ChemBioOffice教程3
化学软件chemoffice安装和基本教程

模板
点击模板工具栏按钮下面的右指三角箭头,单击该按钮, 不松开,就可以弹出模板信息,众多画图的起点。
【Aromatics芳香族模板】 【Bicyclics双环模板】 【Bioart生物艺术模板】
8
总结:共17个模板
绘制与编辑典型化学物质结构式
(1)键工具: 主工具图标板上提供了九个键操作的命令,其中
Chemdraw主界面
(1)菜单栏
菜单栏共有11个下拉菜单,每一个下拉菜单中都包括相应的命令。 其中如果相应的命令前有√,则该条命令已经被执行;若命令后有小 三角表示该指示条有子菜单;指示条灰色表示该命令未激活。
“File”菜单命令
•文件命令对话框
(2)
主 工 具 图 标 板
主 工 具 图 表 板
生重键。 点位为于键中间,单击同样可以产生重键。
②元素符号的输入:
官能团或元素符号的输入
键及元素文本属性的设置:
(2)环工具:主工具面板中提供了10种环
工具命令,其中在模板命令中还有芳香化合物 模板和双环模板可以绘制环状化合物。
•环的绘制
•环的连接
➢面板中选取相应的环命令后,直接在绘制区点击可产 生相应的环。点住鼠标左键不放,拖动鼠标可以对环的 大小,放置的角度进行自由改变。
➢改变元素:
双击需要改变元素符号的原子,弹出输入框,输入元素符号,按回车 键完成原子序号的更改。若原子的三维结构中显示孤对电子,执行关闭 “Tools”菜单中的“Show H’s ans Lp’s”命令,可不显示氢原子和孤对电子。
ChemDraw结构式与3D模型的转换
(1)ChemDraw结构式转换为3D模型 (2)3D模型转换为平面结构式
Chem3D的分子优化
怎样从Word导入ChemDraw分子结构图
怎样从Word导入ChemDraw分子结构图ChemDraw是ChemBioOffice的组件之一,用于画平面分子结构,是国内外普遍采用或指定的分子结构绘图软件。
我们可以在Word中导入ChemDraw化学分子结构,本教程主要介绍以下两种方法:
方法一:将画好的分子结构保存成图片格式插入Word。
1.首先可以将画好的化学分子结构保存成jpg、bmp、png等图片格式。
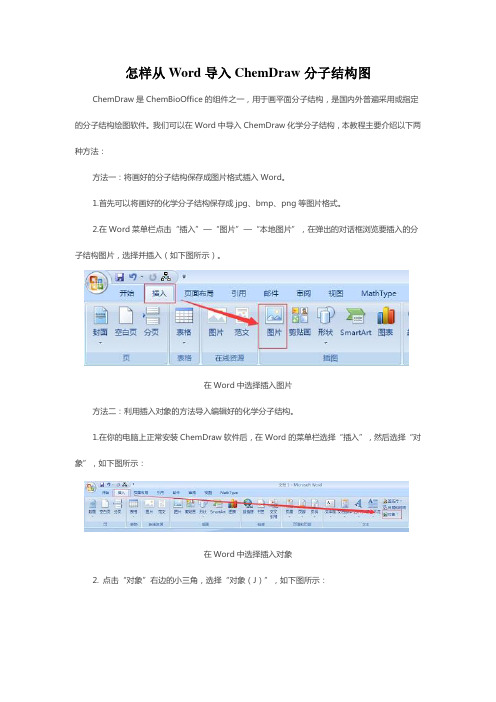
2.在Word菜单栏点击“插入”—“图片”—“本地图片”,在弹出的对话框浏览要插入的分子结构图片,选择并插入(如下图所示)。
在Word中选择插入图片
方法二:利用插入对象的方法导入编辑好的化学分子结构。
1.在你的电脑上正常安装ChemDraw软件后,在Word的菜单栏选择“插入”,然后选择“对象”,如下图所示:
在Word中选择插入对象
2. 点击“对象”右边的小三角,选择“对象(J)”,如下图所示:
在对象选项下选择插入对象
3.弹出“对象”对话框,选择对象类型“CS ChemDraw Drawing”,然后点击确定(如下图所示)。
选择对象类型为CS ChemDraw Drawing
4. 这时会打开ChemDraw Std软件,我们可以在该软件绘制化学分子结构,绘制完成后点击“File”—“Close and Return to 文档”,绘制的化学分子结构就会显示在Word中。
将画好的化学分子结构导入Word中
以上教程介绍了在Word中导入ChemDraw编辑的化学分子结构的两种方法,用户可以根据自己的喜好选择合适的方法。
教你计算ChemBio模型的空间位阻能

教你ChemBio 3D Ultra 14是ChemOffice 中的一模型以及获取空间位阻能等操作,本节内容将教授 在ChemBio 3D 模型中,因分子中靠近反应中称立体效应,重要指分子中某些原子或基团彼此接 获取ChemBio 3D 模型的空间位阻能在交错、重叠的结构组成中,连接在碳上的氢了计算该测量结果,可以计算该结构的空间位阻能 1、本次实例以乙烷模型为例,获取能量之前要在然后选择Calculations 菜单栏下MM2菜单中的2、在对话框中单击Run。
此时乙烷能量信息表教你计算ChemBio 3D 模型的空间位阻能中的一个重要组件,也是一个重要的化学结构演示工具。
在ChemBio 3D 窗口中,你可以将教授各位如何获取ChemBio 3D 模型的空间位阻能。
反应中心的原子和基团各占有一定的空间位置,降低分子反应活性的空间效应就称为“空彼此接近而引起的空间阻碍和偏离正常键角而引起的分子内的张力。
上的氢原子间是以相互保持最大的距离而存在的,体现出整体的能量最小,这也是乙烷模位阻能量,然后与已经确认过的较高能量结构相比较。
空间位阻能的获得过程如下: 前要在ChemBio 3D 窗口建立乙烷模型,如何建立乙烷模型请参考教你利用ChemBio 3中的Compute Properties 命令,此时出现Compute Properties 对话框如下图所示:信息表出现在ChemBio 3D 窗口的最下方,调整后如下图所示: 你可以建立、旋转ChemBio 3D 为“空间位阻能”。
空间位阻又乙烷模型最稳定的结构状态。
为mBio 3D 键工具建立乙烷模型。
示:【温馨提示】这里计算出的能量值是近似的, 以上即是计算ChemBio 3D 模型的空间位阻能习如何操控ChemBio 3D 窗口。
似的,其数值大小也依赖于计算所用的处理器类型。
位阻能的方法,ChemBio 3D 窗口与ChemDraw 窗口有较大改变,就算十分熟悉ChemChemDraw 窗口也需要认真学。
怎样利用子结构建立ChemBio 3D模型

怎样利用子结构建立ChemBio 3D模型ChemBio 3D是一款三维分子结构演示软件,能够轻松快捷地建立化学模型、化
学结构的制作和编辑。
本教程将以十肽菌素分子模型的建立为例向大家讲解如何利用子结构建立ChemBio 3D模型。
ChemBio 3D中已经定义的子结构已超过了200个,这些子结构覆盖了大部分常用的有机结构。
从“View”菜单中选择“Substructures”可查看并使用这些子结构。
肽菌素属于肽类抗生素,是常见的饲料添加剂,具有较强的杀菌免疫作用。
十肽菌素结构的子结构有助于建立蛋白质分子模型。
ChemBio 3D建立十肽菌素分子模型的具体步骤如下:
(1)从“File”菜单中选择“New”。
(2)在Replacement Text框中输入“HAlaArgCysAlaGluLeuLysPheValOH”,如下图所示:
在Replacement Text框中输入“HAlaArgCysAlaGluLeuLysPheValOH”
(3)在模型窗口双击,即可出现一个十肽菌素分子模型(见下图)。
在此十肽菌素分子结构中除了H和OH 都是子结构。
十肽菌素分子模型示例图
【温馨提示】
如果要检索Protein Data Bank文件,可以利用ChemBio 3D的“Open”对话框中的“Protein Data Bank”格式打开。
通过上述教程,相信大家已经掌握如何利用子结构建立ChemBio 3D模型。
利用子结构建立ChemBio 3D模型可以更加简洁高效地建立模型,提高工作效率。
chemical3D教程

Chapter 1 Introduction1.1启动Chem3D在开始(Start)菜单中点击所有程序(P), 选择弹出菜单中ChemOffice2004中的Chem3D Ultra 8.0即可启动程序1.2Chem3D 界面结构Chem3D软件的最大特点就是界面结构简单。
和ChemDraw类似,Chem3D 打开后的窗口包含:工作窗口, 信息窗口, 列表窗口, 工具栏, 菜单栏等, 如图1所示。
1.3图形工具栏图形工具栏包含所有能够在工作窗口绘制结构图形的工具,选择这些工具后,光标将随之改变成相应的工具形状。
工具板见图1-2其它的类似于ChemDraw,有兴趣的同学可以参考FTP所提供的英文参考资料Chapter 2 建模简要教程2.1 使用键工具建模首先打开工作窗口(File菜单– New Model)2.1.1 建模在工具栏选择单键工具将鼠标移动至工作窗口,按住鼠标左键拖动鼠标即可绘制简单的乙烷分子。
(注意:View-Settings-Model Build – Rectify 选择上后会自动为所绘制的分子结构加上氢原子)2.1.2 旋转模型:选择旋转工具可以在任意方向选择所绘制的分子。
1:将鼠标移动至工作窗口,按住鼠标左键2:在任意方向拖动鼠标可以旋转模型注意:当拖动鼠标时,会出现一个圆。
在圆内拖动鼠标使得模型绕X和Y轴旋转。
当鼠标在圆外时,模型绕Z轴旋转。
如图所示2.1.3 查看模型分子信息选择选择工具,将鼠标移动至相应原子位置将显示相应的原子序数,元素标识及原子类型。
如下图所示将鼠标移至C-C键上将显示键长及相应的键级在选择原子的同时,按住Shift键可以同时选择多原子,可以通过这样查看相应的键角和二面角。
1:选择C(1),C(2)和H(7)2:将鼠标移至任意的所选择的原子或键上将显示所选择的键角类似的操作可以用来显示二面角。
2.2 修改模型分子2.2.1 修改键类型1:选择双键工具2:按住鼠标左键,从C(1)的位置拖动鼠标至C(2)的位置将乙烷分子更改为乙烯分子。
ChemBioOffice教程的学习

计算化学实验Chem3D软件指南Chem3D开始:点击桌面“Chem3D Ultra 10.0”图标Chem3D一览菜单和工具栏区组成区坐标区光谱区输出结果区构象区2D分子区3D绘图区Chem3D详解菜单和工具栏区文件打开输入打印设置编辑拷贝粘贴修改选择显示工具结构测量旋转饱和修正重叠计算界面结果密度面轨道电势面分子面电影旋转轨迹数据库窗口排列帮助说明例子Chem3D详解文件新分子模型样本分子打开文件中的分子结构保存Chem3D详解编辑撤消操作重复操作清除当前分子Chem3D详解视图分子显示方式显示取向工具栏分子组成ChemDraw直角坐标Z-矩阵测量列表参数表输出区构象区光谱显示区文件输入原子类型MM2类型Chem3D详解结构测量列表分子位置和取向设置Z-matrix饱和价键(加氢)自动调整分子结构重叠分子对接Chem3D详解计算模块计算结果管理停止计算二面角计算(构象搜索)EHMO计算电荷轨道MM2分子动力学模块分子性质计算模块GAMESS计算界面Gaussian计算界面Jaguar计算界面Mopac计算界面Chem3D详解计算模块二面角计算(构象搜索)扫描一个二面角扫描两个二面角二面角计算(构象搜索) 扫描一个二面角二面角计算(构象搜索) 扫描二个二面角构象过渡态Chem3D详解计算模块EHMO电荷分子面总电子密度面分子轨道Chem3D详解计算模块EHMO电荷Chem3D详解计算模块EHMO总电子密度面等值面图isoval=0.01Chem3D详解计算模块EHMO分子轨道等值面图isoval=0.01MM2分子动力学模块Chem3D详解计算模块运行MM2输入文件优化构型(能量极小化)分子动力学性质计算打印力场参数MM2分子动力学模块Chem3D详解计算模块力场参数M2 Constant Value QualityCubic stretch constant-2.000 4Quarticstretch constant2.333 4X-B,C,N,O-Y Stretch-Bend interaction force constant0.120 4 X-B,C,N,O-H Stretch-Bend interaction force constant0.090 4 Sexticbending constant (* 10**8)7.000 4Cutoff distance for charge/charge interactions35.000 4Cutoff distance for charge/dipole interactions25.000 4Cutoff distance for dipole/dipole interactions18.000 4Cutoff distance for vander Waalsinteractions10.000 4MM2 Atom RadiusEps WeightReduct Lone Pairs Quality 5 1.500 0.047 1.008 0.915 0 4 2 1.940 0.044 12.000 0.000 0 4 Bond KS Bond Length Dipole Quality2-5 4.600 1.100 0.000 42-2 9.600 1.337 0.000 4Angle KB XR2 XRH XH2 Quality 2-2-5 0.360 120.0 120.5 0.0 42-2-2 0.430 120.0 0.0 0.0 4Atoms Force Constant Quality2-2 0.050 42-5 0.050 4Torsional V1 V2 V3 Quality5-2-2-5 0.000 15.000 0.000 42-2-2-5 0.000 9.000 -1.060 42-2-2-2 -0.930 8.000 0.000 4PiAtom Electron Ionization Repulsion Quality 2 1 -11.160 11.134 4PiBond DForce DLength Quality2-2 4.600 0.166 43, 4次键伸缩常数键伸缩-弯曲作用参数截断距离LJ非键参数键伸缩参数键弯曲(角)参数键扭转参数π键(共轭)修正参数MM2分子动力学模块Chem3D详解计算模块优化构型(能量极小化)收敛要求: 最小均方根梯度(力)输出结果能量分解总能量MM2分子动力学模块Chem3D详解计算模块分子动力学模拟步长结构输出间隔轨迹步数升温速率目标温度参数一览MM2分子动力学模块Chem3D详解计算模块分子动力学模拟暂停/开始停止MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果时间(10-15秒)总能势能温度MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果分子平动能= 0分子转动能= 0非平衡平衡态MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果总能守恒!温度控制!MM2分子动力学模块Chem3D详解计算模块π键键级能量Chem3D详解计算模块分子性质计算模块Chem3D详解计算模块GAMESS优化构型优化过渡态分子性质频率分析红外光谱NMR光谱创建输入文件查看输出结果文件运行输入文件Chem3D详解计算模块GAMESS作业类型方法基组波函数类型基组极化函数基组扩散函数基组指数优化方法限制性部分优化坐标自旋多重度= 单电子数+1净电荷Chem3D详解计算模块GAMESS高级设置-1最大SCF循环次数优化最大步数收敛要求(力)温度(仅用于热力学计算)溶剂模型溶剂参数初始猜测方法分子对称性点群内存要求Chem3D详解计算模块GAMESS性质偶极电子密度静电势Lowdin电荷和布居分析Mulliken电荷和布居分析电子势能电子总能量电子动能Chem3D详解计算模块GAMESS计算方法激发态从头算理论密度泛函理论基态Chem3D详解计算模块GAMESS等电子密度面(0.001)HF/STO-3G IR光谱苯甲醇NMR光谱Mopac计算界面Chem3D详解计算模块优化平衡构型优化过渡态分子性质IR光谱Mopac计算界面Chem3D详解计算模块作业类型方法波函数优化方法溶剂模型限制性优化收敛要求坐标Mopac计算界面Chem3D详解计算模块电荷偶极静电势生成焓超精细耦合常数电离能电荷溶剂介电常数Chem3D详解分子面模块分子面探测半径显示模式颜色模式分辨率选择分子轨道等值面值溶剂面总电子密度面总自旋密度面静电势面分子轨道清除所有面Chem3D详解分子面模块SA面总电子密度面isoval=0.01总电子密度面/静电势isoval=0.01静电势isoval=1Connolly面HOMO轨道LUMO轨道甲醇Chem3D详解工具栏新分子打开保存拷贝剪切粘贴恢复重复印模型背景特效红蓝镜像坐标原子符号原子编号残基全屏电影播放选择平移旋转缩放移动选中键型清除饱和调整符输入擦写分子面探测半径显示配色分辨率分子轨道等值状态优化旋转停止Chem3D详解文件格式晶体结构文件Gaussian输入输出文件cif文件pdb文件(蛋白质)Chem3D详解晶体结构文件有机分子晶体: CCDC数据库, 草酸晶体Pbca空间群(61)OXALAC06.datChem3D详解晶体结构文件无机晶体: ICSD, AMCSDAmerican Mineralogist Crystal Structural Database http://rruff.geo.arizona. edu/AMS/amcsd.php不能正确处理周期性的体系!!Chem3D详解生物大分子pdb文件(蛋白质)RSCB PDB蛋白质晶体结构数据库/pdb/home/home.do1AAQChem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件需要把Gamess输出文件修改一下才能用Jmol5.0正确读出!将这些含“REDUCED MASSES”的行全部删除! 然后保存退出此处只留一个空行!Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件旋转分子缩放平移选择测量轨迹/频率Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件打开修改后的GAMESS输出文件*.out打开“extras”→“vibrate ...”Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件选择频率观察“绕CO键的内转动”。
如何使用ChemBioOffice得到物质光谱图
如何使用ChemBioOffice得到物质光谱图
众所周知,ChemBioOffice是化学生物专业重要的软件之一。
其用法简便易学,只要勤于探索,很快就能掌握其基本操作方法。
ChemBioOffice中常用最重要的两个软件是ChemBioDraw和ChemBio3D。
前者主要用于画结构式,书写反应方程式等,后者主要用于观测物质的空间构型。
除此之外,ChemBioOffice的强大计算功能让他们还可以画出物质的光谱图,如红外(IR),紫外(UV),核磁共振(NMR,1HNMR、13CNMR)。
以一下物质为例:
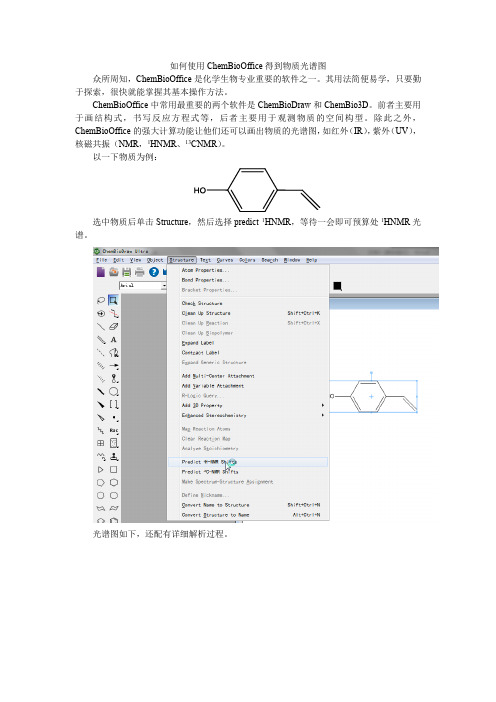
选中物质后单击Structure,然后选择predict 1HNMR,等待一会即可预算处1HNMR光谱。
光谱图如下,还配有详细解析过程。
如果想要得到UV和IR光谱,则需要进入ChemBio3D界面。
新打开一个ChemBio3D,将结构图画在空白处
进入后点击Calculation—MM2---Minimize Energy,计算最低能量的结构。
点击Run,后计算得到最小能量的结构。
(此步骤可以省略,即可以直接得到光谱图)
然后再点击Calculation---GAMESS Interface----Predict IR/Raman Specturm,预测红外光谱,同理可以预测紫外(UV)光谱,也可以预测核磁共振谱图。
红外光谱图
拉曼光谱图
紫外光谱图
(从ChemBioDraw中转化得到的ChemBio3D图可能不能正常计算出光谱图,所以建议直接把图形画在新建的ChemBio3D页面中)。
ChembioOffice及Chembiodraw关系及简介

ChembioOffice及Chembiodraw关系及简介:1、ChembioOffice 是由CambridgeSoft(剑桥软件)开发的综合性科学应用软件。
包2、(ChembioOffice的组成主要有ChembioDraw 化学结构绘图,Chem3D 分子模型及仿真,ChemFinder化学信息搜寻整合系统等部分组成。
)chembioOffice的组成主要有ChembioDraw 化学结构绘图,Chem3D 分子模型及仿真,ChemFinder 化学信息搜寻整合系统,此外还加入了 E-Notebook Ultra ,BioAssay Pro ,量化软件 MOPAC、Gaussian 和 GAMESS 的界面,ChemSAR, Server Excel, CLogP, CombiChem/Excel等。
3、作用或说用途;该软件包是为广大从事化学、生物研究领域的科研人员个人使用而设计开发的产品。
同时,这个产品又可以共享解决方案,给研究机构的所有科技工作者带来效益。
利用ChemBioOffice ,你可以方便的进行化学生物结构绘图、分子模型及仿真、可以将化合物名称直接转为结构图,省去绘图的麻烦;也可以对已知结构的化合物命名,给出正确的化合物名。
目前最新的版本为ChembioOffice 2014。
科学家用 E-Notebook 整理化学信息、文件和数据,并从中取得他们所要的结果。
ChemNMR 可预示分子化学结构的 13C 和 1H NMR 位移。
ChemFinder/Word 通过你的计算机或互联网,可以在 Word, Excel, Powerpoint, ChemDraw, ISIS 等文件中搜索化学结构,以便浏览或修改,并输出到自己的目标文件中。
ChemOffice 支持每一位科学家的日常工作,企业方案制定,建立在ChemOffice 服务器的数据库,有助于各个研究部门的合作,并共享信息。
ChemBioOffice 13.0 十大新功能

ChemBioO ce 13十大新功能全新发布的 ChemBioOf ce ® 13,增加了更多人性化的功能,进一步提高了科学家的工作效率。
新版本通过结合可视化分析,能将生物活性与化学结构关联,协助科学家更深入地了解研究结果。
1. 升级的生物聚合物绘制工具栏,包括 D-型及 L-型氨基酸、β-氨基酸、DNA 及 RNA 模板全新的生物聚合物工具栏可以绘制肽链、DNA 及 RNA 的序列,包括 β-氨基酸及 D-氨基酸、二硫键、内酰胺以及连接基团和保护基团。
用单字母或三字母方式代表多肽及核苷酸序列(DNA 、RNA ),包括 L-氨基酸、D-氨基酸及 β-氨基酸。
用户能方便地在单字母及三字母表示方法之间互换,并可以将缩写字母展开为结构。
可以将连接基团及保护基团预先自定义为字母,简化绘制过程。
使用“键工具”绘制二硫键及内酰胺,以及环肽结构。
2. 在一个氨基酸序列中绘制混合序列结构、连接基团及保护基团为含有氨基或羟基的氨基酸(如 Arg ,Aad )添加保护基团,例如 Trt 及 Fm oc 。
这些保护基团的结构也可以展开或者恢复成缩写形式。
可通过键合序列的末端将多个序列合并为一个序列。
3. 从外部文档中粘贴化学序列支持 FASTA 格式粘贴序列。
将代表聚合物的单字母或者三字母字符串进行粘贴,系统可通过有效的分隔符(space 、tab 、dash 键)自动识别。
4. 升级的 clean -up 功能,可用于分子、反应及生物聚合物的结构修正使用升级的 clean-up 功能修正高质量且结构正确的分子式、反应式及生物聚合物。
序列绘制中新添加的残基会自动调整。
当序列从结构式转变到以字母形式表示,Clean-Up Biopolym er 命令可以调整并重新排布序列,使残基在一条直线上并自动换行。
Richard J. Smith 博士,副主编,Verlag Helvetica Chimica ActaJeremiah J. Gassensmith 博士,美国西北大学ChemBioO ce 长期用户“新的生物聚合物工具栏在绘制多肽及DNA 序列上帮了一个大忙。
chembiodraw使用技巧

ChemBioOffice 2010
ChemDraw界面
菜单栏
通用工具栏 文本工具栏
绘图工具栏
选择工具
透视
化学键
DNA 长链
常用环系
碎片工具 橡皮擦 文本 钢笔 箭头 轨道 绘图 括号 化学符号 查询工具 高级工具 模板
绘图工具栏
基本绘图工具
• 快捷键 • 化学结构的缩写
选择原子, 同时按热键
“选择”指将 鼠标移动到 原子上,或 者左键单击 选择原子
原子热键
原子热键
大小写代表不同元素或官能团
元素符号 A F Ph Ac H Q* Br I R
n-Bu
热键 a f
P or 4 A or 5
h q b i r 1
元素符号 K S
s-Bu Me Si t-Bu N TMS C Na
常见问题
如何改变化学键叠放次序?
常见问题
如何输入化学式中间的分隔点
1)打开文本框 2)输入 Alt + 0183
(小键盘) 3)设置为粗体
(同样适用Word)
常见问题
得出化学结构的命名 从命名得出化学结构
(5R,6R)-5-ethyl-6-((2R,3S,4R,5S,E)-2hydroxy-4-methoxy-3,5-dimethylnon-7-en-
基本绘图工具
绘图 括号 化学符号 查询工具 高级工具 模板
基本绘图工具
常见问题
结构式太大,页面画不下!如何调整页面大小?
常见问题
更改页面 长度和宽 度的页数
常见问题
更改页面长 度和宽度
如何输入字符?
常见问题
如何输入字符?
常见问题
ChembioOffice-软件的操作与使用

2021/3/7
CHENLI
22
4. 立体化学
下图中的四个结构 式是同一种物质
➢第二个图形是使用Object ➢第三个图形是第二个图
菜单的ShowStereochemistry
形水平反转的结构
命令建立的
➢第四个图形是第三 个图形垂直旋转180度 的结果
➢为了出现Rotate180 Vertical,这个菜单项
2021/3/7
CHENLI
4
2.1 简介
美国剑桥(CambridgeSoft)公司() 其最新版本ChemOffice Ultra 2010是目前世界上最优秀、最 重要和最权威的桌面化学办公应用软件,集强大的应用功能 于一身,为化学工作者提供了优秀的化学辅助系统。
单击RUN按钮开始对模型进行 优化,最终给出能量最低状态。
➢它既可以轻松的绘出分子结构图和化学反应方程式、实验 室仪器装置和化学工艺流程图等,并应用到字处理软件中; ➢而且可以将化合物名称直接转为结构图,省却绘图的麻烦; ➢也可以对已知结构的化合物命名,给出正确的化合 物名称。
2021/3/7
CHENLI
5
2021/3/7
CHENLI
6
2021/3/7
次为菜单栏、工具栏和文档区以及状态栏,工具栏提供 了很多常用的化学分子及化学键。 ❖ 使用时只用鼠标点一下工具栏中所需工具,移动鼠标到 编辑区后,鼠标指针变为“+”,再在文档区的合适位置点击 鼠标,则分子结构式就会出现,使用十分方便 ❖ 支持Windows剪贴板,所有结构式可以方便地剪贴到 Word文档中,而当要修改时,只需在Word文档中双击结 构式,即可打开ChemDraw进行修改。
O
利用模板按左图绘制出简式;
ChemBio3D

药物化学Structure Browser:对比化学结构和化学性质作者: Susan M. LeBeau 博士通过对比一组小分子的结构和性质来分析小分子组的功能,对工作在药物开发领域的化学家来说非常有用。
为了让化学家更容易的研究成组小分子的结构和它们的性质,CambridgeSoft 在最新版的ChemBio3D 11.0中新增了药物化学Structure Browser,并引入分子性质的概念。
Structure Browser是一个与Model Explorer很类似的控制列表,但不像Model Explorer那样按等级分层显示。
这个列表能把需要进行分析和对比的分子一一列出,使用非常方便。
同时,最新版的ChemBio3D 11.0可以把ChemBio3D模型中的分子性质和分子结构相互关联起来。
通过Structure Browser生成的列表,浏览查看这些待检查的结构很方便。
这项功能可以通过点击View菜单中的Structure Browser项激活,也可以直接将Model Explorer中列示的分子拖放到Structure Browser里进行激活。
分子性质可以从SD文件或Schrodinger Maestro文件中的数据导入。
另外一种方法是,在ChemBio3D模型上点击右键,选择“Auto Populate”项,Structure Browser就会自动填充当前模型的全部分子。
要使用Structure Browser,用户首先需要点击列表中的某个结构,然后用上下键在列表中的结构间进行选择。
被选定的结构会一直以3D方式显示,等待对比。
同时,如果设定结构为可视的话,它的2D结构会显示在ChemDraw面板中。
结构左边的检验栏标示当前的显示状态。
检查框里是黑色对勾的情况,说明这个结构处于“locked”状态,即使在Structure Browser选中的是另外的结构,这个结构仍旧是可见状态。
Chem3D常用功能使用教程
建立3D模型 1. 利用键工具建立模型
选择【单键】工具,在模型窗口向右按动
圆柱键模型
线状模型
棒状模型
球棍模型
比例模型
X、Y轴旋转指示
选择【轨迹球】工具,使分子模型 在模型窗口沿X、Y轴旋转
Z轴旋转指示
选择【轨迹球】工具并按“Alt”键, 使分子模型在模型窗口沿Z轴旋转
光标位于原子上, 自动显示原子信息
模型结构信息
工具栏显示原子的符号和标号
光标位于键上, 自动显示键信息
模型结构信息
模型的进一步信息
显示键长变化 显示键角变化
显示二面角
显示所有没有 相邻的原子的
距离
模型的键长数据
模型的键长和键角数据
乙烯模型中的双键信息
键级改动
显示氢及孤对电子
不显示氢及孤对电子的环己烷
原子名称及序数
选择工具,双击原子, 按顺序改变原子序号
ChemDraw中 画出的分子结构
复制粘贴到Chem3D中
Chem3D中 画出的分子模型
复制粘贴到 ChemDraw中
计算功能演示
(1)查找原子的范德瓦尔斯半径 • 【View】【Atom Types.TBL】窗口,
找到各种元素在不同环境的半径
(2)观察分子的大小
• 建立苯模型。执行【View】【Connolly Molecular】弹出Suface对话框;【solid】 选项可以选择分子表面的类型; 【Resolution】滑动到右边,其值为100
(4) 检查结构能量
模型旋转 对位交叉
总能量计算
计算结果
空间位阻能:0.977kcal/mol
模型的二面角数据
数据与模型对应
生物化学第三前半-PPT文档资料

酶的催化活性可调节控制
常需要辅助因子
二 酶的化学本质和组成
1、 酶的化学本质---蛋白质或核酸
1926年,James Summer由刀豆中提取出脲酶得到 结晶,证明是蛋白质。 此后,J.Northop等又连续获得胃蛋白酶、胰蛋白 酶和胰凝乳蛋白酶结晶,并证明是蛋白质。 1983年Thomas R.Cech 发现某些RNA分子具有 催化活性,对有催化活性的RNA,命名为核酶 (Ribozyme)
COOH COOH
(CH2)2
H—C—NH2 +
CH3
C O 谷丙转氨酶(CH2源自2H—CCH3COOH
谷氨酸
COOH
丙酮酸
O + NH2 C H— COOH COOH
丙氨酸
酮戊二酸
A· X
+B
A +B· X
(3)
水解酶 hydrolase
• 水解酶催化底物的加水分解反应。 • 主要包括淀粉酶、蛋白酶、核酸酶及脂酶等。 例如,脂肪酶(Lipase)催化的脂的水解反应:
第1亚亚类,H受体为NAD+
该酶在亚亚类中的流水编号
根据反应性质将酶分为6大类
(1) 氧化还原酶类
(2) (3) (4) (5) (6) 转移酶类 水解酶类 裂合酶类 异构酶类 连接酶类
(1) 氧化-还原酶 Oxidoreductase
• 氧化-还原酶催化氧化-还原反应。 主要包括氧化酶(Oxidase)和脱氢酶 (dehydrogenase) 如,乳酸(-羟基丙酸,Lactate)脱氢酶催化乳酸的脱 氢反应。
三 酶的命名和分类
1、习惯命名法 2、系统命名法
3 国际系统分类法和酶的编号
1、习惯命名法-1961年前用的都是此法
Chem3D讲义

这种模型结构一般用于生物大分子
精选课件
26
三、Chem3D的使用
2、3D模型的种类
(7)Cartoons
操作: [View] —>[Model Display] —>[Display Mode] —>[Cartoons]
这种模型结构一精选般课件用于生物大分子
27
精选课件
28
三、Chem3D的使用
Chemoffice讲义 ——Chem3D
化学与环境工程学院 化学工程
李皓宇、杨启泽、官懿、李卓华、李宏程、肖俊宇、韩振、 王泓蛟
2017.09.25
精选课件
1
1
ChemDraw复习
2
Chem3D介绍
3
Chem3D的使用
4
作业
精选课件
2
一、ChemDraw复习
用ChemDraw画出
l-麻黄碱(1R,2S)
• 蓝色是其中的【Other # 5】号颜色。如果用户对这8种颜色不满 意,可以单击最下面的【Other】选项,弹出【颜色】调色板,其 中有更多的颜色选项,并可以自定义颜色。
精选课件
11
快捷菜单和快捷键
• 1)快捷菜单
• 在选中的结构上单击鼠标右键,会弹出快捷菜单,如图所示。 ChemDraw快捷菜单包含了多种选项,使用快捷菜单能完成常用编 辑、属性设置、模板选择等功能。
3、3D模型的建立方法
(1)利用键工具建立模型
1)单击工具栏上的“单键”按钮
2)将椎鼠标移动至模型窗口,按住鼠标左键拖出 一条直线,放开鼠标即成乙烷(C2H6)立体模型。 3)将鼠标移至C(1)原子上,向外拖出一条直线, 放开鼠标即成丙烷(C3H8)立体模型。 4)将鼠标移至C(2)原子上,向外拖出一条直线, 放开鼠标即成丁烷(C4H10)立体模型,如图所示。
Chem3D常用功能使用教程.ppt
扩大(缩小)窗口
模型数据窗口
状态栏
消息窗口
4
选择 轨迹球 大小调整
单键 双键 三键 虚键 文本 橡皮
5
显示设置
6
视频设置 模型显示设置
结构显示
立体显示
弹出信息 配体显示
原子标记
原子显示
色彩选项
7
Model Type
显示设置属性
线状模型 棒状模型
球棍模型 圆柱键模型
比例模型
8
带状模型
9
结构不同显示方式
Molecular】弹出Suface对话框;【solid】 选项可以选择分子表面的类型; 【Resolution】滑动到右边,其值为100
68
69
70
71
72
View/Solvent Accessible 菜单 甲苯结构
73
(3)计算分子体积 • 建立苯模型。执行【Analyze】
【Compute Properties】在【Available Properties】选框中双击【Connolly Solvent-Excluded Volume SVEChemPropStd】选项,使之加入到 【Selected Properties】框中。单击ok,
88
显示轨道时 必须选择
89
选择分子轨道
90
能量随所选轨 道不同而不同
选择HOMO 轨道
轨道选项 HOMO和 LUMO各6个
实心显示 (丝网显示 圆点显示 半透明显示)
HOMO轨道 显示
91
乙烯分子的HOMO轨道
92
选择LUMO 轨道
LUMO轨道 显示
93
乙烯分子的LUMO轨道
生物化学3第三章 维生素与辅酶zjg ppt课件

通常将β-胡萝卜素称为维生素A原。
2021/2/23
ZhangJG-Biochemistry
32
2、功能
(1)维持视觉 维生素A可促进视觉细胞内感光色素的形成。 维生素A可调试眼睛适应外界光线的强弱的能力, 以降低夜盲症和视力减退的发生,维持正常的视觉 反应,有助于对多种眼疾(如眼球干燥与结膜炎等) 的治疗。 (2)促进生长发育 视黄醇也具有相当于类固醇激素的作用,可促进 糖蛋白的合成。促进生长、发育,强壮骨骼,维护 头发、牙齿和牙床的健康。
ZhangJG-Biochemistry
14
三、泛酸和辅酶A(CoA)
1. 结构——特点是含有β-丙氨酸
▪
泛酸又叫遍多酸
▪ 2. 功能
▪
辅酶A——酰基载体
▪
是生物体内代谢反应中乙酰化酶的
辅酶。参与糖、脂类、蛋白质代谢。
2021/2/23
ZhangJG-Biochemistry
15
2021/2/23
生物化学3第三章 维生素与辅酶zjg ppt课件
第三章 维生素与辅酶(2学时)
▪ 第一节 概述 ▪ 第二节 水溶性维生素 ▪ 第三节 脂溶性维生素
2021/2/23
ZhangJG-Biochemistry
2
第一节 维生素概述
▪ 基本营养要素 ▪ 六大要素:糖类、蛋白质、脂类、维生素、水、
无机盐类与微量元素
(1)用拉丁字母A、B、C、D……来命名,但这些字母不 表示发现该种维生素的历史次序(维生素A除外),也不说 明相邻维生素之间存在什么关系。
(2)根据化学结构 和生理功能命名, 如:
从上可知,V命名混乱,1967、1970年国际理论与应 用化学学会、国际营养科学学会先后提出V命名原则的建 议,但一直没被广泛应用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实验三 — 能量计算和模拟 步骤2: 分子间相互作用能量曲线 计算两个甲烷分子间的弱 相互作用
Gamess设置
实验三 — 能量计算和模拟 步骤2: 分子间相互作用能量曲线 两个甲烷分子间的弱 相互作用能数据 R/A ΔE / cm-1 图示
实验三 — 能量计算和模拟 步骤2: 分子间相互作用能量曲线 分子间的弱相互作用能一般用L-J(12-6)函数拟合
“过渡态”
实验三 — 能量计算和模拟
如果你已经阅读到这儿, 恭喜你 已经离一个计算化学研究者很近 了!
6 ⎡⎛ σ ⎞12 ⎛σ ⎞ ⎤ V ( R ) = ε ⎢⎜ ⎟ − 2 ⎜ ⎟ ⎥ ⎝R⎠ ⎥ ⎢⎝ R ⎠ ⎣ ⎦
势阱
平衡间距
实验三 — 能量计算和模拟 步骤2: 分子间相互作用能量曲线 两个甲烷分子间的弱相互作用能数据用L-J(12-6)函数拟合 结论: 两个CH4分子可以形 成弱复合物, 结合能 为73 cm-1, 质心平衡 间距约为4.3 A.
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
• 用Gamess程序 • 用HF, MP2, B3LYP三种方法 • 用6-31G(p)基组 • 计算单重态和三重态两条线 • 用Morse势拟合单重态能量曲线
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 画出H2分子 选中HH键 点右键, 选择 H2 → H(2S) + H(2S)
实验三 — 能量计算和模拟 步骤4: H—H — H共线势能面的创建
实验三 — 能量计算和模拟 步骤4: H—H — H共线势能面的创建 势能数据 R1 R2 E R1 R2 E
实验三 — 能量计算和模拟 步骤4: H—H — H共线势能面的创建 过渡态
3D势能面
投影图
实验三 — 能量计算和模拟 步骤4: H—H — H共线势能面的创建 问题3-6. 仿照H-H-H例子, 绘制H—H—F共线结构 的势能面图 (Gamess, MP2/DZV). H-H: 0.6 ~ 1.2 H-F: 0.8 ~ 1.6
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 HH间距 — 能量数据 H2 → H(2S) + H(2S)
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 类似的方法, 计算三重态能量 H2 → H(2S) + H(2S)
只用MP2方法计算
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 类似的方法, 计算三重态能量 H2 → H(2S) + H(2S)
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
通过点击改变HH距离 (从0.4到2.0, 间隔0.1 A), 扫描获得H2解离的势能曲线
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
平衡解离能 势参数 平衡键长
实验三 — 能量计算和模拟 H2 → H(2S) + H(2S) 步骤1: 分子的解离能量曲线 用Origin软件拟合
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
键能 = 104 kcal/mol 键长 = 0.695 A
排斥态
吸引态
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 一般采用相对能量ΔE = E(H2) - 2E(H) 计算H原子的能量 HF: -312.6456 Kcal/Mol MP2: -312.6456 Kcal/Mol B3LYP: -311.7854 Kcal/Mol 问题3-1. 为什么氢原子的HF 和MP2的能量一样, 而H2的却 不同? H2 → H(2S) + H(2S)
பைடு நூலகம்
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
以此类推, 改变HH距离为0.5, 计算能量
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 获得一系列能量数据 H2 → H(2S) + H(2S)
换成MP2和 DFT-B3LYP 方法, 分别做 同样的计算
实验三 — 能量计算和模拟 步骤3: 分子构象搜索 乙醇构象
(b) 选中二面角的旋转 轴, 扫描这个二面角
实验三 — 能量计算和模拟 步骤3: 分子构象搜索 MM2力场扫描 乙醇构象
实验三 — 能量计算和模拟 步骤3: 分子构象搜索 扫描两个二面角 乙醇构象 构象I 构象II
过渡态
实验三 — 能量计算和模拟 步骤3: 分子构象搜索 成环的分子不能扫描!
与实验值符合很好
问题3-2. 为什么H2的解离曲线不趋向于零? 试分析可 能的原因. 问题3-3. 仿照H2解离的例子, 做出Cl2分子解离的势能曲线, 并 用Morse函数拟合, 确定键能和键长 [Gamess, HF/6-31G(d)].
实验三 — 能量计算和模拟 步骤2: 分子间相互作用能量曲线 计算两个甲烷分子间的弱相互作用 画两个C原子, 设置间距2A, 饱和价键, 点MM2优化, 得 如下图所示的结构. 表明两个甲烷分子可以形成一弱复 合物, 间距3.6A, 相互作用能约0.9 kcal/mol (~ 4 kJ/mol)
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
实验三 — 能量计算和模拟 步骤1: 分子的解离能量曲线 H2 → H(2S) + H(2S)
一般双原子分子的解离势能曲线可以用 Morse函数较好地拟合
V ( R) = De ⎡e 2 β ( R − Re ) − e β ( R − Re ) ⎤ ⎣ ⎦
实验三 — 能量计算和模拟
要求: 1. 掌握计算分子能量的不同方法 2. 掌握分子相互作用能的分析方法 3. 掌握分子构象的分析方法 4. 了解势能面的作用
实验三 — 能量计算和模拟
内容: 1. 计算分子的解离能量曲线 2. 计算分子间弱相互作用能量曲线 3. 分子构象搜索 4. H—H — H共线势能面的创建
实验三 — 能量计算和模拟 步骤2: 分子间相互作用能量曲线 问题3-4. 仿照CH4...CH4的例子, 做出两个苯环平行放置时的相 互作用能曲线, 并用LJ(12-6)函数拟合, 确定势阱和平衡间距 [Gamess, HF/sto-3g].
实验三 — 能量计算和模拟 步骤3: 分子构象搜索 利用扫描一个或两个二面角, 寻找分子的不同构象. 乙醇构象 (a) 选择一个二面角
问题3-5. 计算乙烷的构象, 并确 定其内转动的势垒高度.
实验三 — 能量计算和模拟 步骤4: H—H — H共线势能面的创建 采用两个坐标, 即两个H-H距离, 计算相应的能量, 绘制成 三维图, 即为势能面. 可以直接看出鞍点(过渡态)和谷点 (稳定结构). (a) 画出三个共线的H原子, 沿X轴放置 (b) 改变两个HH的距离{0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2}, 计算共 7 × 7 = 49 个能量. (c) 采用Gamess, MP2/DZV计算, 具体设置见下页
实验三 — 能量计算和模拟 步骤2: 分子间相互作用能量曲线 计算两个甲烷分子间的弱相互作用 (a) 从MM2优化的结构出发, 设置C...C间距离依次为2.0, 2.5, 3.0, 3.5, 4.0, 4.25, 4.5, 4.75, 5.0, 5.25, 5.5, 5.75, 6.0, 7.0, 8.0, 10.0, 15.0, 用Gamess计算MP2/3-21+G(d)的能量. (b) 相互作用能ΔE = E(CH4...CH4) - E(15.0 A)