第一性原理对 Ga n N n ( n = 2~ 5) 小团簇的结构
小尺寸MgO团簇结构与电子性质的第一性原理研究

在选 取 团簇初 始 构型 时 , 仅参 考 了文献 [ ,] 不 8 9 中
的所 有 构 型 , 且 还 参 考 了 C S 的 构 型 和 而 d[
Mg 1 O[ 的构 型. 算 过 程 中 , 用 D L 计 采 Mo 3软 件
过 物理 或 化 学 作 用 结 合 而 成 的 相 对 稳 定 的聚 集
体. 由于团簇 的结构 和性 质 的研 究 对 于理 解 物 质 从 微 观到宏 观 的过 度 有 着 重 要 的作 用 , 此 团簇 因 的稳定 构 型和 电子结 构 随其尺 寸变 化而 变化 的规 律 是 团簇 物 理 学 研 究 的重 要 课 题 之 一[ . 于 1 由 q]
第 2 6卷 第 2期 21 O 2年 3月
甘 肃 联 合 大 学 学报 ( 自然科 学 版 )
J u n l fGa s a h iest Nau a ce c s o r a n uLin eUnv riy( t rlS in e ) o
Vo _ 6 No 2 l2 .
构, 结果 表 明 , 3 6 9 1 、 5 1 z 一 、 、 、 2 1 、 8时 , Mg ( O)
2 结果 及 讨 论
2 几 何结构 . 1
团簇 的结构 最 稳 定 ; h Z u等 利 用 第 一 性 原 理 的 ] 能 带 理 论 , 究 了 Mg 的 电子 结 构 和 相 对 稳 定 研 O 性 , 志雄 等[ 利 用 第 一 性 原 理 对 Mg 的 光 学 陈 7 O 性 质 进 行 了研 究. 管 对 Mg 团 簇 进 行 了大 量 尽 O 的理 论 研 究 , 是 到 目 前 为 止 还 很 少 见 到 用 但 D L 程 序对 该 团簇 的基 态 结 构 和 电 子性 质 进 Mo 3 行计 算 的. 基于 此 , 文 利用第 一性 原理 方法 中的 本 广 义 梯度 近似 对小 尺 寸 Mg 团簇 结 构 与 电子 性 O 质 进 行 研究 , 以便 对 Mg 团簇 的微 观 结 构 有 更 O
什么是第一性原理_

实质“第一性原理”这个词儿被吹得神乎其神,似乎是威力无穷。
大家都知道了伊龙马斯克(Elon Musk)把这个原理用得好,可以把火箭发射成本降低到原先的几十甚至上百分之一。
可是它到底是什么?自从听见这个词儿,我也只是人云亦云,竟然从来也没有认真考察过。
直到今天,品着茶,读老喻的文章。
才看到了马斯克这段话的完整译文:我在想存在一种好的思维框架。
那是物理学的东西,你知道,有点儿像第一原理推理(first principles reasoning)。
总体来讲,我认为存在将事情缩减至其根本实质……你必须能够把那些问题“煮沸”才能从里面找出那些最基本的东西。
看完了我一口水差点儿喷出来。
这令人膜拜的“第一性原理”,不就是“解耦合”(decoupling)吗?协作要搞清楚这个概念,咱们得先看看现代社会的大规模协作方式。
假设你是一名程序员,希望改进一款开源软件产品的功能。
该怎么做?你会打开一个新的空白源代码文件,从头开始,一行行写代码吗?基本上不会。
你该怎么做呢?你会读现有软件的源代码,把新的功能实现补充或更新到对应的位置,提交合并(merge)请求。
注意在这个过程中,你是把前人做的东西,当成基础层。
而你自己,是在这个层次之上,去叠加新的内容。
为了更形象化一点,我拿来个计算机体系结构层级示意图。
许多人弄不明白,既然搞IT的平时都鼓捣个计算机,为什么还得分成搞硬件的、搞架构的、搞通讯的、搞操作系统的、搞软件的、搞算法的、搞应用的?不都是一回事儿吗?因为这种“不理解”,才经常会有人找计算机系的研究生帮着修电脑,觉得这才算是学有所用。
这种分层的架构,使整个儿IT行业从业者,都只需要管好自己这一层的功能,并且为上层提供功能接口。
需要的时候,他会调用下层已经准备好的功能,而不需要去重新发明轮子。
说得通俗一些——铁路警察,各管一段儿。
想想看,为什么现在数据科学那么火? Python、R 和机器学习框架们为何这么受到欢迎?以至于许多非 IT 类人士,都在乐此不疲渴望学习、应用它们?因为许许多多的开发者,已经为你写好了实现数据科学工作的各项基础功能。
CunZn(n=1~12)团簇结构和性质的第一性原理研究

团簇是一种新型的纳米材料概念,由几个到几 千个原子形成,它与小分子和原子相比显得太大,而 和较大的固体物质相比又显得太小。由于量子力学 的影响,团簇的物理与化学性质完全不同于较大的 固体物质,它表现出新的性质,并且随着团簇的大小
而改变[1]。人 们 已 经 较 多 地 研 究 了 纯 铜 和 纯 锌 团 簇各方面的结 构 和 性 能 [2-3],而 且 由 于 铜 基 混 合 团 簇在汽车工业、化学工业、燃料电池及磁性数据存储 等方面存在着巨大的潜力,它在许多方面表现出更 优异的物理和化学性质。有许多关于铜基混合团簇
收稿日期:2018-06-17 基金项目:四川省教育厅重点基金项目资助(14zd1116)。 作者简介:第一作者,冯江平(1994— ),男,硕士研究生,Email:925792110@qq.com;通信作者,邝向军(1967— ),男,教授,研究方向为
团簇的结构和性质,Email:kuangxiangjun@163.com
第 34卷 第 1期 2019年 3月
西 南 科 技 大 学 学 报 JournalofSouthwestUniversityofScienceandTechnology
Vol.34No.1 Mar.2019
CunZn(n=1~12)团簇结构和性质的第一性原理研究
Abstract:AnallelectronscalarrelativisticcalculationonCunZn(n=1-12)clustershasbeenper formedbyusingdensityfunctionaltheorywiththegeneralizedgradientapproximation(GGA)atPW91 level.OurresultsshowthatthelowestenergygeometryofCunZn(n=1,3,4,6-12)clusterscanbe generatedbysubstitutingZnatomforoneCuatom oftheCun+1 clusteroraddingZntotheCuncluster. ThegroundstatestructuresofCu2ZnandCu5Znclustersvarysignificantlyduetotheirelectronicstructure inthatCu2ZnislinearstructureandCu5Znisthreedimensionalstructure,whileCu3 andCu6 areplanar structures.TheCu ZnbondinCunZnclustersisweakerandhasalongerbondlengthwhencompared withtheCu CubondinthecorrespondingpureCucluster,andmostoftheCu CubondsfarfromtheZn atomsinCunZnclustersarestrongerthantheCu CubondinpureCuclusters.DopedwithZnatoms,the secondorderdifferenceofenergyforCunclustersproducesaverysignificantparityconversionphenomenon. Keywords:Copperclusters;Allelectronscalarrelativisticcalculation;DFT;Catalytic
第一性原理对GanNn(n=2~5)小团簇的结构及电子性质的研究

得 到 了 G N (z -5 团簇 的 最稳 定 结 构 . 对 最稳 定 结 构 的 电 子 性 质 、 7=2 ) 并 成键 特 性 和 极 化 率 进 行 分 析 .
结果表 明, 团簇 的最稳定结构为平 面结构 , 且存在着 N 和 单元 , 明 N—N键在 团簇 的形成过程 中起 2 说
( . yL b r t y o c p yi n p r n f h s s Noma C l g ,S ie i i ri ,X- i g8 2 0 ,C i ; 1 Ke a oao f o h s sa dDea t t yi , r l o e e hh  ̄ Unv s y r E c me o P c l e t m a 3 0 3 h a jn n
3.I siueo er t a h sc ,S h o f y i dI fr t nOpteeto i , n t t fTho ei lP y i c o lo s a nomai o lernc t c s Ph c n s o s
He a iest n n Unv ri y,Kafn 7 0 4,Chn ; i g4 5 0 e i a
o lG , , n=2 5 c s r f ma aNl s l l ( - )l t ue s
GE i a EIXu n ,YAN — 3 Gu— n ,L eLi Xi Yu Li,YANG h。 HAO e -i3 Z i,Z W nJe
W ANG n - n ,L QigLi3 UO u Hu 3 Yo - a,
4 S ho f i c,E s hn iesyo i c dT cn l y sm g1i 0 2 7 C i ) . o l e e at i Un ri f e e ehoo , }nl 03 , hn c oS n c C a v t S n a c n g a2 a
过渡金属及氧化物团簇结构、磁性、催化性质的第一性原理计算研究

magnetic moment remarkably increases from 7烛of Scl30 to 131xB of Scl30CO,whereas it reduces
from 199n of Scl3 to 51xs ofScl3CO.
Eley—Rideal㈣and The competition between the
chemical reaction.
II
东南大学博士论文
东南大学博士论文
The performance of Pt/Cu(111)surface alloy catalysts in water gas shift reaction with a few Pt atoms doping in the Cu(111)surface were explored by using spin—polarized DFT.The Pt/Cu(111) surface alloy catalysts not only remarkably decrease the adsorption energy of CO,which Can
sequence in the presence ofCO.
The electronic properties and controlled single atom catalysis of isomorphous substituted bimetallic
oxide TMV3010(TM=Sc,Ti,Cr and Co)and V4010 were studied within the framework of a gradient-corrected DFT calculations.The charged clusters V409。1∥一show different electronic property from isoelectronic subsfitution bimetallic oxide Tiv309J0/CrV309,10 clusters and the terminal TM
第一性原理简介
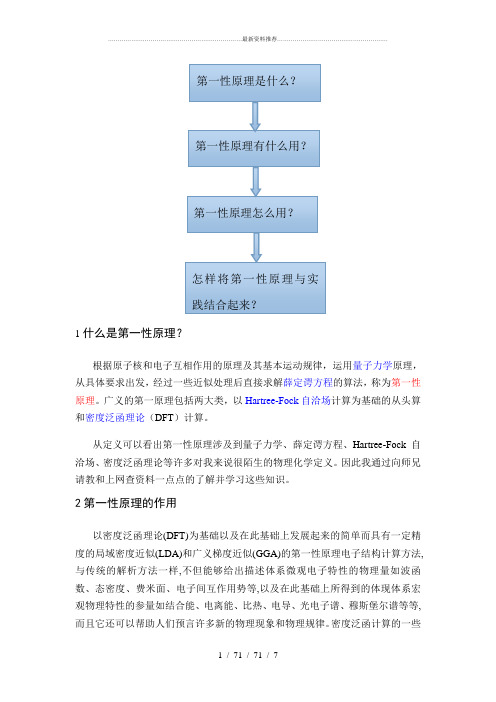
1什么是第一性原理?根据原子核和电子互相作用的原理及其基本运动规律,运用量子力学原理,从具体要求出发,经过一些近似处理后直接求解薛定谔方程的算法,称为第一性原理。
广义的第一原理包括两大类,以Hartree-Fock自洽场计算为基础的从头算和密度泛函理论(DFT)计算。
从定义可以看出第一性原理涉及到量子力学、薛定谔方程、Hartree-Fock自洽场、密度泛函理论等许多对我来说很陌生的物理化学定义。
因此我通过向师兄请教和上网查资料一点点的了解并学习这些知识。
2第一性原理的作用以密度泛函理论(DFT)为基础以及在此基础上发展起来的简单而具有一定精度的局域密度近似(LDA)和广义梯度近似(GGA)的第一性原理电子结构计算方法,与传统的解析方法一样,不但能够给出描述体系微观电子特性的物理量如波函数、态密度、费米面、电子间互作用势等,以及在此基础上所得到的体现体系宏观物理特性的参量如结合能、电离能、比热、电导、光电子谱、穆斯堡尔谱等等,而且它还可以帮助人们预言许多新的物理现象和物理规律。
密度泛函计算的一些结果能够与实验直接进行比较,一些应用程序的发展乃至商业软件的发布,导致了基于密度泛函理论的第一原理计算方法的广泛应用。
密度泛函理论(DFT)为第一性原理中的一类,在物理系、化学、材料科学以及其他工程领域中,密度泛函理论(DFT)及其计算已经快速发展成为材料建模模拟的一种“标准工具”。
密度泛函理论可以计算预测固体的晶体结构、晶格参数、能带结构、态密度(DOS)、光学性能、磁性能以及原子集合的总能等等。
3第一性原理怎么用?目前我所学到的利用第一性原理的软件为Material Studio、V ASP软件。
其中Materials Studio(简称MS)是专门为材料科学领域研究者开发的一款可运行在PC上的模拟软件。
使化学及材料科学的研究者们能更方便地建立三维结构模型,并对各种晶体、无定型以及高分子材料的性质及相关过程进行深入的研究。
第一性原理计算公式

第一性原理计算公式引言第一性原理计算是一种基于量子力学原理的理论和计算方法,可以用于研究和预测材料的物理和化学性质。
它是一种从头开始的计算方法,不依赖于任何经验参数和实验数据,因此被广泛应用于材料科学、化学、物理等领域的研究和设计。
在第一性原理计算中,通过求解薛定谔方程来得到体系的电子结构和能量。
这些计算需要使用一系列的公式和算法,本文将重点介绍一些常见的第一性原理计算公式,帮助读者理解这一领域的基本原理和方法。
基本概念在介绍具体的计算公式之前,我们先来回顾一些基本概念。
哈密顿算符哈密顿算符是量子力学中描述体系总能量和动力学演化的算符。
对于单电子体系,哈密顿算符可以写为:H = T + V其中T表示动能算符,V表示势能算符。
对于多电子体系,哈密顿算符则需要加入电子之间的相互作用算符,形式更加复杂。
波函数和薛定谔方程波函数是描述量子力学体系的状态的函数。
在薛定谔方程中,波函数满足以下的时间无关薛定谔方程:Hψ = Eψ其中H是哈密顿算符,ψ是波函数,E是能量。
求解薛定谔方程可以得到体系的能级结构和波函数。
密度泛函理论密度泛函理论是一种处理多电子体系的方法。
其核心思想是将多电子体系的性质建立在电子密度上。
密度泛函理论的基本方程是:E = T[n] + V[n] + E_{ee}[n]其中E是总能量,T[n]是电子动能的泛函,V[n]是外势能的泛函,E_{ee}[n]是电子之间相互作用的泛函。
第一性原理计算公式赝势方法赝势方法是一种快速计算材料电子结构的方法。
在赝势方法中,原子核和一部分芯层电子对价层电子的作用通过赝势进行描述。
赝势方法的基本方程是:H_{KS}ψ = Eψ其中H_{KS}是Kohn-Sham方程中的赝势哈密顿算符,ψ是波函数,E是能量。
平面波基组展开法平面波基组展开法是一种基于平面波基函数的展开方法。
平面波基组展开法的基本方程是:ψ(r) = ∑ c_k exp(ik·r)其中ψ(r)是波函数,c_k是展开系数,k是波矢。
第一性原理计算简述

第一性原理计算简述第一性原理,英文Firs t Principle,是一个计算物理或计算化学专业名词,广义的第一性原理计算指的是一切基于量子力学原理的计算。
我们知道物质由分子组成,分子由原子组成,原子由原子核和电子组成。
量子力学计算就是根据原子核和电子的相互作用原理去计算分子结构和分子能量(或离子),然后就能计算物质的各种性质。
从头算(ab initio)是狭义的第一性原理计算,它是指不使用经验参数,只用电子质量,光速,质子中子质量等少数实验数据去做量子计算。
但是这个计算很慢,所以就加入一些经验参数,可以大大加快计算速度,当然也会不可避免的牺牲计算结果精度。
那为什么使用“第一性原理”这个字眼呢?据说这是来源于“第一推动力”这个宗教词汇。
第一推动力是牛顿创立的,因为牛顿第一定律说明了物质在不受外力的作用下保持静止或匀速直线运动。
如果宇宙诞生之初万事万物应该是静止的,后来却都在运动,是怎么动起来的呢?牛顿相信这是由于上帝推了一把,并且牛顿晚年致力于神学研究。
现代科学认为宇宙起源于大爆炸,那么大爆炸也是有原因的吧。
所有这些说不清的东西,都归结为宇宙“第一推动力”问题。
科学不相信上帝,我们不清楚“第一推动力”问题只是因为我们科学知识不完善。
第一推动一定由某种原理决定。
这个可以成为“第一原理”。
爱因斯坦晚年致力与“大统一场理论”研究,也是希望找到统概一切物理定律的“第一原理”,可惜,这是当时科学水平所不能及的。
现在也远没有答案。
但是为什么称量子力学计算为第一性原理计算?大概是因为这种计算能够从根本上计算出来分子结构和物质的性质,这样的理论很接近于反映宇宙本质的原理,就称为第一原理了。
广义的第一原理包括两大类,以Hartr ee-Fork自洽场计算为基础的abinitio从头算,和密度泛函理论(DFT)计算。
ScnN(n=2~12)团簇的结构与稳定性研究

摘 要 :基 于第 一 性 原 理 ,在 密度 泛 函理 论 框 架 下 , 广义 梯 度 近 似 ( A) 用 GG 的方 法 研 究 了 团簇 S n ~ 1 ) 几 cN( 一2 2 的
何 构 型 和 电 子 结 构 , 算 了 它 的 束缚 能 、结 合 能 、 高 占据 轨 道 与 最 低 占据 轨道 之 间 的 能 隙 、离 解 能 等 性 质 .结 果 计 最 表 明 , 于 S 2 1 ) 所 有 团 簇 , 以 有 ¨ 种 基 态 构 型 ,但 稳定 的 只有 s 6 和 S N 这 两 种 构 型 , 原 因 对 cN( 一 ~ 2 的 可 cN c o 其 在 于 中 心 原 子参 与 了轨 道 的杂 化 ,中 心原 于 是 否 参 与 轨 道 的 杂 化 , 团簇 稳 定 性 有 重 要 影 响 . 对 关 键 词 : cN 团簇 ;密 度 泛 函 理 论 ;几 何 结 构 ; 电子 结 构 ;稳 定 性 S
2 . 果 与 讨 论 结
2 1 几 何 结 构 .
参 照 A1N( m=2 2 , cA ( =1 , 2 等团簇 的几何 结构 u ,  ̄1 ) S ln ~8 1 ) 并结 合遗 传 算法 采用 密度 泛 函理 论对 S ~1 ) cN( 一2 2 团簇 的所 有可 能构型 进行几 何优 化 ,得 出不 同尺 寸 团簇 的基 态构 型见 图 1 .
文 献 标 识 码 :A 中 图 分 类 号 :04 9 6
掺杂 团簇 由于 电子 结构 、 几何 构 型 、 学反 应活性 等性 质的可 裁剪性 ,近 1 化 O年来成 为人们 探 寻的主要 目标 , 渡金属 由于在理论 研究 中 的重 要性 和在磁 性材料 中的潜 在性 应 用 已经得 到 了广 泛 的研究 .在对 过 掺杂 团簇 的研究 中 , N 作掺 杂物 质的 团簇相关 研究 很 多 ,其 中最 为常 见 的就 是在 A1 用 团簇 中掺 杂 N ,
第一性原理计算LiNBe(N=1~12)团簇的基态结构及其电子性质

第一性原理计算LiNBe(N=1~12)团簇的基态结构及其电子
性质
第一性原理计算LiNBe(N=1~12)团簇的基态结构及其电子性质从第一性原理出发利用密度泛函理论(DFT、)计算了LiNBe(N=1-12)团簇的基态结构及其电子性质.计算结果表明:铍掺杂锂团簇uNBe(N=1-12)的基态结构相当于Be原子取代LiN+1主团簇基态结构中一个LI原子的位置;当团簇尺寸N≥6时,杂质原子Be被束缚在主团簇笼子内;随着团簇尺寸增大,团簇的离解能和二阶能量差分均出现了奇一偶振荡;从结构稳定性上来看,Li6Be是个幻煤田数团簇.
作者:赵文杰闫玉丽杨致雷雪玲葛桂贤王清林罗有华 ZHAO Wen-jie YAN Yu-li YANG Zhi LEI Xue-ling GE Gui-xian WANG Qing-lin LUO You-hua 作者单位:赵文杰,闫玉丽,杨致,雷雪玲,葛桂贤,王清林,ZHAO Wen-jie,YAN Yu-li,YANG Zhi,LEI Xue-ling,GE Gui-xian,WANG Qing-lin(河南大学物理与电子学院理论物理研究所,开封475004)
罗有华,LUO You-hua(河南大学物理与电子学院理论物理研究所,开封475004;华东理工大学理学院,上海,200237)
刊名:原子与分子物理学报 ISTIC PKU英文刊名:JOURNAL OF ATOMIC AND MOLECULAR PHYSICS 年,卷(期):2007 24(4) 分类号:O561 关键词:团簇基态结构密度泛函理论电子性质。
应用第一性原理计算Mg_n(n≤11)团簇的基态结构及稳定性

作者简介 :毛莉萍 , , 女 新疆 乌鲁木齐人 , 硕士 , 新疆医科大学 医学工程技术学院讲师 , 主要研究 物理学 。
第 3期
毛 莉 萍 , 颖 妮 : 用 第 一 性 原 理 计 算 Mg( ≤ 1 )【簇 的 基 态 结 构 及 稳 定 性 段 应 n 1
2 结 果和讨 论
2 1 镁 团 簇 的 几 何 结 构 .
为 了得到 团簇最 低能 量构 型 , 用 了大量 的初 始几 何结构 进行 优化 , 采 得到 n≤ 1 时 中性 Mg 和一 价 1
阴离子 M 2 g 团簇的基态结构, 如图 1 图 2 , 所示 。 g 是哑铃状 的线性结构 , M 键长 347A, .4 结合能O 09 .6 e /tm, Va o 与实验值 3 8 1 .9 A和 0 05e / t .2 V a m接近 。 g 的基态结构为等边三角形 , o M3 平均结合 能为 0 10e / tm, .9 V ao 其异 构 体 , g 线 性 结构 的 能量 比基 态 高 出 00 3e / tm。 M .8 V ao 可见 含原 子 数 目相 同的 镁 团簇 , 平均配位数大的稳定性较高。 这个结论对大尺寸的团簇也同样适用。 g 的基态结构是一个密堆 M4 积 的四面体 结构 , 镁 团簇结 构从 平 面 向三维 立 体演 变 的转折 点 。 Mg 之后 , 团簇 的基 态 结构 都 是 是 从 4 镁 三维 立体 结构 。 于 Mg, 算 结果 表 明稍 微拉 长 了 的三 角双 锥 结构 能 量最 低 。 的基 态结 构 是 具 有 对 计 Mg
团簇 , 增加一个成键电子会使 团簇 的稳定性增强 。 关键词 :镁团簇 ; 第一性原理 ; 几何结构
AlnN(n=1-10)团簇结构和性质的理论研究

第37卷第3期2019年6月凯里学院学报Journal of Kaili UniversityVol.37No.3Jun.2019Aln N(n=1-10)团簇结构和性质的理论研究张颂,岳莉(凯里学院,贵州凯里556011)摘要:利用密度泛函理论对Al n N(n=1-10)团簇结构稳定性和磁性进行比较系统的计算,结果显示,混合团簇变现出与纯铝团簇相似的结构演化趋势,在总原子数为6时,团簇结构由二维平面向三维立体转变.N原子的掺杂不仅增加部分纯铝团簇的稳定性,还保持纯铝团簇的幻数,但却降低5原子铝团簇的稳定性•对磁性而言,除团簇AlN和Al2N的总磁矩分别为1.993加和0.528M夕卜,其余团簇的总磁矩均为零.关键词:团簇;结构;稳定性;第一性原理论文编码:Doi:10.3969/j.issn.1673-9329.2019.03.060引言由几个、几十个乃至上千个原子、分子或离子通过物理或化学的方式结合而得到的微观或亚微观聚集体称为原子分子团簇,简称团簇,它是凝聚态物质的最初状态,不仅被称为是物质第五态,还被认为是研究由单个原子、分子堆积、生长至块体材料的桥梁•由于团簇比较小,所以具有极其显著的量子效应和尺寸效应,其性质不能利用单个原子、分子或离子的性质进行线性组合得到•因此,对团簇结构稳定性、物理化学性质的研究有助于开发、设计新型纳米材料.目前,对单元和多元团簇已有许多研究,但相对单质团簇来说,团簇结构中不同位置、组分的掺杂将对其几何与电子结构产生许多影响,也会使其物理化学性质得到广泛的拓展.众所周知,地壳中含量最多的金属元素是铝,人们已对纯铝团簇做部分研究,发现当铝原子总数为6时,结构由平面向立体转换,而Al?表现出极强的稳定性[1]•早期的研究显示(AlN)n团簇中化学键仅存在于此两种原子间,而且几乎每个原子都成三键,幻数尺寸均是n=4的倍数,但是,对Al-N间相互作用机制却没有详细的讨论[2].另外,金属、非金属原子掺杂铝团簇的研究也比较少,杂质原子的加入对其结构、稳定性、磁性等的影响也还不明白,对单个N原子掺杂铝团簇的结构和性质的研究具有重大意义•因此,本文在密度泛函理论的基础上采用理论计算的方法对Al n N(n=1-10/团簇的结构和性质进行系统的研究,期望能给出掺杂后团簇的结构和性质随尺寸增加的演化规律.1计算方法本文利用局域密度近似(LDA(下的PWC交换关联函数,结构弛豫过程中对所有电子进行优化,为得到更精确的积分及电子的自旋,所有原子都考虑了极化函数、电子自旋和范德瓦尔斯相互作用的贡献•具体能量梯度和总能的收敛精度分别设为1x10"5eV/A和1x10"6eV•初始模型的建立中,每尺寸都考虑几乎所有可能构型,包括Al n N及其同族元素原子团簇,且每个异构体在结构优化的过程中均没有限制对称性•另外,团簇结构中电子态的简并会影响稳定性,因此对所得稳定结构设置不同的自旋多重度进行优化,再通过比较总能,获得能,为,的种性质细的.收稿日期:2019-02-26基金项目:贵州省教育厅青年科技人才成长项目(黔教合KY字[2017]329)作者简介:张颂(1985-/,男,贵州织金人,凯里学院理学院讲师,研究方向为低维纳米材料.-29-表1所有的计算结果TC Sym E b Gap MP NE AlN c,”/v 1.8140.771 1.993-0.606Al2N D,n/h3.1890.3200.528-1.021Al3N D3h3.423 2.4890-1.202A^N C2” 3.180 1.8930-1.359 A^N C s 3.120 1.1150-1.411AhN C2v3.1220.9940-1.450Al7N C3” 3.278 2.1480-2.034Al8N Cs3.1410.9310-1.964Al g N C2” 3.193 1.4050-2.018Al i°N Cs3.1660.9200-1.496图1团簇的基态结构图(图中数字代表键长)2结果分Al n N(n=1~10)团簇结构和性质的计算中,最低能绘1,并将它期关Al的比较•其它统1中,TC,性Sym,吉能E b(eV/原子),最高占据轨未占据轨能量差Gap(eV),性MP(皿(,团簇中N原子电荷NE(e).1中能的看出,AlN的为直线型,Al N原为1.767?,应Al2也为直线型,两原键长为2.858?,Al-N键相比Al-Al短1.091?,对能而言,AlN(1.814eV/原),仏(0.716eV/原子),显然,Al-N间的相互作用比Al -Al间的作用要强,也就是说N原子的加入对Al n团簇的稳定性有一定的贡献•[,]团簇A^N的近直线型,不同于Al3的•过发现,AbN的能构为N原子内嵌的等腰,两腰长3.153?:3.150?,能3.423eV/原子,具有相同原子数的Al4的为棱形能为1.327eV/原,此二者虽形状不一样,但都是-30-平面结构,另外从表1中还可以看出Al g N团簇的能是的,可认为该尺寸为,也许能作为基本单元在中二:米•Al q N Al s N,此二具有较之处‘Al q N的为内嵌N原的Al,Al s N仅仅是在A.N的长边添加Al原成,但A^N是平这点与Al6,均为,同样A^N Al6是 的起点.Al6的能量结构为折叠AJN,具有C”性•从Al7N和Al g的能中能看出变形的Al6,其中N原内,而Al7N可认为是内嵌N原,AhN是在该结构的上添加Al原得,但这两种结构Al8Al9的同,说明N原子的加入对铝的有的影响.Al9N的有Al ioE 能,可以看作是将Al i0中上的铝原子替换为N原子并中心.Al i0N团簇的是四个Al4共用4个铝原一个N原子的,此时的N原子处的一中心,属上内部,但整得比较•总之,对Al n N(n =1~10)),其 随尺寸增加的演化规铝有之处,也有不同之处,特别的不同之处是N原子在n=1~9的范围内都是内部,这主要是N原比Al原的性是判断其物理化学性能及是否独自存在的标准,通常人采用能E b(eV/原子),最高占据轨未占据轨道间能隙Gap(eV)表征•其中,平均结合能的为:E b=(n xE(Al)+E(N)-E(Al n N))/(n+1(,其中,E(Al)、E(N)和E(Al”N)分别为Al原、单个N原Al n N的总能量.1中能看岀,随着总原的增加,能呈现先增加振荡的行为,AhN 的,Al7N次之,二具有极强的稳定性,而在铝的研究中Al3Al7为,N原杂后的还保留了部分纯铝的性,然而,对Al5,它也是,但是杂N原现岀较的能,说明N原子的加入Al5的性.另外,对能隙而言,从表1中可出强烈的振为,但是AhN的Al7N具有比较大的值,分别为2.489eV和2.148eV,这比较大的能隙,导致电子从最高占据轨道向最低未占据轨道跃迁的概率变得比较/J、,能参与化学反应的电子就很少,说明这两个团簇的稳定性很强,这与平均结合能的判据所得结果一致.早期人们对纯铝小团簇结构稳定性与磁性的研究中发现,团簇的总磁矩在1M与2加之间丿并随尺寸的增加出现轻微的振荡行为,而且总原子数为偶数的磁矩是2加,奇数的是1M•⑻然而,根据我们的研究发现,经N原子掺杂后,团簇AlN 和Al2N的总磁矩分别为1.993问和0.528加,且呈减小趋势外,剩余尺寸团簇的磁矩出现淬灭现象,都为0加.这主要是在N原子掺杂的过程中,Al团簇里面未配对的电子逐渐被N原子中不满P 轨道吸收配对所致.3结论本文利用基于密度泛函理论的第一性原理对Al n N(n=1-10)团簇的结构和稳定性进行系统的研究,发现N元素的掺杂对提高Al团簇的稳定性有积极的贡献,但是对个别尺寸(Al s N)却是降低Al团簇的稳定性•另外,N元素的掺杂还保持纯铝元素团簇的幻数对磁性而言,相对于纯铝团簇,掺杂后的团簇仅AlN和Al2N有磁性,其余尺寸团簇的性.参考文献:[1]李东明,温俊青,陈海霞.Al n(n=2-10)团簇结构和性质的密度泛函理论研究[J/西北师范大学学报(自然科学版),2018,54(5):0-55.[2]武海顺丿张聪杰,黄荣彬,等.(AlN/团簇的结构与稳定性[J/中国科学B辑,2001,31(1):42-48.[3]庄琼云,张建华,文玉华.简单金属小团簇Al”(n=2-7)的磁性[J].厦门大学学报(自然科学版),2008,47(6):801-805.[责任编辑:张和平]Theoretical Study on the Structure and Propertiesof Al”N(m=1-10/ClustersZHANG Song,YUE Li(Kaili University,Kaili,Guizhou,556011,China)/Abstract:The structural stability and magnetism of Al n N(n=1-10)clusters were systematically calculated by density functional theory.The results showed that the mixed clusters exhibited a structural evolution trend similar to that of pure aluminum clusters.When the total atomic number was6,the cluster structure changed from two-dimensional to three-dimensional.In addition,the doping of N atom not only increases the stability of some pure aluminum clusters,but also maintains the magic number of pure aluminum clusters,but reduces the stability of5-atom aluminum clusters.In terms of magnetism,the total magnetic moments of clusters are zero except that the total magnetic moments of clusters AlN and A^N are1.993|xB and0.528|xB respectively.Key words:Clusters;structure;stability;first principle・31・。
AlnPn (n = 2~9)团簇结构与性质的理论研究

Modern Physics 现代物理, 2021, 11(3), 41-51Published Online May 2021 in Hans. /journal/mphttps:///10.12677/mp.2021.113006Al n P n (n = 2~9)团簇结构与性质的理论研究彭从一1,马磊1*,马丽2,和一鸣1,王文杰11成都理工大学地球物理学院,四川成都2吉利学院汽车工程学院,四川成都收稿日期:2021年4月16日;录用日期:2021年5月14日;发布日期:2021年5月21日摘要团簇的结构和稳定性具有明显的尺寸效应,研究其性质有助于人们对物质有更深入的认识。
通过结构搜索结合密度泛函方法,我们系统地研究了Al n P n (n = 2~9)团簇的结构、稳定性和电子性质。
随着尺寸的增大,AlP团簇逐渐接近笼状结构,AlP团簇中Al原子和P原子之间交替成键,稳定性增强,Al原子和P原子间的相互作用逐渐减弱。
能隙研究表明Al n P n (n = 2~9)团簇表现为半导体性质。
Al-P原子之间的电荷转移比Al-Al和P-P间更强,表现出离子性质。
成键分析表明,Al-P之间有较强的共价相互作用。
关键词AlP团簇,密度泛函理论,结构与性质Theoretical Study on the Structure andProperties of Al n P n (n = 2~9) ClustersCongyi Peng1, Lei Ma1*, Li Ma2, Yiming He1, Wenjie Wang11Department of Geophysics, Chengdu University of Technology, Chengdu Sichuan2College of Automotive Engineering, Geely University of China, Chengdu SichuanReceived: Apr. 16th, 2021; accepted: May 14th, 2021; published: May 21st, 2021AbstractThe structure and stability of clusters have prominent size effects, and studying their properties is helpful for people to have a deeper understanding of matter. The structures, stability and elec-*通讯作者。
金属团簇的第一性原理计算

金属团簇的第一性原理计算目录一论文正文.1引言 (1)2计算理论和方法 (2)2.1 理论 (2)2.1.1 密度泛函理论 (2)2.1.2 Hohenberg-Kohn 定理 (2)2.1.3 Kohn-Sham方程 (3)2.2 密度泛函近似 (4)2.2.1 局域密度近似 (4)2.2.2 广义梯度近似泛函 (5)2.2.3 杂化密度泛函 (5)2.3 计算方法 (6)2.3.1 GAUSSIAN03 (6)2.3.2 GAUSSIAN形赝势基组 (6)2.4 LUMO和HOMO (6)Au(n=2-4)的计算 (7)3nAg(n=2-4)的计算 (10)4nW(n=4)的计算 (13)5n6 结论 (16)参考文献 (18)谢辞 (19)二附录1 任务书 (20)2 中期检查报告 (22)3 指导教师指导记录表 (23)4 结题报告 (24)5 成绩评定及答辩评议表 (26)6 答辩过程记录 (28)金属团簇的第一性原理计算摘要:本文采用密度泛函理论方法中的杂化泛函B3LYP ,以及结合赝势基组LANL2DZ 对金属团簇n W 、n Au 、n Ag (n=2-4)的所有可能结构进行了研究,得到了这些结构的平均键能,形成能,离解能以及LUMO 、HOMO 。
通过比较离解能的大小,得出这些团簇为3Au 、4Au 、3Ag 、4Ag 、3W 、4W 的最稳定结构。
关键词: Au 团簇;W 团簇;Ag 团簇;密度泛函方法First – Principles calculates of the metal clustersAbstract: In this paper , We employ density functional theory in the hybrid functional B3L YP, with pseudopotential basis sets Lanl2DZ to study the metal clusters n W 、n Au 、n Ag (n=2-4)of all possible structure, get the average bond energy of thosestructures, formation energy , dissociation energy and the LUMO 、HOMO. By comparing the size of dissociation energy . Come to these clusters as 3Au 、4Au 、3Ag 、4Ag 、3W 、4W the most stable structure.Keyword: Au clusters;W clusters;Ag clusters;Density function method (DFT)目录1引言 (1)2计算理论和方法 (2)2.1 理论 (2)2.1.1 密度泛函理论 (2)2.1.2 Hohenberg-Kohn 定理 (2)2.1.3 Kohn-Sham方程 (3)2.2 密度泛函近似 (4)2.2.1 局域密度近似 (4)2.2.2 广义梯度近似泛函 (5)2.2.3 杂化密度泛函 (5)2.3 计算方法 (6)2.3.1 GAUSSIAN03 (6)2.3.2 GAUSSIAN形赝势基组 (6)2.4 LUMO和HOMO (6)Au(n=2-4)的计算 (7)3nAg(n=2-4)的计算 (10)4nW(n=4)的计算 (13)5n6结论 (16)参考文献 (18)谢辞 (19)1引言原子或分子团簇(简称团簇或微团簇)是由几个乃至数千个原子、分子或离子(国际上多数定义含原子数在10-105范围)通过物理或化学结合力组成的相对稳定的微观或亚微观聚集体,其尺寸一般为埃数量级.团簇的各类电子性质和各种量子效应都与其它材料有显著的不同. 而原子团簇的独特性质是源于其结构上的特点,因其尺寸小,处于表面的原子比例极高,而表面原子的几何构型、自旋状态以及原子间作用力都完全不同于体相内的原子。
小团簇结构及其光学性质的第一性原理研究的开题报告

小团簇结构及其光学性质的第一性原理研究的开题报告一、研究背景小团簇是指由少量原子组成的团簇,其尺寸介于单个原子和固体之间,具有介于分子和晶体之间的一些性质。
在材料科学中,小团簇被广泛应用于催化、电子器件等领域。
对于小团簇结构及其光学性质的研究,不仅有助于深入理解小团簇的本质特性,还对于小团簇相关应用的设计和优化有着重要意义。
目前,小团簇的制备方法已经较为成熟,然而,对于小团簇的结构及其特性的理论研究仍受到诸多限制。
因此,采用第一性原理方法对小团簇结构及其光学性质进行研究,可以提供基础的理论支持和指导。
二、研究内容和目标本研究将采用第一性原理方法,基于密度泛函理论(DFT)和紧束缚模型(Tight-binding Model),对小团簇结构及其光学性质进行系统研究。
具体研究内容包括:1. 不同元素组成的小团簇结构的构建和优化。
2. 基于DFT方法,计算小团簇结构的电子结构、能带结构、密度分布等物理性质。
3. 基于Tight-binding Model,研究小团簇的光学性质,包括吸收光谱、电子极化率、光学吸收强度等。
4. 探究小团簇结构和光学性质之间的相互关系。
本研究的主要目标是揭示小团簇结构和光学性质的基本特性,建立小团簇的结构和性质之间的相互关系,并为小团簇相关应用提供理论指导。
三、研究方法和技术路线本研究将采用基于DFT方法的VASP软件包,对小团簇结构进行计算。
首先,根据小团簇中的原子数或者化学成分,提出可能的结构模型;然后,采用优化算法(例如:共轭梯度方法、赝牛顿法等)对模型进行结构优化,得到最稳定的几何构型;最后,利用计算得到的几何构型和能级密度,分析小团簇的某些物理性质,如电子态密度、吸收谱等。
之后,针对小团簇的光学性质,本研究将采用基于Tight-binding Model方法的NanoTCAD ViDES软件包进行计算。
具体来说,我们将采用近似与该软件包相容的耦合扰动方法来计算光学性质。
(CoMn)n(n=1~5)合金团簇的结构和磁性

(CoMn)n(n=1~5)合金团簇的结构和磁性
吕瑾;许小红;武海顺
【期刊名称】《化学物理学报》
【年(卷),期】2005(018)004
【摘要】采用密度泛函理论中的局域自旋密度近似和广义梯度近似对(CoMn)n(n=1~5)团簇的几何构型进行优化、能量、频率和磁性计算,确定了团簇的基态,对其基态的磁性和电子结构进行了系统研究,并与相对应的一元团簇进行了结构和磁性比较.研究表明,两种方法确定的基态构型基本一致,当n=1~4时,等比CoMn二元合金团簇的几何形状仍与一元团簇相同;(CoMn)3和(CoMn)4团簇出现了磁性双稳态,显示铁磁性和反铁磁性耦合;二元CoMn合金团簇中Co、Mn原子磁性仍能保持一元Co、Mn团簇基态的磁性.
【总页数】7页(P552-558)
【作者】吕瑾;许小红;武海顺
【作者单位】山西师范大学化学与材料科学学院,临汾,041004;山西师范大学化学与材料科学学院,临汾,041004;山西师范大学化学与材料科学学院,临汾,041004【正文语种】中文
【中图分类】O664
【相关文献】
1.(CoCr)n(n=1-5)合金团簇的结构和磁性 [J], 吕瑾;许小红;武海顺
2.过渡金属及其合金团簇的磁性检测实验研究进展 [J], 吕瑾;武海顺
3.NixPt(x=-9)合金团簇结构稳定性与磁性的第一性原理研究 [J], 岳莉
4.Pt-Ni合金团簇的结构与磁性的第一性原理研究 [J], 王明阳;宋风忠;张岩星;杨宗献
5.铁铬合金团簇点阵中与成分相关的磁性耦合(英文) [J], 徐钟麒;刘畅;罗维峰;刘飞;韩民
因版权原因,仅展示原文概要,查看原文内容请购买。
znn团簇稳定性和电子性质的第一性原理计算

znn(n=7—14)团簇稳定性和电子性质的第一性原理计算总结(= 2-22)团簇稳定构型、结合能及平均键长的变化规律;③智丽丽等对团簇作了第一性原理的计算,结果发现随着原子数目增加,(213)团簇的稳定性在增强;④张文庆等也对(=2-11)团簇的结构和稳定性作了研究,发现(=2-11)团簇的基态结构均为平而结构。
⑤肖绪洋等用分子动力学方法对(=13-321)团簇研究,结果表明对(=13-321)团簇有球壳层和二维点阵两种原子分布结构。
⑥而对(=7-14)团簇稳定性和电子性质的第一性原理的研究鲜有报道,所以本文基于第一性原理计算了(=7-14)团簇稳定性及电子性质,使之能为金属从原子以及分子状态转变为块体提供一定的理论依据。
1 计算方法本文采用第一性原理数值基组的方法,在量子力学程序dmol3中完成计算。
在计算中,交换关联能采用了广义梯度近似(GGA)的PBE泛函,⑦优化标准为原子间作用力不大于0.01 /,原子的最大位移不大于5.0€祝宓哪谟αΣ淮笥.02 GPa,能量收敛精度不大于 5.0€?eV/atom。
计算中,参与计算的电子组态为[] __,为了验证方法的正确性,本文计算了的键长为3.418,与俞洁等的实验值3.18符合的较好,表明所选方法的可靠性。
2 结果与讨论图1为(=7-14)团簇的结构,所选取(=7-14)团簇的构型都是具有较高的对称性。
从图1中可以看出,从团簇至团簇其结构都为笼状形态;其对称性分别为,,,,,,,。
对于的构型,则是由五边形分别在两边戴帽构成,其最近临的键长为0.2998 nm。
的构型可以看成是下底由一个正三角形,中间由一个不规则的平行四边形,外加一个顶端戴帽构成;最近临的键长为0.2693 nm,最长的键长则为0.3260 nm。
团簇的构型则是由2个四边行空间错位对接,同时在中间位置增加一个原子而成,键长分别为0.2819 nm,0.3618 nm,0.3703 nm。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第25卷 第1期原 子 与 分 子 物 理 学 报Vo l.25 N o.1 2008年2月JOU RNA L OF A T OM IC AN D M OLECUL AR PHYSICS Feb.2008文章编号:1000 0364(2008)01 0143 06第一性原理对Ga n N n(n=2~5)小团簇的结构及电子性质的研究葛桂贤1,雷雪玲2,闫玉丽3,杨 致3,赵文杰3,王清林3,罗有华3,4(1.石河子大学师范学院物理系生态物理重点实验室,新疆832003;2.新疆师范大学数理信息学院,乌鲁木齐830053;3.河南大学物理与信息光电子学院理论物理研究所,开封475004;4.华东理工大学理学院,上海200237)摘 要:利用密度泛函理论的B3LY P方法在6 31G*的水平上对Ga n N n(n=2~5)团簇的结构进行优化,得到了Ga n N n(n=2~5)团簇的最稳定结构.并对最稳定结构的电子性质、成键特性和极化率进行分析.结果表明,团簇的最稳定结构为平面结构,且存在着N2和N3单元,说明N-N键在团簇的形成过程中起着决定性的作用;能隙间隔为1.776~3.563eV,表明Ga n N n(n=2~5)团簇已具有了半导体的性质.关键词:G a n N n团簇;最低能量结构;电子性质中图分类号:O641 文献标识码:AFirst principles study on structure and electronic propertiesof small Ga n N n(n=2~5)clustersGE Gui Xian1,LEI Xue Ling2,YAN Yu Li3,YANG Zhi3,ZHAO Wen Jie3WANG Q ing Lin3,LUO You Hua3,4(1.Key Laboratory of E cophysics and Department of Physics,Normal College,Shihezi University,Xinjiang832003,China;2.School of M aths Physics an d Information Sci ences,Xinjiang Normal Universi ty,U rumqi830053,China;3.Institute of T heoreti cal Physics,School of Physics and Information Optoelectronics,Henan University,Kaifeng475004,China;4.S chool of S cience,East C hi na Un i versity of Science and T echnology,Shanghai200237,China)Abstract:Geometric structure and relative stability of Ga n N n(n=2~5)clusters are studied by using the hy brid functional theory(B3LYP)w ith6 31G*basis sets.For the most stable isomers of Ga n N n(n=2~5) clusters,the electronic properties,bond properties,polarizability are analyzed.T he calculated results show that the optimized Ga n N n(n=2~5)clusters are planar structure.The most stable structures indicate a pref erence for an N2subunit or N3subunit,denoting that N N bonds play a crucial role in stabilizing the cluster. T he energy gaps are from1.776to3.563eV,revealing that these cluster may present semiconductor like properties.Key words:Ga n N n(n=2~5)clusters,the lowest energy structure,electronic properties收稿日期:2006 09 01作者简介:葛桂贤(1977-),女,讲师,研究方向为团簇物理.E mai l:geguixian@1 引 言在过去的十几年中,原子团簇的结构和稳定性得到了广泛的研究[1-3].近几年来, ~族化合物半导体材料奇特的光学性质和潜在的应用价值已引起物理、化学和材料等领域的广泛兴趣,也使得 ~族化合物团簇成为团簇领域的研究热点之一[4 6].对GaN团簇的研究工作主要有:Kan dalam等在密度泛函理论的基础上运用非局域密度近似的方法计算了Ga n N m(n,m=1~2)[7]和Ga n N n(n=3~6)[8,9]团簇的结构.Belbruno[10]等用密度泛函理论研究了Ga n N n(n=2~4)团簇的结构.Song等用全势能线形M uffin T in轨道组合分子动力学(FP LMTO)方法计算了Ga n N n(n=2 ~6)[11]团簇的结构.Costales等用密度泛函理论对Ga n N-n(n=1~3)[12]阴离子团簇进行了研究.以上小组的共同特点是主要集中在结构的研究上,都没有涉及到原子间的成键特性和极化率的计算.众所周知,极化率表征着体系对外场的响应,决定了体系的非线性光学特性,同时它还能够影响分子间如诱导力、色散力等长程相互作用以及碰撞过程中的散射截面等重要的物理量.本文用B3LYP/6 31G*密度泛函方法对Ga n N n(n=2~5)团簇进行了计算,得到了这些团簇的最低能量和一些亚稳态的几何结构,并对最稳定结构的电子性质、成键特性和极化率进行了分析.2 计算方法采用密度泛函理论(DFT)中的B3LYP泛函方法,在6 31G*水平上通过寻找对多维势能面上的极小点,在相同的水平上对振动频率进行了计算.所有的计算都是在Gaussian03程序[13]上进行的.为了验证所选方法的合理性,在相同的条件下计算了二聚体N2的键长和振动频率以及二聚体GaN 的键长.计算结果表明,N-N键长为0.1105nm、振动频率为2457.65cm-1,与实验的N-N键长0.1098nm,振动频率2358.57cm-1[14]符合的很好.Ga-N键长为0.173nm和最近Dsa计算的Ga-N键长(0.188nm)[15]符合的很好.由于B3LYP方法可以很好的描述二聚体N2和GaN,于是我们认为这种方法也适用于Ga n N n(n=2~5)团簇.3 结果与讨论3.1 几何构型Ga n N n(n=2~5)团簇的最低能量结构和一些亚稳态的几何结构如图1所示,原子间距分别小于0.3330nm(Ga-Ga),0.2432nm(Ga-N)及0. 1591nm(N-N)时成键,表1给出了最低能量结构的几何参数(原子括号内的数字代表第几个原图1 Ga n N n(n=2~5)团簇的最低能量结构和一些亚稳态的几何结构(浅色代表Ga原子,深色代表N原子)Fig.1 L owest energ y and some meta stable isomerstructures of Ga n N n(n=2~5)clusters(The w hite circles represent the Ga atoms and the dark circles represent th e N atoms)子).Ga2N2团簇的最低能量结构示于图1(2a),其是不规则的四边形,N-N键长为0.1168nm. Song等人[11]用FP LM TO方法报道的最低能量结构是一个菱形.他们报道的两个氮原子在短轴位置,N-N和Ga-N键长分别为0.1246nm和0.2093nm.我们在优化过程中得到了两种菱形结构,如图1(2b)和图1(2d)所示.其中2b的两个氮原子在短轴位置,N-N和Ga-N键长分别为0 1252nm和0.2151nm,与Song[11]的计算结果符合的很好.其能量仅比最低能量结构高0.007144 原 子 与 分 子 物 理 学 报 第25卷eV,Kandalam[7]和Belbruno[10]在密度泛函理论的基础上运用非局域密度近似的方法均认为其是最低能量结构,没有报道出不规则的四边形.另外在优化过程中得到了三种线性构型,如图1(2c)、图1 (2e)和图1(2f)所示.Ga3N3团簇的最低能量结构如图1(3a)所示,该结构是一个平面,可以看成是一个N3单元与三个镓原子的结合,N3单元中N (3)-N(1)-N(2)的键长分别为0.1296nm和0. 1319nm.这和Song等[11]报道的Ga3N3团簇的最低能量结构以及N3单元中氮原子之间的键长0. 1295nm和0.1314nm符合的很好.Ga3N3团簇的最低能量结构的同分异构体(图1(3b))是一个C2v 的平面结构,其能量比最低能量结构高1.165eV,而Kandalam等[8]认为其是最低能量结构.图1中的3d是一个D3h的环形结构,能量比最低能量结构高2.988eV,Belbruno等[10]认为其是最低能量结构.在优化的所有结构中3c是唯一的一个立体结构,其能量比最低能量结构高1.607eV.Ga4N4团簇的最低能量结构是一个平面,如图3(4a)所示,该结构可以看成是一个N2单元与Ga4N2的结合,结构中的N(3)-N(1)键长为0.1105nm和N2负离子的键长0.1202nm非常接近.立体结构4b的能量比最低能量结构高0.397eV.另两个立体结构如图(4c)和(4d)所示,4c比最低能量结构高0.436eV,4d比最低能量结构高0.437eV,而4c和4d的能量仅差0.001eV,所以二者可看成是简并的.Belbruno[10]提出的Ga4N4的最稳定结构是一个D4h的平面结构,见图1(4f),在我们的计算中这种平面结构比最低能量结构高3.793eV,在所有同分异构体中能量最高.在优化的Ga4N4团簇的结构中,4f中有8个Ga-N键而没有N-N键,可见Ga-N键的增多和N-N键数目的断裂均会降低团簇的稳定性.Ga5N5团簇的最低能量结构如图1(5a)所示,该结构是一个平面结构,存在一个N3单元,N(1)-N(2)-N(3)的键长分别为0 1145nm和0.1221nm,仍和传统的直线N3负离子的键长(0.1183nm)偏离很小,N3单元与Ga(6)原子的键长为0.1948nm.立体结构5b 的能量比最低能量结构高0.379eV.平面结构5c 的能量比最低能量结构高1.022eV,Kandalam 等[8]认为其是最低能量结构.从得到的Ga n N n(n =2~5)团簇的几何结构来看,Ga n N n(n=2~5)的最稳定结构大多是平面结构,结构中存在着N2和N3单元.这说明N-N键在氮化镓团簇的形成中确实起着决定性的作用,Ga Ga之间不易成键,从这一点来看,本文的结果支持了Kandalam等的结果.3.2 能 隙对于半导体材料来说,禁带与导带之间的能量间隔是非常重要的数据,GaN晶体(铅锌矿)的禁带和导带之间的能量差为3.44eV.那么对于半导体团簇来说,这个数据可以用最高占据分子轨道和最低未占据分子轨道的能量间隔(能隙)来反映.能隙差的大小反映了电子从占据轨道向未占据轨道发生跃迁的能力,在一定的程度上代表分子参与化学反应的能力.如表1所示,对于Ga n N n(n=2 ~5)团簇来说,随着团簇尺寸的增加,能隙整体呈增大的趋势,这说明团簇的化学活性逐渐减小.该团簇的能隙在1.776eV~ 3.563eV之间,所以从能隙上来看,Ga n N n(n=2~5)团簇已具有了半导体的性质.在Ga n N n(n=2~5)团簇中,Ga4N4团簇的能隙最大,说明Ga4N4比近邻尺寸的团簇稳定.3.3 分子轨道与成键分子的最高占据轨道(HOM O)和次最高占据轨道(NHOMO)的成键方式与形状直接反映了化学键结构的特点,通过对它们的分析,可以得到分子几何构型稳定性的信息.图2分别给出了Ga n N n(n=2~5)团簇最稳定结构的H OM O和次最高占据轨道(NH OMO),由图2可以看出,Ga2N2的HOMO是主要是由Ga和N原子的s、p轨道组成,有部分的d成分.HOMO可分为三部分:N-N的离域 反键轨道,Ga-N的离域 成键轨道和Ga-Ga的 成键轨道.Ga2N2的NHOMO也可分为三部分:N-N的离域 成键轨道,Ga-N 之间的 成键轨道,Ga-Ga的 反键轨道.从自然键轨道(natural bond orbital,NBO)分析也可以看出Ga、N原子主要发生了sp杂化,且有部分的d成分.由以上分析可以看出HOM O和NHOM O 对分子中的Ga-N和N-N成键都有贡献.Ga3N3的HOM O主要由三部分:N-N的离域 成键轨道,Ga-N的离域 成键轨道,Ga(6)和N(3),Ga(4)和N(3)以及Ga(4)和N(2)的 成键轨道.Ga3N3的NH OM O主要由三部分:Ga(4)和N(2)原子之间的离域 成键轨道,Ga(6)和N (3),Ga(5)和N(2)之间的 成键轨道,N原子之间的离域 成键轨道.从上面的分析可以看出,145第1期 葛桂贤等:第一性原理对Ga n N n(n=2~5)小团簇的结构及电子性质的研究图2 G a n N n(n=2~5)团簇的最高占据分子轨道(HO M O)和次最高占据分子轨道(NHOM O)Fig.2 Contour maps of t he HOM Os and NHO M Os for the lowest energy structur e of Ga n N n(n=2~5)clusters表1 Ga n N n(n=2~5)团簇最稳定结构的对称性,键长( ),结合能E b(eV)和能隙(eV)T able1 Symmetry,bond,length,average binding ener gy(E b)and HOM O LU M O gap for the lo west ener gy structures of Ga n N n(n=2~5)clustersClusters Structure Symmetry Bond length(nm)E b(eV)Gap(eV) Ga2N2(2a)(Cs)N(3) N(4)0.1168 4.267 1.776 Ga2N2(2b)(D2h)N(1) N(2)0.1252 4.265Ga3N3(3a)(Cs)N(3) N(1)0.1296N(2) N(1)0.1319N(2) Ga(5)0.1953 4.403 3.012N(3) Ga(6)0.2015Ga4N4(4a)(Cs)N(1) N(3)0.1105N(2) Ga(8)0.1832N(2) Ga(7)0.1708N(4) Ga(7)0.1826 4.725 3.563N(4) Ga(5)0.1933N(4) Ga(6)0.1931Ga5N5(5a)(Cs)N(2) N(1)0.1145N(2) N(3)0.1221N(3) Ga(6)0.1948N(4) Ga(6)0.1848 4.744 3.229N(4) Ga(5)0.1928N(4) Ga(7)0.1931N(10) Ga(6)0.1834N(10) Ga(8)0.1981HOMO和NH OM O对N-N的成键和Ga-N的成键都有贡献.从Ga4N4的HOM O中可看出Ga、N原子发生了sp杂化,Ga-N是 成键轨道,N-N之间的电子云分布很少,所以HOM O对N-N成键没有贡献.从NHOM O可看出Ga(7),N(2)和Ga(8)形成离域的大 成键轨道,N(4)-Ga(5)和N(4)-Ga (6)都是 成键轨道,N(1)和N(3)之间是三键,N146 原 子 与 分 子 物 理 学 报 第25卷-N之间的电子云分布很少,NHOMO对N-N成键没有贡献.Ga5N5的HOM O和NHOMO的如图2中所示,从图上可以看出H OM O和NHOM O对Ga-Ga的成键都没有贡献,但对Ga-N的成键有贡献,而HOMO对N-N之间的成键有部分贡献. 3.3 Ga n N n(n=2~5)团簇的极化率用B3LYP方法在6 31G*水平上对Ga n N n(n =2~5)团簇的极化率进行了计算,并且由(1),(2)式计算得到了极化率张量的平均值<a>、极化率的各相异性不变量a和每个原子的平均线性极化率<a>/n.由此来衡量分子产生非线性光学性质能力的强弱:<!>=13(!XX+!YY+!ZZ)(1) !=(!XX-!Y Y)2+(!Y Y-!ZZ)2+(!ZZ-!XX)221/2.(2)从表2可以看出,单位原子的平均极化率整体上呈下降的趋势,但变化不大,表明团簇的电子结构随着原子数目的增加略显紧凑;极化率的各相异性不变量单调增加,说明团簇还没有形成密堆积结构,这与优化的团簇最稳定结构都是平面结构相一致.表2 Ga n N n(n=2~5)团簇最稳定结构的极化率张量、极化率张量的平均值<a>、极化率的各相异性不变量a和每个原子的平均线性极化率<a>/nT able2 Polarizabilit y tensor(!ij),po larizability(<!>),stat ic mean polarizabilities(<!>/n) and polarizability anisotro pies( !)of Ga n N n(n=2~5)clustersPolarizabilityClusters!XX!X Y!Y Y!XZ!YZ!ZZ<a><a>/n a Ga2N2142.8237.38382.690.0000.00059.15994.8923.7274.73 Ga3N3176.287 5.717152.9700.0050.00092.377140.5423.4275.020 Ga4N4265.580 5.988172.899-0.005-0.131109.216182.5622.82123.203 Ga5N5262.887-21.468253.9850.0000.003129.798215.5621.56128.8694 结 语本文用DFT在6/31G*的水平上对Ga n N n(n =2~5)团簇的结构、电子性质、成键特性和极化率进行了研究.结果表明,Ga n N n(n=2~5)团簇的最稳定结构为平面结构,还没有形成密堆积结构,结构中存在N2和N3单元,N-N键在团簇的形成过程中起着决定性的作用,Ga-Ga之间不易成键.通过对能隙的分析可以看出Ga n N n(n=2~5)团簇具有半导体性质.参考文献:[1] Zhao W J,Y an Y L,Y ang Z,et al.F irst principlesstudy of the ground state str uctures and electronic propert ies of Li N Be(N=1~12)clusters[J].J.A t.M ol.Phys.,2007,24:716(in Chinese)[赵文杰,闫玉丽,杨致,等.第一性原理计算L i N Be(N=1~12)团簇的基态结构及其电子性质[J].原子与分子物理学报,2007,24:716][2] Lei X L,Yan Y L,Ge G X,et al.Gr ound state g eo metries and stability of Be n L i(n=1~12)clustersw ith density functional theory[J].J.A t.Mol.Phy s.,2007,24:1003(in Chinese)[雷雪玲,闫玉丽,葛桂贤,等.密度泛函理论计算掺杂团簇Be n L i (n=1~12)的基态结构和稳定性[J].原子与分子物理学报,2007,24:1003][3] Yang Z,Y an Y L,Ge G X,et al.Density functio nalstudy of g round structures and electr onic properties ofL i2Be N(N=1~10)clusters[J].J.A t.Mol.Phy s.,accepted(in Chinese)[杨致,闫玉丽,葛桂贤,等.利用密度泛函理论计算L i2Be N(N=1~10)团簇的最低能量结构及其电子性质[J].原子与分子物理学报,已被接受][4] Wang B L,Zhao J J,Shi D,et al.Density functio nalstudy of structural and electro nic properties of Al n N(n=2~12)clusters[J].Phy s.Rev.A,2005,72:023204[5] Guo L,Wu H S,Jin Z H.F irst principles investig atio nof geomet ry and stability of aluminum phosphorous binary clusters A l n P-m(n+m=5)[J].J.A t.Mol.Phy s.,2004,21:335(in Chinese)[郭玲,武海顺,金志浩.第一原理对Al n P-m(n+m=5)团簇结构和稳定性研究[J].原子与分子物理学报,2004,21:147第1期 葛桂贤等:第一性原理对Ga n N n(n=2~5)小团簇的结构及电子性质的研究335][6] L i E L,Chen G C,Wang X W,et al.F irst principlesstudy on structures and photoelectron spectroscopy aboutGa n P-m anions[J].J.A t.M ol.Phys.,2006,23:279(in Chinese)[李恩玲,陈贵灿,王雪文,等.第一性原理对Ga n P-m阴离子团簇结构及其光电子能谱的研究[J].原子与分子物理学报,2006,23:279] [7] Kandalam A K,Randey P,Blanco M A,et al.Firstprinciples study of polyatomic clusters of AlN,GaN,and InN. 1.Structure,stabilit y,vibrations,and ionization[J].J.Phy s.Chem.B,2000,104:4361 [8] K andalam A K,Blanco M A,P andey R.T heoreticalstudy of structural and v ibrational properties of A l3N3,Ga3N3,and In3N3[J].J.Phys.Chem.B,2001,105:6080[9] K andalam A K,Blanco M A,P andey R.T heoreticalstudy of Al n N n,Ga n N n and In n N n(n=4~6)clusters[J].J.Phys.Chem.B,2002,106:1945[10] Belbr uno J J.T he structur e of small gallium nitrideclusters[J].H eter oatom Chemistry,2000,11:281 [11] Song B,Ling L,Cao P L.T heoretical study of thestructure of small GaN Clusters[J].Jour nal of ZhenJ iang University,2004,31:270[12] Costales A,P andey R.Density functio nal calculationsof small anionic clusters of gr oup nitr ides[J].J.Phys.Chem.A,2003,107:191[13] Frisch M J,T rucks G W,Schlegel H B,et al.Gaussian03,Revision C.02.Wallingford CT:Gaussian,Inc.,2004.[14] L ide D R.CRC H andbook of chemistry and p hy sics[M].79th ed.N ew Yor k:CRC Press,1998. [15] Das G P,Rao B K,Jena P.Ferromagnet i sm in M ndoped G aN:from clusters to crystals[J].Phys.Rev.B,2003,68:035207148 原 子 与 分 子 物 理 学 报 第25卷。