ChemBio 3D软件如何添加序列编号
怎样在ChemBio 3D中给分子结构添加序列号

显示序列编号பைடு நூலகம்
(5)在模型窗口中右击,选择“Show Atom Symbols”命令(如下图左),原子上就可以出现 序列编号,如下图右所示:
选择“Show Atom Symbols”命令
(6)在模型窗口单击任意空白区,可消除所有原子和 键的选择:
温馨提示: 系列编号并不反映正常的顺序。这主要由于不是从最小的数值开始建立的。可以通过 以下方法进行重新排列: 选择第一个原子,右击选择“Replace with Text Tool”,在“Replace with Text Tool”框中 输入要开始的数(如图)。
ChemBio 3D能够方便的建立以及编辑各类三维的 分子结构模型,在化学、生物、医药领域有着广泛 的应用!下面就给大家介绍下如何在ChemBio 3D中 给分子结构添加序列编号和原子符号。
(1)单击选择工具。 (2)对角按动鼠标键把要选择的原子和键加上。 (3)此例中释放鼠标键,全部原子和键都将被选上。 (4)在模型窗口中右击,选择“Show Serial Numbers”命令(如下图左), 原子上就可以出现序列编号,如下图右所示:
选择“Replace with Text Tool”命令
通过上述教程,相信大家已经学会给ChemBio 3D模型 添加序列编号和原子符号。如需了解更多ChemBio 3D使用 教程请访问带你认识ChemBio 3D中的常见模型类型、 如何 利用符号建立ChemBio 3D模型、 如何利用键工具建立 ChemBio 3D模型。
综合序列分析软件BioEdit中文说明书

10
3.Features (特征):
想要增加一个特征,如抗生素抵抗标记,从“Vector” 菜单 选择“Add Feature”。 将显示以下对话框:
选择的类型是“Normal Arrow ”、“Wide Arrow ”、“Normal Box” 和、“Wide Box ”。在上面例子中的所有特征是“常规”宽 度的。如果特征是一个箭头,箭头的方向将是从起点位置到终点 位置。
增加特征或酶时,他们各自的标记增加在外面,中心是可能 的尺寸。标记可以被选择工具选择、移动、编辑和缩放。 11
4.General Vector properties
载体属性可通过选 “Vector” 菜单中的“Properties ”来更改 :
12
可以通过指定起点和末端位置,来增加多接头按 钮。多接头显示为“Courier New ”字体。
从“Sequence” 菜单下进入“Protein”, 再进入 “amina acid composition”, 可对序列的氨基酸组成分析, 结果以摘要和图例的形式给出。
图例中的柱形条表示每种氨基酸在序列中的摩尔 比,如下图:
20
以RGDV的minor outer capsid protein-AAS66885为例:
想要从一个DNA序列产生一个质粒,从“Sequence ”菜单中 “Nucleic Acid ”子菜单中选择“Create Plasmid from Sequence ” 选项。选择这个选项时,限制性内切酶图谱将会使用通常商业 化的,储存在存储器中的限制性内切酶。质粒第一次产生时, 它显示成有10个位点标记的圆圈,中央是标题 。
– 组成分析 – 熵图 – 疏水性轮廓 – 联配中搜寻保守区 – 根据密码子的使用翻译核苷酸
chembiodraw使用

ChemBioDraw和ChemBio3D中的NMR、IR 光谱预测功能ChemBioDraw和ChemBio3D软件均包含NMR、IR光谱预测工具作者:Jesse Gordon公司:CambridgeSoft版本:19.2CHEMBIODRAW、CHEMBIO3D软件中均包含NMR(核磁共振图谱)和IR (红外)光谱预测工具。
文本将介绍NMR和IR预测工具的主要功能和预测方法。
与本文相对应的还有一个录制好的视频文件,以实例演示使用技巧,实例中的样本均可下载供用户亲自实践。
本文中涉及的主题:∙ChemNMR:在ChemBioDraw中预测1H-NMR和13C-NMR∙在ChemBioDraw中设置NMR参数∙ChemBio3D中光谱预测的设置∙GAMESS(从头计算量子化学程序):在ChemBio3D中预测NMR、IR图谱CambridgeSoft的ChemBio3D软件在程序编写时整合了多个计算化学包,NMR 和IR预测模块就是已整合的计算化学包的一部分。
这两个模块加上GAMESS和MOPAC均已整合到ChemBio3D软件中,无需另外付费。
此外,ChemBio3D还为其他计算化学软件编写了接口,如Gaussian,Jaugar等。
ChemBio3D还能够整合其它的计算化学包-如果有其他版本的NMR或IR光谱预测,CambridgeSoft会在未来的版本中将其囊括。
在本文的结尾,附上对应的视频文件和ChemBio3D支持的计算化学包的介绍文章链接。
ChemNMR: 在ChemBioDraw中预测1H-NMR和13C-NMRChemNMR模块已整合在ChemBioDraw软件里—在绘制分子时候只需点击一个按键就能看到它的NMR图谱。
要预测1H-NMR和13C-NMR图谱, 首先需要选定目标化学结构式,然后在"Structure"菜单选择"Predict 1H-NMR Shifts"或者"Predict13C-NMR Shifts" 。
如何在NCBI提交序列
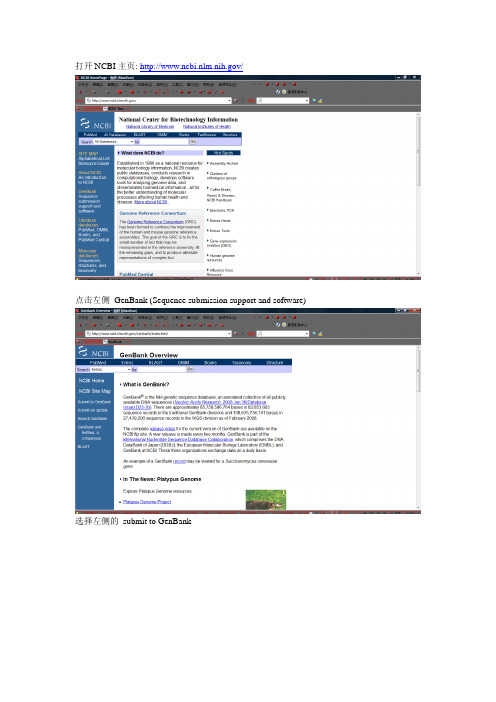
打开NCBI主页: /
点击左侧GenBank (Sequence submission support and software)
选择左侧的submit to GenBank
一般来说,有多种提交方法,Bankit是在线提交的,比较方便,如果不是比较长而且比较复杂的序列的话都可以用这种方法提交。
我们只讲该方法。
我们点击页面中蓝色的Bankit,
然后下拉页面到最底层
把你序列的长度填入这个方框里,然后按“New”按钮。
得到了这个界面,里面内容(contents)包括4点,你可以根据自己序列的情况适当进行选择填写,内容不明就先不要写,提交完后还是可以更新的。
这里提供的序列必须单一填写,每个序列有一个Bankit*******号码,这个要注意。
然后就可以根据下面的内容填写了,完毕后在最低下大框里填入序列,最后你可以save 你的这个格式,然后validate and continue。
如果出错就会有红色的提醒,需要重填,因为我没有序列所以就出现下面情况
不过我想这也说明这些信息是必填的,别的看来都不是很重要,您不妨按着这个思路去填,呵呵,祝您好运。
最后会给您发个邮件,等过几个月后如果没有问题就能得到您的登录号了,如果有问题他会
邮件和您联系,再见。
怎样通过输入原子符号来建立ChemBio 3D模型
怎样通过输入原子符号来建立ChemBio 3D模型
ChemBio 3D作为一款三维分子结构演示软件,能够轻松快捷地绘制各类抽
象的化学模型,更直观的为大家演示分子的立体结构!而在ChemBio 3D中有很多建立化学模型的方法,除了使用键工具和Replacement Text建立模型以外,还可以用Replacement Text框中输入原子符号(元素符号和数字)来建立模型。
今天就以4-甲基-2-戊醇模型来告诉大家如何通过输入原子符号来建立ChemBio 3D模型。
ChemBio 3D建立一个4-甲基-2-戊醇模型的具体操作步骤如下:
(1)点击Replacement Text框
打开Replacement Text框
(2)在 Replacement Text框中输入原子标记“(CH3)CH(CH3)
CH2CH2CH2” ,以碳链作为骨架,如下图所示:
在 Replacement Text框中输入原子标记
(3)在模型窗口双击,此时出现一个4-甲基-2-戊醇模型,见下图:
4-甲基-2-戊醇模型示例图
(4)从“Structure”菜单中选择“Clean Up”命令来定义模型。
Clean Up命令
(5)从“MM2”菜单,选择“Minimize Energy”来确定更准确的最小能量,然后单击“Run”。
精确最小能量
以上就是关于利用原子符号建立ChemBio 3D模型的全部步骤了,通过输入原子符号和数字来建立模型可以有效地降低绘图难度,提高绘图效率。
chemdraw使用说明--使用方法

4. Connection Table(*.ct):一种实例格式,存储关于原子与 元素、系列编号、X和Y轴、键序、键类型等的连接和联系的 列表,是用于多类应用程序间互换信息的通用格式。
5. DARC-F1 (*.fld):Questel DARC系统中存储结构的原本文 件格式。
文件格式介绍
6、DARC-F1 Query(*.flq):Questel DARC系统中存储查询的 原本文件格式。
7、ISIS Reaction(*.rxn):MDL开发的格式,用于存储元素 反应信息。
8、ISIS Sketch(*.skc):在Windows或Macintosh环境下,存 储并传输到另外的ISIS应用程序中。
默认打开 文件格式
默认存盘 文件格式
打开/存盘设置
国内外化学刊物默认文档格式
打开文件
不同文件格式
文件格式介绍
1. CD Template(*.ctp, *.ctr):用于保存模板文档。
2. ChemDraw(*.cdx):ChemDraw原本格式,是一个公共标记的 文件格式,易被其它程序建立和解释。
菜单栏
工具栏
主工具图标板 编辑区
状态/信息栏
滚动栏
主 工 具 图 标 板
套索 实键 双键 虚键 切割键 切割楔键 黑体键 黑体楔键 空心楔键 波浪键 表格 长链 环丙烷环 环戊烷环 环庚烷环 环己烷椅式 环戊二烯环
蓬罩(选取框) 橡皮 文本 笔 箭头 轨道 绘图元素 基元 化学符号 弧形 质询工具 模板 环丁烷环 环己烷环 环辛烷环 环己烷椅式 苯环
12、MSI ChemNote(*.msm):ASCII文本文件,可用于ChemNote 应用程序。
Chem3D使用说明

測量分子幾何形狀:元素及鍵長
將滑鼠移至任何原子後不 動,即可見此原子的基本 資料,如元素,編號 將滑鼠移至任何化學鍵後 不動,即可見此化學鍵的 基本資料,如鍵長, Bond Order
測量分子幾何形狀:鍵角及旋轉角
選按 按著Shift鍵不動,將滑鼠 移至任何連續 3 個原子並 點選之後不動,即可見此 鍵角的大小。 按著Shift鍵不動,將滑鼠 移至任何連續 4 個原子並 點選之後不動,即可見此 旋轉角的大小。
一非常複雜的偏微分方程式
建立分子PF3
選按 ,將滑鼠移至任何位置 按拖曳成一直線後,放開滑鼠 即成C2H6。 選按 選按任一碳原子,請連續按兩次 滑鼠左鍵;於分子模型上方的空 白視窗填寫「P」即磷原子。 選按另外一碳原子,請連續按兩 次滑鼠左鍵;於分子模型上方的 空白視窗填寫「F」即氟原子。 繼續改變其他兩個氫原子。
計算部分電荷
選按「Analyze」 「Extended Huckel Charges」 檢視計算結果必須按一下 ►,如此計算結果才會出 現另一視窗 試比較PF3, PCl3 , PBr3及 PI3 部分電荷的變化趨勢, 看看是否能解釋PX3的鍵 角變化。
改變旋轉角計算單點(定點)能量
將C-C-C-C的旋轉角改為150° 選按「MM2」「Compute Properties…」 「Steric Energy Summary」「Run」 將旋轉角度大小與Total的能量值拷貝至Excel。 依次將C-C-C-C的旋轉角改為180° , 150° , 120° , 90° ,60° , 30° , 0° 。 也將旋轉角度大小與Total的能量值拷貝至Excel。
chemical3D教程

Chapter 1 Introduction1.1启动Chem3D在开始(Start)菜单中点击所有程序(P), 选择弹出菜单中ChemOffice2004中的Chem3D Ultra 8.0即可启动程序1.2Chem3D 界面结构Chem3D软件的最大特点就是界面结构简单。
和ChemDraw类似,Chem3D 打开后的窗口包含:工作窗口, 信息窗口, 列表窗口, 工具栏, 菜单栏等, 如图1所示。
1.3图形工具栏图形工具栏包含所有能够在工作窗口绘制结构图形的工具,选择这些工具后,光标将随之改变成相应的工具形状。
工具板见图1-2其它的类似于ChemDraw,有兴趣的同学可以参考FTP所提供的英文参考资料Chapter 2 建模简要教程2.1 使用键工具建模首先打开工作窗口(File菜单– New Model)2.1.1 建模在工具栏选择单键工具将鼠标移动至工作窗口,按住鼠标左键拖动鼠标即可绘制简单的乙烷分子。
(注意:View-Settings-Model Build – Rectify 选择上后会自动为所绘制的分子结构加上氢原子)2.1.2 旋转模型:选择旋转工具可以在任意方向选择所绘制的分子。
1:将鼠标移动至工作窗口,按住鼠标左键2:在任意方向拖动鼠标可以旋转模型注意:当拖动鼠标时,会出现一个圆。
在圆内拖动鼠标使得模型绕X和Y轴旋转。
当鼠标在圆外时,模型绕Z轴旋转。
如图所示2.1.3 查看模型分子信息选择选择工具,将鼠标移动至相应原子位置将显示相应的原子序数,元素标识及原子类型。
如下图所示将鼠标移至C-C键上将显示键长及相应的键级在选择原子的同时,按住Shift键可以同时选择多原子,可以通过这样查看相应的键角和二面角。
1:选择C(1),C(2)和H(7)2:将鼠标移至任意的所选择的原子或键上将显示所选择的键角类似的操作可以用来显示二面角。
2.2 修改模型分子2.2.1 修改键类型1:选择双键工具2:按住鼠标左键,从C(1)的位置拖动鼠标至C(2)的位置将乙烷分子更改为乙烯分子。
Chem3D常用功能使用教程51822

带状模型(móxíng)常用于生物分子
11
精品PPT
代表用红蓝两种色彩(sècǎi)的立体显示
代表用深色显示模型,就是对于(duìyú)模型的 部分 按代观表察显示者两的个角立度体不结同构着完色全相同的模型
代表模拟现实的角度观察模型,使模型更
接近真实 代表显示模型的时候将后面的部分以阴影
形式表示,这样模型的立体感更强。
35
精品PPT
按【Enter】自动转化(zhuǎnhuà)为模3型6
精品PPT
括号(kuòhào)表示支链 37 精品PPT
38
精品PPT
整理(zhěnglǐ)结构
39
精品PPT
简单优化 观察分子(fēnzǐ)变化
40
精品PPT
4-甲基-2-戊醇模型(móxíng)
41
精品PPT
建立(jiànlì)1,2-双甲基环戊烷模型
12
精品PPT
13
精品PPT
建立3D模型 1. 利用键工具建立模型
选择【单键】工具(gōngjù),在模型窗口向右按动
圆柱(yuánzhù)键模型
14
精品PPT
线状模型(móxíng)
15
精品PPT
棒状模型(móxíng)
16
精品PPT
球棍模型(móxíng)
17
精品PPT
比例(bǐlì)模型
18
精品PPT
X、Y轴旋转 (xuánzhuǎn)指示
选择【轨迹球】工具,使分子模型(móxíng)
在模型(móxíng)窗口沿X、Y轴旋转
19
精品PPT
Z轴旋转(xuánzhuǎn) 指示
选择【轨迹球】工具并按“Alt”键, 使分子(fēnzǐ)模型在模型窗口沿Z轴旋转
chembiodraw使用
ChemBioDraw和ChemBio3D中的NMR、IR 光谱预测功能ChemBioDraw和ChemBio3D软件均包含NMR、IR光谱预测工具作者:Jesse Gordon公司:CambridgeSoft版本:19.2CHEMBIODRAW、CHEMBIO3D软件中均包含NMR(核磁共振图谱)和IR (红外)光谱预测工具。
文本将介绍NMR和IR预测工具的主要功能和预测方法。
与本文相对应的还有一个录制好的视频文件,以实例演示使用技巧,实例中的样本均可下载供用户亲自实践。
本文中涉及的主题:∙ChemNMR:在ChemBioDraw中预测1H-NMR和13C-NMR∙在ChemBioDraw中设置NMR参数∙ChemBio3D中光谱预测的设置∙GAMESS(从头计算量子化学程序):在ChemBio3D中预测NMR、IR图谱CambridgeSoft的ChemBio3D软件在程序编写时整合了多个计算化学包,NMR 和IR预测模块就是已整合的计算化学包的一部分。
这两个模块加上GAMESS和MOPAC均已整合到ChemBio3D软件中,无需另外付费。
此外,ChemBio3D还为其他计算化学软件编写了接口,如Gaussian,Jaugar等。
ChemBio3D还能够整合其它的计算化学包-如果有其他版本的NMR或IR光谱预测,CambridgeSoft会在未来的版本中将其囊括。
在本文的结尾,附上对应的视频文件和ChemBio3D支持的计算化学包的介绍文章链接。
ChemNMR: 在ChemBioDraw中预测1H-NMR和13C-NMRChemNMR模块已整合在ChemBioDraw软件里—在绘制分子时候只需点击一个按键就能看到它的NMR图谱。
要预测1H-NMR和13C-NMR图谱, 首先需要选定目标化学结构式,然后在"Structure"菜单选择"Predict 1H-NMR Shifts"或者"Predict13C-NMR Shifts" 。
生成序列号功能说明

如何生成序列号?标牌制作行业,经常需要制作一组格式相同的编号,为了减少用户排版时间,我们可以使用自动流水号功能,也就是JDPaint4.11及以前的版本提供的“生成流水号”功能。
新增的“生成序列号”功能是该功能的拓展,它具有以下几个特点:●严格保留基准字串的特点,如字体的旋转、弧排、竖排等;●具有选号功能,删除一些不喜欢的编号,如“110、114、13”等;●具有自动前缀功能,可以在所有号码前增加“No.、R、F”等标志;●具有号码预览编辑功能,可以删除、编辑、添加号码;●具有号码输入功能,可以从文本文件中输入号码,比如人名、地名、电话号码都可以用此方式;下面我们详细介绍该功能的几种使用方法。
1.不带基准串的序列号基本的操作过程如下:步骤一、启动菜单功能{专业功能::标牌广告::生成序列号}功能,系统弹出对话框:步骤二、在“排列”一栏输入行列数目及距离。
比如,我们需要排列23个序号,先横排后竖排,总共5列,那么序号的排列此序如下:同样的号码,如果先竖排后横排,总共5行,那么序号的排列此序如下:步骤三、在“序列号设置”一栏输入序列号的产生方法。
比如我们需要生成23个号码,起始号码是1000,相邻号码间隔2,点击“计算序列号”按钮,系统计算获得所需的号码,并显示在最右边的列表框中,如下图所示:步骤四、点击“确定”按钮,系统生成系列号。
如下图所示。
2.带基准串的序列号如果需要按照现有的字串格式排列序列号,必须首先绘制标准字串,如下图所示:基本的操作过程的前三步与不带基准串的完全一致,下面从底然后执行如下步骤:1)选择“生成序列号”功能,设置“排列”、“序列号设置”参数,计算出序列号。
2)点击“选取基准字串”按钮,对话框隐藏,系统提示拾取基准字串,我们选择预先排好的字串,对话框显示;3)点击复选框“保留基准字串”,不要保留基准字串。
4)点击“指定起始位置”按钮, 对话框隐藏,系统提示指定字串左下角的位置。
ChemBioOffice教程的学习

计算化学实验Chem3D软件指南Chem3D开始:点击桌面“Chem3D Ultra 10.0”图标Chem3D一览菜单和工具栏区组成区坐标区光谱区输出结果区构象区2D分子区3D绘图区Chem3D详解菜单和工具栏区文件打开输入打印设置编辑拷贝粘贴修改选择显示工具结构测量旋转饱和修正重叠计算界面结果密度面轨道电势面分子面电影旋转轨迹数据库窗口排列帮助说明例子Chem3D详解文件新分子模型样本分子打开文件中的分子结构保存Chem3D详解编辑撤消操作重复操作清除当前分子Chem3D详解视图分子显示方式显示取向工具栏分子组成ChemDraw直角坐标Z-矩阵测量列表参数表输出区构象区光谱显示区文件输入原子类型MM2类型Chem3D详解结构测量列表分子位置和取向设置Z-matrix饱和价键(加氢)自动调整分子结构重叠分子对接Chem3D详解计算模块计算结果管理停止计算二面角计算(构象搜索)EHMO计算电荷轨道MM2分子动力学模块分子性质计算模块GAMESS计算界面Gaussian计算界面Jaguar计算界面Mopac计算界面Chem3D详解计算模块二面角计算(构象搜索)扫描一个二面角扫描两个二面角二面角计算(构象搜索) 扫描一个二面角二面角计算(构象搜索) 扫描二个二面角构象过渡态Chem3D详解计算模块EHMO电荷分子面总电子密度面分子轨道Chem3D详解计算模块EHMO电荷Chem3D详解计算模块EHMO总电子密度面等值面图isoval=0.01Chem3D详解计算模块EHMO分子轨道等值面图isoval=0.01MM2分子动力学模块Chem3D详解计算模块运行MM2输入文件优化构型(能量极小化)分子动力学性质计算打印力场参数MM2分子动力学模块Chem3D详解计算模块力场参数M2 Constant Value QualityCubic stretch constant-2.000 4Quarticstretch constant2.333 4X-B,C,N,O-Y Stretch-Bend interaction force constant0.120 4 X-B,C,N,O-H Stretch-Bend interaction force constant0.090 4 Sexticbending constant (* 10**8)7.000 4Cutoff distance for charge/charge interactions35.000 4Cutoff distance for charge/dipole interactions25.000 4Cutoff distance for dipole/dipole interactions18.000 4Cutoff distance for vander Waalsinteractions10.000 4MM2 Atom RadiusEps WeightReduct Lone Pairs Quality 5 1.500 0.047 1.008 0.915 0 4 2 1.940 0.044 12.000 0.000 0 4 Bond KS Bond Length Dipole Quality2-5 4.600 1.100 0.000 42-2 9.600 1.337 0.000 4Angle KB XR2 XRH XH2 Quality 2-2-5 0.360 120.0 120.5 0.0 42-2-2 0.430 120.0 0.0 0.0 4Atoms Force Constant Quality2-2 0.050 42-5 0.050 4Torsional V1 V2 V3 Quality5-2-2-5 0.000 15.000 0.000 42-2-2-5 0.000 9.000 -1.060 42-2-2-2 -0.930 8.000 0.000 4PiAtom Electron Ionization Repulsion Quality 2 1 -11.160 11.134 4PiBond DForce DLength Quality2-2 4.600 0.166 43, 4次键伸缩常数键伸缩-弯曲作用参数截断距离LJ非键参数键伸缩参数键弯曲(角)参数键扭转参数π键(共轭)修正参数MM2分子动力学模块Chem3D详解计算模块优化构型(能量极小化)收敛要求: 最小均方根梯度(力)输出结果能量分解总能量MM2分子动力学模块Chem3D详解计算模块分子动力学模拟步长结构输出间隔轨迹步数升温速率目标温度参数一览MM2分子动力学模块Chem3D详解计算模块分子动力学模拟暂停/开始停止MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果时间(10-15秒)总能势能温度MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果分子平动能= 0分子转动能= 0非平衡平衡态MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果总能守恒!温度控制!MM2分子动力学模块Chem3D详解计算模块π键键级能量Chem3D详解计算模块分子性质计算模块Chem3D详解计算模块GAMESS优化构型优化过渡态分子性质频率分析红外光谱NMR光谱创建输入文件查看输出结果文件运行输入文件Chem3D详解计算模块GAMESS作业类型方法基组波函数类型基组极化函数基组扩散函数基组指数优化方法限制性部分优化坐标自旋多重度= 单电子数+1净电荷Chem3D详解计算模块GAMESS高级设置-1最大SCF循环次数优化最大步数收敛要求(力)温度(仅用于热力学计算)溶剂模型溶剂参数初始猜测方法分子对称性点群内存要求Chem3D详解计算模块GAMESS性质偶极电子密度静电势Lowdin电荷和布居分析Mulliken电荷和布居分析电子势能电子总能量电子动能Chem3D详解计算模块GAMESS计算方法激发态从头算理论密度泛函理论基态Chem3D详解计算模块GAMESS等电子密度面(0.001)HF/STO-3G IR光谱苯甲醇NMR光谱Mopac计算界面Chem3D详解计算模块优化平衡构型优化过渡态分子性质IR光谱Mopac计算界面Chem3D详解计算模块作业类型方法波函数优化方法溶剂模型限制性优化收敛要求坐标Mopac计算界面Chem3D详解计算模块电荷偶极静电势生成焓超精细耦合常数电离能电荷溶剂介电常数Chem3D详解分子面模块分子面探测半径显示模式颜色模式分辨率选择分子轨道等值面值溶剂面总电子密度面总自旋密度面静电势面分子轨道清除所有面Chem3D详解分子面模块SA面总电子密度面isoval=0.01总电子密度面/静电势isoval=0.01静电势isoval=1Connolly面HOMO轨道LUMO轨道甲醇Chem3D详解工具栏新分子打开保存拷贝剪切粘贴恢复重复印模型背景特效红蓝镜像坐标原子符号原子编号残基全屏电影播放选择平移旋转缩放移动选中键型清除饱和调整符输入擦写分子面探测半径显示配色分辨率分子轨道等值状态优化旋转停止Chem3D详解文件格式晶体结构文件Gaussian输入输出文件cif文件pdb文件(蛋白质)Chem3D详解晶体结构文件有机分子晶体: CCDC数据库, 草酸晶体Pbca空间群(61)OXALAC06.datChem3D详解晶体结构文件无机晶体: ICSD, AMCSDAmerican Mineralogist Crystal Structural Database http://rruff.geo.arizona. edu/AMS/amcsd.php不能正确处理周期性的体系!!Chem3D详解生物大分子pdb文件(蛋白质)RSCB PDB蛋白质晶体结构数据库/pdb/home/home.do1AAQChem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件需要把Gamess输出文件修改一下才能用Jmol5.0正确读出!将这些含“REDUCED MASSES”的行全部删除! 然后保存退出此处只留一个空行!Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件旋转分子缩放平移选择测量轨迹/频率Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件打开修改后的GAMESS输出文件*.out打开“extras”→“vibrate ...”Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件选择频率观察“绕CO键的内转动”。
CHEMBIO教程:利用键工具建立3D模型

ChemBio教程:利用键工具建立3D模型作为一款专业的三维分子结构演示工具,ChemBio 3D 能够轻松快捷地建立和编辑化学模型,在化学、生物领域应用广泛。
ChemBio 3D可以通过键工具、子结构和符号等方式建立模型,可根据具体情况选用建立方式。
利用键工具建立ChemBio 3D模型主要通过菜单栏中的键工具。
ChemBio 3D Ultra 14是ChemBio 3D的最新版本,增添了新的绘图工具。
本教程将以乙烷模型为例教授大家如何利用键工具建立ChemBio 3D模型。
乙烷(ethane)是烷烃同系列中第二个成员,为最简单的含碳-碳单键的烃。
ChemBio 3D建立乙烷模型的具体步骤如下:(1)选择【单键】工具。
(2)用鼠标键指向模型窗口。
(3)按下鼠标键,再向右按动并释放鼠标键。
一个带有相匹配的氢的乙烷模型即可建立。
如下图所示:
乙烷模型示例图
【温馨提示】氢原子已经自动加入,称做“Automatic Rectification”并被控制在Building 控制板上。
释放鼠标键后,注意模型如何重新适合模型窗口的位置,是由Building控制板上的“Automaticlly Fit Model to Window”复选项控制的。
通过上述教程,相信大家已经掌握ChemBio 3D建立乙烷模型的具体方法,学会如何利用键工具建立ChemBio 3D模型。
chemdraw使用说明--使用方法

主 工 具 图 标 板
箭 头 工 具 轨道工具
键工具
基元 工具
画 图 工 具
符号工具 弧形工具 模板工具17类 原子反应工具
模板工具
氨基酸模板工具
模板工具
芳香化合物模板工具
模板工具
双环模板工具
模板工具
生物模板工具
模板工具
玻璃仪器模板工具(I)
模板工具
玻璃仪器模板工具(II)
模板工具
构象异构体模板工具
套索 结构透视
主 工 具 图 标 板
注:红色为8.0 版本新增工具
实键 双键 虚键
切割键 切割楔键 黑体键 黑体楔键 空心楔键 波浪键 表格 长链 环丙烷环 环戊烷环 环庚烷环 环己烷椅式 环戊二烯环
蓬罩(选取框) 质谱碎片 橡皮 文本 笔 箭头 轨道 绘图元素 基元 化学符号 质询工具 TLC工具 模板 环丁烷环 环己烷环 环辛烷环 环己烷椅式 苯环
光标块
常 用 术 语
拖动: 由三部分动作组成——按下鼠标左键选择对象,移动鼠标, 将被选的对象移动到指定位置后抬起鼠标; “特殊键+单击”:
按下特殊键和单击鼠标左键同时进行。如,“Shift+单击” 的意思是按下”Shift”键和单击鼠标键;
“特殊键+拖动”: 按下特殊键和鼠标左键,移动鼠标。如,“Shift+拖动”的
意思是按下”Shift”键和拖动鼠标光标。
菜单栏
工具栏
主工具图标板
ቤተ መጻሕፍቲ ባይዱ
编辑区
滚动栏
状态/信息栏
主 工 具 图 标 板
套索 实键 双键 虚键 切割键 切割楔键 黑体键 黑体楔键 空心楔键 波浪键 表格 长链 环丙烷环 环戊烷环 环庚烷环 环己烷椅式 环戊二烯环
使用自动编号功能进行列表排版

使用自动编号功能进行列表排版自动编号功能是一项非常实用的排版工具,它可以帮助我们在文章中创建规范的列表,使得文章结构清晰、易于阅读。
本文将介绍如何使用自动编号功能进行列表排版,并为大家提供详细的操作步骤和注意事项。
一、自动编号的基本用法在使用自动编号功能之前,我们首先需要确保打开了适当的软件或编辑器,例如Microsoft Word或Google Docs等。
接下来,按照以下步骤进行操作:1. 插入列表:定位光标到你想要插入自动编号列表的地方,然后点击软件工具栏上的“插入列表”图标。
一般来说,该图标通常是三个数字或者符号组成的列表图标。
2. 选择编号类型:当你点击“插入列表”图标后,会出现一个弹出窗口或下拉列表,其中包含了不同的编号类型供你选择。
这些类型可能涵盖了阿拉伯数字、大写字母、小写字母、罗马数字等多种形式。
3. 调整样式:根据自己的需求,你可以选择调整编号的样式,例如字体大小、颜色、缩进等。
不同的软件或编辑器提供的样式选项会有所不同,可以根据具体情况进行调整。
4. 编辑列表内容:在列表中开始输入文本时,你会发现光标会自动识别并进行编号。
你可以按照需要添加、删除或编辑列表项,自动编号会根据你的操作进行实时调整。
二、自动编号功能的高级用法除了基本的自动编号功能外,一些软件或编辑器还提供了更多的高级用法,帮助我们进一步优化列表排版效果,提高文档可读性和美观度。
以下是一些常见的高级用法:1. 多级编号:有时候我们需要创建多级编号列表,例如分章节的论文、多层次的大纲等。
在这种情况下,我们可以通过调整编号层级来实现。
一般来说,多级编号功能可以在软件或编辑器的工具栏上找到,你可以根据需要进行设置。
2. 使用自定义编号:除了预设的编号类型外,一些软件或编辑器还允许我们自定义编号格式。
这对于特定的排版要求非常有用,例如某些技术文档或法律文件中的特殊编号规范。
你可以在软件或编辑器的设置中找到自定义编号的选项,并按照要求进行设置。
ChemBioDraw“连接点”工具的绘制技巧

ChemBioDraw“连接点”工具的绘制技巧ChemBioDraw是一款专业的化学绘图软件,若想利用ChemBioDraw取代基属性进行搜索查询就需要使取代基框中取代基子结构标记应与父结构用连接点相连接,要标记连接点,使用【连接点】工具,本节ChemBioDraw教程将给您介绍如何使用连接点工具。
更多相关绘制教程请访问chemdraw中文官网获取。
一、子结构的连接点1、选择【连接点】工具;2、在子结构要放置连接点的位置上(下图左一蓝色阴影处)单击,单击后该位置会出现一个连接点编号;3、对所有子结构重复以上操作。
给子结构分配连接点二、连接点的编号如果在Preferences对话框中选择了Building/Display下的Show Attachment Rank Indicators(显示等级指示号),将会在文档窗口看见连接点编号。
这些编号对应ChemBioDraw取代基父结构上的键等级数,指示子结构是如何连接在父结构上。
对于子结构而言,连接点编号与键形成一个组合键。
当建立一个已经含有定义的ChemBioDraw取代基的父结构(化合物),再单击【连接点】工具时,取代基标记的附近会自动出现一个等级数(下图右一红框内)。
键等级数与定义中连接点的编号是一致的。
下图左一为父结构,中间为取代基框,右一为等级数出现。
连接等级指示器号和取代基框内的编号一致三、F1查询文件在ChemBioDraw 查询结构中,完成了的查询可被保存为F1查询文件。
F1查询文件中未在ChemBioDraw取代基框中的将被解释为G0。
以G-基团为例,如果取代基标记不是G1、G2、G3等,在写到F1查询格式时将被重命名。
以上就是如何使用连接点工具内容的ChemBioDraw教程,希望对您的学习过程有帮助,若您能有所获益将不甚欣慰。
ChemOffice使用指导

键完成原子序号的更改。若原子的三维结构中显示孤对电子,执行关闭 “Tools”菜单中的“Show H’s ans Lp’s”命令,可不显示氢原子和孤对电子。
3.3ChemDraw结构式与3D模型的转换
(1)ChemDraw结构式转换为3D模型
(3)链工具
① 链的绘制
选取长链工具,在需要进行的连接原子或环上点击或拖动可产生链; 同时弹出链长对话框,可以选择增长链的长度。
图4-11链长对话框
② 链属性的设置
(4)化合物结构绘制示例
CH3 CH3 CH3 H3C CH3
H3C
CH3
CH3
CH3 H3C
图4-12 胡萝卜素结构
图4-13 连接点及单键的产生
“File”菜单命令
图4-3 文件命令的下拉菜单
•框
(2)
主 工 具 图 标 板
图 4-5主工具图标板
主 工 具 图 表 板
图 4-6主工具图表板的子图标板
2.2模板
图4-7 模板工具图标板示例
2.3绘制与编辑典型化学物质结构式
(1)键工具:
主工具图标板上提供了九个键操作的命令,其中双 键命令板中的子菜单中还包 括12个键命令。利用键工
2 化学结构绘图软件ChemDraw
ChemDraw 软件功能十分强大,可以编辑、绘制与化学
有关的一切图形,如建立和编辑各类分子式、方程式、结 构式、立体图形、对称图形、轨道等,并能对图形进行编
辑、翻转、旋转、缩放、存储、复制、粘贴等多种操作。
用它绘制的图形 可以直接复制粘贴到Word 软件中使用。 最新版本的软件还可以生成分子模型、建立和管理化学信
Chem3D常用功能使用教程

12
13
建立3D模型 1. 利用键工具建立模型
选择【单键】工具,在模型窗口向右按动
圆柱键模型
14
线状模型
15
棒状模型
16
球棍模型
17
比例模型
18
X、Y轴旋转指示
选择【轨迹球】工具,使分子模型 在模型窗口沿X、Y轴旋转
ChemDraw中 画出的分子结构
复制粘贴到Chem3D中 64
Chem3D中 画出的分子模型
复制粘贴到 ChemDraw中
65
计算功能演示
66
67
68
69
70
71
72
73
View/Solvent Accessible 菜单 甲苯结构
74
75
76
77
78
79
80
(4) 检查结构能量
距离
24
模型的键长数据 25
模型的键长和键角数据 26
乙烯模型中的双键信息 27
键级改动
28
显示氢及孤对电子
29
不显示氢及孤对电子的环己烷
原子名称及序数
30
选择工具,双击原子, 按顺序改变原子序号
31
居中 移动到 色彩调节 显示元素符号 显示元素编号 显示实心球 显示点阵表面
调整键长 破坏键 调整键级 就近结合 添加质心 颠倒 调整矩阵
从头算程序:Gamess来自Gaussian需另外安装这两个程序。
半经验或分子力学计算
Mechanics
PM3)
Mopac (Am1, Mindo/3, Mndo,
ChemBioOffice 13.0 十大新功能

ChemBioO ce 13十大新功能全新发布的 ChemBioOf ce ® 13,增加了更多人性化的功能,进一步提高了科学家的工作效率。
新版本通过结合可视化分析,能将生物活性与化学结构关联,协助科学家更深入地了解研究结果。
1. 升级的生物聚合物绘制工具栏,包括 D-型及 L-型氨基酸、β-氨基酸、DNA 及 RNA 模板全新的生物聚合物工具栏可以绘制肽链、DNA 及 RNA 的序列,包括 β-氨基酸及 D-氨基酸、二硫键、内酰胺以及连接基团和保护基团。
用单字母或三字母方式代表多肽及核苷酸序列(DNA 、RNA ),包括 L-氨基酸、D-氨基酸及 β-氨基酸。
用户能方便地在单字母及三字母表示方法之间互换,并可以将缩写字母展开为结构。
可以将连接基团及保护基团预先自定义为字母,简化绘制过程。
使用“键工具”绘制二硫键及内酰胺,以及环肽结构。
2. 在一个氨基酸序列中绘制混合序列结构、连接基团及保护基团为含有氨基或羟基的氨基酸(如 Arg ,Aad )添加保护基团,例如 Trt 及 Fm oc 。
这些保护基团的结构也可以展开或者恢复成缩写形式。
可通过键合序列的末端将多个序列合并为一个序列。
3. 从外部文档中粘贴化学序列支持 FASTA 格式粘贴序列。
将代表聚合物的单字母或者三字母字符串进行粘贴,系统可通过有效的分隔符(space 、tab 、dash 键)自动识别。
4. 升级的 clean -up 功能,可用于分子、反应及生物聚合物的结构修正使用升级的 clean-up 功能修正高质量且结构正确的分子式、反应式及生物聚合物。
序列绘制中新添加的残基会自动调整。
当序列从结构式转变到以字母形式表示,Clean-Up Biopolym er 命令可以调整并重新排布序列,使残基在一条直线上并自动换行。
Richard J. Smith 博士,副主编,Verlag Helvetica Chimica ActaJeremiah J. Gassensmith 博士,美国西北大学ChemBioO ce 长期用户“新的生物聚合物工具栏在绘制多肽及DNA 序列上帮了一个大忙。
如何编辑ChemBioDraw中的列标记

如何编辑ChemBioDraw中的列标记
元素“列标记”可以更加简明地定义搜索查询,通过标记原子为元素“列标记”,可指定“列”中的一个原子与搜索的结构相匹配。
“列标记”中的元素必须用逗号分隔,括号中的元素是可选的。
每个逗号后面的空格也是可选的。
本教程将以ChemBioDraw软件中的元素列标记为例讲解ChemBioDraw查询结构的使用技巧。
被标记的元素只能是原子元素,包括氘和氚。
要建立一个列标记的具体方法如下:
(1)打开一个原子标记的“文本”框。
(2)输入一个左中括号([),接着是以逗号分隔的元素“列标记”(Cl,Br,I),然后是一个有种括号(]),如下图所示:
建立一个列标记
实例:建立一个查询结构,找到符合下列标准的化合物。
(1)非氧硫族元素连接在另一个原子上。
(2)A不一定是碳。
(3)硫族元素和其他原子之间的键类型是单键或双键。
查询结构以列标记表示的实例
温馨提示:A标记表示该原子可以是氢以外的任何原子。
键附近的标记指示这个键有特殊的属性;这里,在键属性对话框中,指定键类型必须是单键或双键,S/D。
最后,通过输入括号包围的S,Se,Te形成的“列标记”,指定目标结构的元素必须有一个与之相匹配。
通过上述教程,相信大家已经学会使用ChemBioDraw软件编辑元素列标记,掌握相关的ChemBioDraw查询结构技巧。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
ChemBio 3D软件如何添加序列编号
ChemBio 3D软件能够轻松快捷地建立和编辑化学模型,在化学、生物、医药领域应用广泛。
本教程将向大家介绍如何给ChemBio 3D模型添加序列编号和原子符号。
ChemBio 3D软件添加序列编号和原子符号的具体步骤如下:
(1)单击选择工具。
(2)对角按动鼠标键把要选择的原子和键加上。
(3)此例中释放鼠标键,全部原子和键都将被选上。
(4)在模型窗口中右击,选择“Show Serial Numbers”命令(如下图左),原子上就可以出现序列编号,如下图右所示:
显示序列编号
(5)在模型窗口中右击,选择“Show Atom Symbols”命令(如下图左),原子上就可以出现序列编号,如下图右所示:
选择“Show Atom Symbols”命令
(6)在模型窗口单击任意空白区,可消除所有原子和键的选择:
温馨提示:
系列编号并不反映正常的顺序。
这主要由于不是从最小的数值开始建立的。
可以通过以下方法进行重新排列:
(1)选择第一个原子,右击选择“Replace with Text Tool”,在“Replace with Text Tool”框中输入要开始的数(如1)。
(2)按需要的顺序单击每一个原子。
即可重新确定系列编号。
选择“Replace with Text Tool”命令
通过上述教程,相信大家已经学会给ChemBio 3D模型添加序列编号和原子符号。
如需了解更多ChemBio 3D使用教程请访问带你认识ChemBio 3D中的常见模型类型、如何利用符号建立ChemBio 3D模型、如何利用键工具建立ChemBio 3D模型。