质粒转染protocol
转染protocol

转染protocol
注意:整个过程要防止293T细胞漂,所以动作一定要轻缓,不要急。
一、细胞准备
转染前12-18小时,铺细胞。
293T 细胞,介于1:2~~1:3传。
二、转染前换液
吸去细胞上层培养基,轻轻地加入PBS(14.5cm皿需要至少5ml培养基),缓慢地晃动培养皿,然后吸去PBS。
轻轻地加入10ml Opti-MEM。
然后放回细胞培养箱。
三、PEI稀释,DNA与PEI混匀
抗体重链与轻链质粒比例按照1:2.5混匀;质粒与PEI按照1:1混匀。
(质量比,PEI 为1ug/ul);每14.5cm皿可以转染40ug质粒。
每皿我们需要1-2mlPEI&DNA混合液(Opti-MEM稀释)。
首先计算好抗体重链、轻链所需质粒,PEI需要量以及二者混合后最终体积。
然后,取一半体积的opti-MEM于15ml离心管中,加入PEI,Vortex,净置5分钟。
取另一半体积的opti-MEM于15ml离心管中,加入DNA,Vortex。
将PEI稀释液逐滴加入DNA稀释液中,混匀。
静置20分钟。
(尽量避光)
四、轻轻地将PEI&DNA混合液加入细胞培养皿中(1-2ml/皿)。
缓慢晃动培养皿,混匀。
然后放回细胞培养箱。
五、一小时后,吸去Opti-MEM,每皿加入20ml 293freestyle培养基。
(动作轻缓)
六、4天后,收上清。
质粒转化、细胞转染步骤

转化:将质粒DNA或以其为载体构建的重组DNA导入细菌体内使之获得新的遗传特性。
转染(transfection):将含有目的gene的载体转到真核cell内
转导:指以噬菌体为载体,在细菌之间转移DNA的过程。
有时也指在真核cell之间通过逆转录病毒转移和获得cellDNA的过程。
1.质粒转染(transfection)
6-well-plate 每well 60-70%时transfection
接种时×105个/ml/well
质粒DNA 转染液优化培养基opti-MEM
步骤:质粒DNA + 200ul opti-MEM + 6ul转染液混匀室温静置15min 待转染cell换优化培养基,切记要轻轻加培养基,勿冲起cell 把transfection complex(200ul)轻轻均匀滴入cell培养基中,十字形摇晃plate,混匀
37℃,CO2培养箱培养
2.
转化:外源DNA(质粒作为载体)导入受体细胞(大肠杆菌感受态cell)步骤:
50ul感受态cell(一管)加入12ul质粒DNA→轻柔混合
↓
冰浴30min
↓
42℃热激90s
↓
迅速冰浴2min
↓
向管中加入培养基,混匀后37℃震荡培养(﹤225rpm)1h,使细菌恢复正常生长状态
↓
涂板正培1h后,倒培过夜。
IP Protocol

IPProtocol1.铺板:按一定细胞数将细胞接种于培养皿内,24h后进行处理。
(转染质粒时,细胞长到90%以上较为合适。
一次IP,使用6孔板足够。
)2.转染:lipo2000转染质粒,质粒转染效果应该提前验证。
(IP转染质粒时,被拽的一个蛋白可以适当加大转染量;如IP FLAG,用FLAG拉HA,总转染体系500ng时,对照组转空载质粒500ng;FLAG组转200ngFLAG+300ng 空载;HA组转300ngHA+200ng空载;共转组300ngHA+200ngFLAG。
)3.细胞裂解:将培养皿置于冰面,弃培养液,预冷PBS洗一遍。
加入事先配置好预冷的lysis buffer +PMSF+cocktail+Na3VO4等(如果是蛋白修饰反应需额外加)(6孔板每孔可加入1ml), 放置4℃摇床摇10min,裂解细胞。
如皿底裂解不完全,可以用枪轻轻吹打几次。
(裂解后皿底可见白色的丝状物,为DNA等成分。
此步最好不要vortex。
)4.离心:4℃12000 rpm离心15min,上清即为裂解后的蛋白样品。
5. Beads准备:取出Beads(交联了IP抗体的Agarose Beads),使用前混合均匀。
另取一只15ml离心管,根据每个样品1ml的用量加入相应的lysis buffer,然后以每个样品10-20ulBeads的量将Beads加入lysis buffer中,充分重悬后,分装至1.5ml EP管内,4℃ 1000-2000rpm离心2min。
弃上清(不要完全吸干,上样枪头)。
6. 将步骤4中离心后上清转移到新的硅化EP管内。
①每个样品各取出80ul做为input。
②IP:每个样品各取800ul上清加入事先备好的Beads中,放置于4℃转子上旋转充分孵育2h。
(孵育的时间需要调整,一般外源过表达IP 2-4H足够;若互作为底物反应,在IP 前需要适当加入的MG132或其它相应的抑制剂。
质粒转染技术

质粒转染技术质粒转染技术是现代生命科学研究中常用的实验手段之一,广泛应用于基因工程、细胞生物学、分子生物学等领域。
本文将从质粒转染技术的原理、方法和应用等方面进行介绍。
一、质粒转染技术的原理质粒转染技术是指将外源质粒DNA导入到细胞内的一种方法。
质粒是一种环状DNA分子,具有自主复制和表达基因的能力。
质粒转染技术通过将质粒DNA导入到细胞内,使其在细胞内复制和表达,从而实现对目标基因的研究和应用。
1. 化学法转染:化学法转染是最常用的质粒转染方法之一。
其基本原理是利用化学物质(如聚合物、脂质体等)与质粒DNA结合,形成复合物后与细胞膜发生相互作用,使质粒DNA进入细胞内。
这种方法操作简单、成本低廉,适用于大多数细胞类型。
2. 电穿孔法转染:电穿孔法利用高压电脉冲的作用,瞬时破坏细胞膜,使质粒DNA能够进入细胞内。
这种方法适用于多种细胞类型,转染效率较高,但操作较为复杂。
3. 粒子轰击法转染:粒子轰击法利用高速粒子(如金属微粒)的撞击作用,将质粒DNA导入细胞内。
这种方法适用于质粒DNA较大、难以通过其他转染方法的细胞。
三、质粒转染技术的应用1. 基因表达研究:质粒转染技术可用于将外源基因导入细胞,使其在细胞内表达,从而研究基因的结构、功能和调控机制。
2. 基因敲除研究:质粒转染技术结合CRISPR/Cas9等基因编辑技术,可以实现对特定基因的敲除,从而研究该基因的功能。
3. 基因治疗:质粒转染技术可用于将治疗基因导入患者的细胞,修复或替代异常基因,实现基因治疗的目的。
4. 细胞药物递送:质粒转染技术可用于将药物敏感基因导入细胞,使其在细胞内表达,从而增强对药物的敏感性,提高治疗效果。
四、质粒转染技术的进展与挑战随着生命科学研究的不断发展,质粒转染技术也在不断改进和完善。
目前,研究人员正在努力提高转染效率、降低细胞毒性和改善转染的特异性等方面。
同时,也面临着一些挑战,如如何选择合适的转染方法、如何提高质粒的稳定性等问题。
质粒大抽protocol(用QIAGENMaxiColumn)

质粒大抽protocol(用QIAGEN Maxi Column)2003/4/28陈俊1. 从转染的细菌平板中挑取中等大小,饱满,而且没有和其他克隆接触的单克隆,接种到3ml选择性培养基,摇床培养14小时,225 rpm。
2. 从小量培养物中取1ml用作小量抽提,并且用小量抽提产物酶切鉴定,选取阳性克隆的培养物,以1:1000接种到100 ml选择性LB培养基中,摇床培养14小时,225 rpm。
3. 从大量培养物中取700微升菌液和300微升甘油(灭菌)加入冻存管,放入液氮中,过夜后,放入-70℃冰箱中。
并且在stocklist中加入菌种的详细信息(stocklist 贴在冻存管的上方)。
4. 把100ml的菌液倒进250ml离心管中,4℃离心,6000g离心15分钟(No41 rotor 8500rpm)。
5. 倒掉上清,并倒扣在吸水纸上片刻.然后加入4ml 4℃预冷的细胞重悬液buffer P1(确定是否加了RNase),反复吹打沉淀至没有悬浮的菌块。
然后将悬浮液转到50ml的离心管中。
6. 加入4ml细胞裂解液 buffer P2,彻底而且轻柔地颠倒混匀4-6次,在室温下孵育不要超过5分钟(从开始加P2时计时,时间太长会使质粒DNA断裂)。
样品不要斡旋,buffer P2用毕也要及时盖好盖子,以免被空气中的CO2酸化。
7. 加入4ml 预冷的PH调节液buffer P3,及时地而且轻柔(避免局部十二烷基磺酸钾沉淀)地颠倒混匀4-6次,在冰上孵育15分钟。
后再一次混匀样品.20000g,4℃离心30分钟(N046 rotor 18000 rpm).若上清不够清需再离心15min. 此步可对上清取样240ul (sample 1)以备检测分析8. 在层析柱的顶端加四层纱布,并且用buffer QBT润湿纱布。
用4ml的buffer QBT来平衡层析柱,让液体自由的流下(尽量将润湿面积减到最小)。
质粒转染技术

质粒转染技术1. 引言质粒转染技术是一种常用的实验手段,用于将外源DNA转移到细胞中。
通过质粒转染技术,研究人员可以研究基因功能、调控基因表达以及制作重组蛋白等。
本文将介绍质粒转染技术的原理、方法和应用。
2. 原理质粒转染技术的原理是通过电穿孔、离子共价结合、化学反应或病毒载体等方法,将外源质粒DNA引入细胞内。
一旦质粒DNA成功转移到细胞内,它可以在细胞中复制和表达。
质粒转染技术可以分为两种类型:转染和转化。
2.1 转染转染是指将质粒DNA引入细胞外的细胞中。
这种方法通常使用化学试剂或物理手段,如钙磷共沉淀、电穿孔、微粒轰击等。
这些方法可以破坏细胞膜,使质粒DNA能够进入细胞。
2.2 转化转化是指将质粒DNA引入细菌细胞中。
这种方法通常使用热冲击或电穿孔等技术,将质粒DNA引入细菌细胞内。
细菌细胞具有天然的转化能力,可以吸收外源DNA并将其整合到细菌染色体中。
3. 方法质粒转染技术的具体方法可以根据实验需求和细胞类型的不同而有所差异。
下面是一般的质粒转染方法的步骤:3.1 培养细胞首先,需要培养细胞以达到适当的细胞密度。
细胞密度的选择取决于实验的目的和细胞类型。
3.2 准备转染体系根据实验要求,制备适当的转染体系。
转染体系通常包括质粒DNA、转染试剂和培养基等。
3.3 转染将质粒DNA与转染试剂混合,并将混合物加入培养细胞中。
转染试剂可以帮助质粒DNA进入细胞。
3.4 培养细胞将转染后的细胞培养在适当的培养条件下,以促进质粒DNA在细胞中的复制和表达。
3.5 分析转染效率使用适当的检测方法,如荧光显微镜观察、PCR、Western blot等,来分析转染效率和质粒DNA在细胞中的表达情况。
4. 应用质粒转染技术在生命科学研究中有广泛的应用。
下面是一些常见的应用:4.1 基因功能研究通过转染外源质粒DNA到细胞中,可以研究基因的功能。
例如,可以将siRNA质粒转染到细胞中,来研究特定基因的沉默效果。
汉恒生物CRISPR protocol-2质粒构建和建系

---基于lentiCRISPR v2应用指南-载体构建和建系
汉恒生物提供CRISPR/Cas9载体的快速构建试剂盒,同时也提供各 种载体构建和病毒包装(慢病毒、腺病毒和AAV)、建系服务
说明
lentiCRISPR v2载体是张锋实验室发布一个慢 病毒载体,cas9和sgRNA表达框在同一各载 体上。而且Cas9的C端带有FLAG融合标签, 载体包含Puromycin抗性基因,适合建系。 验证sgRNA活性的时候也适合顺转;包装成 慢病毒适合常规细胞系的建系。构建非常 方便。
oligo就需要加磷处理!!!(红色框内是FengZhang提供的protocol,设计脱磷,可以参考) )
仅作参考
病毒包装体系
包装病毒使用的包装体系如下(for 100 mm dish, 6x106 293T)
4 μg 3 μg 1 μg lentiCRISPR v2-gDNA plasmid psPAX2 packaging plasmid pMD2.G envelope plasmid
单克隆的挑取---单克隆可以有限稀释到96-well plate(~30 cells/plate)
(293T、HeLa、MEF等细胞系推荐使用)。如果编辑效率50%,推荐分两 块plate即可,最终拿到20-30个单克隆一般可以得到成功KO的细胞系;或 者铺到100 mm dish里,一周后挑取单克隆(这种方法适合单细胞不容易成 活的细胞系如,但是挑取的单克隆偶尔不纯,有时需要重新亚克隆)。
目的细胞系的感染/转染
转染 (2 g plasmid/60 mm dish,细胞密度60-80%)
【v2=lentiCRISPR v2,下同】
PEI转染试剂的Protocol
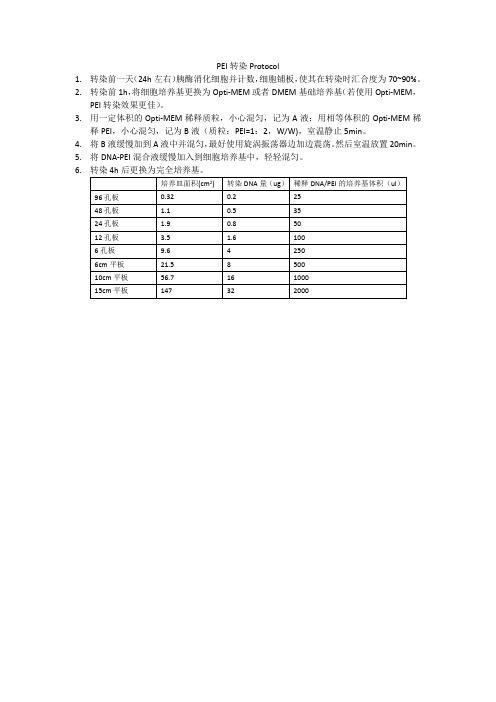
24孔板
1.90.8Fra bibliotek5012孔板
3.5
1.6
100
6孔板
9.6
4
250
6cm平板
21.5
8
500
10cm平板
56.7
16
1000
15cm平板
147
32
2000
4.将B液缓慢加到A液中并混匀,最好使用旋涡振荡器边加边震荡。然后室温放置20min。
5.将DNA-PEI混合液缓慢加入到细胞培养基中,轻轻混匀。
6.转染4h后更换为完全培养基。
培养皿面积(cm2)
转染DNA量(ug)
稀释DNA/PEI的培养基体积(ul)
96孔板
0.32
0.2
25
48孔板
1.1
0.5
PEI转染Protocol
1.转染前一天(24h左右)胰酶消化细胞并计数,细胞铺板,使其在转染时汇合度为70~90%。
2.转染前1h,将细胞培养基更换为Opti-MEM或者DMEM基础培养基(若使用Opti-MEM,PEI转染效果更佳)。
3.用一定体积的Opti-MEM稀释质粒,小心混匀,记为A液;用相等体积的Opti-MEM稀释PEI,小心混匀,记为B液(质粒:PEI=1:2,W/W),室温静止5min。
细胞转染 Protocol

细胞转染是一种常用的生物技术,用于将外源基因或药物引入细胞中。
下面是细胞转染的实验原理、所需试剂和耗材、实验仪器、准备工作、实验方法、注意事项、常见问题及解决方法。
一、实验原理细胞转染是利用细胞膜的通透性,将带有特定基因或药物的载体分子导入细胞内。
常用的载体分子包括病毒载体和非病毒载体。
病毒载体如腺病毒、逆转录病毒等,能将外源基因高效地导入细胞中,但可能对细胞产生一定毒性。
非病毒载体如脂质体、阳离子聚合物等,相对安全,但导入效率较低。
通过细胞转染,可以实现对基因的表达调控,探索基因功能和药物筛选等研究。
二、所需试剂和耗材1.试剂:o培养基:如DMEM、F12等,用于细胞培养。
o血清:如胎牛血清,提供细胞生长所需的营养物质。
o抗生素:如青霉素、链霉素等,用于防止细胞污染。
o转染试剂:如Lipofectamine、Effectene等,用于细胞转染。
o DNA或RNA:携带目的基因的质粒或寡核苷酸。
2.耗材:o细胞培养瓶、板:用于细胞培养。
o离心管:用于细胞转染后洗涤和离心。
o移液器及枪头:用于精确加样。
o过滤器:用于过滤溶液中的杂质。
o无菌水:用于稀释和配制溶液。
三、实验仪器1.实验室搅拌器:用于混合溶液。
2.高速冷冻离心机:用于离心和分离细胞。
3.水浴锅:用于加热溶液。
4.无菌工作台或超净工作台:用于进行无菌操作。
5.分光光度计:用于测量细胞生长状况和转染效率。
6.荧光显微镜:用于观察细胞转染后荧光蛋白的表达情况。
7.CO2培养箱:提供细胞培养所需的气体环境。
四、准备工作1.仔细阅读实验步骤和注意事项,了解所需的试剂和耗材及其使用方法。
2.准备好所需的试剂和耗材,并确保它们处于保质期内。
3.检查实验室内是否具备上述实验仪器,并确保其正常运行。
4.用70%乙醇擦拭实验台面,以确保无菌环境。
5.用高压蒸汽灭菌法灭菌所有的实验器具,包括离心管、移液器等,需在适当的压力和温度下进行灭菌处理,以消除所有潜在的污染源。
质粒转染技术

质粒转染技术质粒转染技术是一种常用的分子生物学技术,用于将外源基因导入细胞中。
本文将从质粒转染的原理、方法和应用等方面进行详细介绍。
一、质粒转染技术的原理质粒转染是指将质粒DNA导入目标细胞中,使其表达外源基因。
质粒是一种环状的双链DNA分子,通常存在于细菌细胞中,具有自主复制的能力。
质粒转染技术利用质粒的特性,将外源基因插入质粒中,然后通过物理或化学手段使质粒进入细胞内,从而实现外源基因的表达。
1. 钙磷法:将质粒DNA与钙盐形成复合物,然后将复合物添加到细胞培养基中,通过细胞摄取复合物实现质粒转染。
2. 电穿孔法:利用电场作用使细胞膜发生短暂通透,从而使质粒DNA进入细胞内。
3. 热激法:将细胞与质粒DNA进行热激处理,使细胞膜通透性增加,从而实现质粒转染。
4. 脂质体转染法:利用脂质体与质粒DNA形成复合物,然后加入细胞培养基中,通过脂质体与细胞膜的相互作用,使质粒DNA进入细胞内。
三、质粒转染技术的应用1. 基因功能研究:通过转染特定的质粒载体,可以实现对目标基因的过表达、沉默或突变,从而研究基因在细胞中的功能。
2. 基因治疗:将治疗相关的基因载体转染到患者的细胞中,通过表达特定的基因来治疗疾病。
3. 转基因生物制备:通过转染特定的质粒载体,将外源基因导入植物或动物细胞中,从而制备转基因生物。
4. 药物筛选:将药物相关的基因载体转染到细胞中,通过观察外源基因的表达情况,评估药物的疗效。
5. 蛋白表达:将目标蛋白基因载体转染到细胞中,使细胞表达目标蛋白,用于蛋白的纯化和功能研究。
质粒转染技术是一种重要的分子生物学技术,通过将外源基因导入细胞中,实现基因功能研究、基因治疗、转基因生物制备、药物筛选和蛋白表达等应用。
随着技术的不断发展和完善,质粒转染技术将在生物医学研究和生物工程领域发挥越来越重要的作用。
质粒共转染与单转步骤

质粒共转染与单转步骤质粒共转染(Co-transfection)和单转(Single transfection)是在分子生物学研究中常用的实验技术,用于将外源DNA导入到细胞中。
下面是它们的步骤解释:质粒共转染:1. 准备质粒:首先,准备包含所需基因或DNA片段的质粒。
质粒通常通过经验性选择或特定实验目的来选择。
2. 细胞培养:细胞株需要预先培养到合适的生长状态,以确保细胞处于最佳转染状态。
通常,细胞会在培养皿中培养至适当的浓度和状态。
3. 转染混合物制备:将所需质粒与适当的转染试剂(例如,转染剂或离子磷脂体)混合,形成转染混合物。
转染试剂有助于促进质粒与细胞之间的相互作用,从而实现转染。
4. 转染:将转染混合物添加到细胞培养皿中。
转染过程中,质粒会进入目标细胞,并在细胞内进行表达或进一步操作。
5. 培养和筛选:将转染后的细胞继续培养,并根据实验需求选择适当的培养基或添加筛选物质(例如抗生素)来选择和筛选转染成功的细胞。
单转:1. 准备质粒:同样,首先准备包含所需基因或DNA片段的质粒。
2. 细胞培养:细胞株需要预先培养到适当的生长状态。
3. 转染混合物制备:将所需质粒与适当的转染试剂混合,形成转染混合物。
4. 转染:将转染混合物添加到细胞培养皿中,与共转染步骤相同。
5. 培养和筛选:与共转染步骤相同,将转染后的细胞继续培养并选择筛选。
区别:- 共转染是将两个或多个不同的质粒同时转染到细胞中,而单转是只转染一个质粒。
- 共转染可以用于研究多个基因的相互作用或共同表达,而单转通常用于研究单个基因的功能。
- 共转染可能需要更多的优化和控制,以确保每个质粒在细胞中的比例适当。
- 单转一般比共转染更简单和直接,因为只涉及一个质粒。
无论是质粒共转染还是单转,都需要根据具体的实验目的和细胞类型进行优化和控制,以确保转染效率和所需结果的实现。
protocol-转染+荧光修改

转染实验:1、消化细胞,加入2ml胰酶,37°孵育5min,4ml完全培养基终止,吹打。
2、收细胞,1000rpm,离心1min.3、细胞接种在24孔板中,每孔20000个,500ul 完全培养基培养,待80%融合开始转染。
4、转染前2小时更换培养基,换成OPTI-MEM,孵育2h,饥饿处理.5、DNA:lipo=0.5ug:2ul(lipo探索4个梯度),分别稀释于25ul MEM中,混合DNA和lipo(50ul),室温孵育15min,加入到细胞中。
做3个副孔。
质粒稀释:Basic:0.3790 ug/ulR:0.3276 ug/ulTFF1:0.3712 ug/ulERE:0.4378 ug/ulAP-1:0.3778 ug/ul双突变:0.4363 ug/ul构建基因与R比例=10:1,取1ul R加入到9ul MEM中,进行10倍稀释Basic+R组:1.19 ul的basic加入到11.31 ul MEM中,1.53ul R加到10.97ul MEM 中,混合,室温15min。
TFF1+R组:1.22ul TFF1加到11.28 ul MEM中,1.53ul R加到10.97ul MEM中,混合,室温15min。
ERE+R组:1.03ul ERE加到11.47ul MEM中,1.53ul R加到10.97ul MEM中,混合,室温15min。
AP1+R组:1.20ul AP-1加到11.3ul MEM中,1.53ul R加到10.97ul MEM中,混合,室温15min。
ERE+AP1+R组:1.04ul 加到11.46ulMEM中,1.53ul R加到10.97ul MEM中,混合,室温15min。
6、37°孵育6h后更换有血清有双抗培养基,继续孵育24h。
雌激素刺激:转染后的细胞孵育24h后,用10-6、10-12两个浓度刺激细胞,每孔500ul,孵育24h。
质粒转染 (2)

质粒转染概述质粒转染是一种常用的实验技术,用于将外源基因导入至细胞内,以研究基因的功能、表达和调控等方面的问题。
质粒转染的方法多种多样,包括瞬时转染、病毒载体转染和电穿孔等。
本文将介绍质粒转染的原理、常用方法和注意事项。
原理质粒是一种环状的双链DNA分子,可在细胞内稳定复制和表达外源基因。
质粒转染的过程主要包括质粒进入细胞、转座和表达等步骤。
质粒进入细胞通常需要越过细胞膜,这可以通过瞬时破坏细胞膜以便质粒进入细胞,或者利用特定的转染试剂辅助质粒进入细胞。
质粒进入细胞后,它可以被随机插入到细胞染色体中,或者停留在高复制活性的细胞结构,如细胞核或线粒体。
一旦质粒转座完成,它可以通过自身的启动子和终止子进行转录和翻译,从而表达相应的外源基因。
外源基因的表达结果可以通过各种方法进行检测和分析。
常用方法1. 瞬时转染瞬时转染是一种将质粒暂时引入细胞内,使之表达外源基因的方法。
常用的瞬时转染方法包括钙磷共沉淀法、电穿孔法和脂质体转染法等。
其中,钙磷共沉淀法是一种经典的转染方法,通过将质粒与钙盐一起处理,形成质粒-钙磷复合物,然后将其加入到细胞培养基中,质粒可通过细胞膜破裂进入细胞内。
电穿孔法利用电场的作用破坏细胞膜,并使质粒进入细胞,脂质体转染法则是利用脂质体与质粒的亲和力,将质粒包裹在脂质体内,通过与细胞膜融合进入细胞。
2. 病毒载体转染病毒载体转染是利用病毒作为载体将质粒导入细胞内。
常用的病毒载体包括腺相关病毒(Adenovirus)、慢病毒(Retrovirus)和腺病毒(Adeno-associated virus)等。
这些病毒载体可以携带外源质粒进入细胞,将质粒插入宿主基因组中,并在细胞内稳定复制和表达。
病毒载体转染方法通常需要通过特定的包装和扩增步骤来制备,以确保足够的载体颗粒用于转染。
3. 电穿孔电穿孔是一种通过电场作用破坏细胞膜,使质粒进入细胞的转染方法。
电穿孔方法通常使用专门的电转染装置,利用高电压脉冲瞬时改变细胞膜的通透性,使质粒进入细胞。
质粒转染-DH5α法

质粒转染-DH5α法
中科院某重点实验室的protocol
质粒转染-DH5α法
1、解冻DH5α:从-80°C冰箱中取DH5α感受态细胞100ul冰上解冻。
2、加质粒:加100ng,1ul质粒(如果是连接产物全都加入)于感受态DH5α中,冰上混匀30min。
(超净工作台里面操作)准备SOC:同时取SOC培养于37°C水箱中预热,待用。
3、热击:将质粒,感受态混合物置于42°C水箱中热击90s
4、恢复:冰上冷却2min。
5、复苏:取预热的SOC培养基于质粒感受态混合物中,摇床摇晃1h.
准备平板:同时取小平板于37°C温箱中预热。
(超净工作台里面操作)
6、涂板子:对于连接产物;低速离心4000rpm×4min,去部分上清,留沉淀约100ul,重悬,把质粒感受态混合物平涂在小平板上。
倒放平板于37°C温箱中过夜。
对于质粒,不用离心,取20ul平涂。
Tips:连接产物不离心去上清的话,涂板液体太多,连接产物容易集中。
7、准备后续的实验:挑板子,小量培养细菌,质粒小抽鉴定。
注意:
1、连接产物比质粒转染效率低,所以加入的量多,热击时间长。
2、用于转染的质粒或者连接产物的体积不能超过感受态细胞体积的15%。
3、用于转化的质粒量在0.1pg-50pg最好,超过50pg转化效率低。
4、热击与离心管的厚度,菌液体积,输入片段大小等直接相关。
一般建议热击45S,时间长,转化效率低。
质粒DNA转化感受态大肠杆菌 protocol

质粒DNA转化感受态大肠杆菌一、实验原理转化(transformation)是将一种生物(供体)的遗传物质(通常为DNA)转入另一种生物(受体)并使其在受体中得以保存和繁殖的过程。
大肠杆菌不是天然感受菌,在低温(0~5℃)环境下经CaCl2处理,细胞壁变松变软后能摄入外源DNA,这种状态称为感受态细胞(competent cell)。
质粒DNA或重组DNA粘附在细菌细胞表面,经过42°C短时间的热击处理,促进吸收DNA.然后在非选择培养基中培养一代,待质粒上所带的抗菌素基因表达,就可以在含抗菌素的培养基中生长。
二、实验材料准备1. 器材微量移液取样器,移液器吸头,恒温水浴锅,制冰机,恒温摇床,培养皿(已铺好固体LB-Amp),超净工作台,酒精灯,玻璃涂棒,1.5 ml Eppendorf管,50 ml离心管,乳胶手套,恒温培养箱2. 试剂LB液体培养基、LB固体培养基、100mg/ml 氨苄青霉素(或卡拉霉素) 、感受态细胞3. 材料处理转化之前超净台紫外照射15-20min;枪头、50ml离心管需提前灭菌,烘箱烘干;液体LB培养基灭菌后放到冷库,防止长菌;三、转化1.从-70℃冰箱中取200μl感受态细胞悬液,置冰上解冻1-2 min。
2.加入质粒DNA溶液(含量不超过50ng,体积不超过10μl),轻轻摇匀,冰上放置30分钟。
3.42℃水浴中热击90秒, 然后迅速置冰上2min,整个过程不要振荡菌液。
4.向管中加入200μl LB液体培养基(不含抗生素),混匀后37℃振荡培养(225 rpm)1小时,使细菌恢复正常生长状态,并表达质粒编码的抗生素抗性基因。
5. 将上述菌液摇匀后涂布于含Amp(或kna)的LB琼脂平板上,正面向上放置半小时,待菌液完全被培养基吸收后,37℃倒置培养12-16小时。
四、注意事项1.加液体LB培养基之前观察其是否长菌2.玻璃涂布棒上的酒精熄灭后稍等片刻,待其冷却后再涂3.提前打开水浴锅,将温度调到42度4.实验目的是表达蛋白,可热击完直接涂平板;目的是质粒扩增或后续要PCR,需在热击后37℃振荡培养复苏1小时5.实验室常用的用于质粒扩增的感受态菌是Top10,用于质粒表达的感受态菌是BL21 Star(DE3)。
质粒转染 (2)

质粒转染引言质粒转染是一种常用的实验方法,用于将外源质粒导入目标细胞中,实现基因转导和表达。
质粒转染广泛应用于生物学研究、基因治疗等领域,成为了现代分子生物学中不可或缺的工具。
质粒转染的原理质粒转染的原理基于细胞膜的通透性和质粒的分子特性。
细胞膜是由磷脂双层构成的,具有一定的通透性。
质粒则是一种环形的DNA分子,可以通过细胞膜的孔隙进入细胞内。
在质粒转染的实验中,通常使用离子交联剂、聚合物或脂质体等载体,将质粒导入细胞。
载体能够与细胞膜相互作用,形成一个复合物,帮助质粒穿越细胞膜。
一旦质粒进入细胞内,它可以在细胞质中进行表达,从而实现基因转导和表达的目的。
质粒转染的方法离子交联法离子交联法是质粒转染的常用方法之一。
在该方法中,使用一种离子交联剂(如聚乙烯亚胺),将质粒与载体(如聚乙烯亚胺-质粒复合物)相互作用。
这种相互作用能够稳定质粒在溶液中的形态,并提高质粒穿越细胞膜的效率。
离子交联法的优点是操作简单、适用于多种细胞类型,但其缺点是质粒入细胞的效率相对较低。
脂质体转染法脂质体转染法是质粒转染中常用的方法之一。
脂质体是一种由磷脂和胆固醇等组成的微小囊泡,具有与细胞膜相似的结构。
在该方法中,质粒与脂质体相结合,形成一个质粒-脂质体复合体。
该复合体能有效地改善细胞膜通透性,增加质粒进入细胞内的概率。
脂质体转染法具有转染效率高、适用于多种细胞类型的优点,但也存在较高的细胞毒性和较高的成本。
聚合物转染法聚合物转染法是质粒转染中的一种新兴方法。
在该方法中,使用合成的阳离子型聚合物,与质粒形成复合物。
这种复合物可以稳定质粒的形态,并提高质粒进入细胞内的效率。
聚合物转染法具有操作简单、转染效率高的优点,但也存在一定的细胞毒性和质粒稳定性不高的问题。
目前,聚合物转染法在科研实验中被广泛应用,但在临床应用中仍需要进一步研究。
质粒转染的应用质粒转染广泛应用于生物学研究和基因治疗等领域。
在生物学研究中,质粒转染可以用于基因敲除、基因表达、蛋白质定位等实验。
质粒转染文档

质粒转染简介质粒转染是一种将外源质粒导入到目标细胞中的技术。
质粒转染广泛应用于分子生物学和基因工程研究中,可以用于基因功能研究、基因治疗以及表达外源蛋白等方面。
本文将介绍质粒转染的原理、方法和一些常用的实验技术。
原理质粒转染主要依靠质粒与细胞之间的相互作用来实现。
一般情况下,质粒会通过不同的转染方法被导入到细胞内,然后在细胞内复制和转录。
而转染后的质粒可以携带外源基因或其他功能序列,使细胞表达这些外源物质。
转染方法常见的质粒转染方法主要包括化学法、电穿孔法和病毒介导法。
化学法化学法是将质粒与转染试剂一起处理,使质粒能够穿过细胞膜进入细胞内。
常见的化学转染试剂包括聚乙烯亚胺(PEI)、脂质体和钙磷共沉淀等。
这些试剂能够与DNA形成复合物,通过与细胞膜相互作用,使质粒进入细胞质。
电穿孔法电穿孔法通过施加高电压脉冲使细胞膜发生短暂的孔穴,从而使质粒能够进入细胞。
这种方法可以通过电击法或电渗透法来实现。
电击法是将细胞悬浮在含有质粒的溶液中,然后施加高压脉冲使细胞膜发生短暂裂开,使质粒进入细胞质。
电渗透法是将细胞与含有质粒的溶液一同置于电穿孔仪中,通过电流作用使质粒进入细胞。
病毒介导法病毒介导法是使用病毒作为质粒的载体,通过病毒感染细胞来实现质粒转染。
常见的病毒载体包括腺病毒、逆转录病毒和整合子病毒等。
这些病毒能够感染细胞并将质粒导入细胞内,使细胞表达质粒中的外源基因。
实验技术质粒提取质粒提取是质粒转染前的一项重要步骤。
质粒提取可以通过碱裂解法或商用试剂盒等方法来实现。
碱裂解法是将菌落或培养物中的质粒与碱性溶液混合,然后进行离心,将质粒沉淀。
商用试剂盒则提供了一种简便快速的质粒提取方法,通过试剂盒中提供的缓冲液和柱子等物品进行操作。
转染优化质粒转染过程中,为了提高质粒转染效率,可以进行一些转染优化的操作。
例如,调整转染试剂与质粒的配比、处理时间和细胞密度等因素,可以进一步提高质粒转染效果。
此外,还可以尝试不同的转染方法并比较它们的转染效果,选择最适合的方法进行转染。
omega质粒小提protocol

Omega 质粒小提试剂盒protocol主要试剂1. 使用前,将RNase A全部加入Solution I中并于4度保存。
2. 按下表用无水乙醇稀释DNA Wash Buffer,并于室温保存。
D6942-00: 加入6 ml无水乙醇D6942-01: 加入120 ml无水乙醇3. 按下表用异丙醇稀释HBC Buffer,并于室温保存。
D6942-00: 加入1.6 ml无水乙醇D6942-01: 加入16 ml无水乙醇D6942-02: 加入32 ml无水乙醇试剂配方及作用Solution I组分浓度25 mM Tris-HCl(pH8.0),10 mM EDTA,50 mM 葡糖糖(Glucose)溶液1的主要作用是将菌体沉淀悬浮起来。
25mM Tris-HCl是缓冲溶液,保证反应体系的pH 恒定。
EDTA是金属离子螯合剂,10 mM EDTA的作用是与微生物体内的金属离子相互结合,抑制DNase的活性。
50 mM葡萄糖的作用是增加溶液的粘度,保证菌体悬浮,延缓菌体沉降的时间。
溶液1在使用前通常要加入RNA酶(RNase A),RNA酶的作用很清楚,降解掉溶液中的RNA。
由于RNA酶属于蛋白质,蛋白质稳定性很差,所以加了RNase A的溶液1需要低温4℃保存。
Solution II组份浓度250 mM NaOH,1%(W/V)SDS(十二烷基硫酸钠)溶液2的主要作用是细胞裂解。
溶液1将细胞悬浮后,就需要添加溶液2了,溶液2的作用就是对细胞进行裂解。
溶液2只包括两种成分:氢氧化钠和SDS。
真正起到到细胞裂解作用的是氢氧化钠,这也是为什么这个方法会被叫做碱裂解法。
氢氧化钠会破坏细胞膜的结构,使之发生bilayer(双层膜)结构向micelle(微囊)结构的相变化,从而导致细胞的裂解。
SDS的作用将在下一步提到。
添加溶液2后需要注意的一个是时间一定不能过长,另一个是不可以剧烈震动离心管。
时间过长以及剧烈震动都将导致氢氧化钠破坏基因组DNA,断裂的基因组DNA碎片将会与相似大小质粒一同被抽提,污染样品。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1. 细胞分盘:通过胰酶消化收集细胞,用适当的完全培养基以1×105 至4×105 细胞/cm2 的密度平铺细胞于60 mm 组织培养皿上(根据实验需要选择培养皿,使细胞贴壁后所占面积达到培养皿总面积的30%)。
将细胞置于含5%
CO2 的37℃温箱中孵育8-24 h,当细胞贴壁完全后即可开始转染。
转染前2 h 换
液(用4 ml 新鲜的完全培养基置换旧的培养基)。
注:为得到较高的转染效率,尽量使用指数生长的细胞,转染时细胞的密度不超过80%。
2. 制备磷酸钙-DNA 沉淀:以60 mm 组织培养皿用480 μl 反应总体积,6孔板培养皿用240ul反应体系。
在灭菌水中加入质粒DNA(总量1-3 μg 为佳),
3.再加入48ul的10*HBS,vortex 3s左右,再加入24ul的5*CaCl2,vortex 10s
,静置12min,将磷酸钙-DNA悬液逐滴加入上述单层细胞的细胞培养基中,轻轻摇动平皿混匀。
注:可以观察到滴入的部位培养基瞬间会出现浑浊的橘黄色,应尽快将其混匀,避免形成过大的颗粒,影响转染效率。
3.若转染的细胞不用转染促进剂处理,则置于含5%CO2 的37℃温箱孵育。
8 h 后吸去培养基与DNA 沉淀,加入5 ml 37℃预热的完全培养基,继续将细胞放
置温箱孵育,16-40 h 观察其转染效率。