DGGE和TGGE的操作步骤
DGGE操作步骤

DGGE操作步骤DGGE(denaturing gradient gel electrophoresis)是一种常用的DNA分析技术,用于研究DNA序列的变异情况和分类等。
DGGE的操作步骤如下:1.准备样品2.PCR扩增将待测DNA进行PCR扩增,以增加DNA的数量。
需要用到一对引物,它们会扩增出感兴趣的DNA片段。
PCR反应体系中含有模板DNA、引物、dNTPs、DNA聚合酶和缓冲液。
引物的选择需要根据研究的目的确定。
3.准备DGGE凝胶将一定比例的聚丙烯酰胺和甲酰胺溶液以一定的比例混合,制备得到DGGE凝胶。
这个比例可以根据需要调整,以获得所需的DNA分离效果。
其中甲酰胺会使凝胶变性,使DNA分子在电泳中以变性形式进行分离。
4.准备DGGE系统将DGGE凝胶置于DGGE装置中。
根据实验需要,在凝胶两端插入电极,接通电源。
将凝胶槽填满电泳缓冲液,用于维持凝胶均匀膨胀,快速散发产热,以及降低电阻。
一般来说,TAE或TBE缓冲液可用于DGGE。
5.准备DNA样品将PCR扩增得到的DNA样品处理成等浓度。
可以用醋酸抽提、酚-氯仿法或商业DNA提取试剂盒等方法进行纯化。
6.电泳分离将等量的DNA样品加入到DGGE样品孔中。
将样品孔上方和下方的空孔填满DNA负载缓冲液,以避免样品在电泳过程中干涸。
开始电泳,一般电泳时间和电场强度要根据平台的规格表决定。
7.停止电泳和染色当DNA样品从顶端到底端通过凝胶后,在适当的时间内停止电泳。
将凝胶取出,根据需要进行染色。
GelRed或者SYBR Green等染料可以用于可视化DNA条带。
8.分析使用相应的软件如Quantity One或ImageJ等,对电泳结果进行分析。
将得到的DGGE电泳图像转化成数值形式,以研究样品中的DNA序列变异情况。
总结:DGGE是一种用于分析DNA序列变异的常用技术。
通过PCR扩增得到的DNA样品,经过一系列处理后,加入到DGGE凝胶中进行电泳分离。
变性梯度凝胶电泳

变性梯度凝胶电泳摘要:变性梯度凝胶电泳(DenaturingGradientGelE1ectrophoresiS,DGGE)是由Fisher 和Lerman发明用于检测DNA突变的技术,其主要是基于突变型和野生型棱酸序列的不同而导致其变性浓度的差异,利用变性梯度凝胶进行分离, 该手段分辨率达一个碱基。
恒定变性凝胶电泳(CDGE)、瞬时温度梯度电泳(TTGE)和温度梯度凝胶电泳(TGGE)是由DGGE经改进而成。
它们在突变分析上都有着重要的作用。
关键词:变性梯度凝胶电泳、恒定变性凝胶电泳、瞬时温度梯度电泳、温度梯度凝胶电泳1、前言:随着结构基因组计划接近尾声,人类基因神秘的面纱已逐步揭开,GenBank中已知基因的数目也与日俱增,各种遗传病的相关基因也了解得越来越多。
但是,在找到了候选基因后,还需要在相关的遗传病病人中进行突变检测,只有找到了相应的突变。
才能真正确定该基因与疾病的关系,从而服务于临床。
变性梯度凝胶电泳就是一种很有实用价值的突变分析方法。
该方法经改进,又发展出了恒定变性凝胶电泳(constant denaturant gel elec—trophoresis,CDGE)、瞬时温度梯度电泳(temporaltemperature gradient electrophoresis,TTGE)和温度梯度凝胶电泳(temperature gradient gel elec—trophoresis,TGGE)。
现在上述方法已经广泛应用于遗传病的诊断、突变分析和肿瘤相关基因的筛查等许多方面。
2、基本原理2.1变性梯度凝胶电泳由Fisher和Izrman口3于1983年创立,以后该技术和PCR技术相结合被广泛应用于各种突变分析。
它主要是利用梯度变性胶来分离DNA片段。
电泳开始时,DNA在胶中的迁移速率仅与分子太小有关,而一旦DNA泳动到某一点时,即到达该DNA变性浓度位置时,使得DNA双链开始分开,从而大大降低了迁移速率。
PCR-DGGE技术

PCR-DGGE技术一、实验原理变性梯度凝胶电泳(denatured gradient gel electrophoresis,DGGE)最早是Lerman 等人于20 世纪80 年代初期发明的,起初主要用来检测DNA 片段中的点突变。
Muyzer 等人在1993 年首次将其应用于微生物群落结构研究。
后来又发展出其衍生技术,温度梯度凝胶电泳(TGGE)。
该技术被广泛用于微生物分子生态学研究的各个领域,目前已经发展成为研究微生物群落结构的主要分子生物学方法之一。
双链DNA分子在一般的聚丙烯酰胺凝胶电泳时,其迁移行为取决于其分子大小和电荷。
不同长度的DNA片段能够被区分开,但同样长度的DNA片段在胶中的迁移行为一样,因此不能被区分。
DGGE技术在一般的聚丙烯酰胺凝胶基础上,加入了变性剂(尿素和甲酰胺)梯度,从而能够把同样长度但序列不同的DNA片段区分开来。
一个特定的DNA片段有其特有的序列组成,其序列组成决定了其解链区域和解链行为。
一个几百个碱基对的DNA片段一般有几个解链区域,每个解链区域有一段连续的碱基组成。
当变性剂浓度逐渐增加达到其最低的解链区浓度时,该区域这一段连续的碱基对发生解链。
当变性剂那浓度再升高依次达到其他解链区域浓度后,最高的解链区域也发生解链,从而双链DNA完全解链。
不同的双链DNA片段因为其序列组成不一样,所以其解链区域和解链区域的解链浓度也是不一样的。
当进行DGGE电泳时,一开始变性剂浓度比较小,不能使双链DNA片段最低的解链区域解链,此时DNA片段的迁移行为和在一般的聚丙烯酰胺凝胶中一样。
然而一旦DNA片段迁移到一特定位置,其变性剂浓度刚好能使双链DNA片段最低的解链区域解链时,双链DNA片段最低的解链区域立即发生解链。
部分解链的DNA片段在胶中的迁移速率会急剧下降。
因此同样长度但序列不同的DNA片段会在胶中不同位置处达到各自最低解链区域的解链变性剂浓度,因此它们会在胶中不同的位置分开。
变性梯度凝胶电泳DGGE实验报告

变性梯度凝胶电泳DGGE刘琳 1131428 环境科学一、 实验目的1. 学习掌握变性梯度凝胶电泳的原理和方法。
2. 练习变性梯度凝胶电泳的操作步骤。
3. 分析并掌握变性梯度凝胶电泳的思路,并了解其在微生物群落研究中的地位。
二、 实验原理双链DNA 分子在一般的聚丙烯酰胺凝胶电泳时,其迁移行为决定于其分子大小和电荷。
不同长度的DNA 片段能够被区分开,但同样长度的DNA 片段在凝胶中的迁移行为一样,因此不能被区分。
DGGE 技术在一般的聚丙烯酰胺凝胶基础上,加入了变性剂(尿素和甲酰胺)梯度,从而能够把同样长度但序列不同的DNA 片段区分开来。
不同的双链DNA 片段因为其序列组成不一样,所以其解链区域及各解链区域的解链温度也是不一样的。
同样长度但序列不同的DNA 片段会在胶中不同位置处达到各自最低解链区域的解链温度,因此它们会在胶中的不同位置处发生部分解链导致迁移速率大大下降,从而在胶中被区分开来。
然而,一旦温度(或变性剂浓度)达到DNA 片段最高的解链区域温度时,DNA 片段会完全解链,成为单链DNA分子,此时它们又能在胶中继续迁移。
因此,如果不同DNA 片段的序列差异发生在最高的解链区域时,这些片段就不能被区分开来。
DNA 片段在DGGE 胶中的迁移行为(如下图):变性剂LowHigh在DNA 片段的一端加入一段富含GC 的DNA 片段(一般30-50个碱基对),使DNA 片段最高的解链区域在GC 夹子这一段序列处,它的解链温度很高,可以防止DNA 片段在DGGE/TGGE 胶中完全解链。
当加了GC 夹子后,DNA 片段中基本上每个碱基处的序列差异都能被区分开(Myers, 1985)。
DGGE 有两种电泳形式:垂直电泳和水平电泳。
垂直电泳是为了确定变性剂梯度;而水平电泳则是对比分析不同样品的微生物差异。
本次试验做的是水平电泳,主要对比分析不同样品的微生物差异。
三、 实验器材与药剂器材:DGGE system (D-Code, Bio-Rad)、移液管、移液枪、枪头、制胶设备、30ml 针筒、聚乙烯细管、电泳仪试剂:16s rDNA V3区PCR 扩增产物、胶浓度为8%变性剂浓度分别为0%和90%丙烯酰胺胶、50×TAE buffer、去离子甲酰胺、尿素、去离子水、10%过硫酸铵(Aps)、TEMED 、sDNA 染料 变性剂Low high四、实验步骤1.首先按照实验要求将50x TAE buffer稀释为1×TAE buffer,并利用90%和0%的变性胶分别配置15.5ml的35%和55%的变性胶溶液。
变性梯度凝胶电泳(PCR-DGGE)

变性梯度凝胶电泳(DGGE): 一种分离相似大小DNA片段 的电泳方法。即双链DNA在变性剂(如尿素和甲酰胺)浓度或 温度梯度增高的凝胶中电泳,随变性剂浓度升高,由于Tm 值不同,DNA的某些区域解链,降低其电泳泳动性,导致 迁移率下降,从而达到分离不同片段的目的。
由于各类微生物(如细菌和古细菌)的16sRNA基因序 列中可变区的碱基顺序有很大的差异,其中不同土壤微生 物的16sRNA基因的V3区扩增的DNA片断在DGGE中的应用最为 广泛,根据电泳条带的多寡和条带的位置可以初步辨别出 样品中微生物的种类多少,粗略分析土壤样品中微生物的 多样性。
In a denaturing gradient acrylamide gel, doublestranded DNA is subjected to an increasing denaturant environment and will melt in discrete segments called "melting domains". The melting temperature (Tm) of these domains is sequencespecific. When the Tm of the lowest melting domain is reached, the DNA will become partially melted, creating branched molecules. Partial melting of the DNA reduces its mobility in a polyacrylamide gel.
The methods of detecting single base mutat ions
DGGE操作手册中文

DGGE安装操作手册1、将电泳槽放在一个水平的位置上;2、安装加热器与搅拌器。
用翼形螺钉固定好位置,确定蒸汽塑胶保护套正确:放置在加热器底部与水箱盒盖之间。
注意:如果水箱里没有注满Buffer时不要运转加热器与搅拌器。
3、安装温度计。
先润湿温度计,然后将温度计安装到水箱盖上的红色硅胶塞里。
4、在电泳槽的架子上的适当位置,安放电源仪。
5、加Buffer至水箱的水平线为止。
把胶架放到槽里,加Buffer至电泳槽上槽底部,盖好盒盖,上槽最大可以装50ml Buffer。
6、连好电源线,并连接到电源插座上7、将加热器与搅拌器的电线连接到电泳插座上注意:如果胶以及水箱还没放好,不要将电泳打开。
8、将加热器与搅拌器开关打到“on”位置并设置温度为60℃胶的准备与灌制1、用dH2O清洗每块玻璃板,风干或者是棉布纸檫干;2、用95%乙醇檫试玻璃板内部(灌胶面),并风干或用棉布纸檫干;3、胶灌制技术:A:胶外套垫圈安装法1、将玻璃板垂直放置在桌面上,开始时将封口胶条沿着玻璃板底角进行;注意:“U”形的封口胶条的一边是平的,另一边有管状的是开始封口位置2、当用封口条围绕玻璃板的一角进行封口时,要确定封条上的凹口与玻璃板平衡,当封条已经固定在玻璃板边角底部边缘后,然后开始进行余下部分的封口;3、然后将有封条的玻璃板放在实验台上,并保持封口向上,然后将两条隔离条沿着封条内边缘插进去,并确定每条隔离条的边角是贴紧玻璃板的底部4、将板的底部放置在桌面上,从底部边缘慢慢放开,并开始检查边缘封条是否封好,然后开始用夹子沿底部进行。
5、将已夹好夹子的板垂直放置,在水平方面,确定板的底部两个夹子完全固定好水平性,然后用夹子夹板的两边,并确定在板之间的封条是平衡的。
B:利用GM-40梯度灌胶器灌制垂直梯度胶方法1. 将GM-40梯度灌胶器放置在一个抬高的磁力搅拌器上,并将一个小型的磁力搅拌子放置靠近出口的圆桶里(C-2)。
梯度灌胶器根据真空管、灌胶阀(V-2)和连接的灌胶针孔将丙烯酰胺自动流到胶板里。
PCR-DGGE操作流程

PCR-DGGE操作流程变性梯度凝胶电泳(Polymerase Chain Reaction-Denaturing Gradient Gel Electrophoresis, PCR-DGGE)和FISH一样,也是一个常用的分子生物学实验,在环境生物技术领域常用于分析微生物群落的多样性。
DGGE实验的大体操作流程如下,比较麻烦,影响因素很多,有一段时间我曾经天天捣鼓PCR-DGGE,反复优化各个条件,非常无聊。
1. PCR扩增与普通PCR不同之处是Primer上要加一个GC夹(GC Clamp),GC夹的序列为:CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGC常用的细菌16S通用引物341fGC/518r的序列如下:341fGC:5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGGCCTACGGGAGGCAGCAG-3′518r: 5′-ATTACCGCGGCTGCTGG-3′2.制作凝胶需要的准备的试剂:(1)新鲜配制的Ammonium Persulfate Solution(APS) 0.1g in 1ml H2O(2)四甲基乙二胺(TEMED)(3)0%和100%变性剂(100%的变性剂不容易溶解,可以用80%的来代替),配制方法见最下面的表格准备四种不同浓度的凝胶:顶层胶: 4ml 0%变性剂+4微升TEMED+40微升APS溶液底层胶: 1ml 80%变性剂+4微升TEMED+40微升APS溶液如果DGGE需要的变性剂梯度为40%~60%,需要配制40%和60%变性剂凝胶溶液各约13ml:40%变性剂凝胶溶液:6.5ml 0%变性剂+6.5ml 80%变性剂+13微升TEMED+130微升APS溶液60%变性剂凝胶溶液:3.25ml 0%变性剂+9.75ml 80%变性剂+13微升TEMED+130微升APS溶液如需其它梯度,请重新计算。
PCR-DGGE操作步骤

16S rDNA - DGGE技术基本路线一样品总DNA的提取1 实验器材: 1.5mL离心管,螺口管,锆珠,搅拌器,4℃离心机。
2 实验试剂:PBS(0.05M, pH=7),水饱和酚,TE饱和酚,TNI50,TE缓冲液,TAE缓冲液,NaAc(pH=5.2),氯仿/异戊醇(24:1)(v/v),96%冷乙醇,70%冷乙醇。
3 实验操作:3.1 细胞的分解称取食糜0.3-0.5g,入含0.3g锆珠的离心管中,加1mL TN150溶解然后加入150μL酸性酚,bead-beater 5000rpm,匀浆180秒,冰上冷却加入150μL氯仿/异戊醇(24/1),vortex几秒,10000rpm离心5min,上清转移至eppendorf管。
3.2 DNA提取加入150 µL氯仿/异戊醇至eppendorf管加入150 µL TE饱和酚,涡旋混匀1 min10000 rpm离心,1 min重复上述步骤直至界面清晰为止加入300 µL氯仿/异戊醇,10000 rpm离心1 min取上清液至另一eppendorf管,加96%冷乙醇1mL加入50 µL3M NaAc(pH=5.2)-20℃沉淀DNA过夜10000 rpm离心20 min去除上清液,加入500 µL 70%冷乙醇溶解沉淀10000 rpm离心5 min去除上清液,风干沉淀加入50µL TE缓冲液溶解DNA-20℃保存提取的DNA4实验注意事项:酚是致癌物质,实验操作应配带手套,且不要将该溶液洒在桌面上或地面上。
实验中使用了挥发性有毒物质,操作应在通风橱中进行。
实验过程中样品应放置冰面上,所有离心操作在4℃下进行。
二DNA的检测—琼脂糖凝胶电泳试剂和器材:电泳缓冲液(1*TAE),溴乙锭(EB)染色贮存液(1 mg/mL),0.25%溴酚兰,琼脂糖,双蒸水。
电泳仪,水平电泳槽,透射紫外灯,胶带纸。
变性梯度凝胶电泳(DGGE)实验

【实验目的】掌握变性梯度凝胶电泳检测新的突变,以及测定高度多态基因的基因型的技术方法。
【实验原理】在现代遗传学中DNA 序列突变的分析占有十分重要的地位。
由于在较大DNA 序列中检测一个细微的突变非常困难,因而现在人们建立了几种方法来解决这一难题。
变性梯度凝胶电泳(DGGE)能把长度相同而核苷酸顺序不同的双链DNA 片段分开。
这种方法利用了DNA 分子从双螺旋型变成局部变性型时电泳迁移率会下降的现象。
不同的DNA 片段发生这种变化所需梯度不同。
DGGE 的凝胶中沿电场方向变性剂(甲醛和尿素)含量递增,当DNA 片段通过这种变性剂递增的凝胶时,不同分子的电泳迁移率在不同区域会发生降低。
这就可使核苷酸顺序不同DNA 片段分开。
此方法可作为测序的初始步骤在杂合个体中分离等位基因。
许多研究表明变性递度凝胶电泳分离能力很强,它可以把相差仅1bp 的DNA 片段分开。
【仪器、材料与试剂】1.50×TAE 缓冲液(2mol/LTris 乙酸盐,0.05mol/L EDTA pH8.0)1L 体积:242gTris碱,57.1mL 冰醋酸,100mL 0.5mol/L EDTA pH8.0,加水至lL。
2.丙烯酰胺贮存液:40%丙烯酰胺(38:2 丙烯酰胺:双丙烯酰胺)。
3.过硫酸铵贮存液(10%):10mL 配制:1g 过硫酸铵加水至10mL。
TEMED(N,N,N',N',—四甲基乙二胺)。
(生物秀实验频道)4.变性剂贮存液:(0%)6%丙烯酰胺TAE 溶液。
250mL 溶液:37.5mL 丙烯酰胺贮存液,5mL50×TAE 缓冲液,加水至250mL,过滤和排气。
5.变性剂贮存液(100%):6%丙烯酰胺,7mol/L 尿素,40%甲醛TAE 溶液。
250mL配制:37.5mL 丙烯酰胺贮存液,5mL50×TAE 缓冲液,105g 尿素,100mL 甲醛,加水至750mL,过滤并排气。
DGGE-TGGE

89 33 39 39
89 32 37 42
84 26 32 26
82 31 36 31
100 100 33 33 100 39 44 89 100 28 33 94 83 100
Among KKAy mice Cs=82%-100%, C57BL/6J mice Cs =83%-94%, and the different genotypes mice Cs =26%-44%
2.目标片段的PCR扩增
采用2次PCR扩增:
第一次扩增用引物不带GC夹子,以粪便总DNA 为 模板,Touchdown反应程序。 第二次扩增引物用带GC夹子,以第一次扩增的产 物为模板,只扩5个循环。叫Reconditioning-PCR,
3. 关于DGGE marker的制作
4. DGGE前的准备工作
• 为了提高DGGE的突变检出率,发明者人为地加入一个高熔点区,
就是在一侧引物的5′端加上一个30~40bp的GC结构,即GC夹 (GC clamp)这样,在PCR产物的一侧可产生一个高熔点区, 使相应的感兴趣的序列处于低熔点区而便于分析。因此,DGGE 的突变检出率可提高到接近于100%。
• 乳杆菌(Lac-1,Lac-GC-2)
8. 测序结果比对、聚类分析
--结果与GenBank和RDP数据库比对
9. DGGE图谱的数字化——Quantity one
10. DGGE图谱的分析
1)DGGE图谱的聚类分析、主成分分析
2)DGGE图谱的相似性分析
• 用Sorenson配对相似性系数(pairwise similarity coefficient,Cs)比较不同样品PCR-DGGE图谱的 相似性 • Cs=2j/(a+b)×100% • 其中a、b分别表示两个比较对象的条带数,j表示a 和b中共有的条带数。完全不同的两个图谱的Cs是
变性梯度凝胶电泳(DGGE)和温度梯度凝胶电泳(TGGE)

实验四变性梯度凝胶电泳(DGGE)和温度梯度凝胶电泳(TGGE)一、实验原理变性梯度凝胶电泳(denatured gradient gel electrophoresis,DGGE)最初是Lerman等人于20世纪80年代初期发明的,起初主要用来检测DNA片段中的点突变(20, 42, 52, 53)。
Muyzer等人在1993年首次将其应用于微生物群落结构研究 (50)。
后来又发展出其衍生技术,温度梯度凝胶电泳(temperature gradient gel electrophoresis,TGGE) (49)。
此后十年间,该技术被广泛用于微生物分子生态学研究的各个领域,目前已经发展成为研究微生物群落结构的主要分子生物学方法之一 (49, 51)。
1.原理双链DNA分子在一般的聚丙烯酰胺凝胶电泳时,其迁移行为决定于其分子大小和电荷。
不同长度的DNA片段能够被区分开,但同样长度的DNA片段在胶中的迁移行为一样,因此不能被区分。
DGGE/TGGE技术在一般的聚丙烯酰胺凝胶基础上,加入了变性剂(尿素和甲酰胺)梯度或是温度梯度,从而能够把同样长度但序列不同的DNA片段区分开来。
一个特定的DNA片段有其特有的序列组成,其序列组成决定了其解链区域(melting domain, MD)和解链行为(melting behavior) (20, 42)。
一个几百个碱基对的DNA片段一般有几个解链区域,每个解链区域有一段连续的碱基对组成(图1)。
当温度逐渐升高(或是变性剂浓度逐渐增加)达到其最低的解链区域温度时,该区域这一段连续的碱基对发生解链。
当温度再升高依次达到各其他解链区域温度时,这些区域也依次发生解链。
直到温度达到最高的解链区域温度后,最高的解链区域也发生解链,从而双链DNA完全解链。
(图1显示的是一个234bp长的DNA片段的解链区域,横坐标是该DNA片段的序列,依次从第一个碱基到第234个碱基;纵坐标是解链温度。
DGGE操作步骤

变性梯度凝胶电泳(DGGE)的溶液配制、操作步骤及注意事项一、实验试剂及配置1、40%丙烯酰胺/双丙烯酰胺(37.5:1)试剂用量丙烯酰胺38.93g双丙烯酰胺 1.07gMilliQ 水加到100mL 溶液采用0.45µm滤膜过滤,储存在4℃2、0%的变性储存液凝胶梯度6%8%10%12%40%丙烯酰胺/双丙烯酰胺15mL 20mL 25mL 30mL 50×TAE缓冲液2mL 2mL 2mL 2mL MilliQ 水83mL 78mL 73mL 68mL总容积100mL 100mL 100mL 100mL3、100%的变性储存液凝胶梯度6%8%10%12%40%丙烯酰胺/双丙烯酰胺15mL 20mL 25mL 30mL 50×TAE缓冲液2mL 2mL 2mL 2mL去离子甲酰胺40mL 40mL 40mL 40mL 尿素42g 42g 42g 42g MilliQ 水加到100mL 加到100mL 加到100mL 加到100mL4、50×TAE缓冲液试剂用量终浓度Tris碱242.0g 2M冰乙酸57.1 1M 0.5M EDTA,pH8.0 100.0mL 50mM MilliQ 水加到1000.0mL将溶液混合溶解,121℃下蒸汽灭菌20-30min,储存在室温5、银染液的配制:原液:8×固定液(250 ml):乙醇200 ml冰乙酸10mlMilliQ 水40ml(A):1×固定液(400 ml):8×固定液50mlMilliQ 水350ml(B)银染溶液(400ml):AgNO3 0.8 g8×固定液50mlMilliQ 水350ml(C):显影剂(500ml):NaOH 7.5gMilliQ 水500 ml甲醛 1.5ml6、10%过硫酸铵(AP):过硫酸胺0.3g超纯水 3.0ml溶解后使用0.45µm微孔滤膜过滤,4℃保存,保存时间为1周。
SEM及PCR-DGGE实验步骤

SEM及PCR-DGGE实验步骤一、扫描电镜实验1、污泥样品预处理方案一:(1)取样:自反应器中取数颗颗粒污泥,放入5ml的离心管中,用去离子水清洗数次,弃去上清液。
(2)固定:加入2.5%,pH为6.8的戊二醛使淹没泥样,并置于4℃冰箱中固定过夜。
(3)冲洗:用1mol/L pH为7的磷酸缓冲溶液冲洗3次,每次15min。
(4)脱水:用浓度为30%,50%,70%,80%,90%的乙醇进行脱水,每次15-30min,再用100%乙醇脱水2次,每次15-30min。
(5)置换:用乙醇:乙酸异戊酯为1:1的溶液,纯乙酸异戊酯各置换一次,每次15min。
(6)干燥:将置换后的样品用针头挑出,放入滤纸叠成的小盒中,置入干燥器中干燥8h。
(7)喷金:用离子溅射镀膜仪(IB-5(Giko)型)在样品表面镀上一层1500nm 厚度的金属膜。
(8)观测:将处理好的待检泥样置于扫描电镜下观察。
方案二:颗粒污泥扫描电镜观察的样品预处理方法如下:(1)取样与清洗:在所需要的不同的反应阶段,从反应器中取出数颗好氧颗粒污泥,放入5mL 的离心管中,用去离子水清洗数次,弃去上清液。
(2)固定:把清洗好的颗粒污泥加入2.5%,pH 为6.8 的戊二醛溶液并淹没样品,并置于4℃冰箱中固定2-12 小时。
(3)冲洗:固定好的好氧颗粒污泥,用0.1mol/L,pH 值为6.8 的磷酸缓冲溶液冲洗3 次,每次10min。
(4)脱水:采用梯度乙醇脱水,将冲洗好的样品依次置于系列浓度50%、70%、80%、90%的乙醇进行脱水,每次10-15min,再用100%的乙醇脱水3 次,每次10-15min。
(5)置换:用乙醇:乙酸异戊酯为1:1 的溶液,纯乙酸异戊酯各置换一次,每次15min。
(6)干燥:将置换后的样品小心取出,放入滤纸叠成的小盒中,置入干燥器中干燥8 小时。
(7)粘样与喷金:将干燥好的颗粒污泥样品,用镊子小心取出干燥好的样品,用导电胶把颗粒样品粘附在铝制托盘上,之后用离子溅射镀膜仪(IB-5(Giko)型)在样品表面镀上一层1500nm 厚度的金属膜。
TGGE电泳系统的基础知识与操作

4.1.2 内容物
包括 预制胶 滤纸片 操作手册 不包括(用户自备) 平衡缓冲液( 1×TAE,8 M 尿素,2 %甘油) 培养皿 摇床
4.1.3 预制胶的复性
1)在培养皿中加入平衡缓冲液,至少 20 ml 。 2)将预制胶放入缓冲液中,胶面朝下。在摇床上轻轻摇动,预制胶的背面不要与缓冲液接触, 同时也不要使胶贴在培养皿底部,以便胶与缓冲液充分接触。 3)平衡大概 90 min。 4)将预制胶从培养皿中取出,用滤纸小心地吸走胶表面残余的缓冲液。请务必尽量除去缓冲液 (如果胶表面已经干了的话,在用滤纸划过时会听到吱吱声)。
3.2 TGGE系统特色
⑴ 高分辨率:温度梯度板由微处理器控制,线性极为严格。2mm的电泳距离最大可对应0.6℃ 的温度差,即使极细微的分子间差异也可以检测出来。 ⑵ 加样量小:2ngDNA或RNA片段可以清晰地相互区分开。 ⑶ 重现性好:易控制的电泳条件可保证电泳的重现性和结果的重复性。 ⑷ 节约时间:小面积温梯板和小尺寸凝胶大大节约电泳时间。 ⑸ 两种模式:垂直TGGE电泳方向与温梯方向垂直,用于优化某个样本的分离条件,也可用于 检测PCR产物的组成。平行TGGE电泳方向与温梯方向平行,可采用垂直TGGE 所优化的电泳条 件,用于分析多个样品 垂直TGGE 温梯方向与电泳方向垂直; 只对一个样品进行分离; 在一个宽的温梯范围内进行分离以分析构象与 温度的关系,确定最佳变性温度; 平行TGGE 温梯方向与电泳方向平行; 同时对多个样品进行分离;
5.1 TGGE电泳的一般考虑因素
与常规电泳(如PAGE 或者琼脂糖凝胶电流)不同,DNA在TGGE电泳中的泳动行为更多地与温 度相关,因而延长电泳时间并不能提高TGGE实验的分辨率。一旦DNA分子开始熔解(解链), 它的移动速度就迅速下降直至停止。因此,延长电泳时间只会导致产生模糊的条带。要提高分 辨率,就必须对温度梯度进行优化。 样品在TGGE的电泳行为与下列因素相关: 温度梯度; 缓冲液; 凝胶浓度; 电压;
DGGE法

变性梯度凝胶电泳变性梯度凝胶电泳原著:Jeroen H. Roelfsema and Dorien J. M. Peters1.翻译:小高1.简介变性梯度凝胶电泳是人们多年来广泛使用的一种健全的检测点突变的方法(1)。
它是一种基于PCR的方法,其原理是,与野生型基因相比,改变了PCR产物中突变序列的解链温度。
通过PCR,致使DNA两个基因其中一个中的点突变变成不同的DNA的混合体。
PCR 后的产物中包括野生型基因和突变型基因。
即同源二聚体。
然而这两种产物的解链温度差别不大。
在PCR反应的最后一个循环,形成另外一种产物,即异源双链DNA,它包括一条野生型的链和一条突变的链。
DGGE的原理就在于同源PCR产物的解链温度远远低于异源PCR的产物,因为异源双链有一个错配(see Fig. 1)。
为了看到同源和异源DNA的解链温度之间的差别,只要让其在含有变性剂尿素和甲酰胺的丙烯酰胺梯度凝胶上电泳即可。
仅仅靠这些变性剂本身是不够的。
另外,跑电泳的胶的温度也比较高,通常在60℃。
电泳过程中,PCR产物在变性以前一直保持双链DNA的形式。
一旦变性后,PCR产物将以单链DNA的形式继续跑,但是片段则会保留在变性的地方。
为了达到这个目的,需要连上一个所谓的GC夹子以阻止完全变性。
这个GC夹子是由40-60个单纯的鸟嘌呤和胞嘧啶核苷酸构成的一条链,然后连接在其中的一种PCR引物上。
加上GC夹子进行PCR后的产物其一端具有很高的变性温度。
因此,PCR产物在DGGE的胶上将部分变性。
GC夹子保持双链。
其余的片段形成“Y”形继续保留在胶上原来的地方。
2.操作步骤2.1.设计PCR引物对于DGGE来说,用于筛选点突变的PCR引物的解链特性是很关键的。
当片段到达凝胶的临界点时,应该及时地变性,而不是在一端开始随着电泳的进行逐渐的慢慢的变性。
这种慢性解链会形成模糊的条带或者斑点,将导致很难检测出突变。
因为解链特性对于实验的成败至关重要,所以选取用来扩增目标产物的引物时必须格外小心。
简化的DGGE操作程序

简化的DGGE操作程序1.将45%和65%变性剂溶液放置至室温2.在吸水纸上装配之前,用70%的酒精洗净玻璃板3.将隔板安放在玻璃板之间,并用平行卡调整凝胶夹心层的位置4.调节吸液管中TEMED为14.4µL,过硫酸铵溶液为112µL,变性剂溶液为5mL5.将调节好的吸液管,合适的枪头,两支50ml 的试管,变性剂溶液,TEMED,10% 的过硫酸铵和带吸管的注射器放入防护罩6.用5ml 的吸液管吸取3次,量取15mL 45%和65%的变性剂溶液,分别加到不同的试管中7.先将14.4µL的TEMED加到每支管子中,然后加入112µL10% APS (10% 过硫酸铵)。
迅速盖上塞子,并翻转几次完全混匀8.吸取混匀的溶液加到对应的注射器中(高浓度的变性剂溶液和低浓度的变性剂溶液)。
在加入前要消除其中的气泡。
按注射器末端驱逐气泡9.将两个注射器安放在梯度轮上对应的架上(有一些标志说明装有变性剂溶液的注射器应该安放在哪里)。
与管子连接并装上19规格的针头10.使针头处于两块玻璃板的中央11.以一个快的恒定速度旋转梯度轮(松开顶部调节控制杆的螺钉,确保轮子平稳旋转)12.安放有14个样孔的梳子,并使之硬化(~1小时)13.用66.5 mL 50X TAE (0.5%) 和DI H2O 配制缓冲液,并用DI H2O添加到刻度线(共7L) (在加到槽中前,用微波炉加热DI HO)214.加热水浴装置并打开搅拌器。
应该预设温度为60ºC15.到达60ºC时关掉加热器,并等待2 min16.用蓝色标记物将梳子的14个齿印在玻璃板上17.将硬化的凝胶放入Dcode 核心装置18.将核心装置放入热缓冲液中,红色阴极对着仪器右边角落的背面19.混匀5ml 50X TAE 并用DI H2O 加到500ml20.加~350mL TAE混合液到热缓冲液中21.用专门的枪头冲洗样孔(2-3次),泵一直开着!22.关掉泵并填充样品23.DNA的准备,取25 µL PCR产物,并加10 µL 6X DNA 填充染料24.将温度控制仪安放在仪器的顶部,设置电压为130V,电泳10-11小时25.用500mL 0.5% 的电泳缓冲液和50µL EB(10mg/mL)配制染色液26.凝胶染色大约10 min (5-15 min)27.在电泳缓冲液中退色10 min (5-20 min)1。
机器人DG单元首件操作流程
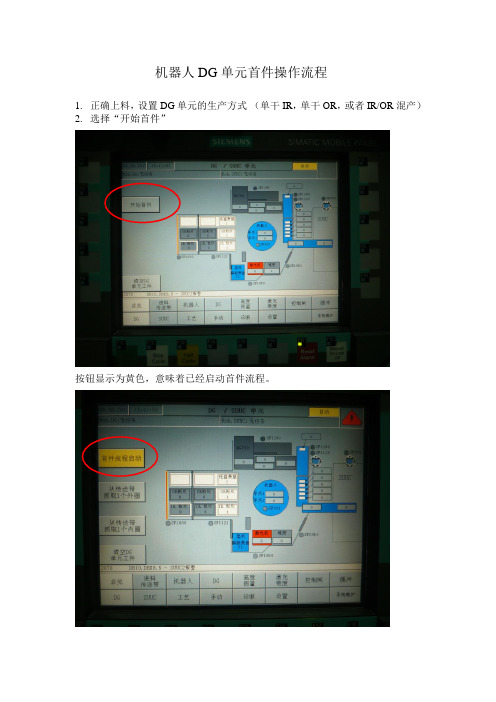
机器人DG单元首件操作流程1.正确上料,设置DG单元的生产方式(单干IR,单干OR,或者IR/OR混产)2.选择“开始首件”按钮显示为黄色,意味着已经启动首件流程。
选择要抓取的工件类型(内圈或者外圈)下图显示的是抓取内圈机器人开始抓取工件,并送在DG设备上加工3.与此同时,操作者需要去控制闸的触摸屏上进行设置:选择“下一个工件从DG到控制闸”按钮,变为黄色,即意味着选中。
同时按下“出料请求”按钮该工件在加工后,会自动测量高度,如果高度超差,机器人将停止, 请将机器人系统设为手动模式并将“测量ok”选框选中。
测量OK 机器人会将工件激光打标,然后送至控制闸4.操作者取出工件,并在触摸屏上进行如下操作:点击“取出工件”按钮,并选择打印。
操作者务必将工件与打印的条码放在一起,一一对应。
首件测量后将工件返回机器人系统如果首件合格,则工件将直接送至SUUC,请进行如下操作:5.将工件放入控制闸取出工件打印条码在控制闸的触摸屏上选中条码输入页面在“搜索ID ”的方框中,通过条码或者手动输入工件的圈号,并回车 如果系统显示“找到工件”并显示正确的工件类型,说明输入正确。
则点击“移至控制闸”按钮。
然后将工件送入系统,点击“送入工件”按钮机器人会自动将工件送入SUUC如果首件宽度超差,需要去DG 返修,请进行如下操作: 6. 将工件放入控制闸在控制闸的触摸屏上选中条码输入页面在“搜索ID ”的方框中,通过条码或者手动输入工件的圈号,并回车 如果系统显示“找到工件”并显示正确的工件类型,说明输入正确。
则点击“移至控制闸”按钮。
接着需要切换到PLC 的移动触摸屏上,选择右侧的“发送到DG 返修”按钮然后将工件送入系统,点击“送入工件”按钮机器人会自动将工件送入SUUC。
变性梯度凝胶电泳(DGGE)实验

变性梯度凝胶电泳(DGGE)实验一.DGGE的实验原理变性梯度凝胶电泳(DGGE)是一种根据DNA片段的熔解性质而使之分离的凝胶系统。
DGGE(denaturing gradient gel electrophoresis)不是将分子量不同的DNA分开,而是通过聚丙烯酰胺凝胶中变性剂浓度梯度的不同,将序列不同的DNA分开。
该方法的原理是根据DNA 的解链特性,不同碱基组成的DNA双螺旋发生变性所要求的变性剂浓度不同,混合双链DNA在变性剂浓度呈线性梯度增加的聚丙烯酰胺凝胶电泳时,当泳动到与DNA变性所需变性剂浓度一致的凝胶位置时,相对应的DNA发生解链变性,导致电泳迁移速率降低。
由于泳动受阻DNA分子在凝胶中的停留位置不同,从而使不同DNA分子得以分离。
根据变性剂梯度方向的不同, DGGE可分为:垂直DGGE,即变性剂梯度与电场方向垂直,常用于试验决定分离型、野生型和变异型的最佳变性剂梯度范围;平行DGGE,其变性剂的梯度和电场方向平行,主要用于解链范围明确的DNA片段的检测。
DGGE已广泛用于分析自然环境中细菌、蓝细菌, 古菌、微型真核生物、真核生物和病毒群落的生物多样性。
这一技术能够提供群落中优势种类信息并同时分析多个样品,具有可重复和操作简单等特点, 适合于调查种群的时空变化, 并且可通过对条带的序列分析或与特异性探针杂交分析鉴定群落组成。
然而一旦变性剂浓度达到DNA片段最高解链区域浓度时,DNA片段会完全解链,成为单链DNA分子,此时他们又能在胶中继续迁移。
因此如果不同DNA 片段的序列差异发生在最高解链区域时,这些片段就不能被区分开来。
在DNA 片段的一端加入一段富含GC的DNA片段(GC夹子,一般30-50个碱基对)可以解决这个问题。
含有GC夹子的DNA片段最高的解链区域在GC夹子这一段序列,它的解链温度很高可以防止DNA片段在胶中完全解链。
当加了GC夹子后,DNA片段中的每个碱基处的序列差异都能被分开。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实验四变性梯度凝胶电泳(DGGE)和温度梯度凝胶电泳(TGGE)一、实验原理变性梯度凝胶电泳(denatured gradient gel electrophoresis,DGGE)最初是Lerman等人于20世纪80年代初期发明的,起初主要用来检测DNA片段中的点突变(20, 42, 52, 53)。
Muyzer等人在1993年首次将其应用于微生物群落结构研究 (50)。
后来又发展出其衍生技术,温度梯度凝胶电泳(temperature gradient gel electrophoresis,TGGE) (49)。
此后十年间,该技术被广泛用于微生物分子生态学研究的各个领域,目前已经发展成为研究微生物群落结构的主要分子生物学方法之一 (49, 51)。
1.原理双链DNA分子在一般的聚丙烯酰胺凝胶电泳时,其迁移行为决定于其分子大小和电荷。
不同长度的DNA片段能够被区分开,但同样长度的DNA片段在胶中的迁移行为一样,因此不能被区分。
DGGE/TGGE技术在一般的聚丙烯酰胺凝胶基础上,加入了变性剂(尿素和甲酰胺)梯度或是温度梯度,从而能够把同样长度但序列不同的DNA片段区分开来。
一个特定的DNA片段有其特有的序列组成,其序列组成决定了其解链区域(melting domain, MD)和解链行为(melting behavior) (20, 42)。
一个几百个碱基对的DNA片段一般有几个解链区域,每个解链区域有一段连续的碱基对组成(图1)。
当温度逐渐升高(或是变性剂浓度逐渐增加)达到其最低的解链区域温度时,该区域这一段连续的碱基对发生解链。
当温度再升高依次达到各其他解链区域温度时,这些区域也依次发生解链。
直到温度达到最高的解链区域温度后,最高的解链区域也发生解链,从而双链DNA完全解链。
(图1显示的是一个234bp长的DNA片段的解链区域,横坐标是该DNA片段的序列,依次从第一个碱基到第234个碱基;纵坐标是解链温度。
该DNA片段共有3个解链区域。
MD1是解链温度最低的一个解链区域,MD3是温度最高的一个解链区域。
)MD1MD2MD3图1:DNA 片段解链区域示意图不同的双链DNA 片段因为其序列组成不一样,所以其解链区域及各解链区域的解链温度也是不一样的。
当它们进行DGGE/TGGE 时,一开始温度(或变性剂浓度)比较小,不能使双链DNA 片段最低的解链区域解链,此时DNA 片段的迁移行为和在一般的聚丙烯酰胺凝胶中一样。
然而一旦DNA 片段迁移到一特定位置,其温度(或变性剂浓度)刚好能使双链DNA 片段最低的解链区域解链时,双链DNA 片段最低的解链区域立即发生解链。
部分解链的DNA 片段在胶中的迁移速率会急剧降低。
因此,同样长度但序列不同的DNA 片段会在胶中不同位置处达到各自最低解链区域的解链温度,因此它们会在胶中的不同位置处发生部分解链导致迁移速率大大下降,从而在胶中被区分开来。
然而,一旦温度(或变性剂浓度)达到DNA 片段最高的解链区域温度时,DNA 片段会完全解链,成为单链DNA 分子,此时它们又能在胶中继续迁移。
因此如果不同DNA 片段的序列差异发生在最高的解链区域时,这些片段就不能被区分开来。
在DNA 片段的一端加入一段富含GC 的DNA 片段(GC 夹子,一般30-50个碱基对)可以解决这个问题。
含有GC 夹子的DNA 片段最高的解链区域在GC 夹子这一段序列处,它的解链温度很高,可以防止DNA 片段在DGGE/TGGE 胶中完全解链。
当加了GC 夹子后,DNA 片段中基本上每个碱基处的序列差异都能被区分开 (52, 53)。
当用DGGE/TGGE 技术来研究微生物群落结构时,要结合PCR (polymerase chain reaction )扩增技术,用PCR 扩增的16S rRNA 产物来反映微生物群落结构组成。
通常根据16S rRNA 基因中比较保守的碱基序列设计通用引物,其中一个引物的5’-端含有一段GC 夹子,用来扩增微生物群落基因组总DNA ,扩增产物用于DGGE/TGGE 分析。
DGGE/TGGE 有两种电泳形式:垂直电泳和水平电泳。
前者是指变性剂梯度或温度梯度的方向和电泳方向垂直;后者是指两个方向是平行的。
在分析微生物群落的PCR 扩增产物时,一般先要用垂直电泳来确定一个大概的变性剂范围或温度范围。
垂直电泳时,胶从左到右是变性剂梯度或温度梯度(图2)。
在胶的左边,变性剂浓度或温度低,DNA片段是双链形式,因此沿着电泳方向一直迁移。
在胶的另一边,由于变性剂浓度或温度高,DNA一进入胶立刻就发生部分解链,因此迁移很慢。
在胶的中间,DNA片段有不同程度的解链。
在变性剂浓度或温度临界于DNA片段最低的解链区域时,DNA的迁移速率有急剧的变化。
因此,DNA片段在垂直胶中染色后呈S形的曲线,如图2所示。
根据垂直电泳确定的范围,再用水平电泳来对比分析不同的样品,如图3所示。
图2:垂直胶(太原焦化厂活性污泥16S rDNA V3区PCR扩增产物)图3:水平胶(太原焦化厂两个嚗气池活性污泥16S rDNA V3区扩增产物)在用水平电泳分析样品之前,先要优化确定电泳所需时间。
一般采用时间间歇(time travel)的方法,即每隔一定时间加一次样品,从而使样品的电泳时间有一个梯度,如图4所示。
根据这个结果,确定最佳的电泳时间。
图4:时间间歇(time travel) (太原焦化厂活性污泥16S rDNA V3区PCR扩增产物)通过各种染色方法可以看到DGGE/TGGE胶中的DNA条带。
最常用的几种染色方法是溴化乙啶(ethidium bromide, EB) (51),SYBR Green I (51), SYBR Gold (68)和银染法 (66)。
EB法染色的灵敏度最低。
SYBR Green I和SYBR Gold相比EB,能更好地消除染色背景,因此它们的检测灵敏度比EB法高很多。
EB和SYBR染色时,双链DNA能很好地显色,单链DNA基本上不能显色。
银染法的灵敏度最高,它不但能染双链DNA,也能染单链DNA,但它的缺点是不能用于随后的杂交分析 (51)。
2.PCR-DGGE/TGGE在微生物生态学中的应用PCR-DGGE/TGGE技术在微生物生态学中的一个主要用途是分析微生物群落结构组成。
Muyzer等人于1993年首次将该技术用于研究微生物菌苔和生物膜系统的群落多样性(50)。
此后,该技术被广泛应用于各微生物生态系统,包括土壤 (24, 27, 72),活性污泥 (13, 87),人体和动物肠道 (71, 94),温泉 (18),植物根系 (72),海洋 (76),淡水湖 (69),油藏(86)等。
绝大部分研究都是通过扩增细菌或古细菌的16S rRNA基因来研究各生态系统中的细菌或古细菌群落多样性。
也有用真菌的通用引物扩增18S rRNA基因 (24),从而研究真菌的群落多样性。
除了通过核糖体RNA基因来分析微生物群落结构外,还可以通过扩增功能基因来研究功能基因及功能菌群的多样性。
氨单加氧酶(ammonia monooxygenase gene,amoA)基因被用来研究氨氧化菌群 (54);[NiFe]氢化酶基因被用来研究硫酸盐还原菌群 (88);多组分苯酚羟化酶大亚基基因(the largest subunit of the multi-component phenol hydroxylase, LmPH)被用来研究降酚菌群 (87, 91)。
PCR-DGGE/TGGE技术的另一大优点是能快速同时对比分析大量的样品。
因此它既可以对比分析不同的微生物群落之间的差异,也可以研究同一个微生物群落随时间和外部环境压力的变化过程 (49, 51)。
除了上述两个主要用途外,PCR-DGGE/TGGE技术还有很多其他用途。
它可以跟踪监测细菌的富集和分离,从而评价不同培养基及培养条件对分离菌种的影响 (67, 85);可以检测单个纯菌rRNA基因的微异质性 (55);可以用来比较不同的DNA提取方法 (51);另外,它还可以辅佐筛选克隆文库 (39)以及检测PCR和克隆过程中的偏差 (37)。
我们实验室用PCR-TGGE技术研究了多个微生物生态系统的微生物群落,包括处理工业焦化废水的活性污泥系统 (92, 95),人体肠道 (89),动物肠道(猪,熊猫,昆虫),人体口腔,纤维素降解土壤,油藏系统等。
另外,我们还研究了处理工业焦化废水的活性污泥系统中苯酚降解菌群 (91)和氨氧化菌群的功能基因多样性。
3.DGGE/TGGE技术的缺陷及优化在用分子生物学方法分析微生物群落结构组成时,有很多偏差。
不同细菌的基因组大小和核糖体RNA拷贝数不同 (16);提取基因组总DNA时细胞的裂解效率不同 (59);DNA 提取和纯化时有偏差 (43, 44);PCR扩增过程中有偏差 (34, 61, 63)。
除了这些,在应用DGGE/TGGE技术分析微生物群落结构组成时还有很多其他的缺陷。
DGGE/TGGE一般只能分析500个碱基对以下的DNA片段 (52),因此得到的系统进化相关的信息就很少。
在研究复杂环境生态系统(如土壤,人体肠道)时,其中微生物种类很多。
但DGGE/TGGE 图谱中一般只有一二十个条带,这些条带反映的是群落中的优势菌群。
一般只有在总的微生物群落中占1%以上的种群才能被检测出来 (50),系统中的弱势菌群不能被检测到。
为了降低分析群落多样性时的复杂度,一种方法是先根据GC含量的不同,将基因组总DNA分成各个部分,再分别PCR-DGGE/TGGE分析 (28)。
另一个方法是使用类群特异性(group-specific)引物对基因组总DNA进行PCR扩增,再用于DGGE/TGGE分析 (39, 56)。
在设计PCR引物去扩增某些特定菌群时,由于某些菌群的序列相互之间同源性不是很高,因此需要设计简并性引物。
此时,相同的微生物会有2个条带,这会对微生物群落结构组成造成高估 (39)。
另外,同一个细菌也可以有多个核糖体RNA操纵单元,它们的序列可以有微小差异 (55),这也会高估微生物群落多样性。
DGGE的电泳条件不一样,即使相同的样品所得的图谱也会有差异 (70)。
在应用DGGE 时,如果所用电压太低,需要的电泳时间就很长,变性剂梯度会有变化,因此导致图谱也会有变动。
另外,DGGE/TGGE的一个很大的优点是可以对胶中所感兴趣的条带进行研究,得到它们的序列,从而可以直接获得和系统生态功能相关的微生物种群的系统进化信息。
以前研究DGGE/TGGE胶中单一条带序列组成的一个比较普遍的方法是先构建整个微生物群落的16S rRNA克隆文库,然后再对文库中的克隆进行扩增,通过DGGE/TGGE和原始样品的条带进行对比,能迁移到相同位置处的克隆的序列就是原始条带所含的序列 (94)。