diseases, autophagy is a particularly interesting target for therapeutic intervention. An important first insight into the role of autophagy in disease came from the observation that Beclin-1, the product of the BECN1gene, is mutated in a large proportion of human breast and ovarian cancers. BECN1 is a homolog of yeast ATG6 that regulates steps in the initiation of autophagy42. This finding generated substantial interest in the role of autophagy in cancer43.Misfolded proteins tend to form insoluble aggregates that are toxic to cells. To cope with this problem the cell depends on autophagy44. In fly and mouse models of neurodegenerative diseases, the activation of autophagy by inhibition of TOR kinase reduces the toxicity of protein aggregates45. Moreover, loss of autophagy in the mouse brain by the tissue-specific disruption of Atg5and Atg7 causes neurodegeneration46,47. Several autosomal recessive human diseases with impaired autophagy are characterized by brain malformations, developmental delay, intellectual disability, epilepsy, movement disorders and neurodegeneration48.Figure 3. Autophagy in health and disease. Autophagy is linked to physiological processes including embryogenesis and cell differentiation, adaptation to starvation and other types of stress, as well as pathological conditions including neurodegenerative diseases, cancer and infections.The capacity of autophagy to eliminate invading microorganisms, a phenomenon called xenophagy, underlies its key role in the activationof immune responses and the control of infectious diseases49,50. Viruses and intracellular bacteria have developed sophisticated strategies to circumvent this cellular defense. Additionally, microorganisms can exploit autophagy to sustain their own growth.ConclusionThe discovery of autophagy genes, and the elucidation of the molecular machinery for autophagy by Yoshinori Ohsumi have led to a new paradigm in the understanding of how the cell recycles its contents. Because of his pioneering work, autophagy is recognized as a fundamental process in cell physiology with major implicationsfor human health and disease.Nils-Göran Larsson and Maria G. Masucci Karolinska InstitutetReferences1. de Duve, C. (2005). The lysosome turns fifty.Nat Cell Biol 7, 847–849.2. de Duve, C., Pressman, B.C., Gianetto, R.,Wattiaux, R., and Appelmans, F. (1955)Tissue fractionation studies. 6. Intracellulardistribution patterns of enzymes in rat-livertissue. Biochem J 60, 604–617.3. Novikoff, A.B, Beaufay, H., and de Duve, C.(1956) Electron microscopy of lysosome-richfractions from rat liver. Journal BiophysBiochem Cytol. 2, 179–190.4. Clark, S.L. (1957) Cellular differentiation in thekidneys of newborn mice studied with theelectron microscope. J Biophys BiochemCytol 3, 349–376.5. Novikoff, A.B. (1959) The proximal tubule cellin experimental hydronephrosis. J BiophysBiochem Cytol 6, 136–138.6. Ashford, T.P., and Porter, K.R. (1962)Cytoplasmic components in hepatic celllysosomes. J Cell Biol 12, 198–202.7. Novikoff, A.B., and Essner, E. (1962)Cytolysomes and mitochondrial degeneration.J Cell Biol 15, 140–146.8. de Duve, C., and Wattiaux, R. (1966)Functions of lysosomes. Annu Rev Physiol 28,435–492.9. Behnke, O. (1963) Demonstration of acidphosphatase-containing granules and cytoplasmic bodies in the epithelium of foetalrat duodenum during certain stages ofdifferentiation. J Cell Biol18, 251–265. 10. Bruni, C., and Porter, K.R. (1965) The finestructure of the parenchymal cell of the normalrat liver: I. General observations. Am J Pathol46, 691–755.11. Hruban, Z., Spargo, B., Swift, H., Wissler,R.W., and Kleinfeld, R.G. (1963) Focalcytoplasmic degradation. Am J Pathol 42,657–683.12. Moe, H., and Behnke, O. (1962) Cytoplasmicbodies containing mitochondria, ribosomes,and rough surfaced endoplasmic membranesin the epithelium of the small intestine ofnewborn rats. J Cell Biol 13, 168–171.13. Napolitano, L. (1963) Cytolysomes inmetabolically active cells. J Cell Biol 18, 478–481.14. Bonneville, M.A. (1963) Fine structuralchanges in the intestinal epithelium of thebullfrog during metamorphosis. J Cell Biol 18,579–597.15. Mortimore, G.E., and Schworer, C.M. (1977)Induction of autophagy by amino-aciddeprivation in perfused rat liver. Nature 270,174–176.16. Pfeifer, U., and Warmuth-Metz, M. (1983)Inhibition by insulin of cellular autophagy inproximal tubular cells of rat kidney. Am JPhysiol 244, E109-114.17. Seglen, P.O., and Gordon, P.B. (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation inisolated rat hepatocytes. Proc Natl Acad SciUSA 79, 1889–1892.18. Arstila, A.U., and Trump, B.F. (1968) Studieson cellular autophagocytosis. The formation ofautophagic vacuoles in the liver after glucagonadministration. Am J Pathol 53, 687–733.19. Takeshige, K., Baba, M., Tsuboi, S., Noda, T.,and Ohsumi, Y. (1992) Autophagy in yeastdemonstrated with proteinase-deficientmutants and conditions for its induction. J CellBiol 119, 301–311.20. Tsukada, M., and Ohsumi, Y. (1993) Isolationand characterization of autophagy-defectivemutants of Saccharomyces cerevisiae. FEBSLett 333, 169–174.21. Klionsky, D.J., Cregg, J.M. Dunn, W.A. Jr.,Emr, S.D., Sakia, J., Sandoval, I.V., Sibirnya,Y.A., Subramani, S., Thumm, M., Veenhuis,M., and Ohsumi, Y. (2003) A unifiednomenclature for yeast autophagy-relatedgenes. Dev Cell 5, 539-545.22. Kametaka, S., Matsuura, A., Wada Y., andOhsumi, Y. (1996) Structural and functionalanalyses of APG5, a gene involved inautophagy in yeast. Gene 178, 139-43.23. Funakoshi, T., Matsuura, A., Noda, T.,Ohsumi Y. (1997) Analyses of APG13 geneinvolved in autophagy in yeast,Saccharomyces cerevisiae.Gene. 192, 207-213.24. Matsuura, A., Tsukada, M., Wada, Y., andOhsumi, Y. (1997) Apg1p, a novel proteinkinase required for the autophagic process inSaccharomyces cerevisiae. Gene 192, 245–250.25. Kamada, Y., Funakoshi, T., Shintani, T.,Nagano, K., Ohsumi, M., and Ohsumi, Y.(2000) Tor-mediated induction of autophagyvia an Apg1 protein kinase complex. J CellBiol 150, 1507–1513.26. Ohsumi, Y. (2014) Historical landmarks ofautophagy research. Cell Res 24, 9–23.27. Kirisako, T., Baba, M., Ishihara, N., Miyazawa,K., Ohsumi, M., Yoshimori, T., Noda, T., andOhsumi, Y. (1999) Formation process ofautophagosome is traced with Apg8/Aut7p inyeast. J Cell Biol 147, 435–446.28. Mizushima, N., Noda, T., Yoshimori, T.,Tanaka, Y., Ishii, T., George, M.D., Klionsky,D.J., Ohsumi, M., and Ohsumi, Y. (1998) Aprotein conjugation system essential forautophagy. Nature 395, 395–398.29. Shintani, T., Mizushima, N., Ogawa, Y.,Matsuura, A., Noda, T., and Ohsumi, Y. (1999)Apg10p, a novel protein-conjugating enzymeessential for autophagy in yeast. EMBO J 18,5234–5241.30. Mizushima, N., Noda, T., and Ohsumi, Y.(1999) Apg16p is required for the function ofthe Apg12p-Apg5p conjugate in the yeastautophagy pathway. EMBO J 18, 3888–3896. 31. Ichimura, Y., Kirisako, T., Takao, T., Satomi,Y., Shimonishi, Y., Ishihara, N., Mizushima,N., Tanida, I., Kominami, E., Ohsumi, M., et al.(2000) A ubiquitin-like system mediatesprotein lipidation. Nature 408, 488–492.32. Hanada, T., Noda, N.N., Satomi, Y., Ichimura,Y., Fujioka, Y., Takao, T., Inagaki, F., andOhsumi, Y. (2007) The Atg12-Atg5 conjugatehas a novel E3-like activity for proteinlipidation in autophagy. J Biol Chem 282,37298–37302.33. Nakatogawa, H., Ichimura, Y., and Ohsumi, Y.(2007) Atg8, a ubiquitin-like protein requiredfor autophagosome formation, mediates membrane tethering and hemifusion. Cell 130,165–178.34. Xie Z., Nair U., Klionsky D.J. (2008) ATG8controls phagophore expansion during autophagosome formation. Mol Cell Biol 19,3290-3298.35. Kabeya, Y., Mizushima, N., Ueno, T.,Yamamoto, A., Kirisako, T., Noda, T.,Kominami, E., Ohsumi, Y., and Yoshimori, T.(2000) LC3, a mammalian homologue of yeastApg8p, is localized in autophagosome membranes after processing. EMBO J 19,5720–5728. 36. Mizushima, N., Yamamoto, A., Matsui, M.,Yoshimori, T., and Ohsumi, Y. (2004) In vivoanalysis of autophagy in response to nutrientstarvation using transgenic mice expressing afluorescent autophagosome marker. Mol BiolCell 15, 1101–1111.37. Kuma, A., Hatano, M., Matsui, M., Yamamoto,A., Nakaya, H., Yoshimori, T., Ohsumi, Y.,Tokuhisa, T., and Mizushima, N. (2004) Therole of autophagy during the early neonatalstarvation period. Nature 432, 1032–1036. 38. Mizushima, N., and Komatsu, M. (2011)Autophagy: Renovation of cells and tissues.Cell 147, 728-741.39. Liu, L., Sakakibara, K., Chen, Q., Okamoto, K.(2014) Receptor-mediated mitophagy in yeastand mammalian systems. Cell Res 24, 787-795.40. Li, W.W., Li, J., Bao, J.K. (2012)Microautophagy: lesser-known self-eating.Cell Mol Life Sci 69, 1125-1136.41. Cuervo, A.M., and Wong, E. (2014)Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24, 92–104.42. Liang, X.H., Jackson, S., Seaman, M., Brown,K., Kempkes, B., Hibshoosh, H., and Levine,B. (1999) Induction of autophagy andinhibition of tumorigenesis by beclin 1. Nature402, 672–676.43. Choi, A.M.K., Ryter, S.W., and Levine, B.(2013) Autophagy in human health anddisease. N Engl J Med 368, 651–662.44. Ravikumar, B., Vacher, C., Berger, Z., Davies,J.E., Luo, S., Oroz, L.G., Scaravilli, F., Easton,D.F., Duden, R., O'Kane, C.J., et al. (2004)Inhibition of mTOR induces autophagy andreduces toxicity of polyglutamine expansionsin fly and mouse models of Huntingtondisease. Nat Genet 36, 585–595.45. Ravikumar, B., Duden, R., and Rubinsztein,D.C. (2002) Aggregate-prone proteins withpolyglutamine and polyalanine expansionsare degraded by autophagy. Hum Mol Genet11, 1107–1117.46. Komatsu, M., Waguri, S., Chiba, T., Murata,S., Iwata, J.-I., Tanida, I., Ueno, T., Koike, M.,Uchiyama, Y., Kominami, E., et al. (2006)Loss of autophagy in the central nervoussystem causes neurodegeneration in mice.Nature 441, 880–884.47. Hara, T., Nakamura, K., Matsui, M.,Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K.,Saito, I., Okano, H., et al. (2006) Suppressionof basal autophagy in neural cells causesneurodegenerative disease in mice. Nature441, 885–889.48. Ebrahimi-Fakhari, D., Saffari, A., Wahlster, L.,Lu, J., Byrne, S., Hoffmann, G.F., Jungbluth,H., and Sahin, M. (2016) Congenital disordersof autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain 139,317–337.49. Nakagawa, I., Amano, A., Mizushima, N.,Yamamoto, A., Yamaguchi, H., Kamimoto, T.,Nara, A., Funao, J., Nakata, M., Tsuda, K., etal. (2004) Autophagy defends cells against invading group A Streptococcus. Science 306,1037–1040. 50. Gutierrez, M.G., Master, S.S., Singh, S.B.,Taylor, G.A., Colombo, M.I., and Deretic, V.(2004) Autophagy is a defense mechanisminhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages.Cell 119, 753–766.Nils-Göran Larsson, MD, PhDProfessor of Mitochondrial Genetics, Karolinska InstitutetAdjunct Member of the Nobel CommitteeMember of the Nobel AssemblyMaria G. Masucci, MD, PhDProfessor of Virology, Karolinska InstitutetAdjunct Member of the Nobel CommitteeMember of the Nobel AssemblyIllustration: Mattias Karlén*FootnotesAdditional information on previous Nobel Prize Laureates mentioned in this text can be found at/The Nobel Prize in Physiology or Medicine 1974 to Albert Claude, Christian de Duve and George E. Palade “for their discoveries concerning the structural and functional organization of the cell”/nobel_prizes/medicine/laureates/1974/claude-facts.html/nobel_prizes/medicine/laureates/1974/duve-facts.html/nobel_prizes/medicine/laureates/1974/palade-facts.htmlGlossary of Terms:Lysosome:an organelle in the cytoplasm of eukaryotic cells containing degradative enzymes enclosed in a membrane.Phagophore: a vesicle that is formed during the initial phases of macroautophagy. The phagophore is extended by the autophagy machinery to engulf cytoplasmiccomponents.Autophagosome:an organelle that encloses parts of the cytoplasm into a double membrane that fuses to the lysosome where its content is degraded. The autophagosome is thekey structure in macroautophagy.Selective autophagy: a type of macroautophagy that mediates the degradation of specific cytoplasmic components. Different forms of selective autophagy are called mitophagy(degrades mitochondria), ribophagy (degrades ribosomes), lipophagy (degradeslipid droplets) xenophagy (degrades invading microorganisms) etc.。
如何在顶级杂志上发表文章1 鲁白教授

• New techniques and methods that can be widely used
人类基因组研究中的自动测序技术 , PCR, Patch clamp
• Discoveries with obvious practical implications
• Support existing idea, “me too”
EGF-R endocytosis requires dynamin, PDGF-R too.
• Follow up
CREB binds to CRE. Working out CRE sequence.
• Incomplete study, preliminary
Procedures for High Profile Journals
You Editors
• • • • • • • •
Pre-submission inquiry Submit/cover letter Initial screen Send out for reviews Reject/soft reject/revise Rebuttal Revise again Accept
563–574
575–585
587–597
599–611
613–623
What Makes Good Science?
• Important and significant • Original and innovative
• Solid and rigorous • Unique and unusual
Novelty is essential Marathon
Interactions Between the Microbiotaand the Immune System

The Gut MicrobiotaREVIEWInteractions Between the Microbiota and the Immune SystemLora V.Hooper,1*Dan R.Littman,2Andrew J.Macpherson 3The large numbers of microorganisms that inhabit mammalian body surfaces have a highly coevolved relationship with the immune system.Although many of these microbes carry out functions that are critical for host physiology,they nevertheless pose the threat of breach with ensuing pathologies.The mammalian immune system plays an essential role in maintaining homeostasis with resident microbial communities,thus ensuring that the mutualistic nature of the host-microbial relationship is maintained.At the same time,resident bacteria profoundly shape mammalian immunity.Here,we review advances in our understanding of the interactions between resident microbes and the immune system and the implications of these findings for human health.Complex communities of microorganisms,termed the “microbiota,”inhabit the body surfaces of virtually all vertebrates.In the lower intestine,these organisms reach extraordi-nary densities and have evolved to degrade a variety of plant polysaccharides and other dietary substances (1).This simultaneously enhances host digestive efficiency and ensures a steady nutrient supply for the microbes.Metabolic efficiency was likely a potent selective force that shaped the evolution of both sides of the host-microbiota lions of years of coevolution,however,have forged pervasive interconnections between the physiologies of microbial commu-nities and their hosts that extend beyond metabolic functions.These interconnections are particularly apparent in the relationship between the microbiota and the immune system.Despite the symbiotic nature of the intestinal host-microbial relationship,the close association of an abundant bacterial community with intesti-nal tissues poses immense health challenges.The dense communities of bacteria in the lower intes-tine (≥1012/cm 3intestinal contents)are separated from body tissues by the epithelial layer (10m m)over a large intestinal surface area (~200m 2in humans).Opportunistic invasion of host tissue by resident bacteria has serious health consequences,including inflammation and sepsis.The immune system has thus evolved adaptations that work to-gether to contain the microbiota and preserve the symbiotic relationship between host and microbiota.The evolution of the vertebrate immune system has therefore been driven by the need to protect thehost from pathogens and to foster complex micro-bial communities for their metabolic benefits (2).In this Review,we survey the state of our understanding of microbiota-immune system in-teractions.We also highlight key experimental challenges that must be confronted to advance our understanding in this area and consider how our knowledge of these interactions might be harnessed to improve public health.Tools for Analyzing the Microbiota –Immune System RelationshipMuch of our current understanding of microbiota –immune system interactions has been acquired from studies of germ-free animals.Such animals are reared in sterile isolators to control their exposure to microorganisms,including viruses,bacteria,and eukaryotic parasites.Germ-free animals can be studied in their microbiologically sterile state or can serve as living test tubes for the establishment of simplified microbial ecosystems composed of a single microbial species or defined species mixtures.The technology has thus come to be known as “gnotobiotics,”a term derived from Greek meaning “known life.”Gnotobiotic ani-mals,particularly rodents,have become critical experimental tools for determining which host immune functions are genetically encoded and which require interactions with microbes.The current impetus for gnotobiotic exper-imentation has been driven by several impor-tant technical advances.First,because any mouse strain can be derived to germ-free status (3),large numbers of genetically targeted and wild-type inbred isogenic mouse strains have become avail-able in the germ-free state.The contribution of different immune system constituents to host-microbial mutualism can thus be determined by comparing the effects of microbial colonization in genetically altered and wild-type mice (4,5).Second,next-generation sequencing tech-nologies have opened the black box of micro-biota complexity.Although advances in ex vivo culturability are still needed,the composition ofhuman and animal microbiotas can be opera-tionally defined from polymorphisms of bacterial genes,especially those encoding the 16S ribo-somal RNA sequences.Such analyses have made possible the construction of defined microbiotas,whose distinct effects on host immunity can now be examined (6).Moreover,these advances allow the study of experimental animals that are both isobiotic and,in a defined inbred host,isogenic.A dominant goal of these efforts is to benefit hu-man health [see Blumberg and Powie (7)].With the developing technology,the species differ-ences can be closed using mice with a defined humanized microbiota (8).On the horizon,there is even the prospect of humanized isobiotic mice that also have a humanized immune system (9).A third advance has been the development of experimental systems that allow the uncoupling of commensal effects on the immune system from microbial colonization.This cannot be achieved by antibiotic treatment alone because a small pro-portion of the targeted microbes will persist.Deletion strains of bacteria lacking the ability to synthesize prokaryotic-specific amino acids have been developed that can be grown in culture but do not persist in vivo,so the animals become germ-free again.This allows issues of mucosal immune induction,memory,and functional protection to be explored without permanent colonization (10).Finally,important insights about the impact of resident microbial communities on mammalian host biology have been acquired by using high-throughput transcriptomic and metabolomic tools to compare germ-free and colonized mice (11,12).These tools include DNA microarrays,which have led to a detailed understanding of how microbiota shape many aspects of host physiology,includ-ing immunity (13,14)and development (15),as well as mass spectrometry and nuclear magnetic resonance spectroscopy,which have provided im-portant insights into how microbiota influence metabolic signaling in mammalian hosts (12).The application of these new approaches to the older technology of gnotobiotics has revolutionized the study of interactions between the microbiota and the immune system.Looking Inside-Out:Immune System Control of the MicrobiotaA major driving force in the evolution of the mammalian immune system has been the need to maintain homeostatic relationships with the microbiota.This encompasses control of micro-bial interactions with host tissues as well as the composition of microbial consortia.Here,we dis-cuss recent insights into how the immune system exerts “inside-out ”control over microbiota local-ization and community composition (see Fig.1).Stratification and compartmentalization of the microbiota.The intestinal immune system faces unique challenges relative to other organs,as it must continuously confront an enormous micro-bial load.At the same time,it is necessary to avoid1The Howard Hughes Medical Institute and Department of Im-munology,The University of Texas Southwestern Medical Center at Dallas,Dallas,TX 75390,USA.2Howard Hughes Medical Institute and Molecular Pathogenesis Program,The Kimmel Center for Biology and Medicine of the Skirball Institute,New York University School of Medicine,New York,NY 10016,USA.3Maurice Müller Laboratories,University Clinic for Visceral Sur-gery and Medicine,University of Bern,Bern,Switzerland.*To whom correspondence should be addressed.E-mail:lora.hooper@8JUNE 2012VOL 336SCIENCE1268 o n M a y 20, 2015w w w .s c i e n c e m a g .o r g D o w n l o a d e d f r o mpathologies arising from innate immune signaling or from microbiota alterations that disturb essential metabolic functions.An important function of the intestinal immune system is to control the expo-sure of bacteria to host tissues,thereby lessening the potential for pathologic outcomes.This oc-curs at two distinct levels:first,by minimizing direct contact between intestinal bacteria and the epithelial cell surface(stratification)and,second, by confining penetrant bacteria to intestinal sites and limiting their exposure to the systemic im-mune compartment(compartmentalization).Several immune effectors function together to stratify luminal microbes and to minimize bacterial-epithelial contact.Intestinal goblet cells secrete mucin glycoproteins that assemble into a~150-m m-thick viscous coating at the intestinal epithelial cell surface.In the colon,there are two structurally distinct mucus layers.Although the outer mucus layer contains large numbers of bacteria,the inner mucus layer is resistant to bacterial penetration (16).In contrast,the small intestine lacks clearly distinct inner and outer mucus layers(17).Here, compartmentalization depends in part on antibac-terial proteins that are secreted by the intestinal epithelium.RegIII g is an antibacterial lectin that is expressed in epithelial cells under the control of Toll-like receptors(TLRs)(18–20).RegIII g limits bacterial penetration of the small intestinal mucus layer,thus restricting the number of bacteria that contact the epithelial surface(5).Stratification of intestinal bacteria on the luminal side of the epithelial barrier also depends on secreted immunoglobulin A(IgA).IgA spe-cific for intestinal bacteria is produced with the help of intestinal dendritic cells that sample the small numbers of bacteria that penetrate the over-lying epithelium.These bacteria-laden dendritic cells interact with B and T cells in the Peyer’s patches,inducing B cells to produce IgA directed against intestinal bacteria(21).IgA+B cells home to the intestinal lamina propria and secrete IgA that is transcytosed across the epithelium and deposited on the apical surface.The transcytosed IgAs bind to luminal bacteria,preventing micro-bial translocation across the epithelial barrier(22).Mucosal compartmentalization functions to minimize exposure of resident bacteria to the sys-temic immune system(Fig.1B).Although bacteria are largely confined to the luminal side of the epithelial barrier,the sheer number of intestinal bacteria makes an occasional breach inevita-ble.Typically,commensal microorganisms that penetrate the intestinal epithelial cell barrier are phagocytosed and eliminated by lamina propria macrophages(23).However,the intestinal im-mune system samples some of the penetrant bac-teria,engendering specific immune responses that are distributed along the length of the intes-tine(21).Bacteria that penetrate the intestinal barrier are engulfed by dendritic cells(DCs)re-siding in the lamina propria and are carried alive to the mesenteric lymph nodes.However,these bacteria do not penetrate to systemic secondarylymphoid tissues.Rather,the commensal-bearingDCs induce protective secretory IgAs(21),whichare distributed throughout all mucosal surfacesby recirculation of activated B and T cells.Thus,distinctive anatomical adaptations in the mucosalimmune system allow immune responses directedagainst commensals to be distributed widely whilestill being confined to mucosal tissues.Other immune cell populations also promotethe containment of commensal bacteria to in-testinal sites.Innate lymphoid cells reside in thelamina propria and have effector cytokine pro-files resembling those of T helper(T H)cells(24).Innate lymphoid cells that produce interleukin(IL)–22are essential for containment of lymphoid-resident bacteria to the intestine,thus preventingtheir spread to systemic sites(25).The compartmentalization of mucosal andsystemic immune priming can be severely per-turbed in immune-deficient mice.For example,mice engineered to lack IgA show priming ofserum IgG responses against commensals,indi-cating that these bacteria have been exposed tothe systemic immune system(22).A similar out-come is observed when innate immune sensingisFig.1.Looking inside-out:immune system control of the microbiota.Several immune effectors function together to stratify luminal microbes and to minimize bacterial-epithelial contact.This includes the mucus layer,epithelial antibacterial proteins,and IgA secreted by lamina propria plasma partmen-talization is accomplished by unique anatomic adaptations that limit commensal bacterial exposure to the immune system.Some microbes are sampled by intestinal DCs.The loaded DCs traffic to the mesenteric lymph nodes through the intestinal lymphatics but do not penetrate further into the body.This compartmentalizes live bacteria and induction of immune responses to the mucosal immune system. There is recirculation of induced B cells and some T cell subsets through the lymphatics and the bloodstream to home back to mucosal sites,where B cells differentiate into IgA-secreting plasma cells. SCIENCE VOL3368JUNE20121269SPECIAL SECTIONThe Gut Microbiotadefective.Mice lacking MyD88or TRIF signal-ing adaptors for TLR-mediated sensing of bacteria also produce serum IgG responses against com-mensals(26).This probably results from the fact that in these settings,large numbers of commensals cross the epithelial barrier and phagocytic cells are less able to eliminate the penetrant organisms.Immune system control of microbiota com-position.The development of high-throughput sequencing technologies for microbiota analysis has provided insight into the many factors that determine microbiota composition.For example nutrients,whether derived from the host diet (27)or from endogenous host sources(28),are critically important in shaping the structure of host-associated microbial communities.Recent evidence suggests that the immune system is also likely to be an important contributor to“inside-out”host control over microbiota composition.Certain secreted antibacterial proteins produced by epithelial cells can shape the composition of in-testinal microbial communities.a-defensins are small(2to3kD)antibacterial peptides secreted by Paneth cells of the small intestinal epithelium.Anal-ysis of the microbiota in mice that were either de-ficient in functional a-defensins or that overexpressed human a-defensin-5showed that although there was no impact on total numbers of colonizing bacte-ria,there were substantial a-defensin–dependent changes in community composition,with reciprocal differences observed in the two mouse strains(29).An interesting question is how far secreted in-nate immune effectors“reach”into the luminal microbial consortia.For example,the impact of hu-man a-defensin-5on luminal community composi-tion contrasts with the antibacterial lectin RegIII g, which limits penetration of bacteria to the epithelial surface but does not alter luminal communities(5). This suggests that some antimicrobial proteins,such as a-defensins,reach into the lumen to shape overall community composition,whereas others,such as RegIII g,have restricted effects on surface-associated bacteria and thus control microbiota location relative to host surface tissues.Questions remain as to ex-actly how a-defensin-5controls luminal community composition,however.In one scenario,these small antimicrobial peptides diffuse through the mucus layer and directly act on bacteria that inhabit the lu-men.Another possibility is that a-defensin-5exerts its antibacterial activity on bacteria that are trapped in the outer reaches of the mucus layer,with those bac-teria acting as reservoirs that seed luminal commu-nities and thus dictate their composition.Answering these questions will require improved tools for fine-mapping microbiota composition and consortia from the surface of the intestine to the interior of the lumen.The impact of the immune system on micro-biota composition is also suggested by several im-mune deficiencies that alter microbial communities in ways that predispose to disease.For example, Garrett et al.studied mice that lack the transcription factor T-bet(encoded by Tbx21),which governs inflammatory responses in cells of both the innate and the adaptive immune system(30).WhenTbx21–/–mice were crossed onto Rag2–/–mice,which lack adaptive immunity,the Tbx21–/–/Rag2–/–progeny developed ulcerative colitis in a microbiota-dependent manner(30).Remarkably,this colitisphenotype was transmissible to wild-type mice byadoptive transfer of the Tbx21–/–/Rag2–/–micro-biota.This demonstrated that altered microbiotawere sufficient to induce disease and could thus beconsidered“dysbiotic.”Similarly,mice lacking thebacterial flagellin receptor TLR5exhibit a syn-drome encompassing insulin resistance,hyper-lipidemia,and increased fat deposition associatedwith alterations in microbiota composition(31).These metabolic changes are transferable to wild-type mice that acquire the Tlr5–/–gut microbiota.A third example of immune-driven dysbiosis isseen in mice deficient for epithelial cell expres-sion of the inflammasome component NLRP6.These mice develop an altered microbiota withincreased abundance of members of the Bacte-roidetes phylum associated with increased intes-tinal inflammatory cell recruitment and susceptibilityto chemically induced colitis.Again,there is evi-dence that dysbiosis alone is sufficient to drive theintestinal inflammation,because conventionallyraised wild-type mice that acquire the dysbioticmicrobiota show similar immunopathology(32).Together,these findings suggest that the im-mune system affords mammalian hosts some con-trol over the composition of their resident microbialcommunities.It is also clear that these commu-nities can be perturbed by defects in the host im-mune system.This leads to the idea of the immunesystem as a form of ecosystem management thatexerts critical control over microbiota compo-sition,diversity,and location[see Costello et al.(33)].However,a number of questions remain.First,although it is apparent that the immune sys-tem shapes community composition at the specieslevel,it is not yet clear whether the immune sys-tem shapes the genetics and physiology of indi-vidual microbial species.Second,how much doesthe immune system combine with gastric acid andintestinal motility to control the longitudinal dis-tribution of microbial species in the gastrointes-tinal tract?Finally,it will be important to determinethe extent to which the immune system also con-trols microbial community composition and loca-tion in other organ systems,such as the respiratorytract,urogenital tract,and skin.Looking Outside-In:How MicrobiotaShape ImmunityThe earliest comparisons of germ-free and colonizedmice revealed a profound effect of microbial colo-nization on the formation of lymphoid tissues andsubsequent immune system development.It wasthus quickly apparent that the microbiota influ-ence the immune system from“outside-in.”Recentstudies have greatly amplified this understandingand have revealed some of the cellular and mo-lecular mediators of these interactions(see Fig.2).The impact of the microbiota on lymphoidstructure development and epithelial function.The tissues of the gastrointestinal tract are rich inmyeloid and lymphoid cells,many of whichreside in organized lymphoid tissues.It has longbeen appreciated that the gut microbiota have acritical role in the development of organized lym-phoid structures and in the function of immunesystem cells.For example,isolated lymphoid fol-licles in the small intestine do not develop ingerm-free mice,and such mice are also deficientin secretory IgA and CD8ab intraepithelial lym-phocytes.The specific microbial molecules en-dowed with this inductive function have not yetbeen described,however.Sensing of commensal microbiota through theTLR-MyD88signaling pathway triggers severalresponses that are critical for maintaining host-microbial homeostasis.The microbiota inducerepair of damaged intestinal epithelium through aMyD88-dependent process that can be rescued inmicrobe-depleted animals by gavage with bacteriallipopolysaccharide(LPS).The innate signals,con-veyed largely through myeloid cells,are required toenhance epithelial cell proliferation(34,35).Asdiscussed above,MyD88-dependent bacterial sig-nals are also required for the induction of epithelialantimicrobial proteins such as RegIII g(5,19).Thisexpression can be induced by LPS(19,20)or flagel-lin(36).The flagellin signals are relayed throughTLR5expressed by CD103+CD11b+dendritic cellsin the lamina propria,stimulating production of IL-23that,in turn,promotes the expression of IL-22by innate lymphoid cells(37).IL-22then stimu-lates production of RegIII g,which is also secretedupon direct activation of MyD88in epithelialcells(5,20).This is one clear example of theimportance of commensals in the induction of hostinnate responses,but it likely represents a tinyfraction of the multitude of effects of microbiota onthe host immune system.Microbiota shaping of T cell subsets.It hasrecently become evident that individual commensalspecies influence the makeup of lamina propria Tlymphocyte subsets that have distinct effector func-tions.Homeostasis in the gut mucosa is maintainedby a system of checks and balances between poten-tially proinflammatory cells,which include T H1cellsthat produce interferon-g;T H17cells that produceIL-17a,IL-17f,and IL-22;diverse innate lymphoidcells with cytokine effector features resemblingT H2and T H17cells;and anti-inflammatory Foxp3+regulatory T cells(T regs).Colonization of mice withsegmented filamentous bacteria(SFB)results inaccumulation of T H17cells and,to a lesser extent,inan increase in T H1cells(38,39).SFB appear able topenetrate the mucus layer overlying the intestinalepithelial cells in the terminal ileum,and they in-teract closely with the epithelial cells,inducing hostcell actin polymerization at the site of interactionand,presumably,signaling events that result in aT H17polarizing environment within the laminapropria.There is little known about host cell8JUNE2012VOL336SCIENCE 1270signaling pathways initiated by SFB.It is possible that SFB influence epithelial gene expression,re-sulting,for example,in expression of antimicro-bial proteins such as RegIII g and of molecules that participate in T H 17cell polarization.SFB may also act directly on cells of the immune sys-tem,either through interactions with myeloid cells that extend processes through the epithelium to the mucus layer or by production of metabolites that act on various receptors expressed by host cells.Other bacteria have been shown to enhance the anti-inflammatory branches of the adaptive immune system by directing the differentiation of T regs or by inducing IL-10expression.For example,coloniza-tion of gnotobiotic mice with a complex cocktail of 46mouse Clostridial strains,originally isolated from mouse feces and belonging mainly to cluster IVand XIV a of the Clostridium genus,results in the expansion of lamina propria and systemic T regs .These have a phenotype characteristic of T regs in-duced in the periphery in response to transforming growth factor (TGF)–b and retinoic acid [in contrast to thymic-derived natural (n)T regs (40)],and manyof these inducible T regs (iT regs )express IL-10.The exact Clostridial strains within the complex exper-imental mixture that drive this regulatory response remain to be defined.Furthermore,polysaccharide A (PSA)of Bacteroides fragilis induces an IL-10response in intestinal T cells,which prevents the expansion of T H 17cells and potential damage to the mucosal barrier (41).In contrast,mutant B.fragilis lacking PSA has a proinflammatory profile and fails to induce IL-10.Production of PSA by B.fragilis has been proposed to be instrumental for the bac-terium ’s success as a commensal.Within the intestine,the balance of effector lym-phoid cells and T reg cells can have a profound in-fluence on how the mucosa responds to stresses that elicit damage.The relative roles of commensal-regulated Tcells differ according to the models used to study inflammation.For example,in mice sub-jected to chemical or pathogen-induced damage to the mucosa,T H 17cells have a beneficial effect that promotes healing.In contrast,T H 1and T H 17cells,as well as IL-23–dependent innate lymphoid cells,promote colitis in models in which T reg cells aredepleted.It is likely that inflammatory bowel dis-eases in humans can be similarly triggered by commensal-influenced imbalance of lymphoid cell subsets.This is supported by numerous observations,including the strong linkage of IL23R polymor-phisms with Crohn ’s disease,a serious condition with relapsing intestinal inflammation and a risk of malignancy,and the severe enterocolitis associated with IL10and IL10R mutations (42,43).Microbiota effects on systemic immunity.The influence of commensal bacteria on the balance of T cell subsets is now known to extend well beyond the intestinal lamina propria.Homeostatic T cell proliferation itself is driven by the microbiota or their penetrant molecules (44).Systemic auto-immune diseases have long been suggested to have links to infections,but firm evidence for causality has been lacking.Recent studies in animal models,however,have reinforced the notion that commen-sal microbiota contribute to systemic autoimmune and allergic diseases at sites distal to the intestinal mucosa.Several mouse models for autoimmunity are dependent on colonization status.Thus,germ-free mice have marked attenuation of disease in models of arthritis and experimental autoimmune encephalomyelitis (EAE),as well as in various colitis models.In models of T H 17cell –dependent arthritis and EAE,monoassociation with SFB is sufficient to induce disease (42,45,46).In all of these models,induction of T H 17cells in the in-testine has a profound influence on systemic dis-ease.Exacerbation of arthritis and EAE is likely the consequence of an increase in the number of arthritogenic or encephalitogenic T H 17cells that traffic out of the lamina propria.The antigen spec-ificity of such cells remains to be examined.Induction of iT regs by the cluster IV and XIV a Clostridia also has a systemic effect on inflamma-tory processes.Colonization of germ-free mice with these bacteria not only results in attenuated disease after chemical damage of the gut epithelium but also reduces the serum IgE response after immuni-zation with antigen under conditions that favor a T H 2response (40).As with pathogenic T H 17cells,the antigen specificity of the commensal-induced iT regs that execute systemic anti-inflammatory func-tions is not yet known,although at least some of the T regs in the gut have Tcell receptors with specificity for distinct commensal bacteria (47).Finally,B.fragilis PSA affects the develop-ment of systemic T cell responses.Colonization of germ-free mice with PSA-producing B.fragilis results in higher numbers of circulating CD4+T cells compared to mice colonized with B.fragilis lacking PSA.PSA-producing B.fragilis also elicits higher T H 1cell frequencies in the circulation (48).Together,these findings show that commen-sal bacteria have a general impact on immunity that reaches well beyond mucosal tissues.Microbiota influences on invariant Tcells and innate lymphoid cells.A recent study extends the role of microbiota to the control of the function invariant natural killer T cells (iNKT cells),whichFig.2.Looking outside-in:how microbiota shape host immunity.Some of the many ways that intestinal microbiota shape host immunity are depicted.These include microbiota effects on mucosal as well as systemic immunity.ILFs,isolated lymphoid follicles.SCIENCEVOL 3368JUNE 20121271SPECIAL SECTION。
The Biology of the Human Immune System

The Biology of the Human Immune System 人类免疫系统的生物学免疫系统是我们身体的一个重要组成部分,能够识别和击败各种细菌、病毒、真菌和寄生虫等入侵的异物。
人类免疫系统的生物学是一个庞大且复杂的领域,涉及许多不同的组织、细胞和蛋白质。
免疫系统的组成免疫系统主要由两个部分组成:先天性免疫系统和后天性免疫系统。
先天性免疫系统是人类生命的早期防线,能够对大多数病原体产生迅速、非特异性的反应。
先天性免疫系统包括皮肤和黏膜、炎症反应、天然杀伤细胞、补体系统和巨噬细胞等。
后天性免疫系统是高度特异性的防御系统,能够对特定的病原体产生精确的、特异性的反应。
后天性免疫系统由T细胞、B细胞、抗体和淋巴组织等组成。
皮肤与黏膜皮肤和黏膜是我们身体最重要的保护屏障,能够防止病原体进入我们的身体。
皮肤由多层角质细胞、汗腺和皮脂腺组成,能够防止水分的流失,并且排泄汗液和皮脂,以防止过度干燥和滋生病原体。
黏膜由多种不同类型的细胞和分泌物组成,能够阻止细菌和病毒进入我们的身体。
例如,鼻腔和喉咙内的细毛能够将病原体从我们的呼吸道移除,而胃酸则能够杀死许多进入我们体内的病菌。
天然杀伤细胞天然杀伤细胞是免疫系统的一种重要组成部分,能够直接识别并杀灭感染的细胞,特别是癌细胞。
天然杀伤细胞在人体内广泛分布,能够通过对目标细胞进行直接杀伤、分泌毒素和介导细胞的凋亡等方式,阻止病原体感染我们的身体。
巨噬细胞巨噬细胞是一种能够摄取和消化病原体的免疫细胞。
巨噬细胞的主要功能是引起炎症反应,并清除细胞碎片、细菌和其他异物。
巨噬细胞能够分泌调节因子,帮助其他免疫细胞定位病原体,并参与各种炎症和免疫过程。
淋巴组织淋巴组织是免疫系统的重要组成部分,包括淋巴结、脾脏和淋巴管等。
这些器官都是淋巴组织的重要部分,能够帮助身体识别和应对各种病原体。
T细胞和B细胞T细胞和B细胞是后天性免疫系统中最重要的组成部分,能够产生特异性的反应,对特定的病原体产生针对性的抗体。
激活小鼠的自噬方法

激活小鼠的自噬方法英文回答:Autophagy is a cellular process that involves the degradation and recycling of cellular components. It plays a crucial role in maintaining cellular homeostasis and has been implicated in various physiological and pathological conditions. Activating autophagy in mice can be achieved through several methods.One common method to activate autophagy in mice is through dietary restriction or caloric restriction. This involves reducing the amount of food intake or restricting the intake of specific nutrients. Caloric restriction has been shown to induce autophagy in various tissues,including the liver, muscle, and brain. For example, studies have shown that reducing calorie intake by 30-40% can significantly increase autophagy levels in the liver of mice.Another method to activate autophagy in mice is through pharmacological interventions. Several compounds have been identified that can induce autophagy. One such compound is rapamycin, which is an inhibitor of the mammalian target of rapamycin (mTOR) pathway. mTOR is a key regulator of autophagy, and inhibiting its activity can lead to autophagy activation. Rapamycin has been widely used to induce autophagy in various animal models, including mice. Other compounds, such as resveratrol and spermidine, have also been shown to induce autophagy in mice.In addition to dietary restriction and pharmacological interventions, exercise has also been shown to activate autophagy in mice. Exercise-induced autophagy has been observed in various tissues, including skeletal muscle and the heart. For example, studies have shown that endurance exercise can increase autophagy levels in skeletal muscle of mice. The exact mechanisms by which exercise induces autophagy are still not fully understood, but it is believed to involve the activation of AMP-activated protein kinase (AMPK) and the inhibition of mTOR signaling.中文回答:自噬是一种细胞过程,涉及细胞成分的降解和再利用。
免疫调节英文介绍作文

免疫调节英文介绍作文Immunomodulation refers to the regulation or modulation of the immune system. It involves the manipulation of the immune response to enhance or suppress immune activity as needed. This can be achieved through various means, such as the use of immunosuppressive drugs, vaccines, or natural remedies.Immunomodulation plays a crucial role in maintaining the balance of the immune system. It helps to prevent excessive immune responses that can lead to autoimmune diseases, allergies, or chronic inflammation. On the other hand, it can also boost the immune response in cases of weakened immunity, such as in cancer patients orindividuals with immunodeficiencies.One way to achieve immunomodulation is through the use of immunosuppressive drugs. These drugs work by suppressing the activity of the immune system, thus reducing inflammation and preventing the immune system fromattacking healthy cells. Examples of immunosuppressivedrugs include corticosteroids, methotrexate, and cyclosporine.Vaccines also play a significant role in immunomodulation. Vaccines contain antigens that stimulate the immune system to produce a protective immune response. This immune response can be targeted towards specific pathogens, such as bacteria or viruses, helping to prevent infections. Vaccines can also be used to boost the immune response in individuals with weakened immunity, such as the elderly or those with chronic illnesses.In addition to conventional medicine, natural remedies and lifestyle changes can also be used for immunomodulation. For example, certain herbs and supplements, such as echinacea or probiotics, have been found to have immunomodulatory effects. Regular exercise, a healthy diet, and stress management techniques can also help to support a balanced immune system.In conclusion, immunomodulation is a vital aspect ofmaintaining a healthy immune system. It involves the regulation and manipulation of the immune response to prevent or treat immune-related disorders. Whether through the use of drugs, vaccines, or natural remedies, immunomodulation aims to achieve a balanced immune response that is appropriate for the individual's needs.。
TLR signaling pathways

Seminars in Immunology 16(2004)3–9TLR signaling pathwaysKiyoshi Takeda,Shizuo Akira ∗Department of Host Defense,Research Institute for Microbial Diseases,Osaka University,and ERATO,Japan Science and Technology Corporation,3-1Yamada-oka,Suita,Osaka 565-0871,JapanAbstractToll-like receptors (TLRs)have been established to play an essential role in the activation of innate immunity by recognizing spe-cific patterns of microbial components.TLR signaling pathways arise from intracytoplasmic TIR domains,which are conserved among all TLRs.Recent accumulating evidence has demonstrated that TIR domain-containing adaptors,such as MyD88,TIRAP,and TRIF,modulate TLR signaling pathways.MyD88is essential for the induction of inflammatory cytokines triggered by all TLRs.TIRAP is specifically involved in the MyD88-dependent pathway via TLR2and TLR4,whereas TRIF is implicated in the TLR3-and TLR4-mediated MyD88-independent pathway.Thus,TIR domain-containing adaptors provide specificity of TLR signaling.©2003Elsevier Ltd.All rights reserved.Keywords:TLR;Innate immunity;Signal transduction;TIR domain1.IntroductionToll receptor was originally identified in Drosophila as an essential receptor for the establishment of the dorso-ventral pattern in developing embryos [1].In 1996,Hoffmann and colleagues demonstrated that Toll-mutant flies were highly susceptible to fungal infection [2].This study made us aware that the immune system,particularly the innate im-mune system,has a skilful means of detecting invasion by microorganisms.Subsequently,mammalian homologues of Toll receptor were identified one after another,and desig-nated as Toll-like receptors (TLRs).Functional analysis of mammalian TLRs has revealed that they recognize specific patterns of microbial components that are conserved among pathogens,but are not found in mammals.In signaling path-ways via TLRs,a common adaptor,MyD88,was first charac-terized as an essential component for the activation of innate immunity by all the TLRs.However,accumulating evidence indicates that individual TLRs exhibit specific responses.Furthermore,they have their own signaling molecules to manifest these specific responses.In this review,we will focus on the recent advances in our understanding of the mechanism of TLR-mediated signaling pathways.∗Correspondingauthor.Tel.:+81-6-6879-8303;fax:+81-6-6879-8305.E-mail address:sakira@biken.osaka-u.ac.jp (S.Akira).2.Toll-like receptorsA mammalian homologue of Drosophila Toll receptor (now termed TLR4)was shown to induce the expression of genes involved in inflammatory responses [3].In addi-tion,a mutation in the Tlr4gene was identified in mouse strains that were hyporesponsive to lipopolysaccharide [4].Since then,Toll receptors in mammals have been a major focus in the immunology field.First,several proteins that are structurally similar to TLR4were identified and named TLRs [5].The TLR family now consists of 10members (TLR1–TLR10).The cytoplasmic portion of TLRs shows high similarity to that of the interleukin (IL)-1receptor family,and is now called the Toll/IL-1receptor (TIR)do-main.Despite of this similarity,the extracellular portions of both types of receptors are structurally unrelated.The IL-1receptors possess an Ig-like domain,whereas TLRs bear leucine-rich repeats (LRRs)in the extracellular do-main.Genetic approaches have mainly been conducted to analyze the physiological function of TLRs,and have revealed essential roles for TLRs in the recognition of pathogens.Each TLR has been shown to recognize spe-cific components of pathogens,thus demonstrating that the mammalian immune system detects invasion by pathogens via the recognition of microbial components by TLRs (Fig.1).1044-5323/$–see front matter ©2003Elsevier Ltd.All rights reserved.doi:10.1016/j.smim.2003.10.0034K.Takeda,S.Akira /Seminars in Immunology 16(2004)3–9Fig.1.TLRs and their ligands.TLR1–TLR7and TLR9have been characterized to recognize microbial components.TLR2is essential for the recognition of microbial lipopeptides.TLR1and TLR6associate with TLR2,and discriminate subtle differences between triacyl-and diacyl lipopeptides,respectively.TLR4recognizes LPS.TLR9is the CpG DNA receptor,whereas TLR3is implicated in the recognition of viral dsRNA.TLR5is a receptor for flagellin.Thus,the TLR family discriminates between specific patterns of microbial components.3.Signaling pathways via TLRsThe activation of TLR signaling pathways originates from the cytoplasmic TIR domains.A crucial role for the TIR domain was first revealed in the C3H/HeJ mouse strain,which had a point mutation that resulted in an amino acid change of the cytoplasmic proline residue at position 712to histidine [4,6].This proline residue in the TIR domain is conserved among all TLRs,except for TLR3,and its substitution to histidine caused a dominant negative effect on TLR-mediated signaling [6,7].In the signaling pathway downstream of the TIR domain,a TIR domain-containing adaptor,MyD88,was first characterized to play a cru-cial role.In addition,recent accumulating evidence indi-cates that TLR signaling pathways consist,at least,of a MyD88-dependent pathway that is common to all TLRs,and a MyD88-independent pathway that is peculiar to the TLR3-and TLR4signaling pathways [8].4.MyD88-dependent pathwayMyD88possesses the TIR domain in the C-terminal por-tion,and a death domain in the N-terminal portion.MyD88associates with the TIR domain of TLRs.Upon stimulation,MyD88recruits IL-1receptor-associated kinase (IRAK)to TLRs through interaction of the death domains of both molecules.IRAK is activated by phosphorylation and then associates with TRAF6,leading to the activation of two dis-tinct signaling pathways,and finally to the activation of JNK and NF-B (Fig.2).4.1.MyD88MyD88knockout mice showed no responses to the TLR4ligand LPS in terms of macrophage production of inflamma-tory mediators,B cell proliferation,or endotoxin shock [9].The cellular responses to the TLR2ligands peptidoglycanand lipoproteins were abolished in MyD88knockout mice [10,11].Furthermore,cells from MyD88knockout mice showed no responses to the TLR9ligand CpG DNA and the TLR7ligand imidazoquinoline [12–14].Finally,MyD88knockout mice did not produce any IL-6in response to the TLR5ligand flagellin [15].These findings demonstrated that the TIR domain-containing adaptor MyD88is essential for the inflammatory responses mediated by all the TLR family members.An alternatively spliced variant of MyD88,MyD88s,which lacks the intermediate domain,has been shown to be induced by LPS stimulation and to inhibit LPS-induced NF-B activation through inhibition of IRAK activity [16,17].Thus,MyD88s may negatively regulate the inflam-matory responses triggered by LPS.4.2.IRAKIRAK was originally identified as a serine/threonine ki-nase associated with the IL-1receptor,which also harbors the TIR domain [18].Four members of the IRAK family have been identified so far:IRAK-1,IRAK-2,IRAK-M,and IRAK-4.IRAK proteins consist of an N-terminal death do-main,which is responsible for interaction with MyD88,and a central kinase domain.IRAK-1and IRAK-4harbor a criti-cal aspartate residue in the kinase domain,but this residue is not conserved in IRAK-2or IRAK-M,which causes them to be catalytically inactive [19].The importance of the IRAK family members in TLR-mediated signaling pathways was first demonstrated in IRAK-1knockout mice,which showed defective LPS-induced responses [20].IRAK-1knockout mice showed defective LPS responses,however,this impair-ment was only partial.In contrast,IRAK-4knockout mice showed almost complete impairment in the response to mi-crobial components that stimulate TLR2,TLR3,TLR4,and TLR9[21].A biochemical study revealed that IRAK-4acts upstream of,and phosphorylates,IRAK-1upon stimulation [22].Thus,IRAK-4is a central mediator of TLR signal-K.Takeda,S.Akira/Seminars in Immunology16(2004)3–95Fig.2.TLR-mediated MyD88-dependent signaling pathway.MyD88binds to the cytoplasmic portion of TLRs through interaction between individual TIR domains.Upon stimulation,IRAK-4,IRAK-1,and TRAF6are recruited to the receptor,which induces association of IRAK-1and MyD88via the death domains.IRAK-4then phosphorylates IRAK-1.Phosphorylated IRAK-1,together with TRAF6,dissociates from the receptor and then TRAF6 interacts with TAK1,TAB1,and TAB2.The complex of TRAF6,TAK1,TAB1,and TAB2further forms a larger complex with Ubc13and Uev1A,which induces the activation of TAK1.Activated TAK1phosphorylates the IKK complex,consisting of IKK␣,IKK,and NEMO/IKK␥,and MAP kinases, such as JNK,and thereby induces the activation of the transcription factors NF-B and AP-1,respectively.ing by activating IRAK-1.In sharp contrast to mice lacking IRAK-1and IRAK-4,IRAK-M knockout mice showed in-creased production of inflammatory cytokines in response to the TLR ligands and exaggerated inflammatory response to bacterial infection,demonstrating that IRAK-M plays a negative inhibitory role in the TLR signaling pathway[23].4.3.TRAF6and downstream moleculesTRAF6is a member of the tumor necrosis factor recep-tor(TNFR)-associated factor(TRAF)family that mediates cytokine signaling pathways[24].TRAF proteins consist of two C-terminal TRAF domains(TRAF-N and TRAF-C), which are responsible for interaction with TRAF proteins and other signaling molecules,N-terminal RINGfinger,and zincfinger domains.Among the TRAF family members, TRAF6has been shown to be involved in the TLR sig-naling pathway in addition to signaling pathways via the OPGL receptor and CD40[25,26].Upon stimulation of TLRs,TRAF6is recruited to the receptor complex,and acti-vated by IRAK-1that binds to the TRAF domain of TRAF6. Then,the IRAK-1/TRAF6complex dissociates from the re-ceptor and associates with TGF--activated kinase1(TAK1) and TAK1-binding proteins,TAB1and TAB2,at the mem-brane portion.IRAK-1stays in the membrane and is de-graded,whereas the complex of TRAF6,TAK1,TAB1,and TAB2moves into the cytoplasm,where it forms a large complex with other proteins,such as the E2ligases Ubc13 and Uev1A[27].The Ubc13and Uev1A complex has been shown to catalyze the synthesis of a Lys63-linked polyubiq-uitin chain of TRAF6and thereby induce TRAF6-mediated activation of TAK1andfinally of NF-B[28].4.4.Other moleculesIn addition to the molecules described above,several other molecules have been implicated in the TLR-mediated signaling pathway.Toll-interacting protein(Tollip)wasfirst identified in an analysis of IL-1signaling[29].Tollip is present in a complex with IRAK-1.Upon stimulation with IL-1,the Tollip-IRAK-1complex is recruited to the IL-1 receptor complex.IRAK-1is then phosphorylated,which leads to the rapid dissociation of IRAK-1from Tollip, thereby inducing activation of TRAF6.Subsequently,Tollip has been shown to negatively regulate the TLR-mediated signaling pathway[30,31].Overexpression of Tollip inhib-ited activation of NF-B in response to IL-1,the TLR2and TLR4ligands.However,it remains unclear how Tollip is physiologically involved in TLR signaling.Pellino was originally identified in Drosophila as a molecule that associates with Pelle,a Drosophila homo-logue of IRAK.In mammals,two Pellino homologues,6K.Takeda,S.Akira/Seminars in Immunology16(2004)3–9Pellino-1and Pellino-2,have been identified.Both Pellino-1 and Pellino-2have been shown to interact with IRAK-1 in response to IL-1stimulation[32,33].Ectopic expres-sion of the Pellino-2antisense construct inhibited IL-1-or LPS-induced activation of the NF-B-dependent promoter, indicating that Pellino-2is involved in the IL-1and TLR4 signaling pathways.Thus,several molecules that may mod-ulate TLR signaling have been identified.5.MyD88-independent pathwayAs described above,MyD88knockout mice did not show any production of inflammatory cytokines,such as TNF-␣and IL-12,in response to any of the TLR ligands. Furthermore,activation of NF-B and JNK in response to the TLR2,TLR7,and TLR9ligands was not observed in MyD88knockout mice.However,in the case of TLR4 stimulation,LPS-induced activation of NF-B and JNK was observed with delayed kinetics,even in MyD88knockout cells,although these cells did not produce any inflam-matory cytokines in response to LPS[9].In an attempt to assess the role of LPS-induced signal activation in a MyD88-independent manner,a subtraction analysis was performed using mRNA extracted from non-stimulated and LPS-stimulated MyD88knockout macrophages[34]. This analysis revealed that IFN-inducible genes,such as IP-10and GARG16,were induced in response to LPS in MyD88knockout cells.Subsequent studies clearly demon-strated that there is a MyD88-independent pathway as well as a MyD88-dependent pathway in TLR signaling.In the MyD88-independent pathway,LPS stimulation leads to activation of the transcription factor IRF-3,and thereby in-duces IFN-.IFN-,in turn,activates Stat1,leading to the induction of several IFN-inducible genes[35–37].In addition to the TLR4ligand,the TLR3ligand dsRNA has been shown to induce activation of NF-B in MyD88 knockout cells[38].Virus and viral-derived dsRNA are po-tent activators of IRF-3,which leads to the initial phase of IFN-induction[39–41].Thus,the TLR3ligand dsRNA also activates the MyD88-independent signaling pathway, in which IRF-3plays a key role.Recently,two independent groups identified kinases responsible for the activation of IRF-3.Hiscott and colleagues tried to identify molecules that interact with IRF-3by two-hybrid screening,and found that IRF-3was associated with IB kinases(IKKs)[42]. IKKs are composed of IKK␣and IKK,both of which phosphorylate Ser32and Ser36of IB␣,thereby inducing NF-B activation.In addition,there are two noncanonical IKKs,TANK-binding kinase1(TBK1)and IKKε/IKK i, which have distinct kinase activities compared with the canonical IKK␣and IKK.They analyzed whether these four IKKs could phosphorylate IRF-3using an in vitro ki-nase assay,and found that TBK1and IKKε/IKK i induced IRF-3phosphorylation.RNAi-mediated ablation of TBK1 and IKKε/IKK i resulted in inhibition of virus-induced phos-phorylation of IRF-3.Maniatis and colleagues also found that overexpression of TBK1and IKKε/IKK i led to acti-vation of IRF-3and induction of IFN-[43].They also showed that reduced expression of TBK1and IKKε/IKK i by RNAi led to impaired induction of IFN-in response to virus and dsRNA.Thus,TBK1and IKKε/IKK i have been shown to be critical regulators of IRF-3activation,leading to the induction of IFN-in response to the TLR3ligand. At present,it remains unclear whether these noncanonical IKKs are involved in TLR4-mediated IRF-3activation. Although TBK1knockout mice have been characterized, involvement of TBK1in the MyD88-independent path-way has not been analyzed in these mice[44].Studies with TBK1and IKKε/IKK i knockout mice will clarify the involvement of these IKKs in the MyD88-independent pathway.6.TIR domain-containing adaptorsDuring analysis of the MyD88-independent pathway,two TIR domain-containing adaptors,TIR domain-containing adaptor protein(TIRAP)/MyD88-adaptor-like(Mal)and TIR domain-containing adaptor inducing IFN-(TRIF)/TIR domain-containing adaptor molecule(TICAM-1),were identified[45–48].Analysis of these two adaptors indi-cated that TIR domain-containing adaptors regulate the TLR-mediated signaling pathways by providing specificity for individual TLR signaling cascades(Fig.3).6.1.TIRAP/MalDatabase search analyses led to the identification of a second TIR domain-containing molecule,which was named TIRAP or Mal[45,46].TIRAP/Mal harbors the TIR domain in the C-terminus.Initial in vitro studies in-dicated that TIRAP/Mal specifically interacts with TLR4, and is involved in the TLR4-mediated MyD88-independent signaling pathway.However,generation of TIRAP/Mal knockout mice revealed an unexpected role of TIRAP/Mal in TLR signaling[49,50].Similarly to MyD88knockout macrophages,TIRAP/Mal knockout macrophages showed impaired inflammatory cytokine production and delayed ac-tivation of JNK and NF-B in response to the TLR4ligand. However,TLR4ligand-induced activation of IRF-3and ex-pression of IFN-inducible genes was normally observed in TIRAP/Mal knockout macrophages.Even in mice lacking both MyD88and TIRAP/Mal,the TLR4ligand-induced expression of IFN-inducible genes was not impaired.Thus, TIRAP/Mal is critically involved in the MyD88-dependent pathway,but not in the MyD88-independent pathway,via TLR4.TIRAP/Mal knockout mice showed normal re-sponses to the TLR3,TLR5,TLR7,and TLR9ligands, but were defective in TLR2ligand-induced inflamma-tory cytokine production.Taken together,these studies clearly established that TIRAP/Mal is essential for theK.Takeda,S.Akira/Seminars in Immunology16(2004)3–97Fig.3.TIR domain-containing adaptors and TLR signaling.MyD88is an essential TIR domain-containing adaptor for the induction of inflammatory cytokines via all the TLRs.TIRAP/Mal is a second TIR domain-containing adaptor that specifically mediates the MyD88-dependent pathway via TLR2 and TLR4.In the TLR4-and TLR3-mediated signaling pathways,a MyD88-independent pathway exists that leads to activation of IRF-3via TBK1and IKKε/IKK i.The TIR domain-containing adaptor TRIF mediates this MyD88-independent pathway.MyD88-dependent signaling pathway via TLR2and TLR4,but not for MyD88-independent signaling.6.2.TRIFA third TIR domain-containing adaptor,TRIF/TICAM-1was identified by a database search and as a TLR3-associatedmolecule by two-hybrid screening[47,48].Unlike MyD88and TIRAP/Mal,TRIF is a large protein consisting of712amino acids in humans.Overexpression of TRIF as wellas MyD88and TIRAP caused activation of the NF-B-dependent promoter in293cells.Furthermore,overexpres-sion of TRIF,but not MyD88or TIRAP,induced activationof the IFN-promoter.Dominant negative TRIF inhibitedthe TLR3ligand-induced activation of the IFN-promoter,and RNAi-mediated knockdown of TRIF caused impairmentin the TLR3ligand-induced IFN-expression.Thus,thesein vitro studies indicated that TRIF is involved in the TLR3-mediated MyD88-independent pathway.Most recently,TRIF knockout mice have been generated.In TRIF knockout mice,TLR3-mediated expression of IFN-and IFN-inducible genes was impaired[51].Furthermore, TRIF knockout mice displayed defective expression of IFN-inducible genes in response to the TLR4ligand.A studyof random germline mutagenesis in mice,using the alkylat-ing agent N-ethyl-N-nitrosourea(ENU),also revealed thatTRIF-mutant mice were defective in the TLR3-and TLR4-mediated responses[52].Thus,TRIF has been demonstratedto be essential for the TLR3-and TLR4-mediated MyD88-independent pathway.These studies clearly established that TIR domain-containing adaptors provide specificity for in-dividual TLR-mediated signaling pathways.In addition to the impaired MyD88-independent pathway,TRIF knock-out mice displayed defective TLR4-mediated inflammatory cytokine production,although activation of the MyD88-dependent pathway,such as IRAK-1phosphorylation and early phase of NF-B activation,was not impaired.There-fore,the TLR4signaling pathway is likely to require ac-tivation of both the MyD88-dependent and-independent pathways to induce inflammatory cytokines.6.3.Other TIR domain-containing adaptorsIn addition to MyD88,TIRAP,and TRIF,a fourth TIR domain-containing adaptor,TIRP,has recently been identi-fied[53].Human TIRP protein consists of235amino acids, and the TIR domain was located in the middle portion of the protein.Although TIRP has been shown to be involved in the IL-1receptor-mediated signaling pathway,it remains unclear whether TIRP mediates the TLR signaling pathway. In addition,there is another TIR domain-containing adap-tor,SARM.This molecule is a large protein consisting of about700amino acids,and the TIR domain is located in the C-terminal portion.At present,we do not know whether this molecule is involved in the TLR-mediated signaling pathway.Generation of knockout mice of all of these adap-tors will provide definite evidence of their roles in TLR signaling.8K.Takeda,S.Akira/Seminars in Immunology16(2004)3–97.Future prospectsSince the discovery of TLRs in mammals,rapid progress has been made on our understanding of the molecular mechanisms of innate immunity.Individual TLRs recognize their specific microbial components and activate signaling pathways.The TLR signaling pathways also have their own cascades for exhibiting their specific responses,which are characterized by several TIR domain-containing adaptors. Elucidation of the physiological roles of these adaptors will provide important clues for understanding how individual TLRs induce their specific innate immune responses. AcknowledgementsWe thank M.Hashimoto for excellent secretarial assis-tance.This work was supported by grants from the Special Coordination Funds of the Ministry of Education,Culture, Sports,Science and Technology,and the Japan Research Foundation for Clinical Pharmacology.References[1]Hashimoto C,Hudson KL,Anderson KV.The Toll gene ofDrosophila,required for dorsal-ventral embryonic polarity,appears to encode a transmembrane protein.Cell1988;52:269–79.[2]Lemaitre B,Nicolas E,Michaut L,Reichhart J-M,Hoffmann JA.Thedorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults.Cell1996;86:973–83.[3]Medzhitov R,Preston-Hurlburt P,Janeway Jr CA.A human homo-logue of the Drosophila Toll protein signals activation of adaptive immunity.Nature1997;388:394–7.[4]Poltorak A,He X,Smirnova I,Liu MY,Huffel CV,Du X,etal.Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutation in Tlr4gene.Science1998;282:2085–8.[5]Rock FL,Hardiman G,Timans JC,Kastelein RA,Bazan JF.A familyof human receptors structurally related to Drosophila Toll.Proc Natl Acad Sci USA1998;95:588–93.[6]Hoshino K,Takeuchi O,Kawai T,Sanjo H,Ogawa T,Takeda Y,et al.Cutting edge:Toll-like receptor4(TLR4)-deficient mice are hyporesponsive to lipopolysaccharide:evidence for TLR4as the Lps gene product.J Immunol1999;162:3749–52.[7]Underhill DM,Ozinsky A,Hajjar AM,Stevens A,Wilson CB,Bassetti M,et al.The Toll-like receptor2is recruited to macrophage phagosomes and discriminates between pathogens.Na-ture1999;401:811–5.[8]Akira S,Takeda K,Kaisho T.Toll-like receptors:critical proteinslinking innate and acquired immunity.Nat Immunol2001;2:675–80.[9]Kawai T,Adachi O,Ogawa T,Takeda K,Akira S.Unresponsivenessof MyD88-deficient mice to endotoxin.Immunity1999;11:115–22.[10]Takeuchi O,Takeda K,Hoshino K,Adachi O,Ogawa T,AkiraS.Cellular responses to bacterial cell wall components are me-diated through MyD88-dependent signaling cascades.Int Immunol 2000;12:113–7.[11]Takeuchi O,Kaufmann A,Grote K,Kawai T,Hoshino K,MorrM,et al.Cutting edge:preferentially the R-stereoisomer of the My-coplasmal lipopeptide macrophage-activating lipopeptide-2activates immune cells through a Toll-like receptor2-and MyD88-dependent signaling pathway.J Immunol2000;164:554–7.[12]Hacker H,Vabulas RM,Takeuchi O,Hoshino K,Akira S,Wagner H.Immune cell activation by bacterial CpG-DNA through myeloid dif-ferentiation marker88and tumor necrosis factor receptor-associated factor(TRAF)6.J Exp Med2000;192:595–600.[13]Schnare M,Holt AC,Takeda K,Akira S,Medzhitov R.Recognitionof CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88.Curr Biol2000;10:1139–42.[14]Hemmi H,Kaisho T,Takeuchi O,Sato S,Sanjo S,Hoshino K,et al.Small antiviral compounds activate immune cells via TLR7 MyD88-dependent signalling pathway.Nat Immunol2002;3:196–200.[15]Hayashi F,Smith KD,Ozinsky A,Hawn TR,Yi EC,Goodlett DR,et al.The innate immune response to bacterialflagellin is mediated by Toll-like receptor-5.Nature2001;410:1099–103.[16]Janssens S,Burns K,Tschopp J,Beyaert R.Regulation of interleukin-1and lipopolysaccharide-induced NF-B activation by alternative splicing of MyD88.Curr Biol2002;12:467–71.[17]Burns K,Janssens S,Brissoni B,Olivos N,Beyaert R,Tschopp J.Inhibition of IL-1receptor/Toll-like receptor signaling through the alternatively spliced,short form of MyD88is due to its failure to recruit IRAK-4.J Exp Med2003;197:263–8.[18]Cao Z,Henzel WJ,Gao X.Irak:a kinase associated with theinterleukin-1receptor.Science1996;271:1128–31.[19]Janssens S,Beyaert R.Functional diversity and regulation of differentinterleukin-1receptor-associated kinase(IRAK)family members.Mol Cell2003;11:293–302.[20]Swantek JL,Tsen MF,Cobb MH,Thomas JA.IL-1receptor-associated kinase modulates host responsiveness to endotoxin.J Im-munol2000;164:4301–6.[21]Suzuki N,Suzuki S,Duncan GS,Millar DG,Wada T,Mirtsos C,et al.Severe impairment of interleukin-1and Toll-like receptor signalling in mice lacking IRAK-4.Nature2002;416:750–6.[22]Li S,Strelow A,Fontana EJ,Wesche H.IRAK-4:a novel memberof the IRAK family with the properties of an IRAK-kinase.Proc Natl Acad Sci USA2002;99:5567–72.[23]Kobayashi K,Hernandez LD,Galan JE,Janeway Jr CA,MedzhitovR,Flavell RA.IRAK-M is a negative regulator of Toll-like receptor signaling.Cell2002;110:191–202.[24]Arch RH,Gedrich RW,Thompson CB.Tumor necrosis fac-tor receptor-associated factors(TRAFs)—a family of adapter pro-teins that regulates life and death.Genes Dev1998;12:2821–30.[25]Lomaga MA,Yeh WC,Sarosi I,Duncan GS,Furlonger C,Ho A,et al.TRAF6deficiency results in osteopetrosis and defective interleukin-1,CD40,and LPS signaling.Genes Dev1999;13:1015–24. [26]Naito A,Azuma S,Tanaka S,Miyazaki T,Takaki S,Takatsu K,et al.Severe osteopetrosis,defective interleukin-1signalling and lymph node organogenesis in TRAF6-deficient mice.Genes Cells 1999;4:353–62.[27]Deng L,Wang C,Spencer E,Yang L,Braun A,You J,et al.Acti-vation of the IÉ B kinase complex by TRAF6requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain.Cell2000;103:351–61.[28]Wang C,Deng L,Hong M,Akkaraju GR,Inoue J-I,Chen ZJ.TAK1is a ubiquitin-dependent kinase of MKK and IKK.Nature 2001;412:346–51.[29]Burns K,Clatworthy J,Martin L,Martinon F,Plumpton C,MascheraB,et al.Tollip,a new component of the IL-1RI pathway,links IRAK to the IL-1receptor.Nat Cell Biol2000;2:346–51.[30]Bulut Y,Faure E,Thomas L,Equils O,Arditi M.Cooperation of Toll-like receptor2and6for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1receptor signaling molecules in Toll-like receptor2signaling.J Immunol2002;167:987–94. [31]Zhang G,Ghosh S.Negative regulation of toll-like receptor-mediatedsignaling by Tollip.J Biol Chem2002;77:7059–65.K.Takeda,S.Akira/Seminars in Immunology16(2004)3–99[32]Yu KY,Kwon HJ,Norman DA,Vig E,Goebl MG,Harrington MA.Cutting edge:mouse Pellino-2modulates IL-1and lipopolysaccharide signaling.J Immunol2002;169:4075–8.[33]Jiang Z,Johnson HJ,Nie H,Qin J,Bird TA,Li X.Pellino1isrequired for interleukin-1(IL-1)-mediated signaling through its in-teraction with the IL-1receptor-associated kinase4(IRAK4)-IRAK-tumor necrosis factor receptor-associated factor6(TRAF6)complex.J Biol Chem2003;278:10952–6.[34]Kawai T,Takeuchi O,Fujita T,Inoue J,Muhlradt PF,Sato S,etal.Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IRF-3and the expression of a subset of LPS-inducible genes.J Immunol2001;167:5887–94.[35]Doyle SE,Vaidya SA,O’Connell R,Dadgostar H,Dempsey PW,Wu T-T,et al.IRF3mediates a TLR3/TLR4-specific antiviral gene program.Immunity2002;17:251–63.[36]Toshchakov V,Jones BW,Perera PY,Thomas K,Cody MJ,ZhangS,et al.TLR4,but not TLR2,mediates IFN--induced STAT1␣/-dependent gene expression in macrophages.Nat Immunol2002;3: 392–8.[37]Hoshino K,Kaisho T,Iwabe T,Takeuchi O,Akira S.Differentialinvolvement of IFN-in Toll-like receptor-stimulated dendritic cell activation.Int Immunol2002;14:1225–31.[38]Alexopoulou L,Holt AC,Medzhitov R,Flavell RA.Recognition ofdouble-stranded RNA and activation of NF-B by Toll-like receptor3.Nature2001;413:732–8.[39]Weaver BK,Kumar KP,Reich NC.Interferon regulatory factor3andCREB-binding protein/p300are subunits of double-stranded RNA-activated transcription factor DRAF1.Mol Cell Biol1998;18:1359–68.[40]Yoneyama M,Suhara W,Fukuhara Y,Fukuda M,Nishida E,FujitaT.Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3and CBP/p300.EMBO J1998;17:1087–95.[41]Sato M,Suemori H,Hata N,Asagiri M,Ogasawara K,Nakao K,et al.Distinct and essential roles of transcription factors IRF-3and IRF-7in response to viruses for IFN-␣/gene induction.Immunity 2000;13:539–48.[42]Sharma S,tenOever BR,Grandvaux N,Zhou GP,Lin R,Hiscott J.Triggering the interferon antiviral response through an IKK-related pathway.Science2003;300:1148–51.[43]Fitzgerald KA,McWhirter SM,Faia KL,Rowe DC,Latz E,Golen-bock DT,et al.IKKεand TBK1are essential components of the IRF3signaling pathway.Nat Immunol2003;4:491–6.[44]Bonnard M,Mirtsos C,Suzuki S,Graham K,Huang J,Ng M,et al.Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-B-dependent gene transcription.EMBO J2000;19: 4976–85.[45]Horng T,Barton GM,Medzhitov R.TIRAP:an adapter molecule inthe Toll signaling pathway.Nat Immunol2001;2:835–41.[46]Fitzgerald KA,Palsson-McDermott EM,Bowie AG,Jefferies C,Mansell AS,Brady G,et al.Mal(MyD88-adaptor-like)is required for Toll-like receptor-4signal transduction.Nature2001;413:78–83.[47]Yamamoto M,Sato S,Mori K,Hoshino K,Takeuchi O,Takeda K,et al.Cutting edge:a novel Toll/IL-1receptor domain-containing adapter that preferentially activates the IFN-promoter in the Toll-like receptor signaling.J Immunol2002;169:6668–72.[48]Oshiumi H,Matsumoto M,Funami K,Akazawa T,Seya T.TICAM-1,an adaptor molecule that participates in Toll-like receptor3-mediated interferon-induction.Nat Immunol2003;4:161–7.[49]Horng T,Barton GM,Flavell RA,Medzhitov R.The adaptormolecule TIRAP provides signalling specificity for Toll-like recep-tors.Nature2002;420:329–33.[50]Yamamoto M,Sato S,Hemmi H,Sanjo H,Uematsu S,Kaisho T,et al.Essential role of TIRAP/Mal for activation of the signaling cascade shared by TLR2and TLR4.Nature2002;420:324–9. [51]Yamamoto M,Sato S,Hemmi H,Hoshino K,Kaisho T,Sanjo H,et al.Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway.Science2003;301:640–3.[52]Hoebe K,Du X,Georgel P,Janssen E,Tabeta K,Kim SO,et al.Identification of Lps2as a key transducer of MyD88-independent TIR signalling.Nature2003;424:743–8.[53]Bin LH,Xu LG,Shu HB.TIRP:a novel TIR domain-containingadapter protein involved in Toll/interleukin-1receptor signaling.J Biol Chem2003;278:24526–32.。
cAMP调节剂对卵母细胞体外成熟效果的调节机制研究进展

cAMP调节剂对卵母细胞体外成熟效果的调节机制研究进展齐雅天1,房晓欢1,李飒1,李俊杰1,2*(1.河北农业大学动物科技学院,河北保定 071000;2.河北省牛羊胚胎技术创新中心,河北保定 071000)摘 要:卵母细胞体外成熟是家畜体外胚胎生产关键技术之一。
卵母细胞的环磷酸腺苷(cAMP)水平能够通过调节核质同步成熟进程影响卵母细胞发育能力,因此,cAMP对卵母细胞体外成熟质量的影响受到广泛关注。
本文针对常用的cAMP调节剂及其调节卵母细胞体外成熟的机制进行综述,以期为改善卵母细胞体外成熟效果提供依据。
关键词:卵母细胞;体外成熟;cAMP;进展中图分类号:S814.8 文献标识码:A DOI编号:10.19556/j.0258-7033.20200627-02卵母细胞体外成熟(IVM)是体外胚胎生产的关键技术之一,对于人类辅助生殖技术和优良种畜快速扩繁具有重要意义[1],其成熟效果直接影响着后续体外受精、体细胞核移植、转基因、胚胎干细胞与胚胎移植等技术的效果,对于胚胎基因组激活和早期胚胎发育至关重要。
卵母细胞成熟包括细胞核与细胞质成熟2个方面[2],但哺乳动物卵母细胞在常规IVM培养时,易导致细胞核成熟先于细胞质成熟,造成核质成熟不同步,影响卵母细胞成熟质量[3]。
研究发现,卵母细胞体内成熟时高水平的环磷酸腺苷(cAMP)是抑制核成熟的关键调节因子[4]。
因此,常在IVM前增加预成熟阶段升高cAMP,抑制核成熟,使细胞质充分发育,有助于提高卵母细胞发育能力[5]。
Zhang等[6]研究发现,使用cAMP调节剂可维持小鼠卵母细胞内高cAMP水平,从而抑制核成熟,提高卵母细胞核质同步成熟效果。
本文通过对卵母细胞内cAMP的动态变化、常用cAMP调节剂及其调节卵母细胞成熟的机制进行综述,以期为提高卵母细胞体外成熟效果提供依据。
1卵母细胞成熟过程中cAMP变化及对卵母细胞体外成熟的影响哺乳动物卵母细胞来源于胚胎期出现的卵原细胞,卵原细胞经多次有丝分裂发育为初级卵母细胞,后经2次减数分裂得到成熟的卵母细胞。
3.2生物性污染(final细菌、病毒、寄生虫和虫鼠)

二、细菌性食物中毒
食用富有大量病原菌的食物引起的中毒称为感 染性食物中毒。
食用由于细菌大量繁殖而产生毒素的食物中毒 称为毒素型食物中毒。
细菌的致病性和毒力
致病性: 一定种类的病原菌在一定的条件下,能 在宿主体内引起感染的能力称为致病性。
致病性是细菌种的特征之一。
毒力:病原菌致病力的强弱程度,是量的概念,同 一细菌的不同菌株,其毒力不一样。
Inhibit protein synthesis - N-glycosidase 糖苷酶 Activate 2nd messenger pathways - ADP-ribosyltra
nsferase 核糖基转移酶 Protease - Zinc-metalloprotease 金属蛋白酶 3. 激发免疫反应 - Superantigen
透明质酸酶,能分解结缔组织的透明质酸,如葡萄球 菌、链球菌等可产生。
胶原酶,主要分解结缔组织中的胶原蛋白,见之于梭 菌、气单胞菌等。
神经氨酸酶,主要分解肠黏膜上皮细胞的细胞间质, 如霍乱弧菌及志贺菌等可产生。
三﹑细菌毒素
toxigenesis:细菌产生毒素的能力。细菌性疾病的一种 机制。
第三章 生物性污染对食品安全的影响
生物性污染是指微生物、寄生虫、 昆虫等生物对食品的污染。
2020/3/4
一、微生物安全
食品安全
据国内外统计,在各种食物中毒中,以细菌性食物 中毒最多。
化学安全 – 三鹿﹑双汇 微生物安全 - 召回制度 (recall)
2007, Topps Meat,美国最大冷冻hamburgers的生产商, Escherichia coli O157:H7, 召回 980万公斤牛肉 – 自动破 产
炎症小体与自噬相互调控关系研究进展

㊃综 述㊃D O I 10 3760 c m a ji s s n 1673-436X 2018 13 011作者单位:510280广州,南方医科大学珠江医院呼吸内科通信作者:于化鹏,E m a i l h u a p e n g y u @a l i yu n c o m 炎症小体与自噬相互调控关系研究进展吴玲玲 于化鹏 陈丽嫦 曾冠盛ʌ摘要ɔ 炎症小体作为重要的固有免疫成分,可通过外源性病原体相关分子模式(P AM P s )和内源性损伤相关分子模式(D AM P s)激活㊂炎症小体的激活涉及蛋白复合物的形成和寡聚化,引起c a s p a s e -1激活,导致促炎因子I L -1β和I L -18释放㊂目前,自噬被认为是炎症小体的主要调节器㊂细胞自噬参与细胞稳态,清除损伤的细胞器(例如线粒体)和胞内病原体的重要的细胞内过程㊂深入研究炎症小体与自噬相互调控机制,对认识炎症性疾病的发生发展非常重要㊂ʌ关键词ɔ 炎症小体;自噬;c a s pa s e -1基金项目:广东省科技计划项目(2014A 020212395)R e l a t i o n s h i p b e t w e e ni n f l a m m a s o m ea n da u t o p h a g y Wu L i n g l i n g Y u H u a p e n g C h e n L i c h a n g Z e n g G u a n s h e n g D e p a r t m e n to f R e s p i r a t o r y M e d i c i n e Z h u j i a n g H o s p i t a l S o u t h e r n M e d i c a l U n i v e r s i t y G u a n gz h o u510280 C h i n a C o r r e s p o n d i n g a u t h o r Y u H u a p e n g E m a i l h u a p e n g y u @a l i yu n c o m ʌA b s t r a c t ɔ I n f l a mm a s o m e a s a n i m p o r t a n ti n n a t ei mm u n e c o m p o n e n t c a n b e a c t i v a t e d b ye x o g e n o u s p a t h o g e n -a s s o c i a t e d m o l e c u l a r p a t t e r n s P AM P s a n de n d o g e n o u sd a m a g e -r e l a t e d m o l e c u l a r p a t t e r n s D AM P s A c t i v a t i o nof t h e i n f l a mm a s o m e i n v o l v e s t h e f o r m a t i o n a n d o l i go m e r i s a t i o n o f p r o t e i n c o m p l e x e s t r i g g e r i n g t h ea c t i v a t i o no fc a s p a s e -1 l e a d i n g t ot h er e l e a s eo f p r o i n f l a mm a t o r y c y t o k i n e s i n t e r l e u k i n1β I L -1β a n dI L -18 A t p r e s e n t a u t o p h a g y i sc o n s i d e r e da st h e m a i nr e g u l a t o ro ft h e i n f l a mm a s o m e A u t o p h a g y i s i n v o l v e di nc e l l u l a rh o m e o s t a s i s a ni m po r t a n t i n t r a c e l l u l a r p r o c e s s e st h a t r e m o v e s d a m a g e do r g a n e l l e s s u c ha s m i t o c h o n d r i a a n di n t r a c e l l u l a r p a t h o g e n s F u r t h e rs t u d y i nt h e m e c h a n i s mo fm u t u a l r e g u l a t i o ni n f l a mm a s o m ea n da u t o p h a g y i sv e r y i m p o r t a n t f o ru n d e r s t a n d i n g th e o c c u r r e n c e a n dd e v e l o p m e n t o f i n f l a mm a t o r y di s e a s e s ʌK e y wo r d s ɔ I n f l a mm a s o m e A u t o p h a g y C a s p a s e -1F u n d p r o gr a m P r o v i n c i a l S c i e n c e a n dT e c h n o l o g y P r o j e c t o fG u a n g d o n g 2014A 020212395 天然免疫反应在宿主防御病原体起着重要的作用[1-2]㊂组织损伤或感染刺激机体时,产生的炎症反应可能导致器官和组织损伤从而对机体有害,引起炎症性疾病㊂炎症小体激活是导致疾病的主要炎症反应途径之一㊂近年来大量研究表明,自噬在炎症性疾病中受到抑制或削弱,机体表现为炎症过度或炎症小体过度激活[3]㊂为保护宿主避免过度的炎症反应,炎症小体可上调自噬过程[4]㊂本文就炎症小体与自噬相互调控关系的研究进展作一综述㊂1 炎症小体的组成与激活炎症小体是由病原体相关分子模式(p a t h o ge n a s s o c i a t e dm o l e c u l a r pa t t e r n ,P AM P )和损伤相关分子模式(d a m a g ea s s o c i a t e d m o l e c u l a r pa t t e r n s ,D AM P s )[2]等多种因素诱导激活的细胞内的多蛋白复合物㊂典型的炎症小体是由核苷酸结合寡聚化结构域样受体(n u c l e o t i d e -b i n d i n g o l i g o m e r i z a t i o nd o m a i n -l i k e r e c e p t e r ,N L R )家族或黑素瘤缺乏因子2样受体家族[a b s e n t i n m e l a n o m a2(A I M 2)-l i k e r e c e p t o r ,A L R ]受体蛋白㊁衔接蛋白凋亡相关点样蛋白(a p o p t o s i s -a s s o c i a t e d s pe c k -l i k e p r o t e i n ,A S C )㊁以及效应分子c a s p a s e -1前体(p r o -c a s p a s e -1)构成[5]㊂炎症小体激活后,p r o -c a s p a s e -1自身水解切割成活性c a s p a s e -1,进一步将p r o -I L -1β和pr o -I L -18转变为具有生物学活性的I L -1β和IL -18成熟体[1]㊂目前研究较多炎症小体包括N L R s 家族中的典型家族成员N L R P 1㊁N L R P 3㊁N L R C 4以及核酸感受器A I M 2[1]㊂炎症小体N L R P 1和N L R C 4分别被特定的P AM P 激活,如胞壁酰二肽和鞭毛蛋白[2]㊂N L R P 3炎症小体则可被多种刺激因素激活,包括病原微生物和内源性介质,如活性氧(r e a c t i v e o x y g e n s pe c i e s ,R O S ),线粒体损伤相关分子和三磷酸腺苷(a d e n o s i n et r i p h o s p h a t e ,A T P ),以及尿酸㊁β-淀粉样蛋白和二氧化硅等[6]㊂A I M 2炎症小体被双链D N A(d o u b l e -s t r a n d e dD N A ,d s D N A )特异性激活,其d s D N A㊃1101㊃国际呼吸杂志2018年7月第38卷第13期 I n t JR e s p i r ,J u l y 2018,V o l .38,N o .13Copyright ©博看网. All Rights Reserved.可能来自宿主的核酸和线粒体或病原体[7]㊂最近研究表明,大多数情况下N L R炎症小体的激活途径需要通过特定的蛋白介质结合㊂神经元凋亡抑制蛋白(n e u r o n a la p o p t o s i s i n h i b i t o r yp r o t e i n,N A I P)特异性结合鞭毛蛋白和Ⅲ型分泌系统蛋白[8],从而激活N L R C4炎症小体㊂类似的, N e k7与N L R P3结合,使细胞内钾水平下降,导致激活N L R P3炎症小体[9]㊂然而,炎症小体的激活受到内源性或外源性刺激因素水平㊁以及炎症小体成分的调控,这些复合物成分在固有免疫应答细胞中一直保持在低水平表达[1]㊂2自噬对炎症小体激活的调节作用自噬在细胞代谢㊁细胞发育与分化㊁退化性疾病,以及与衰老相关的病理过程发挥重要作用[3]㊂产生过度或错误折叠的蛋白㊁损伤细胞器(如线粒体)或细胞内细菌,被包装成双层包膜的自噬体,并被运送至溶酶体以降解㊁循环利用氨基酸㊂自噬的过程受内体和吞噬体途径中的重要蛋白㊁以及自噬相关基因(A t g)编码的自噬蛋白调控[4,10]㊂S a i t o h等首次发现自噬可以负性调节炎症小体激活,其后多项实验研究显示类似结论[11-12]㊂最初的研究显示A t g16L1缺陷的巨噬细胞在内毒素刺激后,c a s p a s e-1大量活化,I L-1β和I L-18产生增加[11]㊂类似地,在A t g7缺陷或抑制剂3-甲基腺嘌呤(3-m e t h y l a d e n i n e,3-MA)处理后的巨噬细胞中,I L-1β产生显著增加[11]㊂后来的研究进一步表明,自噬可通过多种方式对炎症小体的激活进行调节㊂21清除线粒体来源的D AM P s自噬清除受损的线粒体,使线粒体来源D AM P s释放减少,从而抑制炎症小体的激活[13-15]㊂Z h o u等[15]的研究证明,随着线粒体复合物Ⅰ和Ⅲ的药物抑制作用,线粒体R O S产生增加,促进N L R P3炎症小体介导的c a s p a s e-1活化和单核细胞中I L-1β的释放,而R O S抑制剂可逆转这种作用㊂同样,自噬的减少导致产生R O S的线粒体累积,并随后引起刺激信号A T P,尿酸钠晶体㊁棕榈酸或流感A病毒[14-17]触发促进N L R P3炎症小体的激活㊂S h i m a d a等[13]和N a k a h i r a等[14]的另外两项研究提出,线粒体完整性丧失之后,胞质释放的线粒体D N A(m i t o c h o n d r i aR N A,m t D N A)负责激活N L R P3炎症小体㊂此外,氧化的m t D N A比普通的m t D N A能更大程度活化N L R P3炎症小体,这表明线粒体R O S和m t D N A 对于N L R P3炎症小体的活化具有重要的作用[13]㊂自噬过程的破坏会损害线粒体内稳态,并促进线粒体D N A的胞质转运,导致c a s p a s e-1大量活化和下游细胞因子大量释放[14]㊂目前,对于线粒体R O S促进N L R P3炎症小体的激活方式以及自噬对该过程的调节机制尚未完全明确,其中几方面仍有待进一步阐明㊂第一,在N L R P3诱导因子缺少的情况下,细胞自噬的减少似乎不足以触发炎症小体激活[14-15]㊂其次,线粒体R O S是线粒体呼吸的正常副产物,线粒体代谢增加导致完整的线粒体产生的R O S能否激活N L R P3炎症小体尚未得知㊂此外,R O S寿命短,可被细胞内的抗氧化剂(如谷胱甘肽和硫氧还蛋白)清除,所以抗氧化剂有可能是另一重要的调节因子[18]㊂另外,半胱天冬酶(c a s p a s e,包括c a s p a s e-1)可以在翻译后修饰(例如通过S-亚硝基化和泛素化)[19-20],这些修饰不仅可以影响c a s p a s e的活性[21-22],还可以影响其降解[20],从而减少炎症小体激活后的细胞因子切割和释放㊂R O S介导的N L R P3炎症小体激活的机制尚未明确,目前存在几种假说㊂第一个假设表明,在尼日利霉素㊁尿酸盐晶体或明矾激活N L R P3炎症小体后,N L R P3易位到线粒体和线粒体相关的内质网膜(m i t o c h o n d r i a-a s s o c i a t e d e n d o p l a s m i c r e t i c u l u m m e m b r a n e s,MAM s),使其接近新产生的线粒体R O S[15]㊂另一个假说涉及硫氧还蛋白相互作用蛋白(t h i o r e d o x i n-i n t e r a c t i n gp r o t e i n,T X N I P)和N L R P3之间的相互作用[23]㊂在这种情况下,T X N I P在R O S依赖性N L R P3活化之后转位到MAM s/线粒体,使其与N L R P3潜在结合[15]㊂由此看来,自噬可通过去除功能异常的线粒体来降低N L R P3炎症小体的过度活化㊂22选择性自噬降解自噬调节炎症小体的另一个机制是通过p62依赖性炎症复合体和线粒体的降解㊂在A I M2和N L R P3炎症小体的刺激物中,A S C的K63(L y s63)相关多聚泛素化被触发,然后被泛素传感器p62识别,从而自噬体靶向降解A S C[24]㊂单核细胞在A I M2诱导因子多聚(d A:d T)[24]的刺激条件下,自噬的药物抑制和p62的减少可以极大程度地促进c a s p a s e-1激活和炎性因子的大量产生㊂这种刺激条件下,A I M2也可以进行泛素化,并在巨噬细胞中通过p62依赖性选择性自噬被降解[25]㊂三结构域蛋白11(t r i p a r t i t em o t i f11,T R I M11)属于E3泛素连接酶,有研究证明T R I M11与A I M2炎症小体有关,可促进A I M2募集到p62选择性自噬降解[25]㊂为了证明炎症小体泛素化导致降解作为一种自噬调节方式,有项独立研究显示T R I M20作为巨噬细胞特异性自噬炎症复合体的主要组成部分,其靶向自噬成分包括干扰素-γ(i n t e r f e r o nγ, I F N-γ)刺激条件下反应的N L R P3㊁N L R P1和p r o-c a s p a s e-1[26]㊂另外,功能失调的线粒体也可被泛素化并标记为自噬㊂在这个过程中,p a r k i n是泛素化必不可少的线粒体外膜蛋白,用于招募自噬受体,随后介导受损线粒体的选择性降解或线粒体自噬[27]㊂Z h o n g等[28]最近研究表明,p a r k i n依赖的p62结合线粒体自噬可以抑制巨噬细胞中N L R P3的激活和I L-1β的释放,这可能是自噬介导抑制炎症小体激活的另一种机制㊂23I L-1β信号途径自噬机制直接调节I L-1β的激活㊁释放和信号转导途径㊂H a r r i s等[29]研究表明,雷帕霉素是一种抑制哺乳动物雷帕霉素靶蛋白(m a mm a l i a nt a r g e to f r a p a m y c i n,m T O R)的自噬诱导剂,利用其处理巨噬细胞后,p r o-I L-1β可被自噬体隔离进行降解,从而使N L R P3炎症小体激活下I L-1β分泌量减少㊂然而,另外两项研究表明自噬实际上是非经典途径分泌I L-1β所必需的,抑制自噬可使巨噬细胞中对A I M2和N L R P3激活物应答中I L-1β的释放减少[30-31]㊂在其中一项研究中,W a n g等[30]阐明A I M2炎症小体激活所需的微管末端结合蛋白1(e n d-㊃2101㊃国际呼吸杂志2018年7月第38卷第13期I n t JR e s p i r,J u l y2018,V o l.38,N o.13Copyright©博看网. All Rights Reserved.b i n d i n gp r o t e i n1,E B1)将A I M2与自噬依赖性分泌联系起来㊂我们发现抑制蛋白激酶AM P K可以调节E B1介导的炎症小体激活诱导I L-1β的分泌,破坏自噬能够阻断I L-1β分泌㊂D u p o n t等[31]也证明饥饿状态启动细胞自噬,可使N L R P3炎症小体激动剂尼日利霉素引起的反应应答中I L-1β分泌增加㊂类似的非传统分泌途径也有助于胞外传递炎症小体激活的产物I L-18[31]㊂自噬对I L-1β释放的调节作用复杂,可能依赖于细胞类型和炎症小体诱导剂或激活剂,目前仍需要进一步研究㊂3炎症小体对自噬的调节作用31 N L R与自噬目前研究最多的是N L R炎症小体,包括N L R P1㊁N L R P3和N L R C4[1]㊂大多数N L R蛋白含有C-端富含亮氨酸重复结构域(l e u c i n e-r i c hr e p e a t s,L R R s),中间为核苷酸结合结构域(N A C H T或N A I P,C I I T A, H E T-E和T P1),N-端为C A R D,P Y D结构域㊂这些结构域允许N L R寡聚化并与其他具有类似结构域的炎症小体蛋白相互作用,形成大炎症复合体超微结构,具有类似朊病毒的外观,甚至有时可以通过光学显微镜观察到[32]㊂自噬蛋白与N L R结构域之间的联系也有相关报道,有研究阐明N L R可以直接调节自噬㊂J o u n a i等[33]最近提出,N L R可以通过N A C H T结构域与b e c l i n1(自噬启动蛋白)相互作用㊂其中N L R P4对b e c l i n1具有强烈的亲和力,R N A干扰抑制N L R P4可导致生理条件下的自噬过程和侵入性细菌感染的上调㊂A群链球菌感染后,N L R P4从b e c l i n1瞬时分离,使其与其他自噬蛋白相互作用启动b e c l i n1介导的自噬㊂N L R P3炎症小体也已被证明通过下调线粒体自噬的启动蛋白来负向调节自噬[34]㊂Z h a n g等[34]证明N L R P3基因缺陷的小鼠可免受高氧暴露的影响,这种作用是由线粒体自噬的启动蛋白表达增加和小鼠肺内皮细胞中自噬保护所引起㊂除N L R P3外,N L R C4也可抑制自噬㊂在志贺菌感染条件下, N L R C4缺失的巨噬细胞中自噬显着增加,并且N L R C4介导的抑制作用依赖于c a s p a s e-1[35],但目前N L R C4/ c a s p a s e-1调节自噬的确切机制仍然未知㊂也有证据表明N L R可刺激自噬体形成㊂N L R P6是新近发现的N L R蛋白,在肠上皮细胞中高表达,并参与炎症小体信号途径㊂N L R P6缺陷导致肠道自噬受损,黏液分泌受损,这使小鼠不能从黏膜表面清除病原体,从而易于持续感染[36]㊂另外,铜绿假单胞菌感染后N L R P3炎症小体的活化促进巨噬细胞发生自噬[37],而N L R P3介导的自噬对这些细胞中的细菌清除很重要㊂综上,N L R对自噬的调节作用可能因参与的细胞类型和诱导自噬或炎症小体激活的条件不同而存在差异㊂32 A L R与自噬 A L R炎症小体在体内外实验中也有研究,主要包括A I M2和I F I16㊂与N L R不同的是,A L R可以通过H I N200结构域直接与其配体d s D N A相连㊂通过A I M2和I F I16识别微生物D N A导致促炎因子I L-1β和I L-18的分泌,并促进了宿主对弗朗西斯菌㊁李斯特菌㊁分枝杆菌属等胞内微生物发生免疫防御反应㊂另一方面,宿主来源的d s D N A也可被A I M2识别,导致银屑病㊁关节炎和其他自身免疫性疾病和炎症疾病的发生发展[38]㊂S h i等[24]首次利用合成的d s D N A多聚(d A:d T)诱导A I M2炎症小体,增加了巨噬细胞中的自噬体形成㊂A I M2激活后, R a s样小G蛋白R a l B被激活,从而诱导组装含b e c l i n1的自噬启动复合物㊂在这种情况下,自噬的诱导均不依赖于c a s p a s e-1或A S C,因为在c a s p a s e-1或A S C-缺陷的巨噬细胞中自噬体形成不受影响[24]㊂A I M2介导的自噬激活目前已在两个独立的模型中进行了描述㊂在第一个模型中,缺血灌注损伤后的肝细胞中A I M2炎症小体激活,导致肝保护反应,引起自噬量增加[39]㊂同时小鼠体内炎症小体激活后诱导自噬则通过清除肝脏中产生R O S的线粒体来防护㊂与先前的R a l B研究类似,b e c l i n1在该调节途径中扮演重要介质的角色㊂相反,在肝细胞中依赖A I M2的自噬调节由c a s p a s e-1介导的,这表明A I M2介导的自噬调节机制可能在免疫细胞与非免疫细胞类型间有所不同㊂在第二个模型中,重组卡介菌(r e c o m b i n a n t B a c i l l u sC a l l m e t t e-G u e r i n, r-B C G)是一种针对结核病的活减毒疫苗,S a i g a等[40]证明巨噬细胞中诱导自噬激活部分依赖于A I M2㊂因此,微生物或内源性D N A激活A I M2炎症小体可能导致宿主适应性自噬反应,旨在限制过度炎症和恢复细胞内稳态㊂33 C a s p a s e-1与自噬 C a s p a s e-1是炎症小体的重要组成成分,目前认为c a s p a s e-1通过对其底物的水解切割来调节自噬过程㊂Y u等[41]表明c a s p a s e-1可介导自噬调节剂p a r k i n的切割,从而抑制线粒体自噬㊂受损的线粒体质膜通透性增强,R O S产生增加和线粒体肿胀,进一步促进炎症小体激活及促炎性细胞死亡㊂另一项研究中,c a s p a s e-1激活后被证实直接切割β-干扰素T I R结构域衔接蛋白(T I R-d o m a i n-c o n t a i n i n g a d a p t o r i n d u c i n g i n t e r f e r o n-β, T R I F),从而导致铜绿假单胞菌感染后T R I F介导的自噬下调[42]㊂此外,c a s p a s e1抗性T R I F突变体的表达显着增加感染巨噬细胞的自噬㊂所以在免疫细胞中,c a s p a s e-1水解㊁切割自噬调节蛋白,可导致这些底物的功能丧失和自噬缺陷㊂然而,c a s p a s e-1的调节作用可能存在细胞类型特异性,还可能与实验模型或c a s p a s e-1激活剂有关㊂34炎症小体和自噬激活的非经典途径最近的证据表明,对革兰阴性菌免疫应答反应中,脂多糖(l i p o p o l y s a c c h a r i d e,L P S)对c a s p a s e-1的激活不依赖于经典的炎症小体途径㊂相反,c a s p a s e-11对于感染大肠杆菌的巨噬细胞中炎症小体和c a s p a s e-1激活起关键作用㊂C a s p a s e-11直接与胞内L P S结合,使其自身寡聚化和激活,随后导致c a s p a s e-1激活,以及巨噬细胞的死亡[43]㊂这个通路已被称为炎症小体激活的非经典途径㊂这途径已有研究阐明并涉及c a s p a s e-1和c a s p a s e-11的底物蛋白g a s d e r m i nD㊂g a s d e r m i nD的N-末端一旦被切割可在自身胞膜上寡聚形成穿孔,这种炎症细胞死亡称为凋亡㊂N末端也被证明与N L R P3直接相关,并导致非经典途径的N L R P3-c a s p a s e-1激活[44]㊂与典型的炎症小体激活途径相似,c a s p a s e-11活化受㊃3101㊃国际呼吸杂志2018年7月第38卷第13期I n t JR e s p i r,J u l y2018,V o l.38,N o.13Copyright©博看网. All Rights Reserved.自噬的调控㊂抑制剂3-MA可增加了巨噬细胞中非经典途径的炎症小体激活[45]㊂这些结果在自噬蛋白5缺陷的巨噬细胞中得到了进一步的证实,表明自噬是c a s p a s e-11活化的负性调节因子㊂但自噬对c a s p a s e-11活化的调节机制并未清楚,目前认为自噬通过去除过量的线粒体R O S来抑制c a s p a s e-11的表达和活化㊂另一方面,非典型途径的炎症小体激活似乎也调节机体的自噬水平㊂R o b e r t s等[46]描述了c a s p a s e-11促进溶酶体与吞噬体融合的能力,尤其在细菌感染时促进吞噬体周围的聚合肌动蛋白的形成,起着抗菌自噬的作用㊂这些研究表明,c a s p a s e-11和自噬的相互调节作用,在维持细胞内稳态和抵御胞内细菌的固有免疫应答中也起着重要的作用㊂4展望大多数情况下,这种双向调控在宿主防御反应,和预防组织损伤以及过度炎症中起着监控和平衡作用㊂目前已证实炎症小体与自噬之间发挥着重要的相互调控作用,但其具体机制和途径之间的相互影响㊁相互联系尚未完全清楚㊂揭开两者之间复杂的调控网络机制,尤其是在炎症性疾病中的作用,可为寻找新的治疗方法提供理论基础㊂参考文献1 B r o zP D i x i tVM I n f l a mm a s o m e s m e c h a n i s m o f a s s e m b l yr e g u l a t i o na n d s i g n a l l i n g J N a tR e v I mm u n o l2016167407-420D O I101038n r i2016582 V a n a j aS K R a t h i n a m V A K F i t z g e r a l d K A M e c h a n i s m so fi n f l a mm a s o m e a c t i v a t i o n r e c e n ta d v a n c e sa n dn o v e l i n s i g h t sJ T r e n d sC e l l B i o l2015255308-315D O I101016jt c b2014120093 D e r e t i c V S a i t o h T A k i r a S A u t o p h a g y i n i n f e c t i o ni n f l a mm a t i o na n d i mm u n i t y J N a tR e vI mm u n o l20131310722-737D O I101038n r i35324 C a d w e l lK C r o s s t a l kb e t w e e na u t o p h a g y a n di n f l a mm a t o r ys i g n a l l i n gp a t h w a y s b a l a n c i n g d e f e n c ea n dh o m e o s t a s i s JN a tR e v I mm u n o l20161611661-675D O I101038n r i20161005 K a n n e g a n t i T D T h e i n f l a mm a s o m e f i r i n g u p i n n a t ei mm u n i t y J I mm u n o lR e v201526511-5D O I101111i m r122976 D a v i sB K W e n H T i n g J P T h ei n f l a mm a s o m e N L R si ni mm u n i t y i n f l a mm a t i o n a n da s s o c i a t e d d i s e a s e s J A n n uR e vI mm u n o l201129707-735D O I101146a n n u r e v-i mm u n o l-031210-1014057 M a l t e z V I T u b b s A L C o o k K D e t a l I n f l a mm a s o m e sc o o rd i n a te p y r o p t o s i sa n dn a t u r a lk i l l e rc e l lc y t o t o x i c i t y t oc l e a r i n f e c t i o nb y au b i q u i t o u se n v i r o n m e n t a lb a c t e r i u m JI mm u n i t y2015435987-997D O I101016j i mm u n i2015100108 Z h a oY S h a oF T h eN A I P-N L R C4i n f l a mm a s o m e i n i n n a t ei mm u n e d e t e c t i o no f b a c t e r i a l f l a g e l l i na n dt y p e I I I s e c r e t i o na p p a r a t u s J I mm u n o lR e v2015265185-102D O I101111i m r122939 H eY Z e n g MY Y a n g D e t a l N E K7i s a n e s s e n t i a lm e d i a t o ro fN L R P3a c t i v a t i o n d o w n s t r e a m o f p o t a s s i u m e f f l u x JN a t u r e 2016 5307590 354-357 D O I 10 1038n a t u r e1695910 Y uL C h e n Y T o o z eS A A u t o p h a g yp a t h w a y c e l l u l a ra n dm o l e c u l a rm e c h a n i s m s J A u t o p h a g y2018142207-215D O I101080155486272017137883811S a i t o hT F u j i t a N J a n g MH e ta l L o s so ft h ea u t o p h a g y p r o t e i n A t g16L1e n h a n c e s e n d o t o x i n-i n d u c e d I L-1b e t a p r o d u c t i o n J N a t u r e20084567219264-268D O I101038n a t u r e0738312S h i b u t a n iS T S a i t o h T N o w a g H e ta l A u t o p h a g y a n da u t o p h a g y-r e l a t e d p r o t e i n si nt h ei mm u n es y s t e m J N a tI mm u n o l201516101014-1024D O I101038n i3273 13S h i m a d a K C r o t h e r T R K a r l i n J e t a l O x i d i z e d m i t o c h o n d r i a l D N A a c t i v a t e s t h e N L R P3i n f l a mm a s o m ed u r i n g a p o p t o s i s J I mm u n i t y2012363401-414D O I101016j i mm u n i20120100914 N a k a h i r a K H a s p e lJ A R a t h i n a m V A e ta l A u t o p h a g yp r o t e i n s r e g u l a t e i n n a t e i mm u n er e s p o n s e sb y i n h i b i t i n g t h e r e l e a s e o f m i t o c h o n d r i a l D N A m e d i a t e d b y t h e N A L P3i n f l a mm a s o m e J N a t I mm u n o l2011123222-230D O I101038n i198015 Z h o uR Y a z d iA S M e n uP e t a l Ar o l e f o rm i t o c h o n d r i a i nN L R P3i n f l a mm a s o m e a c t i v a t i o n J N a t u r e20114697329221-225D O I101038n a t u r e0966316 L u p f e rC T h o m a sP G A n a n dP K e t a l R e c e p t o r i n t e r a c t i n gp r o t e i nk i n a s e2-m e d i a t e dm i t o p h a g y r e g u l a t e s i n f l a mm a s o m ea c t i v a t i o nd u r i n g v i r u s i n f e c t i o n J N a t u r eI mm u n o l2013145480-488D O I101038n i256317 W e nH G r i sD L e iY e t a l F a t t y a c i d-i n d u c e dN L R P3-A S Ci n f l a mm a s o m e a c t i v a t i o n i n t e r f e r e sw i t h i n s u l i n s i g n a l i n g JN a t u r eI mm u n o l2011125408-415D O I101038n i202218 A o n MA S t a n l e y B A S i v a k u m a r a n V e ta l G l u t a t h i o n et h i o r e d o x i ns y s t e m sm o d u l a t em i t o c h o n d r i a lH2O2e m i s s i o na ne x p e r i m e n t a l-c o m p u t a t i o n a ls t u d y J J G e n P h y s i o l20121396479-491D O I101085j g p20121077219S e n g u p t a R B i l l i a r T R K a g a n V E e ta l N i t r i co x i d ea n d t h i o r e d o x i nt y p e1m o d u l a t et h e a c t i v i t y o fc a s p a s e8i nH e p G2c e l l s J B i o c h e m B i o p h y R e s C o mm u n201039111127-1130D O I101016j b b r c20091203620 Z a m a r a e v A V K o p e i n a G S P r o k h o r o v a E A e t a l P o s t-t r a n s l a t i o n a l m o d i f i c a t i o n o f c a s p a s e s T h e o t h e r s i d e o fa p o p t o s i s r e g u l a t i o n J T r e n d sC e l lB i o l2017275322-339D O I101016j t c b20170100321S e n g u p t aR B i l l i a r T R A t k i n sJ L e ta l N i t r i co x i d ea n dd i h y d r o l i p o i c a c i dm o d u l a te t h e a c t i v i t y of c a s p a s e3i nH e p G2c e l l s J F E B S L e t t2009583213525-3530D O I101016j f e b s l e t20091001622 L i J B i l l i a rT R T a l a n i a n R V e ta l N i t r i co x i d er e v e r s i b l yi n h i b i t s s e v e n m e m b e r s o f t h e c a s p a s e f a m i l y v i a S-n i t r o s y l a t i o n J B i o c h e m B i o p h y sR e sC o mm u n1997240㊃4101㊃国际呼吸杂志2018年7月第38卷第13期I n t JR e s p i r,J u l y2018,V o l.38,N o.13Copyright©博看网. All Rights Reserved.2419-424D O I101006b b r c1997767223 Z h o uR T a r d i v e lA T h o r e n sB e t a l T h i o r e d o x i n-i n t e r a c t i n gp r o t e i n l i n k s o x i d a t i v e s t r e s s t o i n f l a mm a s o m e a c t i v a t i o n JN a t u r eI mm u n o l2010112136-140D O I101038n i183124S h i C S S h e n d e r o v K H u a n g N N e t a l A c t i v a t i o n o fa u t o p h a g yb y i n f l a mm a t o r y s i g n a l s l i m i t s I L-1b e t a p r o d uc t i o nb y t a r g e t i n g u b i q u i t i n a t e d i n f l a mm a s o m e s f o r d e s t r uc t i o n JN a t I mm u n o l2012133255-263D O I101038n i221525 L i u T T a n g Q L i u K e ta l T R I M11s u p p r e s s e s A I M2i n f l a mm a s o m e b y d e g r a d i n g A I M2v i a p62-d e p e n d e n ts e l e c t i v e a u t o p h a g y J C e l lR e p20161671988-2002D O I101016j c e l r e p20160701926 K i m u r aT J a i n A C h o iS W e ta l T R I M-m e d i a t e d p r e c i s i o na u t o p h a g y t a r g e t s c y t o p l a s m i c r e g u l a t o r so f i n n a t e i mm u n i t yJ JC e l lB i o l20152106973-989D O I101083j c b20150302327 L a z a r o u M S l i t e rD A K a n eL A e ta l T h eu b i q u i t i nk i n a s eP I N K1r e c r u i t s a u t o p h a g y r e c e p t o r s t o i n d u c em i t o p h a g y JN a t u r e 2015 5247565 309-314 D O I 10 1038n a t u r e1489328 Z h o n g Z Um e m u r a A S a n c h e z-L o p e z E e t a l N F-κBr e s t r i c t s i n f l a mm a s o m e a c t i v a t i o nv i a e l i m i n a t i o no f d a m a g e d m i t o c h o n d r i a J C e l l20161645896-910D O I101016j c e l l20151205729 H a r r i s J H a r t m a n M R o c h eC e t a l A u t o p h a g y c o n t r o l s I L-1b e t a s e c r e t i o nb y t a r g e t i n gp r o-I L-1b e t a f o r d e g r a d a t i o n JJB i o lC h e m 2011286119587-9597D O I101074j b cM11020291130 W a n g L H u a n g H H u a n g M e t a l T h e m i c r o t u b u l e-a s s o c i a t e d p r o t e i n E B1l i n k s A I M2i n f l a mm a s o m e s w i t ha u t o p h a g y-d e p e n d e n ts e c r e t i o n J JB i o lC h e m 20142894229322-29333D O I101074j b c M11455915331 D u p o n t N J i a n g S P i l l i M e t a l A u t o p h a g y-b a s e du n c o n v e n t i o n a l s e c r e t o r y p a t h w a y f o r e x t r a c e l l u l a r d e l i v e r y o fI L-1βJ E M B OJ201130234701-4711D O I101038e m b o j201139832 L e c h t e n b e r g B C M a c e P D R i e d l S J S t r u c t u r a lm e c h a n i s m s i nN L Ri n f l a mm a s o m es i g n a l i n g J C u r r O p i n S t r u c t B i o l20142917-25D O I101016j s b i20140801133J o u n a iN K o b i y a m a K S h i i n a M e ta l N L R P4n e g a t i v e l y r e g u l a t e sa u t o p h a g i c p r o c e s s e st h r o u g ha na s s o c i a t i o n w i t hb ec l i n1J JI mm u n o l201118631646-1655D O I104049j i mm u n o l100165434 Z h a n g Y S a u l e r M S h i n n A S e t a l E n d o t h e l i a l P I N K1m e d i a t e s t h e p r o t e c t i v ee f f e c t so f N L R P3d e f i c i e n c y d u r i n g l e t h a l o x i d a n t i n j u r y J JI mm u n o l2014192115296-5304D O I104049j i mm u n o l140065335S u z u k iT F r a n c h i L T o m aC e t a l D i f f e r e n t i a l r e g u l a t i o no fc a s p a s e-1a c t i v a t i o n p y r o p t o s i s a n da u t o p h a g y v i aI p a fa n dA S Ci n S h i g e l l a-i n f e c t e d m a c r o p h a g e s J P L o S P a t h o g200738e111D O I101371j o u r n a l p p a t003011136 W l o d a r s k a M T h a i s s C A N o w a r s k i R e t a l N L R P6i n f l a mm a s o m e o r c h e s t r a t e s t h e c o l o n i c h o s t-m i c r o b i a li n t e r f a c eb y r e g u l a t i n gg o b l e t c e l lm u c u ss e c r e t i o n J C e l l201415651045-1059D O I101016j c e l l201401026 37 D e n g Q W a n g Y Z h a n g Y e ta l P s e u d o m o n a sa e r u g i n o s at r i g g e r sm a c r o p h a g e a u t o p h a g y t oe s c a p e i n t r a c e l l u l a rk i l l i n gb y ac t i v a t i o n o f t h e N L R P3i n f l a mm a s o m e J I n f e c tI mm u n201584156-66D O I101128I A I00945-1538 M a nS M K a r k iR K a n n e g a n t iT A I M2i n f l a mm a s o m ei ni n f e c t i o n c a n c e r a n da u t o i mm u n i t y R o l ei n D N A s e n s i n gi n f l a mm a t i o n a n d i n n a t e i mm u n i t y J E u r J I mm u n o l2016462269-280D O I101002e j i20154583939S u n Q L o u g h r a n P S h a p i r o R e t a l R e d o x-d e p e n d e n t r e g u l a t i o no f h e p a t o c y t e a b s e n t i nm e l a n o m a2i n f l a mm a s o m ea c t i v a t i o n i n s t e r i l e l i v e r i n j u r y i nm i c e J H e p a t o l o g y2017651253-268D O I101002h e p2889340S a i g a H N i e u w e n h u i z e n N G e n g e n b a c h e r M e t a l T h e R e c o m b i n a n tB C GΔu r e C h l y V a c c i n e T a r g e t st h e A I M2I n f l a mm a s o m e t o I n d u c eA u t o p h a g y a n dI n f l a mm a t i o n J JI n f e c tD i s2015211111831-1841D O I101093i n f d i sj i u67541 Y u J N a g a s uH M u r a k a m i T e t a l I n f l a mm a s o m e a c t i v a t i o nl e a d s t oC a s p a s e-1-d e p e n d e n tm i t o c h o n d r i a l d a m a g e a n d b l o c k o fm i t o p h a g y J P r o cN a t iA c a dS c iU S A 20141114315514-15519D O I101073p n a s141485911142J a b i rM S R i t c h i eN D L iD e ta l C a s p a s e-1c l e a v a g eo f t h e T L R a d a p t o r T R I F i n h i b i t s a u t o p h a g y a n dβ-i n t e r f e r o n p r o d u c t i o nd u r i n g P s e u d o m o n a s a e r u g i n o s a i n f e c t i o n J C e l lH o s tM i c r o b e2014152214-227D O I101016j c h o m20140101043S h i J Z h a o Y W a n g Y e ta l I n f l a mm a t o r y c a s p a s e sa r ei n n a t e i mm u n er e c e p t o r sf o r i n t r a c e l l u l a rL P S J N a t u r e20145147521187-192D O I101038n a t u r e1368344 H eWT W a nH H uL e t a l G a s d e r m i nDi sa ne x e c u t o ro fp y r o p t o s i s a n dr e q u i r e df o r i n t e r l e u k i n-1βs e c r e t i o n J C e l l R e s201525121285-1298D O I101038c r2015139 45 M e u n i e rE D i c k M S D r e i e rR F e ta l C a s p a s e-11a c t i v a t i o nr e q u i r e s l y s i s o f p a t h o g e n-c o n t a i n i n g v a c u o l e s b y I F N-i n d u c e dG T P a s e s J N a t u r e20145097500366-370D O I101038n a t u r e1315746 R o b e r t sJ S Y i l m a zÖ D a n g e r o u s L i a i s o n s C a s p a s e-11a n dr e a c t i v eo x y g e n s p e c i e sc r o s s t a l ki n p a t h o g e n e l i m i n a t i o nJ I n t JM o l S c i2015161023337-23354D O I103390i j m s161023337收稿日期2017-12-12㊃5101㊃国际呼吸杂志2018年7月第38卷第13期I n t JR e s p i r,J u l y2018,V o l.38,N o.13Copyright©博看网. All Rights Reserved.。
The Inflammasomes

Leading EdgeReviewThe InflammasomesKate Schroder1,2and Jurg Tschopp1,*1Department of Biochemistry,University of Lausanne,CH-1066Epalinges,Switzerland2Monash Institute of Medical Research,Monash University,Melbourne,Victoria3800,Australia*Correspondence:jurg.tschopp@unil.chDOI10.1016/j.cell.2010.01.040Inflammasomes are molecular platforms activated upon cellular infection or stress that trigger the maturation of proinflammatory cytokines such as interleukin-1b to engage innate immune defenses.Strong associations between dysregulated inflammasome activity and human heritable and acquired inflammatory diseases highlight the importance this pathway in tailoring immune responses.Here,we comprehensively review mechanisms directing normal inflammasome func-tion and its dysregulation in disease.Agonists and activation mechanisms of the NLRP1,NLRP3, IPAF,and AIM2inflammasomes are discussed.Regulatory mechanisms that potentiate or limit inflammasome activation are examined,as well as emerging links between the inflammasome and pyroptosis and autophagy.Traditionally,innate immunity has been viewed as thefirst line of defense discriminating‘‘self’’(e.g.,host proteins)from‘‘nonself’’(e.g.,microorganisms).However,emerging literature suggests that innate immunity actually serves as a sophisticated system for sensing signals of‘‘danger,’’such as pathogenic microbes or host-derived signals of cellular stress,while remaining unre-sponsive to nondangerous motifs,such as normal host mole-cules,dietary antigens,or commensal gutflora.The notion that innate immunity functions as a danger sentinel has similarities to Matzinger’s‘‘danger hypothesis,’’proposed for adaptive immune responses(Matzinger,1994).Such a model for recog-nizing situations of host danger allows for coordinate activation of immune system antimicrobial and tissue repair functions in response to infection or injury,while avoiding collateral damage in situations in which harmless nonself is present.The innate immune system engages an array of germline-encoded pattern-recognition receptors(PRRs)to detect invari-ant microbial motifs.PRRs are expressed by cells at the front line of defense against infection,including macrophages,mono-cytes,dendritic cells,neutrophils,and epithelial cells,as well as cells of the adaptive immune system.PRRs include the membrane-bound Toll-like receptors(TLRs)and C-type lectins (CTLs),which scan the extracellular milieu and endosomal compartments for pathogen-associated molecular patterns (PAMPs).Intracellular nucleic-acid sensing PRRs cooperate to provide cytosolic surveillance,including the RNA-sensing RIG-like helicases(RLHs),RIG-I and MDA5,and the DNA sensors, DAI and AIM2.The outcome of PAMP recognition by PRRs depends upon the nature of both the responding cell and the invading microbe.However,signal transduction from these receptors converges on a common set of signaling modules, often including the activation of the NF-k B and AP-1transcrip-tion factors that drive proinflammatory cytokine/chemokine production and members of the IRF transcription factor family that mediate type I interferon(IFN)-dependent antiviral responses.A further set of intracellular PRRs,distinct from those described above,are the NOD-like receptors(NLRs)that recog-nize PAMPs,as well as host-derived danger signals(danger-associated molecular patterns,DAMPs).Microbial detection by PRRs such as TLRs is reviewed elsewhere(see Review by O.Takeuchi and S.Akira et al.on page805of this issue).This Review focuses on those PRRs that assemble into high-molec-ular weight,caspase-1-activating platforms called‘‘inflamma-somes’’that control maturation and secretion of interleukins such as IL-1b and IL-18,whose potent proinflammatory activities direct host responses to infection and injury.The NLR FamilyThe NLRs are comprised of22human genes and many more mouse genes because of gene expansion since the last common ancestor.The NLR family is characterized by the presence of a central nucleotide-binding and oligomerization(NACHT) domain,which is commonlyflanked by C-terminal leucine-rich repeats(LRRs)and N-terminal caspase recruitment(CARD)or pyrin(PYD)domains.LRRs are believed to function in ligand sensing and autoregulation,whereas CARD and PYD domains mediate homotypic protein-protein interactions for downstream signaling.The NACHT domain,which is the only domain com-mon to all NLR family members,enables activation of the signaling complex via ATP-dependent oligomerization.Phyloge-netic analysis of NLR family NACHT domains reveals3distinct subfamilies within the NLR family:the NODs(NOD1-2,NOD3/ NLRC3,NOD4/NLRC5,NOD5/NLRX1,CIITA),the NLRPs (NLRP1-14,also called NALPs)and the IPAF subfamily,consist-ing of IPAF(NLRC4)and NAIP(Figure1A).The phylogenetic rela-tionships between subfamily members(Figure1A)are also sup-ported by similarities in domain structures(Figure1B).This is particularly clear for the NLRPs,which all contain PYD,NACHT, and LRR domains,with the exception of NLRP10,which lacks LRRs.This Review uses the most common NLR family nomen-clature;however,an alternative nomenclature based on NLR family member domain structure was proposed(Ting et al., 2008),and a full list of alternative gene names for NLRP1, NLRP3,and IPAF are given in Table S1available online.Cell140,821–832,March19,2010ª2010Elsevier Inc.821The class II transactivator (CIITA)was the first NLR to be char-acterized,and is a key regulator of class II MHC genes that is mutated in bare lymphocyte syndrome (Steimle et al.,1993).The transcriptional coactivator factor function of CIITA appears to be distinct among NLRs,as no other NLRs have been shown to exert transcriptional regulator activity or other nuclear func-tions.Other members of the NLR family are generally considered to perform cytoplasmic surveillance for PAMPs or DAMPs.NOD1and NOD2both recognize breakdown products of bacte-rial cell walls (mesodiaminopimelic acid and muramyl dipeptide [MDP],respectively)and,upon ligand sensing,oligomerize and recruit RIP2via CARD-CARD interactions.Assembly of NOD1and NOD2signalosomes ultimately culminates in the activation of the NF-k B transcription factor,which drives proinflammatory gene regulation (reviewed in Kufer et al.,2006).Mutations in NOD2are associated with human inflammatory diseases such as Crohn’s disease and Blau syndrome (Hugot et al.,2001;Miceli-Richard et al.,2001;Ogura et al.,2001).The functions of NOD3and NOD4await clarification.The function of NOD5(NLRX1)is a matter of debate;recent reports position NOD5within the mitochondrial matrix,or,alternatively,as recruited to the outer mitochondrial membrane,and propose functions in either suppressing MAVS-dependent antiviral pathways or promoting the generation of reactive oxygen species (ROS)(Arnoult et al.,2009;Moore et al.,2008;Tattoli et al.,2008).Many of the remaining NLR family members are poorly charac-terized at present;however,we describe below the function of those NLR family members that regulate caspase-1activity through inflammasome formation.Inflammasomes:Platforms for Caspase-1Activation and IL-1b MaturationCaspases are cysteine proteases that initiate or execute cellular programs,leading to inflammation or cell death.TheyareFigure 1.Human and Mouse NLR Family Members(A)Phylogenetic relationships between NACHT domains of each human (uppercase)and mouse (lowercase)NLR (NOD-like receptor)protein show 3distinct subfamilies within the NLRs:the NOD,NLRP,and IPAF subfamilies.(B)Domain structures for human NLRs reveal commonalities within the subfamilies.Domains are classified according to the NCBI domain annotation tool for the longest human protein product,with the exception of the FIIND domain that was identified independently of NCBI (Tschopp et al.,2003).It should be noted that CIITA is often annotated as harboring a CARD domain,because a splice variant expressed in dendritic cells contains a domain with homology to CARD domains (Nickerson et al.,2001);however,the translated transcript variant is not classified as containing a classical CARD domain by typical approaches (NCBI conserved domains,Simple Modular Architecture Research Tool [SMART]).Likewise,these domain prediction approaches do not classify NOD3and NOD4as CARD-containing and experimental evidence for a CARD domain function has yet to be reported.Domains:BIR,baculoviral inhibition of apoptosis protein repeat domain;CARD,caspase recruitment domain;FIIND,domain with function to find;LRR,leucine-rich repeat;NACHT,nucleotide-binding and oligomerization domain;PYD,pyrin domain.822Cell 140,821–832,March 19,2010ª2010Elsevier Inc.synthesized as inactive zymogens,and their potent cellular activities are tightly controlled by proteolytic activation.Cas-pases are categorized as either proinflammatory or proapop-totic,depending upon their participation in these cellular programs.The proinflammatory caspases are comprised of cas-pase-1,-11and-12in mouse and caspase-1,-4,and-5in human(Martinon and Tschopp,2007).Caspase-12is mutated to encode a nonfunctional protein in most human populations (Xue et al.,2006).Of the proinflammatory caspases,caspase-1 is the most fully characterized.Its catalytic activity is tightly regu-lated by signal-dependent autoactivation within multiprotein complexes called‘‘inflammasomes’’that mediate caspase-1-dependent processing of cytokines such as IL-1b(Martinon et al.,2002).IL-1b is an important proinflammatory mediator that is gener-ated at sites of injury or immunological challenge to coordinate programs as diverse as cellular recruitment to a site of infection or injury and the regulation of sleep,appetite,and body temper-ature(see Review by C.A.Dinarello on page935of this issue). IL-1b activity is rigorously controlled by expression,maturation, and secretion;proinflammatory stimuli induce expression of the inactive IL-1b proform,but cytokine maturation and release are controlled by inflammasomes.An endogenous IL-1receptor antagonist(IL-1RA)also regulates IL-1b action.Most reports characterizing inflammasomes have focused on cells of the myeloid lineage,such as macrophages or dendritic cells; however,cells outside the myeloid compartment can activate inflammasomes.For example,keratinocyte exposure to skin irri-tants or ultraviolet B(UVB)irradiation triggers NLRP3inflamma-some activation(Feldmeyer et al.,2007;Watanabe et al.,2007). Inflammasomes are assembled by self-oligomerizing scaffold proteins.A number of NLR family member have been reported to exhibit inflammasome activity in vitro;however,few NLR family members have clear physiological functions in vivo.NLRP1, NLRP3,and IPAF are danger sentinels that self-oligomerize via homotypic NACHT domain interactions to form high-molecular weight complexes(probably hexamers or heptamers)that trigger caspase-1autoactivation.The HIN-200family member, AIM2,also mediates inflammasome assembly.Inflammasome components and activation mechanisms depend on the nature of the individual protein scaffolds(Figure2).Domain structure conservation between NLRPs(Figure1B)suggests that unchar-acterized family members may also mediate or regulate inflam-masome activation.The NLRP3InflammasomeThe NLRP3inflammasome is currently the most fully character-ized inflammasome and consists of the NLRP3scaffold,the ASC (PYCARD)adaptor,and caspase-1.NLRP3is activated upon exposure to whole pathogens,as well as a number of structurally diverse PAMPs,DAMPs,and environmental irritants(Table S1). Whole pathogens demonstrated to activate the NLRP3inflam-masome include the fungi Candida albicans and Saccharomyces cerevisiae that signal to the inflammasome via Syk(Gross et al., 2009),bacteria that produce pore-forming toxins,including Listeria monocytogenes and Staphylococcus aureus(Mariatha-san et al.,2006),and viruses such as Sendai virus,adenovirus, and influenza virus(Kanneganti et al.,2006;Muruve et al., 2008).In some cases,the individual microbial components (PAMPs,virulence factors)that activate the inflammasome have been identified(for instance,the alpha-toxin of S.aureus; Craven et al.,2009).The unexpectedfinding that the NLRP3inflammasome can be activated by host-derived molecules forms part of an emerging literature supporting a model in which the innate immune system detects endogenous indicators of cellular danger orstress, Figure2.Minimal NLRP1,NLRP3,IPAF,and AIM2Inflammasomes For simplicity,the unoligomerized inflammasome complexes are depicted. Removal of the CARD domain and processing of the caspase domain of cas-pase-1by autocleavage at the indicated sites results in the formation of the active caspase-1p10/p20tetramer.It should be noted that although human NLRP1contains a PYD,mouse NLRP1proteins do not harbor functional PYDs.Human NLRP1can also recruit a second caspase,caspase-5,to the complex(not shown).Maximal caspase-1activation in response to IPAF agonists can require ASC or NAIP,depending on the stimulus.The interaction of these proteins with the IPAF inflammasome activation is currently unclear. Domains:CARD,caspase recruitment domain;FIIND,domain with function tofind;HIN,HIN-200/IF120x domain;LRR,leucine-rich repeat;NACHT,nucle-otide-binding and oligomerization domain;PYD,pyrin domain.Cell140,821–832,March19,2010ª2010Elsevier Inc.823a hypothesis with similarities to the‘‘danger model’’proposed for adaptive immune responses in place of the more simplistic self/nonself recognition model(Matzinger,1994).A number of host-derived molecules indicative of injury activate the NLRP3 inflammasome,including extracellular ATP(Mariathasan et al., 2006)and hyaluronan(Yamasaki et al.,2009)that are released by injured cells.Fibrillar amyloid-b peptide,the major compo-nent of Alzheimer’s disease brain plaques,also activates the NLRP3inflammasome(Halle et al.,2008).The NLRP3inflamma-some also detects signs of metabolic stress,including elevated extracellular glucose(Zhou et al.,2010)such as that occurring in metabolic syndrome,and monosodium urate(MSU)crystals that form as a consequence of hyperuricemia in the autoinflamma-tory disease gout(Martinon et al.,2006).Uric acid can also be released during cell injury,and uric acid-dependent pathways in this context also activate the inflammasome(Gasse et al., 2009;Griffith et al.,2009).Additionally,the NLRP3inflamma-some drives inflammation in response to a number of environ-mental irritants,including silica(Cassel et al.,2008;Dostert et al.,2008;Hornung et al.,2008),asbestos(Cassel et al., 2008;Dostert et al.,2008),UVB irradiation(Feldmeyer et al., 2007),and skin irritants such as trinitrophenylchloride,trinitro-chlorobenzene,and dinitrofluorobenzene(Sutterwala et al., 2006;Watanabe et al.,2007).NLRP3inflammasome activation in response to these insults has been linked to pathology asso-ciated with silicosis,asbestosis,sunburn,and contact hypersen-sitivity reactions,respectively.Models for NLRP3ActivationPrior to agonist treatment,the NLRP3LRR domains are thought to mediate autoinhibition in a manner similar to that described for IPAF(Poyet et al.,2001).This may be mediated by the SGT1and HSP90chaperones that appear to hold NLRP3in an inactive,but signal-competent state;these chaperones also interact with IPAF(Mayor et al.,2007).Upon NLRP3activation,NLRP3oligo-merization leads to PYD domain clustering and presentation for homotypic interaction with the PYD-and CARD-containing adaptor ASC,whose CARD domain in turn recruits the CARD of procaspase-1.Procaspase-1clustering permits autocleavage and formation of the active caspase-1p10/p20tetramer,which then processes cytokine proforms such as IL-1b to generate the active molecules.Mature IL-1b is secreted alongside cas-pase-1by an unconventional protein secretion pathway that is currently unclear.Mechanisms leading to NLRP3inflammasome activation are intensely debated.Three models that may not be exclusive are widely supported in the literature(Figure3).Extracellular ATP stimulates the purogenic P2X7ATP-gated ion channel(Kahlen-berg and Dubyak,2004),triggering K+efflux and inducing gradual recruitment of the pannexin-1membrane pore(Kanne-ganti et al.,2007).Thefirst model posits that pore formation allows extracellular NLRP3agonists to access the cytosol and directly activate NLRP3(Kanneganti et al.,2007).However,the structural diversity within NLRP3agonists argues against direct interaction between NLRP3and all of its activators.A second model was proposed for activators that form crystal-line or particulate structures,such as MSU,silica,asbestos, amyloid-b,and alum,wherein engulfment of these agonists by phagocytes leads to lysosomal damage,resulting incytosolic Figure3.NLRP3Inflammasome ActivationThree major models for NLRP3inflammasome activation are favored in the field,which may not be exclusive:(1)The NLRP3agonist,ATP,triggers P2X7-dependent pore formation by the pannexin-1hemichannel,allowing extracellular NLRP3agonists to enter the cytosol and directly engage NLRP3.(2)Crystalline or particulate NLRP3agonists are engulfed,and their physical characteristics lead to lysosomal rupture.The NLRP3inflammasome senses lysosomal content in the cytoplasm,for example,via cathepsin-B-dependent processing of a direct NLRP3ligand.(3)All danger-associated molecular patterns(DAMPs)and pathogen-associated molecular patterns (PAMPs),including ATP and particulate/crystalline activators,trigger the generation of reactive oxygen species(ROS).A ROS-dependent pathway trig-gers NLRP3inflammasome complex formation.Caspase-1clustering induces autoactivation and caspase-1-dependent maturation and secretion of proin-flammatory cytokines,such as interleukin-1b(IL-1b)and IL-18.824Cell140,821–832,March19,2010ª2010Elsevier Inc.release of lysosomal contents that are somehow sensed by the NLRP3inflammasome(Halle et al.,2008;Hornung et al.,2008).A role for the lysosomal protease,cathepsin B,in activation of a direct NLRP3ligand was suggested in this model(Halle et al.,2008;Hornung et al.,2008).However,cathepsin B-defi-cient macrophages exhibit normal caspase-1activation and IL-1b maturation in response to particulate NLRP3agonists (Dostert et al.,2009),implicating off-target effects of the cathepsinB inhibitor,as was recently suggested for NLRP1 (Newman et al.,2009).Under the third model,all NLRP3agonists trigger the genera-tion of ROS,and this common pathway engages the NLRP3 inflammasome(Cassel et al.,2008;Cruz et al.,2007;Dostert et al.,2008).The production of ROS represents one of the most evolutionarily conserved pathways of response to infection or injury;for example,a gradient of ROS is the apical signal directing wound healing in zebrafish(Niethammer et al.,2009), and ROS are antimicrobial effectors in plants(Bolwell,1999). In support of this model,all NLRP3agonists tested,including ATP and particulate activators,induce ROS and ROS blockade by chemical scavengers suppresses inflammasome activation (Cassel et al.,2008;Cruz et al.,2007;Dostert et al.,2008;Gross et al.,2009;Pe´trilli et al.,2007;Shio et al.,2009).The source of ROS is currently unclear,but one or several NADPH oxidases are implicated,as suppression of the common p22subunit inhibits inflammasome activation(Dostert et al., 2008);alternatively,ROS may be of mitochondrial origin.Mech-anisms directing ROS-dependent NLRP3inflammasome activa-tion remain to be characterized in detail;however,a recent report implicates a ROS-sensitive NLRP3ligand,thioredoxin-interact-ing protein(TXNIP/VDUP1),in NLRP3activation(Zhou et al., 2010).Despite strong evidence for the ROS model,a number of aspects of this model require resolution.For example,some ROS-inducing agents(such as cytokines)do not engage the NLRP3inflammasome,suggesting that while necessary,ROS alone is insufficient for triggering NLRP3activity.Alternatively, a very specific ROS location or nature may be required.Addition-ally,superoxide directly inhibits caspase-1activity by modifying redox-sensitive cysteines(Meissner et al.,2008);whether such a mechanism provides temporal-or dose-dependent negative feedback to limit caspase-1function triggered by a ROS-depen-dent NLRP3pathway requires clarification.The manner by which cytoplasmic K+concentration modulates NLRP3activity is also currently unclear.Macrophages cultured in medium containing a high concentration of K+show decreased capacity for NLRP3-dependent caspase-1activation in response to a range of agonists,suggesting that K+efflux is necessary upstream of NLRP3activation(Dostert et al.,2008;Fernandes-Alnemri et al.,2007;Franchi et al.,2007a;Gross et al.,2009;Pe´trilli et al.,2007;Shio et al.,2009).Future studies are required to determine whether ionicflux and ROS pathways are inter-regu-lated or independently required for NLRP3activation.The NLRP1InflammasomeThe NLRP1inflammasome was thefirst to be described.Human NLRP1has three orthologs in mouse(Nlrp1a-c,Figure1)that are highly polymorphic between inbred mouse strains(Boyden and Dietrich,2006).Strain variation in the mouse Nlrp1b locus appears to underlie susceptibility to Bacillus anthracis lethal toxin(LeTx),as macrophages from susceptible,but not resis-tant,mouse strains activate caspase-1after LeTx exposure (Boyden and Dietrich,2006).The NLRP1inflammasome can also be activated by MDP(Faustin et al.,2007).As a consequence of domain structure differences between NLRP1and NLRP3,the minimal components of the NLRP1 inflammasome are somewhat different to its NLRP3counterpart (Figure2).NLRP1contains a C-terminal extension that harbors a CARD domain,which can interact directly with procaspase-1 and bypass the requirement for ASC,although ASC inclusion in the complex augmented human NLRP1inflammasome activ-ity(Faustin et al.,2007).Unlike human NLRP1,murine NLRP1 orthologs lack functional PYD domains and are predicted to be unable to interact with ASC.Indeed,ASC is dispensable for cas-pase-1activation by NLRP1b in mouse macrophages(Hsu et al., 2008).In addition to caspase-1,NLRP1also interacts with cas-pase-5,which may contribute to IL-1b processing in human cells (Martinon et al.,2002).The exact mechanisms of NLRP1activa-tion remain obscure,but,as for NLRP3,K+efflux appears to be essential(Fink et al.,2008;Wickliffe et al.,2008).The IPAF InflammasomeThe IPAF inflammasome is activated by gram-negative bacteria possessing type III or IV secretion systems,such as Salmonella typhimurium,Shigellaflexneri,Legionella pneumophila,and Pseudomonas aeruginosa(Amer et al.,2006;Franchi et al., 2007b;Mariathasan et al.,2004;Miao et al.,2008;Sutterwala et al.,2007;Suzuki et al.,2007).As IPAF contains a CARD domain,it could be expected to interact directly with procas-pase-1,which is indeed the case(Figure2)(Poyet et al.,2001). Maximal caspase-1activation in response to S.typhimurium, S.flexneri,and P.aeruginosa requires the ASC adaptor(Franchi et al.,2007b;Mariathasan et al.,2004;Suzuki et al.,2007).The role for ASC in the IPAF inflammasome remains unclear;it is presumed that these proteins do not interact directly as IPAF does not contain a PYD domain.It is possible that IPAF collabo-rates with a PYD-containing protein(such as an NLRP)for responses to these pathogens.ASC is dispensable for IPAF-dependent caspase-1activation in response to L.pneumophila (Case et al.,2009),but protection against this pathogen appears to require IPAF collaboration with another NLR family member, NAIP(Ren et al.,2006;Zamboni et al.,2006).IPAF-dependent caspase-1activation is accompanied by rapid cell death(Fink and Cookson,2006).Interestingly,the pathways downstream of IPAF appear to be independently regulated;maximal cas-pase-1activation by the IPAF inflammasome requires ASC,but IPAF-dependent cell death is independent of ASC upon macro-phage infection with S.flexneri(Suzuki et al.,2007;Suzuki and Nu´n˜ez,2008)and P.aeruginosa(Sutterwala et al.,2007).The exact mechanisms directing IPAF inflammasome activa-tion and the participation of ASC and NAIP in this process remain elusive.The LRR domain of IPAF is likely to autorepress in the absence of ligand,as removal of the LRR domain from IPAF results in a spontaneously active mutant(Poyet et al.,2001). The IPAF inflammasome is activated by cytosolicflagellin (Miao et al.,2007).It is likely that other PAMPs modulate IPAF function,as P.aeruginosa appears to activate IPAF through flagellin-dependent and-independent pathways(Miao et al., Cell140,821–832,March19,2010ª2010Elsevier Inc.8252008;Sutterwala et al.,2007),and nonflagellated bacteria such as S.flexneri trigger IPAF inflammasome activity(Suzuki et al., 2007).IPAF activation depends upon virulence factor injection into the cytosol via bacterial type III and IV secretion systems (Ren et al.,2006;Sun et al.,2007;Suzuki et al.,2007).Cytosolic localization offlagellin by other means(liposomes,expression systems)is sufficient for IPAF-dependent caspase-1activation, suggesting that the sole function of bacterial secretion systems in IPAF activation is cytoplasmic injection of bacterial compo-nents(Franchi et al.,2006;Lightfield et al.,2008;Miao et al., 2006).Unlike NLRP3,IPAF inflammasome activity is not inhibited by high extracellular K+,suggesting that IPAF is not a sensor for ionicflux(Pe´trilli et al.,2007).Direct interaction between IPAF and an activating ligand has not been demonstrated,so it is possible that IPAF senses a common pathway induced by cyto-solic PAMPs,analogous to the ROS pathway proposed for NLRP3.The AIM2InflammasomeThe recent identification of the HIN-200family member,AIM2,as a cytosolic double-stranded DNA(dsDNA)sensor that induces caspase-1-dependent IL-1b maturation is an important advance in the inflammasomefield(Bu¨rckstu¨mmer et al.,2009;Fer-nandes-Alnemri et al.,2009;Hornung et al.,2009;Roberts et al.,2009).It is thefirst identification of a non-NLR family mem-ber forming an inflammasome scaffold,and oligomerization of the complex is suggested to be mediated not by a central oligomerization domain within the inflammasome scaffold pro-tein(as for the NLR NACHT domain)but by clustering upon multiple binding sites in the ligand,dsDNA,to which AIM2binds via its C-terminal HIN domain(Bu¨rckstu¨mmer et al.,2009;Fer-nandes-Alnemri et al.,2009;Hornung et al.,2009).The AIM2 inflammasome is composed of AIM2,ASC,and caspase-1. AIM2contains a PYD domain that,as for NLRP3,interacts with ASC via homotypic PYD-PYD interactions,allowing the ASC CARD domain to recruit procaspase-1to the complex(Figure2). As for other inflammasomes,upon autoactivation,caspase-1 directs proinflammatory cytokine maturation and secretion (such as IL-1b and IL-18).Ligand requirements for AIM2are quite permissive,as cytosolic dsDNA from virus,bacteria,or the host itself can activate the AIM2inflammasome(Hornung et al.,2009; Muruve et al.,2008).Further studies are required tofirmly estab-lish the physiological relevance of this pathway,but AIM2is proposed to function in cytosolic surveillance for DNA viruses and may contribute to autoimmune responses against self-DNA in systemic lupus erythematosus.Regulation of Inflammasome Activityand IL-1b SecretionTLRs and Other Proinflammatory Signaling Pathways Both inflammasome activity and pro-IL-1b availability are highly influenced by integration with proinflammatory signaling path-ways such as those triggered by TLR ligation.For this reason, experimental protocols examining inflammasome activation commonly include‘‘priming’’with a TLR agonist(such as lipo-polysaccharide,LPS)or a proinflammatory cytokine(such as tumor necrosis factor,TNF).Most importantly,although inflam-masome-dependent caspase-1activation can be observed in the absence of priming,IL-1b secretion is minimal,because most cells do not constitutively express pro-IL-1b.Pro-IL-1b is potently induced by proinflammatory signals such as LPS or TNF that activate the NF-k B transcription factor and allow IL-1b promoter activation(Hiscott et al.,1993).Although the primary function of priming in experimental protocols is to induce pro-IL-1b,it also potentiates NLRP3inflammasome activity through NF-k B-dependent induction of NLRP3,which may be a limiting component of the complex(Bauernfeind et al.,2009). Whether other inflammasomes are similarly subject to synergy from priming signals has yet to be determined.The IFN-inducible nature of AIM2(DeYoung et al.,1997)suggests that it could also be primed in such a manner by TLR agonists that induce auto-crine type I IFN or indeed by IFNs themselves.A recent report suggests that inflammasome activity is modu-lated by antiviral pathways(Poeck et al.,2010).RIG-I ligation by vesicular stomatitis virus or synthetic RNA triggered caspase-1 activity and IL-1b maturation,in a manner independent of the MAVS and CARD9adaptors that mediate RIG-I-dependent tran-scriptional responses(e.g.,NF-kB and IFN pathways).The exact mechanisms directing RIG-I-dependent caspase-1activation are currently unclear.RIG-I-dependent caspase-1cleavage is ASC dependent but occurs independently of NLRP3,suggesting either that RIG-I can form its own inflammasome or that it regu-lates the activity of a known or uncharacterized NLRP3-indepen-dent inflammasome.Antiviral pathways triggered by the related RIG-I-like helicase,MDA5,also modulate caspase-1cleavage; MDA5collaborates with NLRP3for inflammasome responses to encephalomyocarditis virus(Poeck et al.,2010).In this case, augmented NLRP3inflammasome activity may be mediated at least partially through MDA5-dependent‘‘priming’’of inflamma-some activity,for instance by sensitizing cells to NLRP3agonists via NLRP3induction.Negative Regulation of Inflammasome ActivationThe importance of the inflammasome in controlling infection is highlighted by microbial evolution of inflammasome inhibi-tors.These include viral PYD proteins and various bacterial viru-lence factors that inhibit caspase-1activation.Such factors are reviewed extensively elsewhere(Martinon et al.,2009).A number of host mechanisms also suppress inflammasome activation and presumably function to inhibit the extent of potentially dangerous immune activation.Most recently,mouse CD4+effector and memory T cells were demonstrated to suppress NLRP1and NLRP3,but not IPAF, inflammasome-mediated caspase-1activity and IL-1b secretion (Guarda et al.,2009).Inhibition is dependent on cell-cell contact and is mediated by signaling by specific TNF family ligands (Guarda et al.,2009).Such an inhibitory pathway is likely to aid in‘‘switching off’’innate immune responses once the adaptive arm of the immune system is engaged.A number of CARD-and PYD-containing proteins have been proposed to suppress inflammasome activity by blocking inflam-masome component recruitment.Such PYD-containing proteins include pyrin,POP1(PYDC1),and POP2(PYDC2).The biological relevance of POP1-and POP2-dependent inflammasome inhibi-tion is difficult to assess,as these genes are only present in primates.Conversely,modulation of inflammasome activity by pyrin has clear physiological relevance,as human pyrin mutations are responsible for familial Mediterranean fever826Cell140,821–832,March19,2010ª2010Elsevier Inc.。
人类的免疫系统英语作文

人类的免疫系统英语作文The Human Immune System。
The human immune system is a complex network of cells, tissues, and organs that work together to defend the body against harmful invaders such as viruses, bacteria, and other pathogens. It is a highly sophisticated defense mechanism that is constantly working to keep us healthy and free from illness.The immune system is made up of a variety of cells, including white blood cells, antibodies, and other specialized cells that work together to identify and destroy harmful pathogens. When a pathogen enters the body, the immune system quickly goes to work to identify it and mount a defense. This process involves a series of complex interactions between different types of immune cells, which work together to neutralize the threat and protect the body from harm.One of the key components of the immune system is the production of antibodies, which are proteins that are produced by the body in response to specific pathogens. These antibodies are designed to recognize and bind to specific antigens on the surface of the pathogen, markingit for destruction by other immune cells. This process is known as the adaptive immune response, and it is a crucial part of the body's ability to fight off infections and keep us healthy.In addition to the adaptive immune response, the body also has a built-in defense mechanism known as the innate immune system. This system is always active and provides a rapid, non-specific response to any potential threat. It includes physical barriers such as the skin and mucous membranes, as well as a variety of cells and proteins that work together to detect and neutralize pathogens beforethey can cause harm.The immune system is also capable of forming a memoryof past infections, which allows it to respond more quickly and effectively to future encounters with the same pathogen.This is the basis of vaccination, which involves exposing the body to a harmless form of a pathogen in order to stimulate the immune system to produce a protective response. This helps to prevent future infections and is a key tool in the fight against infectious diseases.While the immune system is a powerful defense mechanism, it is not infallible, and there are many factors that can weaken its ability to protect the body. Poor nutrition,lack of sleep, stress, and certain medical conditions canall have a negative impact on the immune system, making the body more susceptible to infection and illness. It is important to take care of our immune system by maintaininga healthy lifestyle, eating a balanced diet, getting enough sleep, and managing stress.In conclusion, the human immune system is a remarkable and complex defense mechanism that works tirelessly to protect the body from harm. It is a highly sophisticated network of cells, tissues, and organs that work together to identify and destroy harmful pathogens, keeping us healthy and free from illness. By understanding the immune systemand taking steps to support its function, we can help to keep ourselves healthy and resilient in the face of potential threats.。
免疫系统
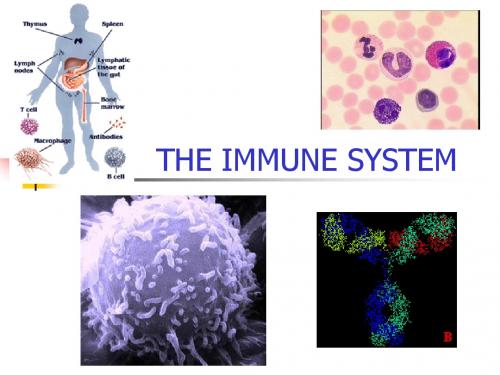
免疫系统 (immune system)的组成
T细胞发育成熟 的场所
胸腺
免疫器官
淋巴结,扁桃体 支气管相关淋巴组织
骨髓
淋巴结 骨髓 脾脏
所有血细 胞产生和 除T细胞外 的所有血 细胞发 育成熟的 场所
固有膜 肠系膜淋巴结
最大的淋巴 器官,具造 血、贮血和 过滤作用, 是成熟T、B 细胞移居和 抗原刺激后 发生免疫应 答的重要场 所。
A scanning electron microscope (SEM) image of a single human lymphocyte. From
MHC/抗原肽
IL-2
运铁蛋白 CD71
TCR CD4 or CD8 B7-2 CD28
CD2
CD58 (LFA-3) FcmR 抗体 FcgR CD35
又称初级淋巴器官。是各类免疫细胞产生、分化和成熟的场 所,对外周免疫器官的发育起主导作用。 包括: 骨髓(bone marrow):所有血细胞和免疫细胞发生和分化的 场所,B淋巴细胞分化成熟的场所,抗体产生的主要场所。 胸腺(thymus): T细胞发育成熟的场所。 腔上囊(bursa fabricius,法氏囊):鸟类特有,B细胞发育 成熟的中枢免疫器官。
T cell的分化
T cell的分化
(1) positive selection 阳性选择 T细胞的TCR 基质细胞表面的MHC;
不识别,凋亡;识别的,继续发育;
→获得可识别自身MHC分子的单阳性T细胞(MHC的限制性)。
(2) negative selection 阴性选择 T细胞的TCR 复合体; 基质细胞表面的自身多肽-自身MHC分子
什么是免疫系统英语作文

The immune system is an intricate network of cells, tissues, and organs that work together to protect our bodies from harmful invaders like bacteria, viruses, and other pathogens. Its a fascinating subject that has always piqued my interest, and Ive always been curious about how it functions and its importance in maintaining our health.Growing up, I was often sick, which led me to learn more about the immune system. I remember the countless trips to the doctor and the various medications I had to take. It was during these visits that I first heard about the immune system and its role in fighting off infections. The doctor explained that a strong immune system could help prevent illnesses and recover more quickly from them. This sparked my curiosity, and I began to research the topic further.The immune system is like a welltrained army, constantly on the lookout for potential threats. It has two main components: the innate immune system, which provides a rapid, nonspecific response to invaders, and the adaptive immune system, which is more specific and can remember pathogens for a quicker response in the future. This dual approach allows our bodies to combat a wide range of threats effectively.One of the most intriguing aspects of the immune system is its ability to recognize and remember specific pathogens. This is achieved through the use of lymphocytes, which are specialized white blood cells. There are two main types of lymphocytes: B cells, which produce antibodies, and T cells, which can directly attack infected cells. When a pathogen enters the body, these cells spring into action, identifying and neutralizing the threat.A good example of the immune system in action is the common cold. When a cold virus enters the body, the immune system responds by producing antibodies that bind to the virus and prevent it from infecting cells. At the same time, T cells are activated to destroy any cells that have already been infected. This coordinated response helps to limit the spread of the virus and, over time, the body is able to clear the infection.However, the immune system is not infallible. Sometimes, it can become overactive and attack the bodys own tissues, leading to autoimmune diseases like rheumatoid arthritis or lupus. In other cases, the immune system may not respond strongly enough to a particular pathogen, resulting in a weakened ability to fight off infections. This is often the case in individuals with compromised immune systems, such as those withHIV/AIDS or certain genetic disorders.Understanding the immune system is crucial for developing effective treatments and vaccines. By studying how it works and identifying its weaknesses, scientists can create therapies that boost its ability to fight off infections. For example, vaccines work by introducing a harmless version of a pathogen or its components into the body, stimulating the immune system to produce a response without causing the disease. This primes the immune system for future encounters with the real pathogen, allowing it to respond more quickly and effectively.In conclusion, the immune system is a remarkable and complex defense mechanism that plays a vital role in our health. Its a topic that hasfascinated me for years, and I hope to continue learning more about it as I pursue my studies. By understanding how it works and how we can support its function, we can better protect ourselves and others from the myriad of threats that exist in our world.。
医学免疫学名解英文综述

免疫Immune response: the response made by the host to defend itself against the introduction of foreign substances.Antigen: An antigen is any agent capable of binding specifically to components of immune system, such as BCR and soluble antibodiesImmunogen - A substance that induces a specific immune response.(All immunogens are antigens, but not all antigens are immunogens)Antigenicity: The ability of a compound to bind with antibodies or cells of the immune system. This binding is highly specific.ImmunogenicityImmunogenicity is the ability of a particular substance, such as an antigen or epitope, to provoke a specific immune response in the body of a human or animal.Hapten半抗原:A hapten is a small molecule which can elicit an immune response only when attached to a large carrier such as a protein; the carrier may be one which also does not elicit an immune response by itself.EpitopeEpitope is the portion of the antigen that binds specifically with the binding site of an antibody or a receptor on a lymphocyte.TI-AgThymus -independent antigens are antigens which can directly stimulate the B cells to produce antibody without the requirement for T cell help in general.TD-AgThymus -dependent antigens are those that do not directly stimulate the production of antibody without the help of T cellsSuper antigenAn antigen which polyclonally activates some subtypes of the T cells (up to 20%).Adjuvants:A substance that when mixed with an immunogen, enhances the immune response against the immunogen.ImmunoglobulinThe Immunoglobulins are globulin which function as antibodies or similar to antibodies in chemical structure.Complementarity determining region (CDR)互补决定区:A complementarity determiningregion (CDR) is a short amino acid sequence found in the variable domains of antigen receptor (e.g. immunoglobulin and T cell receptor) proteins that complements an antigen and therefore provides the receptor with its specificity for that particular antigen.Complement:A group of serum proteins involved in the control of inflammation, the activation of phagocytes and the lytic attack on cell membranes. It belongs to the innate immune system, and can be recruited and brought into action by the adaptive immune system.Common receptor subunitThere is same receptor subunit for cytokine signaling among the different cytokine receptors.e.g. common γ chainCytokine (CK) : Small soluble proteins that mediate immune and inflammatory reactions and are responsible for communications between leukocytes and other cells.Soluble cytokine receptorSoluble cytokine receptor is the extracellular part of the receptor, which can competitively bind to cytokineCytokine storm: Under certain circumstances (e.g. septic shock), large amounts of CKs (such as TNF) are produced, they may be active distant from their site of secretion.Leukocyte differentiation antigen白细胞分化抗原:The cell surface markers which express or disappear on the different cells in the different stages of differentitation and activation.Cluster of differentiation (CD): Cell surface molecules of leucocytes that are distinguishable with monoclonal antibodies as an immunologic marker.Cell adhesion molecules, CAM: A group of proteins involved in adhesion of cell to cell or cell to extracellular matrix (ECM), such as ICAM-1, ICAM-2, ICAM-3, VCAM-1 and PECAM etc.IntegrinIntegrins are transmembrane receptors that mediate the attachment between a cell and the tissues that surround it, such as other cells or the extracellular matrix (ECM)SelectinsSelectins (CD62) are a family of cell adhesion molecules.MHC主要组织相容性复合物:The major histocompatibility complex (MHC) is a large genomic region or gene family found in most vertebrates. It is the most gene-dense region of the mammalian genome and plays an important role in the immune system, autoimmunity, and reproductive success.PolymorphismThe phenomenon of having multiple alleles at given genetic locus in the populationSomatic hypermutation体细胞高度突变:Somatic hypermutation (or SHM) is a mechanism inside cells that is part of the way the immune system adapts to the new foreign elements which confront it (for example, microbes).ITAMAn immunoreceptor tyrosine-based activation motif (ITAM) is a conserved sequence of four amino acids that is repeated twice in the cytoplasmic tails of certain cell surface proteins of the immune system.Negative selection负选择:The death of autoimmune lymphocytes shortly after they develop. Also known as clonal deletion.Positive selection: Double positive cells that bind, with moderate affinity, to MHC-Ag on thymic stroma cells survive. DP cell acquire MHC restriction though positive selection.Foxp3A member of the FOX protein family, FOXP3 appears to function as a master transcription factor in the development and function of regulatory T cells.APC:A variety of cell types specialized in the presentation of peptide-MHC to lymphocytes, causing either tolerance or immunity.Cross-presentationClass I MHC molecules present exogenous Ags to CD8+ T cells.Immunological synapseWhen the TCR complex recognizes MHC-associated peptides on an APC, several T cell surface proteins and intracellular signaling molecules are rapidly mobilized to the site of T cell-APC contact. This region of physical contact between the T cell and the APC has been called the immunological synapseAnergy无反应性:Anergy is a term in immunobiology that describes a lack of reaction by the body's defense mechanisms to foreign substances, and consists of a direct induction of peripheral lymphocyte tolerance.Regulatory T cell调节性T细胞:Regulatory T cells (sometimes known as suppressor T cells) are a specialized subpopulation of T cells that act to suppress activation of the immune system and thereby maintain immune system homeostasis and tolerance to self-antigens.AICD激活诱导的细胞死亡:activation-induced cell death (AICD) recognition and deletion of T lymphocytes that have been induced to proliferate by receptor-mediated activation, preventing their overgrowth.Class switchingClass switching is a biological mechanism that changes a B cell's production of antibody from one class to anotherCentral toleranceis the mechanism by which newly developing T cells and B cells are rendered non-reactive to self during their development in thymus and bone marrow.Secondary Antibody: An antibody that binds to primary antibodies or antibody fragments. They are typically labeled with probes that make them useful for detection, purification or cell sorting applications.Affinity(亲和力)Strength of the reaction between a single antigenic determinant and a single Ab combining siteAvidity(亲合力)The overall strength of binding between an Ag with many determinants and multivalent AbsELISA (Enzyme linked immuno-sorbent assay) An immunological test, using an enzyme as a label to determine presence of target protein.ELISPOT (Enzyme-linked immuno-sorbent spot) A common method for monitoring immune responses in humans and animals. At appropriate conditions the ELISPOT assay allows visualization of the secretary product of individual activated or responding cells.Immuno-labeling techniquesSpecific Abs (or Ags ) labelled with fluorescein, enzymes or radioisotopes are used as probes for the detection of Ags (or Abs).Artificial active immunization: Administration of an antigen for active production of immunity. Active immunization results in the production of antibodies directed against the infecting agent or its toxic products; it may also initiate cellular immunity.Artificial passive immunization:Immunization may be accomplished passively by administering either performed immunoreactive serum (Abs, CKs) or cells.Vaccine: Administration of an antigen for active production of immunity is called artificial active immunization. The agent used for artificial active immunization is called vaccine.Planned immunization: A rational program of childhood immunization against infectious disease, when many of the most damaging and preventable infections normally appear.ImmunotherapyImmunotherapy is the approach to balance or intervene the immunologic function in order to fight against the disease by the principle of immunology.Pattern recognition receptors (PRRs)Pattern recognition receptors (PRRs) are a primitive part of the immune system. They are proteins expressed by cells of the innate immune system to identify pathogen-associated molecular patterns (PAMPs), which are associated with microbial pathogens or cellular stress, as well as damage-associated molecular patterns (DAMPs), which are associated with cell components released during cell damage.antigen,Ag 抗原immunogenicity 免疫原性immunoreactivity 免疫反应性complete antigen 完全抗原incomplete antigen,hapten 不完全抗原,半抗原antigenic specificity 抗原特异性epitope,antigenic determinant 抗原表位,抗原决定基antigenic valence 抗原结合价sequential epitope,linear epitope 顺序表位,线性表位conformational apitope 构象表位common apitope 共同抗原表位cross-reaction 交叉反应cross antigen 交叉抗原conformation 分子构象accessibility 易接近性thymus dependent antigen,TD-Ag 胸腺依赖性抗原thymus independent antigen,TI-Ag 非胸腺依赖性抗原heterophilic antigen 异嗜性抗原xenogenic antigen 异种抗原allogenic antigen 同种异型抗原autoantigen 自身抗原idiotypic antigen 独特型抗原endogenous antigen 内源性抗原exogenous antigen 外源性抗原allergen 变应原tolerogen 耐受原stimulator 免疫刺激剂superantigen 超抗原adjuvant 佐剂mitogen 丝裂原antibody 抗体immunoglobilin 免疫球蛋白class 类type 型variable region 可变区,V区constant region 恒定区,C区hypervariable region,HVR 高变区complementarity determining region,CDR 互补决定区antigen-binding site 抗原结合部位framework region,FR 骨架区hinge region 铰链区joining chain J链secretory piece,SP,secretory component,S C 分泌片,分泌成分papain 木瓜蛋白酶pepsin 胃蛋白酶fragment of antigen binding,Fab 抗原结合片段fragment crystallizable,Fc 可结晶片段isotype 同种型allotype 同种异型idiotype,Id 独特型idiotope 独特位anti-idiotype antibody,AId 独特性抗体opsonization 调理作用antibody-dependent cell-mediated cytotoxicity,ADCC 抗体依赖的细胞介导的细胞毒作用macroglobulin 巨球蛋白polyclonal antibody,pAb 多克隆抗体monoclonal antibody,mAb 单克隆抗体complement,C 补体complement regulatory protein 补体调节蛋白complement receptor,CR 补体受体classical pathway 经典途径C5 convertase C5转化酶membrane attack complex,MAC 攻膜复合物alternative pathway 旁路途径,替代激活途径lectin pathway,MBL pathway 凝集素途径mannose-binding lectin,MBL 甘露糖结合凝集素ficolin ,FCN 纤维胶原素MBL-associated serine protease,MASP MBL相关丝氨酸蛋白酶C1 inhibitor,C1INH C1抑制物C4 binding protein,C4bp C4结合蛋白decay-accelerating factor ,DAF 衰变加速因子immune adherence 免疫黏附cytokine 细胞因子autocrine 自分泌paracrine 旁分泌endocrine 内分泌pleiotropism 多效性redundancy 重叠性synergy 协同性antagonoism 拮抗性interleukin,IL 白细胞介素colony-stimulating factor,CSF 集落刺激因子interferon,IFN 干扰素tumor necrosis factor,TNF 肿瘤坏死因子growth factor,GF 生长因子chemokine 趋化因子class1 cytokine receptor family 1类细胞因子受体家族class 2 cytokine receptor family 2类细胞因子受体家族tumor necrosis factor receptor肿瘤坏死因子受体家族superfamily,TNFRSFIg superfamily receptor,Ig SFR 免疫球蛋白超家族受体chemokine receptor family 趋化因子受体家族cytokine storm 细胞因子风暴cell surface marker 细胞表面标记human leukocyte differentiation antigen,HLDA 人白细胞分化抗原lineage 谱系cluster of differentiation,CD 分化cell adhension molecule,CAM 细胞黏附分子extracellular matrix,ECM 细胞外基质immunoglobulin superfamily,IgSF 免疫球蛋白超家族integrin family 整合素家族selectin family 选择素家族lymphocyte homing receptor,LHR 淋巴细胞归巢受体HEV 高内皮微静脉major histocompatibility complex 主要组织相容性复合体human leukocyte antigen 人类白细胞抗原B2 microglobulin,b2m 微球蛋白polymorphism 多态性HLA genotyping HLA基因分型haplotype 单体型linkage disequilibrium 连锁不平衡anchor position 锚定位anchor residue 锚定残基MHC restriction MHC限制性cross-matching 交叉配合B lymphocyte B淋巴细胞bursa of Fabricius 禽类法氏囊B cell receptor,BCR B细胞受体gene rearrengement 基因重排gene segment 基因片段recombinase 重组酶recombination activating gene,RAG 重组激活酶基因recombination signal sequence,RSS 重组信号序列terminal deoxynucleotidyl transferase,TdT 末端脱氧核苷酸序列allelic exclusion 等位排斥isotype exclusion 同型排斥combinational diversity 组合多样性junctional diversity 连接多样性receptor editing 受体编辑somatic hypermutation 体细胞高频突变pro-BCR 前B细胞受体pro-B cell 祖B细胞pre-B cell 前B细胞immature B cell 未成熟B细胞mature B cell 成熟B细胞clone deletion 克隆清除anergy 失能immunoreceptor tyrosine-based activation免疫受体酪氨酸激活基序motif,ITAMcoreceptor 共受体co-stimulatory molecule 共刺激分子self-renewal 自我更新polyreactivity 多反应性natural antibody 天然抗体plasma cell 浆细胞memory B cell 记忆B细胞regulatory B cell 调节性B细胞T lymphocyte T淋巴细胞thymus 胸腺hematopoietic,HSC 骨髓多能造血干细胞lymphoid progenitor cell 淋巴样祖细胞double negative cell,DN cell 双阴性细胞double positive cell,DP cell 双阳性细胞sigle positive cell,SP cell 单阳性细胞positive selection 阳性选择negative selection 阴性选择immunoreceptor tyrosine-based inhibitory免疫受体酪氨酸抑制基序motif,ITIMphytohemagglutinin,PHA 植物血凝素naive T cell 初始T细胞memory T cell,Tm 记忆T细胞effector T cell 效应T细胞helper T cell,Th 辅助T细胞cytotoxic T lymphocyte,CTL 细胞毒性T细胞regulatory T cell,Treg 调节性T细胞antigen-presenting cell,APC 抗原提呈细胞profession APC 专职性APCdendritic cell,DC 数突状细胞conventional DC,cDC 经典DCplasmacytoid DC,pDC 浆细胞样DCregulatory DC 调节性DCfollicular DC,FDC 滤泡DCimmature DC 未成熟DCmature DC 成熟DCLangerhans cell,LC 朗格汉斯细胞interstitial DC 间质DCveiled cell 隐蔽DCperipheral blood DC 外周血DC interdigitating DC,IDC 并指状DCmonocyte 单核细胞macrophage 巨噬细胞antigen processing 抗原加工proteasome 蛋白酶体transporter associated with antigen抗原加工相关转运物processing ,TAPchaperone 伴侣蛋白ER resident aminopeptidase,ERAP 氨基肽酶endosome 内体phagosome 吞噬体MHC class 2 compartment,M2C MHC2类小室Ia-associated invariant chain,Ii Ia相关恒定链class 2-associated invariant chain peptide,CLIP MHC2类相关的恒定链多肽cross-presentation,cross-priming 交叉提呈,交叉致敏pMHC,peptide-MHC2 complex 抗原肽-MHC分子复合物antigen recognition 抗原识别immunological synapse 免疫突触perfotrin 穿孔素granzyme 颗粒酶activation-induced cell death,AICD 活化诱导的细胞死亡humoral immune response 体液免疫应答germinal center 生发中心centroblast 中心母细胞centrocyte 中心细胞follicular helper T cell,Tfh 滤泡辅助T细胞somatic hypermutation 检查是否重复,体细胞高频突变affinity maturation 抗体亲和力成熟class switching 类别转换isotype switching 同种型转换switching region 转换区antibody forming cell,AFC 抗体形成细胞primary response 初次免疫secondary response,anamnestic response 再次免疫lag phase 潜伏期log phase 对数期plateau phase 平台期decline phase 下降期。
sensitive immune system阅读理解

sensitive immune system阅读理解Allergies (过敏) cause loads of trouble. Some people suffer from seasonal hay fever as pollen (花粉) flws through the air. Others react tomaterials at their very touch. And some sorry souls go into shock at the mere presence of certain foods, particularly peanuts and shellfishThe cause in each case is an overensitive immune system.This creates annoying and sometimes life-threatening symptoms.Over-reactive immunsystems may not, though, be an entirely bad thing. Another role played by the immune system is to destroy malignant tumors (恶性肿瘤) before theytake hol-work carried out recently by Annete Wigertz of the Karolinska Institute, in Stockholm, and her colleague suggests that the immune systemsof those with allergies may be particularly good at thisThis finding came after Doctor Wigertz and her team interviewed 1, 527 people with gliomas (a type of brain tumor)in Denmark, FinlandNorway, Sweden and the southeast of England. The researchers asked the patients in guestion whether they had a history of allergies, and then comparedhe results with those for 3.309 otherwise similar individuals who did not have brain tumors. As Doctor Wigertz reports in the Americamn Joumnal ofEpidemiology, the tumor-free were more likely to suffer from allergies. The presence of an alergy was associated with a 30% reduction in the likelihood of having a glioma.This was not al that suroisng. Previous research nad detected simi ar correlatons. suggesting that a we come side eftet of alergy was resistanceto cancer.But this new study went further. It looked carefully at the time in the patients' lives when their allergies were active, and it found that thistiming was crucial. Doctor Wigertz noted that the absence of allergy was correlated with the time when the glioma first formedAwkwardly, this result is open to two rather difterent interpretations. The timistic explanation is that the hyperactive immune syvstem associatedwith aergy does protect against fumors.The other interpretation is that tumors are doing something as they grow that stops the immune system and thusallergic reactions. Either way tumor and lack of allergy coincide. And either way. something interesting is going on.1.What accounts for most allergies?A.Pollen in-the airB.Presence of some foodC .Sensitivity of the immune system.D .Overreacting to the world2. How is this finding different from those from the previous research?A. It indicates a benefit of allergyB. It uncovers the cause of allergiesC. It proves the correlations between allergy and cancersD It discovers the cancer started when there was no allergy.3. What's the last paragraph about?A.What explains the finding of the researchB. Why the research on allergy is of significance.C.Whether cancer is related to the absence of allergyD.How allergy protects people against cancers.4. What can we learn from the research findings?A.Bad news has wingsB .When we win some, we lose someC.Robbing Peter to pay Paul.D. One man's meat, another man's poison.。
人类的免疫系统英语作文

人类的免疫系统英语作文英文回答:The human immune system is a complex network of cells, tissues, and organs that work together to defend the body against infection. It is a remarkable system that has evolved over millions of years to protect us from a wide range of pathogens, including bacteria, viruses, and parasites.One of the most important components of the immune system is the white blood cells, also known as leukocytes. White blood cells are produced in the bone marrow and circulate throughout the body in the blood and lymph fluid. There are several different types of white blood cells, each with a specific function in the immune response.Neutrophils are the most common type of white blood cell. They are phagocytic, which means that they can engulf and destroy foreign particles. Neutrophils are particularlyimportant in the early stages of an infection, when they help to clear away bacteria and other microorganisms.Macrophages are another type of phagocytic white blood cell. They are larger than neutrophils and are found in tissues throughout the body. Macrophages are important for clearing away dead cells and debris, and they also play a role in the immune response to cancer.Lymphocytes are a type of white blood cell that is responsible for the body's adaptive immune response. Lymphocytes are produced in the bone marrow and mature in the thymus gland. There are two main types of lymphocytes: B cells and T cells.B cells produce antibodies, which are proteins that bind to specific antigens. Antigens are molecules that are found on the surface of foreign particles, such as bacteria and viruses. When a B cell binds to an antigen, it produces an antibody that is specific for that antigen.Antibodies help to neutralize pathogens and target them for destruction by other immune cells.T cells are responsible for cell-mediated immunity. They recognize and destroy infected cells and cancer cells. T cells also play a role in regulating the immune response.The immune system is a complex and dynamic system that is constantly adapting to new threats. It is a remarkable example of the body's ability to protect itself from harm.中文回答:人类的免疫系统是一个复杂的细胞、组织和器官网络,它们共同作用以保护身体免受感染。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
NATURE BIOTECHNOLOGY VOLUME 23 NUMBER 11 NOVEMBER 20051399Activation of the mammalian immune system by siRNAsJoao T Marques & Bryan R G WilliamsInhibition of gene expression through RNA interference (RNAi) is emerging as a powerful experimental tool for gene function and target validation studies. The potential uses of this technology seem unlimited, extending to the prevention and therapy of human diseases. However, recent work demonstrating that there are unanticipated, different nonspecific effects associated with the use of small interfering RNAs in mammals has raised concerns about the safe use of RNAi in vivo . These nonspecific effects include activation of the immune system, potentially harming the individual. The application of screening assays for nonspecific activation of both innate and acquired immunity will be necessary for further development of RNAi as a therapeutic tool.RNA interference (RNAi) was first described in Caenorhabditis ele-gans 1 in 1998, where long double-stranded RNA (dsRNA) injected into adult animals silenced the expression of genes with sequence homol-ogy to the dsRNA 2. Subsequently, similar phenomena were described in Drosophila melanogaster plants and undifferentiated mammalian cells 2–4. When dsRNA is introduced into the cytoplasm, it is processed by an RNase III enzyme, Dicer, into 21-nucleotide (nt) dsRNAs with 2-nt 3′ overhangs, termed small interfering RNAs (siRNAs) (see Fig. 1 and Box 1 for glossary)5,6. SiRNAs are incorporated into the RNA-induced silencing complex (RISC), which mediates the degradation of complementary mRNAs, silencing gene expression 7. Initially, it was believed that nonspecific activation of the antiviral response by dsRNA longer than 30 nt would preclude the use of RNAi as an experimental or therapeutic tool in mammals 3,8,9. However, in 2001, Tuschl and col-leagues 8 and Caplen et al.9 described the delivery to the cytoplasm of mammalian cells of siRNAs that efficiently mediated gene silencing. Small interfering RNAs could be easily chemically synthesized allowing exploration of their use to regulate gene expression. Importantly, this was different from previous strategies such as antisense and ribozymes, in that RNAi exploited an endogenous mechanism 10. However, dur-ing attempts to develop techniques to efficiently deliver siRNAs and trigger RNAi in vivo, it was noted that siRNAs could activate cells of the immune system and induce the production of cytokines both in vivo and in vitro 11,12. Together with earlier reports of activation of the interferon (IFN) system in cell culture by siRNA 13–16, this has raised questions about the specificity of RNAi. This review describes what is known about nonspecific activation of the immune system by siRNAs and suggests future directions that may allow use of RNAi in vivo by bypassing or controlling these unwanted effects.Activation of the immune system by siRNAsThe pathways responsible for the recognition of siRNAs by immune cells were initially not clear 11,12. Mammalian immune cells express a family of toll-like receptors (TLRs), which recognize pathogen-associated molec-ular patterns including unmethylated CpG DNA and viral dsRNA 17. TLR3, the receptor for dsRNA, was a logical candidate for the recogni-tion of the siRNAs in the context of immunostimulation. Indeed, TLR3 overexpressed in cultured human embryonic kidney (HEK) 293 cells was capable of recognizing siRNAs 12. However, the activation of immune cells by siRNAs is sequence dependent and sense or antisense strands separately can induce cytokine production as efficiently as duplex siR-NAs 18–20. Therefore, as TLR3 neither recognizes single-stranded RNA (ssRNA) nor requires sequence specificity 17, it is unlikely involved in activation of the immune system by siRNAs. It has now become clear that TLR7 and TLR8 mediate the recognition of siRNAs in a sequence-dependent manner; using the appropriate knockout mice, it has been shown that TLR7 is absolutely required for induction of cytokines in murine immune cells in response to siRNAs 18–20.TLR7 and TLR8 were initially shown to mediate the recognition of RNA viruses 21,22 and small synthetic antiviral compounds referred to as imidazoquinolines, such as imiquimod and its derivative resiquimod (R-848)23. Interestingly, not all ssRNA molecules are capable of activating TLR7 and TLR8 but rather U- and G-rich ssRNAs seem to be preferen-tially recognized 21,22. Despite these prior observations, recent reports of the activation of the immune system by siRNAs in a sequence-dependent manner have surprised the field 18–20.Studies of HEK 293 cells overexpressing TLRs have indicated that ssRNAs are recognized by mouse TLR7 and human TLR8 but not by mouse TLR8 or human TLR7 (ref. 21). However, siRNAs can be recog-nized by human plasmacytoid dendritic cells (pDCs) through TLR7 (refs. 18,19,24) and human monocytes likely via TLR8 (refs. 24,25). The vari-ability in the immunostimulatory activity of different siRNA sequences observed in immune cells from human and mice may result from siRNA recognition by TLR7 or TLR8 homo or heterodimers as determined byDepartment of Cancer Biology, Lerner Research Institute, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, Ohio, 44195, USA. Correspondence should be addressed to B.R.G.W. (williab@)Published online 4 November 2005; doi:10.1038/nbt1161R E V I E W©2005 N a t u r e P u b l i s h i n g G r o u p h t t p ://w w w .n a t u r e .c o m /n a t u r e b i o t e c h n o l o g ytheir expression patterns in different cell types. It is also possible that siRNA recognition by TLR7 and/or TLR8 requires accessory proteins. Although such cofactors have not been identified for TLR7 or 8, this pos-sibility is supported by the finding that autologous plasma enhances the induction of cytokines by siRNA in peripheral blood mononuclear cells (PBMC 19) (Fig. 1). Furthermore, precedent for a cofactor requirement exists since recognition of bacterial lipopolysaccharide by TLR4 requires the CD14 and MD-2 proteins 17. There has also been speculation about possible coreceptors for TLR9, which recognize unmethylated CpG DNA (Box 2) and could modulate different responses 26.TLR7, TLR8 and TLR9 all recognize nucleic acids, are expressed in T o further complicate the issue of TLR7, TLR8 and TLR9 expression in immune cell subsets, the data differ between mice and humans 24,25,28. Therefore, it remains unclear which TLRs are expressed in each cell type of the immune system in humans and mice.All vertebrate species analyzed possess a single copy of the TLR7 and TLR8 genes present in tandem on the same chromosome 36. This indi-cates that TLR7 and TLR8 likely originated through duplication of an ancestral gene before the evolution of vertebrates and that there was selective pressure for the maintenance of both receptors 36.Therefore, despite the fact that TLR7 and TLR8 are closely related, it is likely that they fulfill nonoverlapping functions. Indeed, synthetic agonists that R E V I E Wn o l o g yNATURE BIOTECHNOLOGY VOLUME 23 NUMBER 11 NOVEMBER 2005 1401humans that complicate functional analy-sis of siRNA signaling using mouse models. Furthermore, a recent report that a poly(G) oli-godeoxynucleotide was stimulatory to human CD4+ T cells in a TLR8-dependent, TLR7- and TLR9-independent manner further illustrates the functional differences between TLR7 and TLR8 in humans 38.Endosomal recognition of siRNAsAs mentioned above, TLR7, TLR8 and TLR9 are expressed in endosomes and require endo-somal maturation for efficient signaling 27. Given the similarities among TLR7, TLR8 andTLR9, it is possible to apply the large body of knowledge about signaling by CpG DNA through TLR9 (refs. 26,27,39,40) (Box 2) and speculate about TLR7- and TLR8-dependent signaling. Endosomal recognition by TLR9 is not bypassed by combining the nucleic acid with cationic lipids or polymers and, in fact, this has been shown to extend the time that CpG oligodeoxynucleotides (ODNs) migrate through the endo-somes 41 (Box 2). Similarly, several studies have demonstrated that the method of delivery is critical for the immunostimulatory activity of siRNAs 18–20. Injection of siRNAs in complex with cationic lipids into mice induces a potent cytokine response that is debilitating to the organ-ism 18,19. On the other hand, injection of naked, unmodified siRNAs or siRNAs conjugated to cholesterol has no significant effect on immune system activation 42,43.There are two possible explanations for the absence of immunos-timulation by naked siRNAs. First, unmodified siRNAs have a short half-life in serum and may be degraded before being recognized by specific receptors. Second, siRNA in complex with lipids, perhaps by resembling a viral particle, might be more readily recognized by immune cells than would naked siRNA (Figs. 1 and 2). In favor of the second option, Sioud 20 reported that naked nuclease-resistant siRNAs contain-ing a phosphothioate backbone were not immunostimulatory. However, these results should be interpreted carefully considering that naked CpG ODNs can be immunostimulatory independent of backbone modifica-tions 26 (Box 2).Recognition of siRNAs by TLRs takes place in the endosome, before the siRNAs enter the cytoplasm (Fig. 1). Therefore, it would be expected that if siRNAs could enter the cytoplasm avoiding the endosome, they should bypass the activation of immune cells but still mediate gene silencing. Although electroporation should provide direct cytoplasmic delivery of siRNAs without their migrating through the endosome, con-flicting results have been obtained using this technique. Sioud 20 did not observe cytokine production after siRNA delivery through electropora-tion of monocytes, but Hornung et al.18 showed that a naked siRNA still induced IFN-α when pDCs were electroporated. Unfortunately, these studies did not assess the subcellular localization of the siRNAs or their silencing efficiency in either monocytes or pDCs. The discrep-ancies between Sioud 20 and Hornung et al .18 could reflect differential trafficking of the siRNAs in the two cell types. For example, the IFN induction observed by Hornung et al.18 after siRNAs entered the cell by electroporation might be due to trafficking of small amounts of siRNA through the endosome in pDCs.Notably, when siRNAs are coated with poly-L -lysine, significantly more IL-6 and tumor necrosis factor are induced in PBMCs than when they are in complex with cationic lipids or polyethylenimine 19. In con-trast, more IFN-α is induced by siRNA in complex with cationic lipids or polyethylenimine compared with the same sequence coated with poly-L -lysine 19. It is possible that different siRNA complexes could either modify the fate of the siRNA in a given cell type or change which cell types are preferentially stimulated. In accordance with the latter hypothesis, TNF induction by siRNAs is abrogated in PBMCs depleted of monocytes, whereas IFN-α induction is abrogated only in pDC-depleted PBMCs 19. Of note, there is a difference in the cytokine profile induced in myeloid dendritic cells (mDCs) and pDCs by the TLR7 and TLR8 ligand, R-848, with IL-12 being induced only in mDCs and IFN-α being induced only in pDCs 28. Similar differential responsiveness to different sequences or complexes also occurs with the immune responses to CpG ODNs medi-ated by TLR9 (ref. 26) (Box 2).The importance of sequence, size and structureA complete understanding of the sequence requirements for siRNA-mediated immune activation and gene silencing is necessary before attempts can be made to engineer siRNAs that shut down gene expres-sion without inducing deleterious side effects. U- and G-rich regions seem to be a common feature among the immunostimulatory motifs;R E V I E W©2005 N a t u r e P u b l i s h i n g G r o u p h t t p ://w w w .n a t u r e .c o m /n a t u r e b i o t e c h n o l o g y1402 VOLUME 23 NUMBER 11 NOVEMBER 2005 NATURE BIOTECHNOLOGYGUCCUUCAA and UGUGU are present in the immunostimulatory ssRNAs 18,19 (Table 1). However, as mentioned above, several immuno-stimulatory siRNAs do not contain either of these motifs, whereas oth-ers are not stimulatory despite being U- and G-rich 20. Therefore, other characteristics, such as the position or proportion of UG dinucleotides within the molecule or the composition of flanking sequences, may play a role in determining the nature and potency of the cytokine response induced by ssRNA.In addition to sequence composition, the size of ssRNAs is also important for activation of the immune system. Hornung et al .18 observed that 12-nt ssRNAs containing the immunostimulatory motif (GUCCUUCAA) were poor inducers of IFN-α in pDCs but that increas-ing their size to 16 nt or 19 nt completely restored cytokine induction. It remains unclear if the recognition of immunostimulatory motifs in the siRNA can occur in the context of the duplex. Sioud 20 has observed that the single strands separately tend to be stronger stimuli than the duplex, indicating that the presence of single strands is critical. An excess of one of the strands during annealing can lead to the presence of single strands in a siRNA preparation, but in most cases denaturation of the duplex in the endosome will release the single strands. This was likely the case where a short hairpin RNA containing the immunostimulatory motif was still capable of activating the immune response 18.There might be qualitative differences among siRNA sequences that are recognized by TLR7 and TLR8 as has been demonstrated for CpG ODNs. The latter can be divided into groups based upon sequences flanking the CpG motif that determine the cytokine profile they induce 26 (Box 1). In humans, substitution of U with A in the siRNA immunos-timulatory sequence abrogates TNF and IL-6 induction in PBMCs, but the substitution of G with A abrogates only induction of IFN-α in pDCs without affecting the induction of TNF, IL-6 and IL12 in PBMCs 21. Moreover, siRNAs with the same UGUGU motif embedded in differ-ent sequence contexts induce qualitatively different cytokine profiles in human PBMCs 19. This suggests that there are important qualitative differences in responses to different ssRNAs, especially in humans, where both TLR7 and TLR8 are functional. It remains to be determined if het-erodimerization between TLR7 and/or TLR8 and possibly other acces-sory proteins determines sensitivity to different immunostimulatorysequences. Interestingly, a ssRNA containing a CpG motif and a 6 nt poly-(G) run at the 3′ end has been shown to activate monocytes, but the receptors responsible for recognition of this molecule remain unknown(Table 1). When TLR3, TLR7, TLR8 and TLR9 are overexpressed sepa-rately in HEK 293 cells, they are not able to provide responsiveness to this sequence 44. It is important to note that for recognition of CpG ODNs by TLR9, the sequences flanking the CpG motif that induce the strongest immunostimulatory activity vary between mice and humans 26. Therefore, human and mouse TLR7s likely have different preferences in terms of immunostimulatory sequences. Purified recombinant TLR9 binds CpG ODNs directly in a sequence- and pH-dependent manner, implying that all of the in vivo requirements for recognition of unmeth-ylated CpG DNA are provided by the receptor itself 40. The crystal struc-ture of TLR3 also suggests a mechanism for direct binding of dsRNA 45. Despite these precedents, it is not yet known whether TLR7 and TLR8 can bind siRNAs directly.Because the sequence and chemical nature of siRNAs can determine not only their gene silencing potential, but also their ability to induce immune responses, interest has been intense in identifying chemical modifications that might preserve silencing while bypassing immune system activation 18,46. Locked nucleic acid (LNA) modifications of the 3′ or 5′ termini of the sense strand of siRNAs can abrogate their immunostimulatory activity 18. LNA nucleotides contain a methylene linkage between the 2´ oxygen and the 4´ carbon of the ribose ring in the ribonucleotides 47,48. However, depending on the extent and loca-tion of the LNA modifications, they can block not only the inductionR E V I E W©2005 N a t u r e P u b l i s h i n g G r o u p h t t p ://w w w .n a t u r e .c o m /n a t u r e b i o t e c h n o l o g yNATURE BIOTECHNOLOGY VOLUME 23 NUMBER 11 NOVEMBER 2005 1403of IFN but also the gene silencing activity of the siRNA 18. Different modifications of the siRNA backbone have also been tested and several identified that do not affect silencing activity, at least in cell culture systems 49. The inclusion of DNA bases in siRNAs is well tolerated and may even increase silencing activity 50. In vivo , these DNA-RNA hybrids could potentially activate not only TLR7 and TLR8, but also TLR9 if they contain CpG motifs 51.In contrast to unmodified molecules, siRNAs containing such back-bone modifications, such as 2´ F, 2´ O-methyl and 2´ H, do not induce cytokines when injected into mice while maintaining silencing activ-ity 46. More studies are necessary to determine the optimal modifications that abrogate immunostimulatory activity without loss of silencing. The effects of these modifications, however, have to be determined empiri-cally on a case-by-case basis because their immunostimulatory proper-ties are not completely understood and careful studies will be required before modified siRNA can be safely used in therapeutic applications.acids are known 53cytosine is usually methylated bacterial DNA has a much higher frequency of CpG dinucleotides and they are not methyl-ated 54for the lack of immunostimulatory activity of mRNAs 56. However, an in vitro –transcribed mRNA encoding green fluo-rescent protein (GFP) has been shown to trigger production of IFN-α by pDCs, regardless of the presence of either a short poly(A) tail or the 7-methyl-guanosine cap characteristic of eukaryotic mRNAs 22. These data do not rule out the possibility that a long poly(A) tail might act as a decoy preventing recognition of immunostimulatory motifs. Notably, human mRNAs, when in complex with protamine 57 or a cationic lipid 58, are also immunostimulatory to dendritic cells. Therefore, as mentioned above for CpG DNA, multiple levels of control are likely necessary to modulate recognition of ssRNAs. Compartmentalization of cellular RNAs may account at least in part for their failure to be recognized as immunostimulatory under normal conditions 21. Coated siRNA may resemble a viral particle and recognition of ssRNAs in such a context may be required for activation of the immune system, in addition to the presence of the immunostimulatory motifs. The notion that, as for CpG ODNs, particular sequence motifs in ssRNAs influence immunostimula-tion 18,19,21 is intriguing and indicates that to avoid immune recognition protein; OAS, 2´5´-oligoadenylate synthetase.R E V I E W©2005 N a t u r e P u b l i s h i n g G r o u p h t t p ://w w w .n a t u r e .c o m /n a t u r e b i o t e c h n o l o g y1404 VOLUME 23 NUMBER 11 NOVEMBER 2005 NATURE BIOTECHNOLOGYSequence-independent recognition of siRNAsNonimmune cells and immune cells can also recognize siRNAs indepen-dent of the sequence leading to the induction of type I IFNs 13–16. The signaling pathways that mediate this recognition are complex and involve a number of dsRNA-sensing receptors, both intracellular and membrane bound (Figs. 1 and 2). The intracellular pathways include the dsRNA-activated protein kinase (PKR) and the 2´5´-oligoadenylate synthetases that can differentially induce general protein synthesis inhibition 61, and signaling pathways that activate transcription factors including NF-κB and IFN regulatory factor (IRF)-3, leading to induction of genes, such as those encoding the IFNs 62 (Fig. 2). TLRs are primarily expressed in cells of the immune system, but can also be present in other cell types, includ-ing endothelial cells, fibroblasts and hepatocytes 30,35,63–65. In cultured cells, PKR and TLR3 have been shown to mediate the induction of IFN by siRNAs 12,14 (Figs. 1 and 2). Recognition of siRNAs by these pathways could potentially synergize with TLR7 and TLR8 in the activation of the immune system. It is known, for example, that dsRNA and CpG ODN can synergize in the activation of innate immune responses leading to enhanced tumor clearance 66. In addition, viruses can induce high levels of IFN in pDCs in the absence of both TLR7 and PKR signaling, presum-ably through production of intracellular dsRNA that is recognized by other RNA sensors 67. Such molecules might include caspase recruitment domain (CARD)-containing helicases, such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene-5 (MDA-5) proteins 68–70. Although their activation is capable of triggering IFN production, these helicases have not been implicated in recognition of siRNAs. However, experiments with TLR7-knockout mice indicate that TLR3, PKR and CARD-containing helicases all have at least minor roles in siRNA recognition in vivo 18. Additional experiments in TLR3- and PKR-knockout mice are needed to define the role of these proteins in response to siRNAs in vivo . It is also important to keep in mind that TLR8 is functional in humans but not in mice and until the differences between TLR7- and TLR8-mediated recognition of siRNAs are eluci-dated, studies in mice must be interpreted with caution.Implications for siRNA-mediated therapyThe immunostimulatory ‘side effects’ of siRNAs must be taken into account when considering possible applications of siRNAs for in vivo therapy. The potentially deleterious results of these side effects are illustrated by experiments in which mice injected with immunostim-ulatory siRNAs present signs of toxicity, including elevated levels of serum alanine and aspartate aminotransferases and reduced numbers of lymphocytes and platelets 19,52. As detailed above, there may be ways to experimentally bypass siRNA-induced side effects on the immune system while preserving gene-silencing activity. These might include modification of the siRNA backbone 18,52 or avoidance of immunos-timulatory sequence motifs 19. Furthermore, naked siRNAs conjugated to cholesterol or in complex with atelocollagen have been successfully used to promote gene silencing in vivo 42,71 and as discussed previously, the delivery of naked siRNAs might bypass activation of the immune system 43. Notably, endogenous dsRNAs that work through the RNAi pathway, called micro RNAs 72, likely present base modifications (e.g., methylation) or secondary structures that preclude the activation of the immune system, which could be incorporated into synthetic siRNAs to avoid nonspecific effects.M oreover, in some scenarios, activation of the immune system together with siRNA-mediated gene silencing might actually be desired, leading to enhanced therapeutic effects. Therefore, in the treatment of viral infections and tumors, siRNAs containing potent immunostimu-latory motifs could prove useful. However, it will be important to first understand whether there are distinct immunostimulatory sequencesthat determine qualitative differences in cytokine induction as well as the effect of different delivery methods on immune responses to siRNAs.Although the use of siRNA-mediated therapy in vivo is more complex than originally expected because of nonspecific effects on the immune system, there remains great potential for RNAi as a tool for in vitro experimentation as well as in vivo disease treatment.ACKNOWLEDGMENTSWe would like to thank Patricia Stanhope-Baker, Anthony Sadler, Mark Whitmore and Michelle Holko for helpful discussion and suggestions. Work in the Williams laboratory is supported by National Institutes of Health grants RO1 AI34039 and PO1 CA 62220.COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.Published online at /naturebiotechnology/Reprints and permissions information is available online at /reprintsandpermissions/1. Fire, A. et al. Potent and specific genetic interference by double-stranded RNA inCaenorhabditis elegans. Nature 391, 806–811 (1998).2. Mello, C.C. & Conte, D. Jr. Revealing the world of RNA interference. Nature 431,338–342 (2004).3. Yang, S., Tutton, S., Pierce, E. & Yoon, K. Specific double-stranded RNA interferencein undifferentiated mouse embryonic stem cells. Mol. Cell. Biol. 21, 7807–7816 (2001).4. Baulcombe, D. RNA silencing in plants. Nature 431, 356–363 (2004).5. Meister, G. & Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature431, 343–349 (2004).6. Elbashir, S.M., Lendeckel, W. & Tuschl, T. RNA interference is mediated by 21- and22-nucleotide RNAs. Genes Dev. 15, 188–200 (2001).7. Tomari, Y. & Zamore, P .D. Perspective: machines for RNAi. Genes Dev. 19, 517–529(2005).8. Elbashir, S.M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference incultured mammalian cells. Nature 411, 494–498 (2001).9. Caplen, N.J., Parrish, S., Imani, F ., Fire, A. & Morgan, R.A. Specific inhibition of geneexpression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl. Acad. Sci. USA 98, 9742–9747 (2001).10. Robinson, R. RNAi therapeutics: how likely, how soon? PLoS Biol. 2, E28 (2004).11. Sioud, M. & Sorensen, D.R. Cationic liposome-mediated delivery of siRNAs in adultmice. Biochem. Biophys. Res. Commun. 312, 1220–1225 (2003).12. Kariko, K., Bhuyan, P ., Capodici, J. & Weissman, D. Small interfering RNAs mediatesequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J. Immunol. 172, 6545–6549 (2004).13. Sledz, C.A. & Williams, B.R. RNA interference in biology and disease. Blood 106,787–794 (2005).14. Sledz, C.A., Holko, M., de Veer, M.J., Silverman, R.H. & Williams, B.R. Activationof the interferon system by short-interfering RNAs. Nat. Cell Bio l. 5, 834–839 (2003).15. Kim, D.H. et al. Interferon induction by siRNAs and ssRNAs synthesized by phagepolymerase. Nat. Biotechnol. 22, 321–325 (2004).16. Persengiev, S.P., Zhu, X. & Green, M.R. Nonspecific, concentration-dependentstimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA 10, 12–18 (2004).17. Takeda, K. & Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 17, 1–14(2005).18. Hornung, V. et al. Sequence-specific potent induction of IFN-alpha by short interferingRNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 11, 263–270 (2005).19. Judge, A.D. et al. Sequence-dependent stimulation of the mammalian innate immuneresponse by synthetic siRNA. Nat. Biotechnol. 23, 457–462 (2005).20. Sioud, M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J. Mol. Biol. 348, 1079–1090 (2005).21. Heil, F . et al. Species-specific recognition of single-stranded RNA via toll-like receptor7 and 8. Science 303, 1526–1529 (2004).22. Diebold, S.S., Kaisho, T., Hemmi, H., Akira, S. & Reis e Sousa, C. Innate antiviralresponses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004).23. Hemmi, H. et al. Small anti-viral compounds activate immune cells via the TLR7MyD88-dependent signaling pathway. Nat. Immunol. 3, 196–200 (2002).24. Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immuneresponses. Nat. Immunol. 5, 987–995 (2004).25. Liu, Y.J. IPC: professional type 1 interferon-producing cells and plasmacytoid dendriticcell precursors. Annu. Rev. Immunol. 23, 275–306 (2005).26. Verthelyi, D. & Zeuner, R.A. Differential signaling by CpG DNA in DCs and B cells: notjust TLR9. Trends Immunol. 24, 519–522 (2003).27. Heil, F . et al. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncoversa strong relationship within the TLR7, 8 and 9 subfamily. Eur. J. Immuno l. 33, 2987–2997 (2003).R E V I E W©2005 N a t u r e P u b l i s h i n g G r o u p h t t p ://w w w .n a t u r e .c o m /n a t u r e b i o t e c h n o l o g y。