连锁不平衡和关联研究默认分类 2
生物学新研究方法

一、图位克隆相关汇总1、用于QTL初定位群体的类型有哪些以及各群体的特点F2群体易于配制,需要时间短,所提供的遗传信息最为丰富,可以估算加性效应及显性效应。
但F2群体由单株组成且尚未达到纯合,提供的材料有限,很难对其进行连续性研究。
由于每个基因型只有一株,由此得到的数量性状数据可靠性差。
补救办法是利用F2代单株衍生的F2:3家系,选取同一家系中的若干个体进行分析,但这样做不仅加大了工作量,而且容易造成抽样误差。
回交(BC)群体:低代回交群体重组交换的信息量比F2少,为了弥补回交群体的不足现在多采用先回交再自交的方式,对回交后代进行自交。
DH系群体:加倍单倍体群体,是通过诱导F1单倍体并加倍形成的群体,群体内基因完全纯合,群体内的差异构成了分离群体的遗传特性,是永久群体,但重组交换的信息较少。
RIL系:RIL群体基因基本纯合,群体结构稳定,也是一个永久性分离群体,重组程度高于F2群体,因此,RIL 群体构建的图谱比F2的有着更高的解析度。
但建立一套RIL需要多年的工作。
而且,在基因组的某些区域的纯合比理论预期需要更长的时间,而且不能估计显性效应。
2、简答图位克隆常见的群体(至少三个)及其特点。
根据分离群体的特点,图位克隆作图群体分为临时性分离群体、永久性分离群体和回交近交系群体三大类。
临时性分离群体:包括单交组合产生的F2及其衍生的F3、F4家系回交群体。
其显著特征时群体中每一个个体后代均可发生分离,除非自交不亲和性,F2易配制。
而且提供给遗传分析的信息最为丰富,可以同时估计加性效应和显性效应。
但由于F2存在分离,很难进行多年多点研究。
永久性分离群体:主要包括重组自交系群体(RIL)和加倍单倍体群体(DH)。
其显著特征是群体中每一个体其后代稳定,不发生分离,可重复进行试验,将区组效应、重复效应和随机误差最小化或分解,增加检测QTL准确性。
但构建RIL 群体需很长时间;构建DH群体受基因型限制,难度较大,且它们不能估计显性效应。
疾病相关基因SNP的分析与验证

疾病相关基因SNP的分析与验证随着技术的不断发展,生物信息学研究也日渐深入。
其中,SNP(单核苷酸多态性)成为研究生物学、药理学和医学中最重要的基因变异类型之一。
SNP分析已经成为了检测疾病和药物代谢的重要方法,而在研究人类遗传学和疾病相关基因中,SNP的应用更是不可或缺。
1. SNP的概念和分类SNP,即单个核苷酸的变异,也被称为基因突变或是基因多态性。
SNP是由单个碱基的变异所引起,通常在全基因组中有约1%的概率。
SNP被广泛应用于评估个体对疾病的易感性、药物代谢和肿瘤发生等领域。
SNP按照其在基因组中的位置分类,可分为外显子SNP、内含子SNP和调控SNP。
外显子SNP指的是存在于基因的外显子区域,可以直接影响蛋白质序列的结构和功能;内含子SNP存在于外显子和调节区域之间,通常对基因功能的影响较小;调控SNP存在于基因调节区域,可以影响基因的转录和表达,进而影响基因的功能。
2. SNP的分析SNP的分析通常包括三个步骤:SNP检测、基因型鉴定和统计分析。
其中SNP 检测是最为关键的一步,目前主要的检测技术有PCR-RFLP法、MassARRAY、SNP-PCR等。
在SNP检测的基础上,需要对检测结果进行基因型鉴定。
常见的基因型鉴定方法有PCR引物延伸分析、限制性片段长度多态性分析、基因芯片以及测序等。
最后,需要进行统计分析。
在统计分析中,最常用的是卡方检验和连锁不平衡分析。
卡方检验被广泛应用于检测基因型频率和疾病之间的关联性,而连锁不平衡分析则可以确定SNP之间的互连性。
3. SNP的验证SNP验证是保证SNP检测结果准确可靠的重要步骤。
SNP验证通常包括三个方面:测序验证、多样性验证和遗传流行病学验证。
测序验证是指通过测序对SNP检测结果进行验证。
这种验证方式直接检测SNP并确定其具体的位置和变异。
然而,测序验证的成本较高,时间较长,因此不适合高通量的SNP检测。
多样性验证是指将SNP检测结果与其他不同个体的SNP检测结果进行比较,以此确认SNP检测结果的可靠性。
【小工具】ACMG评级指南

【⼩⼯具】ACMG评级指南简介2015年,美国权威机构——美国医学遗传学与基因组学学会(ACMG)编写和发布了《ACMG 遗传变异分类标准与指南》。
该指南将变异位点的致病、良性证据列为具体的28条评判标准。
⾸先将证据按类型分类(如⼈群数据、计算预测数据、功能数据等),并将证据的⽀持度分为⼏类(⽀持,中等,强,⾮常强以及独⽴);然后使⽤“标准组合”的形式来评估致病性。
不同组合将产⽣五个类别的致病性分类:致病,可能致病,临床意义不明,可能良性,良性。
该指南是多学科专家基于⼤量临床案例和丰富经验建⽴的,主要⽤处在于⼤体思路指导,具体案例还需具体分析,新的证据出现时,其评级可上下调整。
变异的命名HGVS命名为标准命名,临床报告应该包含参考序列以确保该变异在DNA⽔平上的明确命名,并提供编码和蛋⽩质命名法来协助功能注释(如“g”为基因组序列,“c”为编码DNA序列,“p”为蛋⽩质,“m”为线粒体)。
编码命名应该使⽤翻译起始密码⼦ATG中的“A”作为位置编号1来描述。
基因组坐标应根据标准基因组版本(如hg19)或覆盖整个基因(包括5'和3'⾮翻译区以及启动⼦)的基因组参考序列来界定。
当描述编码变异时,应该在报告中使⽤和提供每个基因的⼀个参考转录本。
该转录本应该是最长的已知转录本或者是最具临床相关性的转录本。
展开剩余95%ACMG⽀持HGVS命名规则之外的三种特殊例外:►除了当今HGVS推荐的“*”和“Ter”,“X”仍然被认为⽤于报告⽆义变异;►建议根据指定变异选择的参考转录本对外显⼦进⾏编号;►通常因为临床解释直接评估致病性,所以推荐使⽤术语“致病性”⽽不是“影响功能”。
数据库的使⽤基因组数据库收录不断被发现的变异,当我们需要对某⼀变异分类并报告,可在已有的数据库中找到有价值的信息。
⼈群数据库适⽤于获取某变异在⼤规模⼈群中发⽣频率的相关信息。
需要注意的是,⼈群数据库中的信息不仅来源于健康个体,也包含致病性的变异。
连锁不平衡分析

连锁不平衡分析连锁不平衡分析,简称ChA,是一种多元素数据分析技术,可以从多个维度准确地分析出一组相关或不相关事物之间的关系。
它的目的是为了指出过度关联的变量,分析平衡以及不平衡的指标,并可以提供有价值的结论。
为了实现这一目标,学者们提出了许多不同的方法,包括主成分分析(PCA)、相关分析(CA)、因子分析(FA)、结构方程模型(SEM)等,而最常用的是ChA方法。
连锁不平衡分析的基本思路是:在观察到的数据中,建立一个多元素的变量模型,并计算这丛变量之间的关联度。
这些变量可以是连续变量,也可以是类别变量。
经过ChA的运算,可以得出变量之间的抑制,协同,不平衡以及平衡的程度。
通过这一过程,可以提出建议,以提高业务的整体效率,以及指出可能出现紊乱情况的可能性。
连锁不平衡分析方法在多元统计分析中有着重要的地位,因其对研究对象之间的关系具有更深刻而准确的认识。
通过ChA方法,可以更完整地把握研究对象之间的关系,发掘研究对象内部的多种潜在关系,分析关系的不平衡性,得出有效的结论。
实施ChA的关键是正确构建变量模型,计算出变量之间的关联度。
首先,要对研究对象的变量进行分类,依据变量的构成属性以及变量的状态,选择合适的算法以计算其关联度,并绘制出关联关系图。
其次,需要计算每个变量的平衡度,分析出变量之间的抑制,协同,不平衡以及平衡关系,为后续的分析提供数据支持。
最后,分析出变量之间的相互关系,推导出有价值的结论。
连锁不平衡分析的应用范围非常广泛,主要用于经济、管理和社会等领域的定量性研究,也可用于分析数据中的复杂因素,并从中挖掘出可以改善现状的有效策略。
从技术上讲,连锁不平衡分析具有优势:首先,它可以一次衡量多个变量之间的关联度,并可以分析出变量之间的抑制,协同,不平衡以及平衡关系;其次,它可以在冗长的数据中探索研究对象的关系;最后,它可以进行多因素的综合考虑,从中发现研究对象的内在规律以及有价值的结论。
总之,连锁不平衡分析是一种重要的多元素数据分析技术,它可以从多个维度更深刻地分析出一组相关或不相关变量之间的关系,发掘研究对象内部的复杂关系,并提供有价值的结论。
系统与进化生物学名词解释完整版

第一章:绪论进化生物学Evolutionary Biology:是研究生物进化的科学,不仅研究进化的过程,更重要的是研究进化的原因、机制、速率和方向。
(研究生物进化的科学,包括进化的过程、证据、原因、规律、演说以及生物工程进化与地球的关系等。
)系统学Taxonomy:is the science of defining groups of biological organisms on the basis of shared characteristics and giving names to those groups.根据生物体显现出的的基本特征定义并确定其群体名称的学科。
系统生物学Systematic Biology:研究生物系统组成成分的构成与相互关系的结构、动态与发生,以系统论和实验、计算方法整合研究为特征的生物学。
系统与进化生物学Systematic and Evolutionary Biology:分类Classification:provide a convenient method of identification and communication.为生物的辨识与交流提供更便捷方法的学科。
系统发育Phylogeny:the evolutionary relationships among a group of species,provide a classification which as far as possible expresses the natural relationships of organism.研究种群之间进化的联系,尽可能地为解读生物体之间的自然关系提供一种分类方式的科学。
进化Evolution:detect evolution at work,discovering its processes and interpreting its results.(PPT)进化指食物由低级的、简单的形式向高级的、复杂的形式转变过程。
打不开下载的数据库解决办法--针对NCBI下载SNP不被Haploview识别
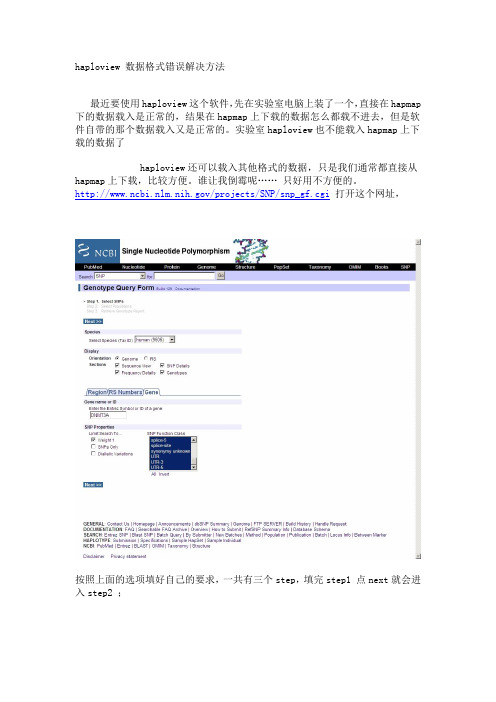
haploview 数据格式错误解决方法最近要使用haploview这个软件,先在实验室电脑上装了一个,直接在hapmap 下的数据载入是正常的,结果在hapmap上下载的数据怎么都载不进去,但是软件自带的那个数据载入又是正常的。
实验室haploview也不能载入hapmap上下载的数据了haploview还可以载入其他格式的数据,只是我们通常都直接从hapmap 上下载,比较方便。
谁让我倒霉呢…… 只好用不方便的。
/projects/SNP/snp_gf.cgi打开这个网址,按照上面的选项填好自己的要求,一共有三个step,填完step1 点next就会进入step2 ;在step2 选择好所需的人种,点display results 就会进入step3 ;在step3 下方的四个蓝色按钮的上面那一行小字里有download haploview files,点击就可以下载压缩文件。
解压这个文件里面有info 和ped 两个文件,这是对应于haploview 里linkage format 的,ped 对应data file,info 对应 locus information file,选择好后点ok 就能载入了。
换了个格式总算是把数据给载进去了,我的脑细胞呀~都损失在之前的极度焦虑之中了……其实这个事情已经成为过去式有一小段时间了,一直在纠结要不要写出来呢(之所以纠结,其实是因为我懒,脸红一下)。
虽然换一种数据格式是我自己想到的,可筒子们要理解,数据库的使用是复杂滴,探索使用方法的道路是艰辛滴,所以那个下载数据的方法是本人在探索无果的情况下经过长时间的谷歌找到滴。
既然下载的方法是借鉴的别人的劳动成果,所以我决定也把我的经验提供给大家,希望能帮到需要的筒子。
帮助说明/projects/SNP/docGenotype.html Detailed information for SNP GenotypesThe URL for the genotype pages is/projects/SNP/snp_gf.cgiThe HTML interface has 3 pages:The first page is a form for selecting species and SNPs.The second page is a form for selecting populations.The third page contains the search results in HTML, text, or XML.All form parameters can be specified on any page and will be reflected in theExamples∙Perl program for retrieving a genotype report in XML.∙XSLT for converting an XML report to TAB separated text and an additional required XSLT file.第一,要知道所选的基因上都有哪些SNP位点。
基于SNP的连锁不平衡分析

基于SNP的连锁不平衡分析SNP(单核苷酸多态性)是一种常见的遗传变异形式,它在人类基因组中广泛存在。
SNP的分析对于研究基因和疾病之间的关联以及个体遗传多样性具有重要意义。
连锁不平衡是指不同位点上的SNP之间存在非随机的关联。
基于SNP的连锁不平衡分析是一种研究人类遗传变异和疾病相关性的重要方法。
在过去的几十年中,研究人员已发现SNP在人类基因组中的定位,并建立了全球多种族的SNP数据库。
借助这些数据库,研究人员可以对不同个体和个体群体中的SNP进行分析,并研究这些SNP与特定疾病之间的关联。
连锁不平衡分析基于人类基因组中SNP之间的非随机关联进行。
通常,SNP之间的连接表现为连锁不平衡区块。
通过对大量SNP的分析,研究人员可以确定这些区块,并评估它们与疾病之间的相关性。
在研究中,研究人员通常使用统计学方法来确认这些关联,如皮尔逊卡方检验和Fisher确切概率检验。
连锁不平衡分析的目标是确定SNP与疾病之间的关联,以便进一步研究疾病的遗传机制。
通过分析SNP之间的连锁不平衡,研究人员可以发现一些SNP与特定疾病之间的高度关联,从而提供了潜在的遗传变异标记。
这些标记可以用于疾病风险评估、个体遗传多样性研究、个性化医疗和药物研发等领域。
在连锁不平衡分析中,研究人员通常需要考虑多个因素,如样本大小、个体群体的遗传背景和其他环境因素。
此外,SNP之间的连锁不平衡关系可能存在种族和地理差异。
因此,在连锁不平衡分析中,研究人员需要对样本进行严密的筛选和分类,并考虑这些因素的影响。
需要注意的是,虽然连锁不平衡分析可以揭示SNP与疾病之间的关联,但它并不能确定因果关系。
因此,研究人员在进行连锁不平衡分析时需要谨慎解读结果,并结合其他实验证据来确定SNP与疾病之间的具体关系。
总之,基于SNP的连锁不平衡分析是一种有效的研究人类遗传变异和疾病相关性的方法。
通过分析SNP之间的非随机关联,研究人员可以鉴定潜在的遗传变异标记,并进一步研究这些标记与疾病之间的关系。
连锁分析原理

连锁分析原理
连锁分析原理是一种用于解决复杂问题的方法,它通过将问题划分为一系列相互关联的小问题来进行分析。
每个小问题都通过解决它们的解题过程被链接在一起,最终得到问题的整体解决方案。
连锁分析的基本原理是将一个大问题分解成若干个较小的子问题,并将这些子问题的解答逐步连接起来,以得到最终的解决方案。
在这个过程中,每个子问题的解答都会对其他子问题产生影响,并相互关联。
通过连锁分析,我们能够更好地理解问题的本质和关键因素,从而能够更加全面地考虑解决问题的方法。
它能够帮助我们从整体的角度来看待问题,而不仅仅是关注细节。
连锁分析的具体步骤可以根据实际情况进行调整,但通常包括以下几个基本步骤:
1. 确定问题:明确问题的背景和目标,明确要解决的具体问题。
2. 划分子问题:将问题分解成多个子问题,每个子问题关注不同的方面。
3. 分析子问题:对每个子问题进行深入分析,寻找解决方案。
这可以包括使用各种分析工具和方法,如SWOT分析、鱼骨
图等。
4. 连接子问题:将子问题的解答连接起来,理解各个子问题之间的关系和相互作用。
5. 综合整体解决方案:综合所有子问题的解答,得出一个整体的解决方案。
通过连锁分析,我们能够利用系统思维的方式来解决问题,更好地理解问题的全貌和各个因素的相互关系。
这种方法可以帮助我们更加全面地考虑问题,并提供更有效的解决方案。
群体遗传学和分子生态学软件介绍

附录3分子生态学统计软件介绍分子生态学是研究生命系统与环境系统相互作用的分子基础与分子机理的崭新的分子生物学与生态学的交叉学科,是从基因、蛋白质、酶等生物分子活动规律来阐释生态规律进化、生态过程、适应和演变历程(Burke et al ,1992; Bachmann et al ,1994)。
这些研究通常会产生大量而复杂的分子数据,选择合适的统计方法对正确的解释科学现象是非常重要的。
以下就介绍几类常用的分子生态学软件。
3.1 遗传多样性与遗传结构分析软件遗传多样性是生物多样性的基础,丰富的遗传多样性可以提供很多宝贵的遗传资源。
因此为了对天然群体的遗传多样性研究,分子生态学专家开发出了一系列的评估软件,用于计算和检测生物群体基因变异的度量和遗传指标,其中用得比较广泛的有POPGENE 、STRUCTURES 、GENEPOP 、GenAlEx 6、NTSYSpc 、FSTAT 等。
POPGENE 是由Francis Yeh 等人开发的用共显性和显性标记来研究群体内和群体间的遗传多样性。
这个软件操作较简单,功能也比较全,主要包括计算广泛的遗传学数据如等位基因频率、遗传多样性、遗传距离、G -statistics 、F -statistics 等以及复杂的遗传学数据基因流、中性检测、连锁不平衡、多位点结构等。
新版本的POPGENE 还可用来分析数量遗传变异以及提供更高质量的系统聚类图。
POPGENE 下载地址:http://www.ualberta.ca/~fyeh/download.htmFSTAT 软件包是Jérôme Goudet 开发的用于计算共显性标记的遗传多样性和遗传分化参数。
主要功能如下:检测样本和总体水平上的基因频率,观察和期望基因型,等位基因数,基因丰富度;检测整体水平上以及每个样本或位点是否处于哈温平衡; Nei's (1987)的遗传多样性和遗传分化的估计值和 Weir & Cockerham (1984)每个等位基因,每个位点以及总体上的Capf (Fit), theta (F st )和smallf (F is)的估计值;检测R- statistics (Slatkin , 1995),5 将原始数据转化成Genepop 的格式等。
(完整word版)连锁不平衡原理

生物信息——连锁不平衡 Linkage Disequilibrium 不同基因座位的各等位基因在人群中以一定的频率出现。
在某一群体中,不同座位某两个等位基因出现在同一条染色体上的频率高于预期的随机频率的现象,称连锁不平衡 (linkage disequilibrium) 由于HLA 不同基因座位的某些等位基因经常连锁在一起遗传,而连锁的基因并非完全随机地组成单体型,有些基因总是较多地在一起出现,致使某些单体型在群体中呈现较高的频率,从而引起连锁不平衡。
例如两个相邻的基因A B, 他们各自的等位基因为a b. 假设A B相互独立遗传,则后代群体中观察得到的单倍体基因型 AB 中出现的P(AB)的概率为 P(A) * P(B).实际观察得到群体中单倍体基因型 AB 同时出现的概率为P(AB)。
计算这种不平衡的方法为:D = P(AB)- P(A) * P(B).连锁不平衡又称等位基因关联(allelic association),其原理其实很简单。
假定两个紧密连锁的位点1,2,各有两个等位型(A,a;B,b),那么在同一条染色体上将有四种可能的组合方式:A—B,A—b,a—B,和a—b。
假定等位型A的频率为Pa,B的频率为Pb,那么如果不存在连锁不平衡(如组成单倍型的等位型间相互独立,随机组合)单倍型A—B的频率就应为PaPb。
而如果A与B是相关联的,单倍型A—B的频率则应为PaPb+D,D是表示两位点间LD程度的值。
如果位点2上的等位型B与疾病易患性有关,那么将会观察到等位型A的频率在病人群体中高于对照群体。
换句话说,等位型A与该疾病性状相关。
事实上,可以检测遍布基因组中的大量遗传标记位点,或者候选基因附近的遗传标记来寻找到因为与致病位点距离足够近而表现出与疾病相关的位点,这就是等位基因关联分析或连锁不平衡定位基因的基本思想。
植物基因组中的连锁不平衡

mapping is an effective approach to discovering novel genes and a bridge for connecting structural genomics to phenomics. LD mapping was first applied in plants in 2001. Since then, researches on the structure and extent of LD and LD mapping have been reported in a wide range of plant species. The basic theory of LD and its application in LD mapping, haplotype diversity analysis, htSNP identification and population genetics were reviewed in this paper. And advances of LD research in plants including influences of population structure, gene conversion, epistasis and G×E interactions, and future prospects were also presented. China has abundant germplasm resources, but gene discovery lags behind. Intensive researches on LD will certainly accelerate rapid development of plant genomics, especially the progress of gene discovery based on germplasm resources in China. Keywords: linkage disequilibrium (LD); LD structure; LD mapping
遗传学中的连锁不平衡分析方法

遗传学中的连锁不平衡分析方法遗传学是生物学的一个分支学科,研究的是基因在自然界中的传递与变化。
自然界中的生物均通过基因来传递其遗传信息。
基因位于染色体上,由不同的基因型组成,决定了生物的各种性状。
遗传学家们通过研究基因型来探究生物遗传规律,并应用到实践中,帮助人类解决了许多遗传性疾病和农业生产中的问题。
在普通的遗传学研究中,经常会使用连锁分析法,来确定染色体上不同基因间的相互作用关系和相互距离。
但是,连锁分析法存在着种种局限,例如只能确定近亲间的遗传关系,而无法确定非近亲间的遗传关系。
这时,连锁不平衡分析法就派上用场了。
连锁不平衡分析法,又称关联分析法,是一种用于研究基因型遗传规律的方法,其基本思想是:通过统计分析在某个疾病或性状的家系中,某一基因座的两个等位基因的组合是否有偏离随机组合的趋势,从而确定这两个基因座之间的相互关系。
连锁不平衡分析法所研究的物质基础是基因型,通过分析基因型的组合,可以探究同一染色体上不同基因的相互作用关系。
连锁不平衡分析法的应用十分广泛。
首选应用是在不了解疾病原因的情况下,通过分析相关基因型的分布和遗传规律,来确定某些基因是否与疾病发生有关,并了解其发生机制和遗传方式。
此外,连锁不平衡分析法也可以用于基因组关联研究、种群遗传分析、选择育种等领域。
在实际应用中,连锁不平衡分析法也存在一些问题。
首先是需要对样本进行充分筛选,以减小结果的误差。
其次是需要建立一套完整的模型,来分析不同基因座之间的相互作用和影响,确保结果的准确性。
最后,由于基因组的复杂性,连锁不平衡分析法只能探讨单个基因与疾病发生之间的关系,而无法全面了解基因与疾病之间的关联。
总之,连锁不平衡分析法作为一种常用的遗传学研究手段,其应用范围十分广泛,可以有效的帮助研究人员探讨基因与疾病之间的关联,但其在应用中也存在一些局限性和问题。
随着科技的发展和研究方法的不断拓展,相信连锁不平衡分析法也会不断的完善和改进,为人类疾病防治和遗传学研究带来更大的助力。
连锁不平衡分析

连锁不平衡分析连锁不平衡分析(ChainUnbalanceAnalysis,简称CUA)是一种统计学方法,用于研究社会经济不平等,这也是社会学研究的经典理论之一。
这种方法首先假设可能会引起不平等的各种因素形成一条“不平衡链”,然后将其逐级拆分,使各级因素获得清晰的认识,从而把握整个状况。
通常,一般拆分到六级,用以描述不平等的整个过程。
此外,连锁不平衡分析的核心是社会结构的分类,也就是指在一个社会环境中,人们根据职业、社会地位或其他因素被分成不同的层级。
这些层级的区分很重要,因为不同的层级可能存在不同的条件权利和受支配的可能。
为此,连锁不平衡分析首先要明确定义这些层级,以便可以把握不同层级之间的不平衡性。
连锁不平衡分析的实践可以追溯到20世纪60年代以来,其理论框架发展到20世纪90年代。
它是社会学相关理论的重要组成部分,被用来分析社会、文化与经济等领域之间的关系,它被认为是“综合整个社会出现不平等的过程”,是一种有力的分析方法。
连锁不平衡分析主要分为四个步骤:首先,要明确定义不同层级之间的不平衡程度;其次,要分析不同层级之间的直接影响关系;再次,要分析不同级别之间可能发生的曲折或反向变化;最后,要综合分析不同层级之间的相互关系,由此推动其变化的动力。
在实践中,连锁不平衡分析尤其可以用于检测和研究社会发展、地区发展、教育发展, 以及社会结构、社会群体之间的不平等状况。
比如,在研究中国收入分配不平衡时,国内的学者根据国家制定的参考指标,将收入的起源、中间过程、社会支配等各个环节拆分,使从宏观到微观的不平等关系都得以清楚地分析出来,从而更好地理解“社会不平等”的真实状况。
总体来说,连锁不平衡分析是一种重要的统计学方法,它可以帮助我们深刻理解社会中不同层级之间的不平衡现象,从而有助于开发一种更加公平合理的社会结构。
它在检测和研究不平等问题方面发挥着巨大的作用,也是社会学研究的重要理论和方法之一。
进化生物学中基因多态性的建模与预测

进化生物学中基因多态性的建模与预测进化生物学是研究生命体系中各种生物在漫长的进化过程中的形态、生理和行为的变异的学科。
基因是生命的基本单位,也是进化的基本物质基础。
基因多态性指的是同一基因在不同个体中出现的各种不同形式,是基因进化和适应的反映。
基因多态性是生物学和人类学研究中的一个重要问题,对于疾病的发生发展、物种的起源和分化等方面有着重要的意义。
因此,建立基因多态性的模型、预测基因多态性变化趋势具有重要的理论意义和实践意义。
1. 基因多态性的分类基因多态性的类型非常丰富,主要包括下面几类:(1)等位基因的多态性。
等位基因是指对于同一基因座而言,有两个或两个以上的不同形态,在个体群体中存在。
(2)基因型频率的多态性。
基因型频率是指某一个基因型在总的个体中的出现次数与个体总数之比。
当一个基因型频率高于0.01时,就认为是多态的。
(3)单核苷酸多态性(SNP)。
单核苷酸多态性是指基因序列某一位点上出现的单一核苷酸在个体中的多样性。
(4)重复长度多态性(STR)。
重复长度多态性是指基因序列中某一特定区域内出现的短序列重复次数在个体中的多样性。
2. 基因多态性建模方法基因多态性的模型主要有两种:一种是基于统计的模型,另一种是基于遗传算法的模型。
(1)统计模型统计模型包括遗传频率分析、连锁不平衡分析、关联分析等方法。
其中,遗传频率分析是预测个体表现型的遗传基础的方法,能够比较准确地预测与明确定位的基因座相关的表现型。
连锁不平衡分析是近几十年来被广泛应用于复杂疾病遗传学研究的一种分析方法,能够在非明确定位的基因座上发现与疾病易感性相关的多态性标记。
而关联分析则是用来探讨基因型与表现型之间的相关性及其机制的一种分析方法。
(2)遗传算法模型遗传算法是一种仿生学优化算法,可以用于迭代搜索和优化问题的通用解决方案。
遗传算法模型则是以这种算法为基础,利用遗传算法优化基因多态性模型结构的一种方法。
3. 基因多态性预测方法基于遗传算法的模型可以实现对基因多态性的预测。
群体遗传学数据的分析方法及应用

群体遗传学数据的分析方法及应用随着基因组学技术的发展,很多人类疾病的研究越来越依赖于大规模群体遗传学数据的收集和分析。
通过对这些数据进行分析,人们可以发现基因对人类疾病的风险的影响程度,从而提高对疾病的认识,并有望发现新的治疗方法。
本文介绍了群体遗传学数据的主要分类、分析方法及其应用。
一、群体遗传学数据的分类遗传学数据有很多类别,其中最常见的包括:(1)基因型数据。
基因型数据是指人类或其他生物在几个位点的基因型信息。
这种数据是通过对DNA进行分子分析来进行收集的,其精度可以高达99.99%。
(2)表型数据。
表型数据是指个体的可观测特征,比如身高、体重、血压、血糖水平等。
表型数据需要进行标准化处理,以便进行群体遗传学研究。
(3)环境数据。
环境数据是指影响特定表型的各种环境因素。
这些因素包括生活方式、饮食、药物、外部因素等。
二、群体遗传学数据的分析方法(1)单点关联分析。
单点关联分析是一种常用的群体遗传学分析方法,其思想是通过比较某个基因型与特定表型之间的关系,寻找具有统计学意义的位点。
由于这种方法是基于每个位点独立的,因此可以准确地判断每个位点对疾病的风险的影响程度。
(2)连锁不平衡分析。
连锁不平衡分析利用基因型中的连锁不平衡信息来寻找与表型相关的位点。
这种方法比单点关联分析更加准确,因为它可以利用不同位点之间的信息相互作用。
(3)基因组关联分析。
基因组关联分析是一种全基因组的分析方法,通过比较整个基因组与表型之间的关系,寻找与疾病相关的位点。
由于这种方法可以同时分析所有位点,因此可以确保发现尽可能多的相关位点。
三、群体遗传学数据的应用通过群体遗传学数据的分析,人们可以获得许多有关人类疾病的重要信息。
以下是一些应用:(1)寻找疾病相关的位点。
通过对基因型和表型数据的联合分析,可以发现与某些疾病相关的基因。
(2)预测和诊断疾病的风险。
通过对遗传和环境数据的综合分析,可以准确地预测和诊断个体对某种疾病的风险。
基于关联或连锁不平衡的分析方法

基于关联或连锁不平衡的分析方法中山大学公共卫生学院医学统计与流行病学系李彩霞博士licx@(020)87330673-83(引用时请注明资料来源以及作者信息)如果两个基因座上的等位基因是随机关联的,即不独立,这种情况就叫做等位基因关联(allelic association)或者连锁不平衡(linkage disequilibrium,LD)。
关联通常反映了分子标记与性状功能突变之间在统计学上的非独立性(连锁不平衡),但并不一定意味着因果关系。
如果一个群体在初始状态下连锁不平衡(δ0≠0),在随机婚配条件下,在n代以后,有δn=(1-θ)nδ0。
因此连锁不平衡状态随着代数增加逐渐演变为平衡状态。
当连锁很弱,即重组率θ很大(接近1/2)时,连锁不平衡参数将随着代数的增加而迅速减小。
如果两个基因座紧密连锁,重组率θ很小(接近0),则不平衡状态将持续很多代。
连锁分析考察重组,因此,考察连锁必须有家庭数据,而由等位基因关联性(或连锁不平衡性)可以由一般的群体数据观察到,有的连锁不平衡现象可能是因为群体混杂造成的,但过大的连锁不平衡通常被视为紧密连锁的证据。
传统的连锁分析的结果通常是将基因定位在较大(例如~30cM)的基因组区域,而连锁不平衡被视为一种精细定位的方法。
Ott(1999)指出,对于那些远系繁殖的大群体,连锁不平衡通常只能延伸到0.3cM。
群体关联分析传统的病例-对照研究是基于群体而非家系的疾病关联分析,它通过随机选择病例和对照,然后比较其在标记等位基因和基因型频率上的差异来说明位点与疾病的关联性。
其缺点是:阳性结果可能由混杂因素造成,如不同分层人群(stratified populations)混杂在一起造成的虚假联系。
为了克服不同分层人群混杂的影响,相应产生了基于家庭的病例-对照研究方法。
单倍型相对风险分析(HRR,haplotype relative risk)单倍型相对风险分析是基于家系的病例-对照研究方法。
structure-2.3---中文使用手册

s t r u c t u r e-2.3---中文使用手册work Information Technology Company.2020YEARStructure 2.3中文使用手册Jonathan K. Pritchard aXiaoquan Wen aDaniel Falush b 1 2 3a芝加哥大学人类遗传学系b牛津大学统计学系软件来自/structure.html2010年2月2日1我们在Structure项目中的其他的同事有Peter Donnelly、Matthew Stephens 和Melissa Hubisz。
2开发这个程序的第一版时作者(JP、MS、PD)在牛津大学统计系。
3关于Structure的讨论和问题请发给在线的论坛上:structure-software@。
在邮递问题之前请查对这个文档并搜索以前的讨论。
1 引言程序Structure使用由不连锁的标记组成的基因型数据实施基于模型的聚类方法来推断群体结构。
这种方法由普里查德(Pritchard)、斯蒂芬斯(Stephens)和唐纳利(Donnelly)(2000a)在一篇文章中引入,由Falush、斯蒂芬斯(Stephens)和普里查德(Pritchard)(2003a,2007)在续篇中进行了扩展。
我们的方法的应用包括证明群体结构的存在,鉴定不同的遗传群体,把个体归到群体,以及鉴定移居者和掺和的个体。
简言之,我们假定有K个群体(这里K可能是未知的)的一个模型,每个群体在每个位点上由一组等位基因频率来刻画。
样本内的个体被(按照概率)分配到群体,或共同分配到两个或更多个群体,如果它们的基因型表明它们是混和的。
假定在群体内,位点处于哈迪-温伯格平衡和连锁平衡。
不精确地讲,个体被按达到这一点那样的方法指定到群体。
我们的模型不假定一个特别的突变过程,并且它可以应用于大多数通常使用的遗传标记,包括微卫星(microsatellites)、SNP和RFLP。
关联研究方法

关联研究方法是一种分析不同数据项之间关系的方法,主要包括关联规则挖掘和灰色关联分析。
具体来看:
- 关联规则挖掘:这是一种在数据集中发现频繁项集并基于这些项集生成关联规则的分析方法。
它包括两个主要步骤:首先寻找频繁项集,即经常出现在一起的对象集合;其次从这些频繁项集中生成关联规则。
关联规则通常表示为A→B的形式,其中A是前提或左部(LHS),B是结果或右部(RHS)。
例如,在零售业中,关联规则可以用来发现哪些商品经常一起被购买,从而帮助商家制定营销策略。
- 灰色关联分析:这是一种通过灰色关联度来分析和确定系统因素间的影响程度或因素对系统主行为的贡献测度的方法。
它的基本思想是根据序列曲线几何形状的相似程度来判断其联系是否紧密。
灰色关联分析的优点是对样本量的多少和样本有无规律都同样适用,计算量小,方便实用。
此外,全基因组关联研究(GWAS)也是一种重要的关联研究方法,它通过分析大规模样本的基因型和表型数据,识别与特定性状或疾病风险相关的基因变异。
GWAS的结果不仅有助于理解疾病的遗传基础,还为个体化医疗和药物开发提供了重要信息。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
由于同一染色体上位点间紧密连锁或其他原因,在同一配子中某些等位基因的组合可能增加,这种遗传现象称为等位基因的连锁不平衡,也叫关联(association)。这种非随机分布可通过比较患者与正常人群中有关遗传标记的等位基因频率测出,以此找到连锁不平衡位点中的相关基因标记。造成连锁不平衡的原因有多种,如随机遗传漂移、始祖效应(foundereffect)、重组、突变、选择和种群的混合等。与连锁分析一样,连锁不平衡同样是以物理距离与重组频率为依据,因此连锁不平衡也能用于定位易感基因,而且尚可用于研究基因变换(conversion)、人类的进化以及推断交换(crossing—over)的分布等。由于任何连锁不平衡状态是历经整个群体历史中重组事件的“磨损”而保存下来的,因此连锁不平衡信号相比于利用连锁分析得出的区域要小得多,连锁不平衡基因定位(1inkage disequilibriumzygositymapping)相似,连锁不平衡方法亦是基于血缘同一性。事实上,若某一突变是来自一个祖先,经携带者传递,则可形成一个巨大的拥有很多代数的家系。当然家系创始初期的几代人肯定不复存在,在世的多数是最晚的1~2代人,这使得大量的减数分裂无法直接观察到(传统连锁分析方法高度依赖于分析群体所能提供的关于遗传标记与定位目标基因座间的有效减数分裂的数量)。但随着一系列重组事情的发生,突变携带者共有的染色体片段不断缩短,以致含致病位点的染色体区域亦得以大为缩小,因此通过关联研究所定位的区域往往较小,甚至只含一个基因或基因的片段。如Laitinen等人定位的哮喘相关区域只有133kb,为哮喘相关基因GPRA第2内含子与第5内含子之间的片段。关联研究已被成功地应用于含较多新近的祖先突变的隔离群体以及更多古老突变的致病相关基因的研究中。
关联研究是基于群体中无亲缘关系的病例组和表型正常的对照组在某一个遗传位点上会出现不同的频率而设计的。遗传标记与疾病关联的原因有两种:一是致病基因与遗传标记存在很强的连锁不平衡;另一原因是遗传标记位点本身与疾病的发生有关。某个等位基因A和B之间相关测量的经典公式为:Δ值=AB基因频率一A基因频率XB基因频率,并且Δ值以1—r的速率递减,直到为零(r为两点之间的重组值)。若Δ值为零,则A,B随机关联;若Δ值为1,则A,月完全关联;若0<Δ<1,则非随机关联,即连锁不平衡。假如A位点代表易感基因,月位点代表遗传标记,如两者处于连锁不平衡,患者中遗传标记的频率将高于对照组人群。一旦在人群进化的某个阶段形成特定位置基因的连锁不平衡,由于致病基因与被研究遗传标记间的紧密连锁,连锁不平衡状态可以观察到很多代。但由于遗传漂变和选择产生的不平衡在不连锁的基因座将很快消失,而紧密连锁基因座之间的连锁不平衡消失很慢,因而研究一个遗传标记与疾病相关基因座之间的连锁不平衡,将有助于疾病基因的精细定位。
在定位克隆中,利用连锁可检查到那些产生连锁信号的变异。而在关联研究中,经由邻近位点形成的关联状态,我们将能检测到疾病的致病位点。因此,连锁不平衡在基因定位中具有非常重要的作用。多数情况下,即使有足够的统计学可信度的连锁证据,往往并无任何的染色体异常以及确凿的候选基因可供利用。对于这些疾病,往往都是利用连锁不平衡,进一步缩小定位区域,并在很多疾病研究中取得了成功。