RNAseq (lncRNA) -materials and methods
Tn5转座酶建库原理

Tn5引爆极速建库和长片段之战近10年间,NGS(Next Generation Sequencing)技术高速发展,测序仪器不断更新迭代,形成规模化。
在测序模式产业化的大环境下,测序样本制备成为其中很重要的一环。
样本起始量要求过高、建库流程繁琐等都会限制NGS技术的应用。
科学家们一直在不断探索,试图突破此类限制,其中转座酶(Transposase)技术引入建库中就是很大的进步,不仅解决了上述问题,转座酶也在NGS上开发了多个技术应用,拓展了NGS的适用范围。
20世纪40年代,美国遗传学家芭芭拉.麦克林托克在玉米的研究中发现了转座子(Transposon),随后,她花了整整6年的时间来研究转座子的奥秘。
在50年代,当麦克林托克向世人公布她的发现时,学术界并没有多少人认可,但是真理是经得起时间检验的,随后的几十年间科学家们在生物界各个领域证实了转座子系统的广泛存在。
1983年,瑞典皇家科学院诺贝尔奖金评定委员会终于把该年度的生理学和医学奖授予这位81岁高龄的、不屈不挠的女科学家。
麦克林托克是在遗传学研究领域第一位独立获得诺贝尔奖的女科学家,她的名字将和转座基因一起被载入科学史册。
转座基因俗称跳跃基因,它的发现改变了人们对基因组序列稳定性的认识。
理论上的突破也带来了应用上的拓展,科学家们群策群力,将转座子系统开发成基因研究的各种工具,这又反过来促进了遗传学理论的研究。
转座子标签(transposon tagging)技术是研究功能基因的有效工具之一。
比如模式植物大多都有很好的突变体库,其中玉米的两个应用最为广泛的突变体库(uniformMu和Ac/Ds突变体库)就是利用转座子创建的。
转座子标签技术克隆基因的基本原理:转座子是染色体上一段可移动的DNA片段,它可从染色体的一个位置跳到另一个位置。
当转座子跳跃而插入到某个功能基因时,就会引起该基因的失活,并诱导产生突变型。
通过遗传分析可确定某基因的突变是否由转座子引起,由转座子引起的突变便可以转座子DNA为探针,从突变株的基因组文库中钓出含该转座子的DNA片段,并获得含有部分突变株DNA序列的克隆,进而以该DNA为探针,筛选野生型的基因组文库,最终得到完整的基因。
Tn 转座酶建库原理

Tn5引爆极速建库和长片段之战近10年间,NGS(Next Generation Sequencing)技术高速发展,测序仪器不断更新迭代,形成规模化。
在测序模式产业化的大环境下,测序样本制备成为其中很重要的一环。
样本起始量要求过高、建库流程繁琐等都会限制NGS技术的应用。
科学家们一直在不断探索,试图突破此类限制,其中转座酶(Transposase)技术引入建库中就是很大的进步,不仅解决了上述问题,转座酶也在NGS上开发了多个技术应用,拓展了NGS的适用范围。
20世纪40年代,美国遗传学家芭芭拉.麦克林托克在玉米的研究中发现了转座子(Transposon),随后,她花了整整6年的时间来研究转座子的奥秘。
在50年代,当麦克林托克向世人公布她的发现时,学术界并没有多少人认可,但是真理是经得起时间检验的,随后的几十年间科学家们在生物界各个领域证实了转座子系统的广泛存在。
1983年,瑞典皇家科学院诺贝尔奖金评定委员会终于把该年度的生理学和医学奖授予这位81岁高龄的、不屈不挠的女科学家。
麦克林托克是在遗传学研究领域第一位独立获得诺贝尔奖的女科学家,她的名字将和转座基因一起被载入科学史册。
转座基因俗称跳跃基因,它的发现改变了人们对基因组序列稳定性的认识。
理论上的突破也带来了应用上的拓展,科学家们群策群力,将转座子系统开发成基因研究的各种工具,这又反过来促进了遗传学理论的研究。
转座子标签(transposon tagging)技术是研究功能基因的有效工具之一。
比如模式植物大多都有很好的突变体库,其中玉米的两个应用最为广泛的突变体库(uniformMu和Ac/Ds突变体库)就是利用转座子创建的。
转座子标签技术克隆基因的基本原理:转座子是染色体上一段可移动的DNA片段,它可从染色体的一个位置跳到另一个位置。
当转座子跳跃而插入到某个功能基因时,就会引起该基因的失活,并诱导产生突变型。
通过遗传分析可确定某基因的突变是否由转座子引起,由转座子引起的突变便可以转座子DNA为探针,从突变株的基因组文库中钓出含该转座子的DNA片段,并获得含有部分突变株DNA序列的克隆,进而以该DNA为探针,筛选野生型的基因组文库,最终得到完整的基因。
要了解单细胞测序技术,这8篇经典综述一定要看哦!

要了解单细胞测序技术,这8篇经典综述⼀定要看哦!今天我们继续介绍单细胞测序技术,这是“技术篇——单细胞测序”系列⽂章的第6期了,前⾯5期我们分别从:(1)科研热度(⽤这个技术发表的⽂章中,每4篇中就有1篇10分+,还不赶快了解⼀下!);(2-3)使⽤单细胞测序发表⾼分杂志的两个案例(⼀篇肿瘤领域的Science和⼀篇⼼⾎管领域的Circulation);(4)单细胞测序技术的常见问题(关于这个⾼⼤上技术的问题,这篇⽂章早就总结好了!);以及(5)如果我们经费预算不够,可以使⽤的两个数据库:⾃⼰做不起单细胞测序,别担⼼有数据库可以⽤啊!。
今天我们把中间遗漏的⼀期:单细胞测序的经典综述补上。
正好是假期,如果你不看球,可以静下⼼来看看⽂章。
废话不多说,我们直接上⽂献:1. Unravelling biology and shifting paradigms in cancer with single-cell sequencing.Nat RevCancer. 2017 Aug 24;17(9):557-569.这篇去年8⽉份发表在Nat Rev Cancer杂志上的综述主要从单细胞测序在肿瘤研究中的应⽤来展开,综述了单细胞测序的技术、在研究肿瘤异质性和基因组进化中的作⽤,并以肺癌为例说明了单细胞测序的应⽤场景:Single-cell sequencing for the decomposition of heterogeneous cellular populations and theanalysis of rare cells in lung cancerSingle-cell sequencing can be leveraged to study cancergenetics and biology at all stages ofdisease development.2. Single-cell RNA sequencing for the study of development, physiology and disease.Nat Rev Nephrol. 2018 May 22. doi: 10.1038/s41581-018-0021-7.今年5⽉22号发表在Nat Rev Nephrol杂志上的综述,主题是单细胞RNA测序在发育、⽣理和疾病研究中的应⽤,本⽂介绍了单细胞测序技术的流程、不同单细胞捕获⽅法、数据分析流程和软件,以及应⽤等等。
微生物-扩增子测序

· 属水平物种进化树
为了进一步研究属水平物种的系统进化关系,通过多序列比对得到 top100属的代 表序列的系统发生关系,并结合每个属的相对丰度进行整合展示,由此可以得到这 些属的系统进化关系[3]。
组间群落结构差异显著性检验 Adonis 和 amova 分析
组间群落结构差异显著性检验在 Anosim 和 MRPP 分析的基础上,新增 adonis[4] 和 amova[5] 分析, 深度探索专属于您的样本类型的组间差异检验方法,全方位为您的数据保驾护航。
加量不加价;
福利 二
环境因子关联分析由高级分析免费 升级为标准分析,同时标准分析里 更有众多主流分析内容的增加;
升级内容 数据量
物种注释与统计 组间群落结构差异显著性检验
环境因子关联分析 (高级分析升级为标准分析)
Network 分析
升级点展示 分析点 3W免费升级为5W 5W 免费升级为10W GraPhlAn 图 ternaryplot(三元相图)分析 属水平物种进化树 Adonis 和 amova 分析 Spearman 相关性分析 Mantel test 分析 CCA/RDA 分析 VPA 分析 共发生网络图
升级亮点 深入挖掘低丰度新奇、稀有的物种信息。
让您样本中的优势物种直观显现。 三个样本(分组)间优势物种差异的最佳选择。
前列腺癌转录组亚型(中文)

前列腺癌转录组亚型(中文)Prostate Cancer Transcriptomic Subtypes虽然肿瘤的 DNA 通常等同于指纹或其独特的遗传识别,但肿瘤的RNA 谱代表了一种复杂的动态状态,更类似于肿瘤的个性或独特的行为。
在 11 种 RNA 中,前列腺癌的转化和临床重点主要集中在 mRNA 和 lncRNA。
基于 RNA 的生物标志物最常见的用途是评估肿瘤的侵袭性或治疗敏感性。
然而,已经开发了多个基因表达特征来捕获由规范DNA 改变导致的功能状态,包括 ERG 融合、SPOP 突变和 Rb 丢失。
更常见的是,这些生物标志物已被用于开发 30 多种预后基因表达特征,其中三种现已商业化,并越来越多地纳入临床试验。
在平行下,使用微阵列和RNAseq 技术的能力允许使用高通量方法进行全转录组分析。
这使得能够发现和训练植根于生物学信息通路的众多预测性生物标志物特征,以确定哪些肿瘤对各种治疗(包括雄激素剥夺治疗、放疗、化疗和 PARP 抑制)更敏感或更耐药。
本章将回顾各种类型的 RNA、可用于评估基因表达的技术,并描述前列腺癌的可用基因表达特征。
这使得能够发现和训练植根于生物学信息通路的众多预测性生物标志物特征,以确定哪些肿瘤对各种治疗(包括雄激素剥夺治疗、放疗、化疗和 PARP 抑制)更敏感或更耐药。
本章将回顾各种类型的 RNA、可用于评估基因表达的技术,并描述前列腺癌的可用基因表达特征。
这使得能够发现和训练植根于生物学信息通路的众多预测性生物标志物特征,以确定哪些肿瘤对各种治疗(包括雄激素剥夺治疗、放疗、化疗和 PARP 抑制)更敏感或更耐药。
本章将回顾各种类型的 RNA、可用于评估基因表达的技术,并描述前列腺癌的可用基因表达特征。
转录组学概述转录组学是对 RNA 分子的研究,用于通过测量其 RNA 组成来询问细胞或肿瘤中基因组的活性。
尽管已知的RNA 至少有11 种类型(例如mRNA、rRNA、tRNA、snRNA、snoRNA、siRNA、hnRNA、gRNA、tmRNA、端粒酶 RNA、催化 RNA),但目前肿瘤学中最感兴趣的 RNA 是信使 RNA(mRNA ),它从 DNA 中主动转录并最终转化为蛋白质。
长链非编码RNA在癌症中作用机制的研究进展及临床应用

长链非编码RNA在癌症中作用机制的研究进展及临床应用艾贻伟;刘洋;魏云巍【期刊名称】《西南国防医药》【年(卷),期】2017(027)004【总页数】3页(P419-421)【关键词】长链非编码RNA;癌症;作用机制;研究进展【作者】艾贻伟;刘洋;魏云巍【作者单位】150000 哈尔滨,哈尔滨医科大学第一附属医院普外科;150000 哈尔滨,哈尔滨医科大学第一附属医院普外科;150000 哈尔滨,哈尔滨医科大学第一附属医院普外科【正文语种】中文【中图分类】R730.231在人类基因组当中,只有2%的序列编码蛋白质[1],并且至少有75%会被活跃地转录为mircoRNA和被称为长链非编码RNA(long noncoding RNA,lncRNA)等非编码RNA[2]。
lncRNA被定义为长度大于200个碱基,缺乏蛋白质编码能力的RNA[3-5]。
癌症是由于基因突变和环境因素共同作用所导致的一类疾病。
虽然很多基因突变都位于缺乏编码蛋白质能力的区域,但其突变所产生的效应却可以通过被转录为非编码RNA来实现。
microRNA在癌症中起到重要作用,相比于microRNA,lncRNA的研究才刚刚起步。
lncRNA的表达具有组织特异性。
虽然microRNA已经被证实与特定种类的癌症相关,但是关于lncRNA的研究以及临床应用还很少[6]。
lncRNA不仅可以改变细胞周期、存活、免疫反应、增殖等功能,也决定了癌细胞的表型[7]。
一些lncRNA在转录水平上受到癌基因或抑癌基因的控制[8-9]。
功能研究也从另一个方面证实了lncRNA在癌症发展中的作用。
lncRNA的作用机制之间也存在很大差异,他们通过多种不同的机制来调节基因表达。
在肿瘤中,lncRNA的差异表达直接影响到正常细胞是否会转变为肿瘤细胞。
例如,lncRNA HOX转录反义RNA(HOTAIR)是一种已经被广泛研究的lncRNA[10]。
HOTAIR的过度表达可以通过表观遗传修饰,从而沉默HOXD簇,促进乳腺癌转移。
人类单细胞基因组测序
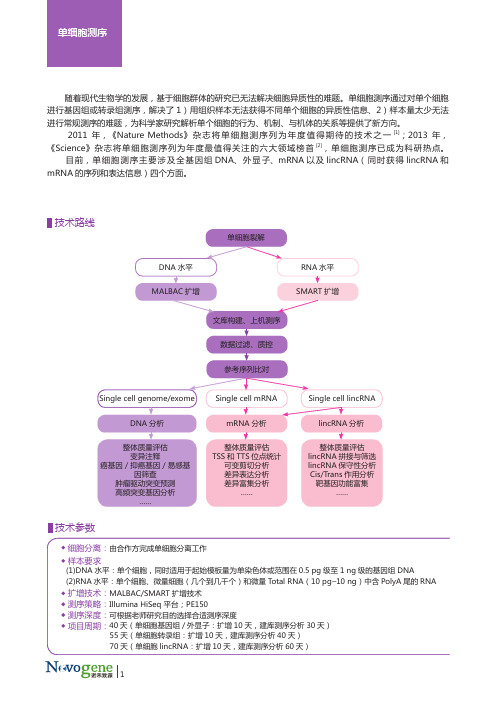
随着现代生物学的发展,基于细胞群体的研究已无法解决细胞异质性的难题。
单细胞测序通过对单个细胞进行基因组或转录组测序,解决了1)用组织样本无法获得不同单个细胞的异质性信息、2)样本量太少无法进行常规测序的难题,为科学家研究解析单个细胞的行为、机制、与机体的关系等提供了新方向。
2011 年,《Nature Methods》杂志将单细胞测序列为年度值得期待的技术之一[1];2013 年,《Science》杂志将单细胞测序列为年度最值得关注的六大领域榜首[2],单细胞测序已成为科研热点。
目前,单细胞测序主要涉及全基因组DNA、外显子、mRNA以及lincRNA(同时获得lincRNA和mRNA的序列和表达信息)四个方面。
技术路线技术参数产品优势最严格的数据指控标准诺禾建立了新一代高通量测序平台,包括10台Hiseq X 测序仪,10台Hiseq 2500,从样品检测到文库构建采用最严格的指控标准,从源头保证了数据的可靠性。
采用国际认可的全基因组扩增技术DNA 水平:诺禾致源采用多次退火环状循环扩增技术(MALBAC),MALBAC 扩增具有低偏倚性。
最新《Science》文章比较MALBAC 与MDA 扩增技术,发现MALBAC 扩增后的数据与混合建库的数据质量更为接近(如图1)[3]。
该技术应用于单精子测序,研究成果[4]已在《Science》杂志发表。
RNA 水平:诺禾致源采用SMARTer 扩增技术(美国Clontech 公司原装试剂盒),能够对单个细胞、微量细胞(几个到几千个)和微量Total RNA(10 pg-10 ng)中含PolyA 尾的RNA 全长进行准确而高效地扩增,扩增后的cDNA 片段适用于Illumina 建库测序平台,为单细胞样本的RNA-seq 分析提供了利器。
图2 SMARTer 扩增无3'和5'端偏好性定制化信息分析针对不同项目,除了进行标准信息分析外,通过和合作伙伴共同探讨,制定可行的个性化信息分析方案,整合各种主流软件,优化分析结果,以确保结果的准确性及创新性。
基因组学-Genomics-知识考点汇总

基因组学-Genomics-知识考点汇总•基因组(Genome:Gene+chromosome)细胞或生物体中一套完整的单倍体遗传物质•基因组学(Genomics)最早Thomas Roderick在1986年提出,包括基因组作图、测序和分析。
可分为结构基因组学和功能基因组学。
一、结构基因组学1.遗传图(Genetic Mapping Genomes) : Based on the calculation of recombination frequencyby linkage analysis .通过亲本的杂交,分析后代的基因间重组率,并用重组率来表示两个基因之间距离的线形连锁图谱每条染色体组成一个连锁群,所有染色体的连锁群组成的图谱即构成基因组遗传图。
重组率代表基因位点之间的相对距离。
在遗传作图中,人们把一个作图单位定义为1厘摩(cM),1cM等于1%的重组率。
提高遗传作图的分辨率:选用不同的杂交群体;增加杂交群体的数目;增加分子标记的数目;扩大分子标记的来源分子标记:绘制基因组遗传图需要的坐标点。
分子标记的主要来源是染色体上存在的大量等位基因。
在DNA水平上,两个基因间一个碱基的差异就足以形成等位基因。
2.物理图(physical map):指DNA序列上两点的实际距离,它是以DNA的限制酶片段或克隆的大片段的基因组DNA分子为基本单位,以连续的重叠群为基本框架,通过遗传标记将重叠群或基因组DNA分子有序排列于染色体上。
物理图的绘制: Based on molecular hybridization analysis and PCR techniques杂交法;指纹法;荧光原位杂交技术。
3.基因组序列测定: Sequencing methods: the chain termination procedure;Map-based clone by clone strategy;Whole genome shotgun (WGS) strategy;Sequence assembly;•传统基因组测序的方法:克隆步移法(BAC-by-BAC Strategy)和全基因组鸟抢法(Whole Genome Shotgun Strategy)。
生物信息学Bioinformatics

Analysis!
Definition Bioinformatics
an intersecting discipline
----is an interdisciplinary scientific field that develops methods and software tools for storing, retrieving, organizing and analyzing biological data.
Kir2.1
/
常用蛋白质三维结构观察和修改工具
工具
网站
备注
SwissPdbViewer Jmol
MolMol PyMol Rasmol VMD
/spdbv/
/
NCBI BLAST: Understand the Output
Raw score (S) Calculated by summing scored for individual aligned position. Scores for each position are calculated using a substitution matrix BLOSUM or PAM).
Biological Data
Biological Data
Simpleroject, HGP ----The challenge of huge data
RNA-seq研究方法与策略-zzz

SNP/SNV biomarker Gene fusion
RNA-Seq
Quantification
Re-sequencing
mRNA
mRNA
50SE
90PE
√
√
√
√
-
√
-
√
√
√
-
√
Small RNA
Small RNA
miRNA 50SE
√
√ √ √ √ -
complex (RISC) and that recognize partially complementary target mRNAs to induce translational
repression, which is often linked to degradation.
Long non-coding RNAs (long ncRNAs, lncRNA) are non-protein coding transcripts longer than 200
from DNA to the ribosome, where they specify
mRNA Coding
RNA
snRNA circRNA
the amino acid sequence of the protein products of gene expression. A non-coding RNA (ncRNA) is a functional
mRNA的特点
5’端帽子结构和3’端Poly A尾巴 分子长度一般介于500-10000nt 有前体,包含内含子 能翻译成功能蛋白
原核生物mRNA缺少cap和Poly-A tail的结构!
长链非编码RNA的作用机制及其研究方法

Acting mechanisms and research methods of long noncoding RNAs
XIA Tian, XIAO Bing-Xiu, GUO Jun-Ming
Department of Biochemistry and Molecular Biology, Ningbo University School of Medicine, Zhejiang Provincial Key Laboratory of Pathophysiology, Ningbo 315211, China Abstract: Long noncoding RNAs (lncRNAs) play biological roles through a variety of mechanisms, including genetic imprinting, chromatin remodeling, cell cycle control, splicing regulation, mRNA decay, and translational regulation. LncRNAs are involved in the regulation of gene expression through the above mechanisms in different levels. Establishment and application of research technologies are important in understanding of lncRNAs functions. Microarray, RNA sequencing, Northern blot, real time quantitative reverse transcription-polymerase chain reaction, fluorescence in situ hybridization, RNA interference, and RNA-binding protein immunoprecipitation are major tools of exploring biological functions of lncRNAs. Here, we highlighted three advanced methods, i.e., fast predictions of RNA and protein interactions and domains (catRAPID), chromatin isolation by RNA purification (ChIRP), and combined knockdown and localization analysis of noncoding RNAs (c-KLAN). Keywords: long noncoding RNAs (lncRNAs); acting mechanisms; gene expression regulation; research methods
RNAi片段siRNA设计原则

RNAi片段siRNA设计原则RNAi target selection rules:1.Targeted regions on the cDNA sequence of a targeted gene should be located 50-100 nt downstream of the start codon (ATG).2.Search for sequence motif AA(N19)TT or NA(N21), or NAR(N17)YNN, where N isany nucleotide, R is purine (A, G) and Y is pyrimidine (C, U).3.Avoid targeting introns, since RNAi only works in the cytoplasm and not within the nucleus.4.Avoid sequences with > 50% G+C content.5.Avoid stretches of 4 or more nucleotide repeats.6.Avoid 5URT and 3UTR, although siRNAs targeting UTRs have successfully induced gene inhibition.7.Avoid sequences that share a certain degree of homology with other related or unrelated genes.How to obtain a cDNA sequence for target selectionBefore finding a RNAi target on the gene of your interest, first you have to get its mRN A sequence or sequence accession number as some siRNA design tools can take accessionnumber as input. It is recommended to use the gene's RefSeq from NCBI, since the Ref Seq represents non-redundant, curated and validated sequences. RefSeq mRNA sequences h ave unique accession numbers which start with NM or XM, followed by 6 digits. For exa mple, NM_123456 (curated mRNA sequence) or XM_0123456 (model mRNA sequence pr edicted by genome sequence analysis). There are several ways of querying RefSeq.1.Search LocusLink by gene name or symbol at /LocusLink/. Once the locus of your gene is found, scroll down to the "NCBI Reference Sequen ce (RefSeq)" section and look for mRNA.2.Search Entrez Gene at /entrez/query.fcgi?db=gene, and select the right gene of desired organism. Once the page for the gene is shown, scroll d own to the "NCBI Reference Sequence (RefSeq)" and look for mRNA.3.Search Nucleotide database using Entrez query tool at /entrez/query.fcgi?db=Nucleotide and use Entrez Limits settings to restrict your query to t he RefSeq database onlyo select "RefSeq" from the "Only from" menu, this restricts the query to the RefSeq collectiono select "mRNA" from the "Molecule" menu, this restricts the query to mR NA RefSeq recordsHomology searchThe RNAi targeted region on the mRNA sequence of a gene should not share significant homology with other genes or sequences in the genome, therefore, homology search is ess ential to minimize off-target effects. Although most siRNA design tools provide BLAST o ption, some simply use NCBI BLAST tools which sometimes are quite slow. Here are so me BLAST tools for homology search.∙NCBI Blast tool: Nucleotide-nucleotide BLAST (blastn) or Search for short, nearly exact matches∙Blat tool on UCSC Genome Website /cgi-bin/hgBlat∙Ensembl Blast /Multi/blastviewExamples of RNAi target selectionReferences1. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucl eotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001 May 24;411 (6836):494-8.2. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleoti de RNAs. Genes Dev. 2001 Jan 15;15(2):188-200.更多信息:siRNA Design Guidelines (Ambion) - Using siRNA for gene silencing is a rapid ly evolving tool in molecular biology. There are several methods for preparing siRNA, s uch as chemical synthesis, in vitro transcription, siRNA expression vectors, and PCR ex pression cassettes. Irrespective of which method one uses, the first step in designing a s iRNA is to choose the siRNA target site. This guidelines for choosing siRNA target site s are based on both the current literature, and on empirical observations by scientists at Ambion. Using these guidelines, approximately half of all siRNAs yield>50% reduction in target mRNA levels.siRNA Design Rules (Protocol Online) - General siRNA design rules and rational siRNA design.相似一篇General Guidelines1.siRNA targeted sequence is usually 21 nt in length.2.Avoid regions within 50-100 bp of the start codon and the termination codon3.Avoid intron regions4.Avoid stretches of 4 or more bases such as AAAA, CCCC5.Avoid regions with GC content <30% or > 60%.6.Avoid repeats and low complex sequence7.Avoid single nucleotide polymorphism (SNP) sites8.Perform BLAST homology search to avoid off-target effects on other genes or sequences9.Always design negative controls by scrambling targeted siRNA sequence. The control RNA should have the same length and nucleotide composition as the siRNA but have at least 4-5 bases mismatched to the siRNA. Make sure the scrambling will not createnew homology to other genes.Tom Tuschl's rules1.Select targeted region from a given cDNA sequence beginning 50-100 nt downstream of start condon2.First search for 23-nt sequence motif AA(N19). If no suitable sequence is found, then,3.Search for 23-nt sequence motif NA(N21) and convert the 3' end of the sense siRNA to TT4.Or search for NAR(N17)YNN5.Target sequence should have a GC content of around 50%A = Adenine; T = Thymine; R = Adenine or Guanine (Purines); Y = Thymine or Cytosi ne (Pyrimidines); N = Any.Rational siRNA designBy experimentally analyzing the silencing efficiency of 180 siRNAs targeting the mRNA of two genes and correlating it with various sequence features of individual siRNAs, Reyn olds et al at Dharmacon, Inc identified eight characteristics associated with siRNA functio nality. These characteristics are used by rational siRNA design algorithm to evaluate poten tial targeted sequences and assign scores to them. Sequences with higher scores will have higher chance of success in RNAi. The table below lists the 8 criteria and the methods o f score assignment.A sum score of 6 defines the cutoff for selecting siRNAs. All siRNAs scoring higher tha n 6 are acceptable candidates.*Tm = 79.8 + 18.5*log10([Na+]) + (58.4 * GC%/100) + (11.8 * (GC%/100)2) - (820/Leng th)For example, the Tm can be calculated as follows for the siRNA UUCUCCAGCUUCUA AAAUATm = 79.8 + 18.5*log10(0.05) + (58.4 * 31.6/100) + (11.8 * (31.6/100)2) - (820/19)Tm = 32.19There are two siRNA design tools which implement this siRNA design algorithm: one is offered by Dharmacon, Inc; the other is a downloadable Excel template, written by Mauric e Ho at t.hk/RNAi/siRNA.References1.Elbashir SM et al. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494-498.2.Elbahir SM et al. (2001). Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 20:6877-6888.3.Elbashir SM et al. (2002). Analysis of gene function in somatic mammalian cellsusing small interfering RNAs. Methods. 26:199-213.4.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. RationalsiRNA design for RNA interference. Nat Biotechnol. 2004 Mar;22(3):326-30.5./biotools/oligocalc.html6.Maurice Ho, Rational siRNA Design。
非编码RNA来源的小肽:“微不足道”却“功能强大”

第 62 卷第 3 期2023 年 5 月Vol.62 No.3May 2023中山大学学报(自然科学版)(中英文)ACTA SCIENTIARUM NATURALIUM UNIVERSITATIS SUNYATSENI非编码RNA来源的小肽:“微不足道”却“功能强大”*陈晓彤,赵文龙,孙林玉,王文涛,陈月琴中山大学生命科学学院,广东广州 510275摘要:非编码RNA(ncRNA, non-coding RNA)长久以来被认为不具有编码能力。
近年来随着研究技术和生物信息学工具的迅速发展,研究发现在基因组的非编码区域上存在大量小开放阅读框(sORFs,small/short open read‐ing frames),其翻译产物被称作小ORF编码肽(SEPs,sORF encoded peptides)或小肽(micropeptides)。
部分小肽被证实在细胞内稳定存在并独立于其来源RNA发挥重要作用。
本文系统总结了非编码RNA来源小肽的鉴定方法、可编码小肽的RNA类型以及其研究困难和瓶颈,并重点回顾了疾病和植物中发现的功能小肽,以期对小肽的筛选鉴定提供思考,对小肽作为药物研发或者农作物增产的关键靶点提供新的思路和方向。
关键词:非编码RNA;小肽;非经典翻译;鉴定方法;调控机制中图分类号:Q71 文献标志码:A 文章编号:2097 - 0137(2023)03 - 0001 - 13 Micropeptides derived from non-coding RNAs: Tiny but powerful CHEN Xiaotong, ZHAO Wenlong, SUN Linyu, WANG Wentao, CHEN Yueqin School of Life Sciences, Sun Yat-sen University, Guangzhou 510275,ChinaAbstract:It was long presumed that non-coding RNAs (ncRNAs) are lacking in protein-coding poten‐tial. However, recent advances in technology and tools have led to an important finding that a number of small open reading frames (sORFs) were found in different kind of ncRNAs, and their translated products have been termed sORF encoded peptides (SEPs) or micropeptides. Some micropeptides have been confirmed to exist stably in cells and play important roles independently of their source RNA. In this review,we summarize the identification methods of micropeptides derived from ncRNAs,the types of RNA that can encode micropeptides,and focus on the functional micropeptides found in diseases and plants. The purpose of the review is to provide a thought on the screening and identifica‐tion of micropeptides, and provide new ideas for micropeptides as potentials for drug development or crop yield improvement.Key words: non-coding RNA; micropeptide; non-canonical translation; identification methods; regula‐tion mechanism随着人类基因组计划的完成以及ENCODE计划的开展,科学家发现,约75%的基因组可以产生转录本(Derrien et al.,2012;Djebali et al.,2012)。
lncrna的转录因子研究方法

lncrna的转录因子研究方法英文回答:Methods for Studying Transcription Factors of lncRNAs.Long non-coding RNAs (lncRNAs) are a class of non-coding RNAs that have been increasingly recognized fortheir important roles in various biological processes. LncRNAs can act as transcriptional regulators byinteracting with transcription factors (TFs), thereby influencing gene expression. Studying the TFs that regulate lncRNA expression is crucial for understanding the molecular mechanisms underlying lncRNA function.Chromatin Immunoprecipitation (ChIP)。
ChIP is a widely used technique for identifying proteins that bind to specific DNA sequences. In the context of lncRNA research, ChIP can be used to identify TFs that associate with lncRNA promoters or regulatoryregions. This involves crosslinking proteins to DNA, immunoprecipitating the DNA-protein complexes using antibodies specific to the TF of interest, and then sequencing the DNA to identify the bound regions.RNA Immunoprecipitation (RIP)。
基于生物信息学方法构建肝细胞癌SNHG17-miRNA-CDK4预后预测模型

第59卷 第6期2023年12月青岛大学学报(医学版)J O U R N A LO FQ I N G D A O U N I V E R S I T Y (M E D I C A LS C I E N C E S)V o l .59,N o .6D e c e m b e r 2023[收稿日期]2023-01-18; [修订日期]2023-09-13[基金项目]国家自然科学基金青年科学基金项目(81802458)[第一作者]刘建东(1993-),男,硕士研究生㊂[通信作者]徐慧荣(1986-),男,硕士,主治医师㊂E -m a i l :332699352@q q.c o m ㊂基于生物信息学方法构建肝细胞癌S N H G 17-m i R N A -C D K 4预后预测模型刘建东1,韩翠云2,钟敬涛3,徐慧荣3(1 山东大学齐鲁医院急诊科,山东济南 250000; 2 山东第一医科大学第一附属医院(山东省千佛山医院)护理部;3 山东省肿瘤防治研究院(山东省肿瘤医院)介入三病区)[摘要] 目的 探讨长链非编码R N A (l n c R N A )小核仁R N A 宿主基因17(S N H G 17)在肝细胞癌(H C C )发生发展中的作用㊂方法 利用基因表达谱交互分析(G E P I A )数据库,分析H C C 中S N H G 17的表达及其与生存的关系;利用基因表达综合(G E O )数据库的数据进行验证,使用G E O 2R ㊁l n C A R 和M e t a s c a p e 软件分析S N H G 17相关性基因㊁构建可视化网络并进行G O 和K E G G 富集分析㊂根据通路利用多个G E O 数据集筛选出细胞周期蛋白依赖性激酶4(C D K 4);利用数据库平台S t a r B a s e ㊁m i R B D 以及HM D D 筛选出同时以S N H G 17和C D K 4为靶基因的m i c r o R N A (m i R N A )㊂结果 l n c R N AS N H G 17在H C C 中的表达显著上升(P <0.05),其表达高的病人预后显著变差(P <0.05)㊂G O 和K E G G 富集分析结果显示,S N H G 17与核蛋白复合物以及细胞周期相关㊂筛选出高度相关基因C D K 4并筛选出以S N H G 17和C D K 4为靶标的m i R N A s ㊂结论 S N H G 17在H C C 中表达增高,并可通过结合多种m i R N A 发挥竞争性内源R N A 的功能,对C D K 4发挥调控作用㊂[关键词] 癌,肝细胞;R N A ,长链非编码;微R N A s;细胞周期蛋白依赖激酶4;预后;计算生物学[中图分类号] R 730.261;R 342.2 [文献标志码] A [文章编号] 2096-5532(2023)06-0850-05d o i :10.11712/jm s .2096-5532.2023.59.204[开放科学(资源服务)标识码(O S I D )][网络出版] h t t ps ://l i n k .c n k i .n e t /u r l i d /37.1517.R.20240104.1606.005;2024-01-05 20:16:24E S T A B L I S H M E N T OF A P R OG N O S T I C P R E D I C T I O N M O D E L F O RH E P A T O C E L L U L A R C A R CI N O M A B A S E D O NS N H G 17-M I R N A -C D K 4A N DB I O I N F O R M A T I C SM E T H O D S L I UJ i a n d o n g ,HA N C u i y u n ,Z H O N GJ i n g t a o ,X U H u i r o n g (D e p a r t -m e n t o fE m e r g e n c y M e d i c i n e ,Q i l uH o s p i t a l o f S h a n d o n g U n i v e r s i t y,J i n a n250000,C h i n a )[A B S T R A C T ] O b je c t i v e T o i n v e s t i g a t e t h e r o l e of t h e l o ng n o n -c o d i n g R N A (l n c R N A )s m a l l n u c l e o l a rR N Ah o s t g e n e 17(S N H G 17)i n t h e d e v e l o p m e n t a n d p r o g r e s s i o n o f h e pa t o c e l l u l a r c a r c i n o m a (H C C ). M e t h o d s G E P I Ad a t ab a s ew a s u s e d t o a n a -l y z e t h e e x p r e s s i o no fS N H G 17i n H C Ca n di t sa s s oc i a t i o n w i t hs u r v i v a l ;t h ed a t a i n G E O d a t a b a s ewe r eu s e df o rv a l i d a t i o n ;G E O 2R ,l n C A R ,a n dM e t a s c a p ew e r e u s e d t o a n a l y z e S N H G 17-r e l a t e dg e n e s ,c o n s t r u c t a v i s u a l i z e d n e t w o r k ,a n d p e r f o r m G Oa n d K E G Ge n r i ch m e n t a n a l y s e s .B a s e do nr e l a t e d p a t h w a y s ,m u l ti pl eG E O d a t a s e t sw e r eu s e dt os c r e e nf o rC D K 4,a n dS t a r B a s e ,m i R B D ,a n d HM D Dd a t a b a s e p l a t f o r m sw e r eu s e dt oo b t a i nt h e m i c r o R N A s (m i R N A s )t a r g e t i n g bo t hS N H G 17a n d C D K 4.R e s u l t s T h e r ew a s a s i g n i f i c a n t i n c r e a s e i n t h e e x p r e s s i o no f l n c R N AS N H G 17i n H C C (P <0.05),a n dt h e p a t i e n t sw i t hh i gh S N H G 17e x p r e s s i o n t e n d e d t oh a v e a p o o r e r p r o g n o s i s (P <0.05).G Oa n dK E G Ge n r i c h m e n t a n a l ys e s s h o w e d t h a t S N H G 17w a s a s s o c i a t e dw i t hn u c l e a r p r o t e i n c o m p l e xa n d c e l l c y c l e .T h e h i g h l y r e l a t e d g e n e C D K 4w a s o b t a i n e d ,a sw e l l a s t h em i R N A s t a r ge -t i n g S N H G 17a n d C D K 4. C o n c l u s i o n T h e r e i s a n i n c r e a s e i n t h e e x p r e s s i o no fS N H G 17i n H C C ,a n dS N H G 17c a n p l a y t h e r o l e of c o m p e t i t i v e e n d og e n o u sR N Ab y b i n d i n g t o a v a r i e t y o fm i R N A s ,th e r e b y e x e r ti n g a r e g u l a t o r y ef f e c t o nC D K 4.[K E Y WO R D S ] c a r c i n o m a ,h e p a t o c e l l u l a r ;R N A ,l o ng n o n c o d i n g ;m i R N A s ;c y c l i n -d e p e n d e n t k i n a s e 4;p r o g n o s i s ;c o m pu -t a t i o n a l b i o l o g y肝细胞癌(H C C )作为最常见的恶性肿瘤之一,已成为全球死亡率第二高的肿瘤[1]㊂由于H C C 在确诊时往往已经处于晚期,并且由于远处转移和术后复发等因素,H C C 病人的预后很差[2-3]㊂因此,探究H C C 发生发展的分子机制,寻找与H C C 发生有关的新型生物标志物对H C C 病人的早期诊断和治疗十分重要㊂长链非编码R N A (l n c R N A )是长度超过200个核苷酸的无蛋白质编码能力的R N A ,与肿瘤发生发展密切相关[4]㊂小核仁R N A 宿主基因17(S N H G 17)是新发现的位于20q11的l n c R N A ,在多种肿瘤中表达上调,发挥癌基因的功能[5-7]㊂相关研究显示,S N H G 17过表达通过竞争性内源R N A6期刘建东,等.基于生物信息学方法构建肝细胞癌S N H G17-m i R N A-C D K4预后预测模型851(c e R N A)网络调控肺腺癌等恶性肿瘤细胞的增殖和迁移[8-9]㊂然而,S N H G17在H C C中的表达水平及其临床意义研究较少㊂本研究利用生物信息学方法分析S N H G17在H C C中的表达水平及其对生存的影响,并通过基因表达综合(G E O)数据库数据集信息的深度挖掘预测靶基因细胞周期蛋白依赖性激酶4(C D K4),为后期S N H G17在H C C中的功能研究提供理论依据㊂1资料和方法1.1S N H G17在H C C中的表达及生存分析本文研究利用基因表达谱交互分析(G E P I A)数据库(h t t p://g e p i a.c a n c e r-p k u.c n/)分析T C G A H C C数据集中S N H G17在癌与癌旁组织中表达水平差异,分析S N H G17表达与病人总生存期(O S)和无病生存期(D F S)的相关性,以及S N H G17表达与肿瘤分级之间的关系㊂1.2S N H G17在癌和癌旁组织中表达验证从G E O数据库中下载H C C的临床数据,对G S E36376㊁G S E14520数据集中S N H G17在癌与癌旁组织中的表达水平进行验证㊂1.3S N H G17相关靶基因生物学功能及通路分析本文利用l n C A R数据库(h t t p s://l n c a r.r e n l a b. o r g/)分析G E O数据集中与S N H G17相关的基因并构建可视化网络,利用M e t a s c a p e平台(h t t p s:// m e t a s c a p e.o r g/g p/i n d e x.h t m l#m a i n/s t e p1)进行靶基因的G O和K E G G富集分析㊂1.4S N H G17及其下游靶基因C D K4相关性分析采用韦恩图取交集的方法在不同G E O数据集中取得高度相关的基因的交集,参考基因富集通路挑选出最有意义的靶基因C D K4㊂同时在G E P I A 数据库中采用P e r a r s o n相关性分析进行验证㊂1.5C D K4基因在H C C中的表达及预后分析通过U A L C A N数据库(h t t p://u a l c a n.p a t h. u a b.e d u/)分析C D K4在癌与癌旁组织中的表达水平,分析C D K4表达与病人O S和D F S的相关性,以及与肿瘤分级之间的关系㊂1.6与S N H G17和C D K4相互作用m i R N A的筛选及其调控关系分析通过S t a r B a s e(h t t p://s t a r b a s e.s y s u.e d u.c n/)㊁m i R B D(h t t p://w w w.m i r d b.o r g/)筛选可能同时结合S N H G17和C D K4的m i R N A,并分析S N H G17㊁m i R N A和C D K4之间的调控关系㊂利用HM D D v3.0(h t t p://w w w.c u i l a b.c n/h m d d)检索并分析与H C C有关的m i R N A,通过S t a r B a s e进行P e r a r s o n 相关性分析,以进一步证实S NH G17通过m i R N A 发挥对C D K4的调控㊂1.7统计学分析采用S P S S软件进行统计学分析㊂两组之间比较采用双尾未配对的t检验或配对的t检验,通过K a p l a n-M e i e r曲线检验S N H G17和C D K4表达对H C C病人的影响,并使用对数值检验分析了病人的O S和D F S㊂以P<0.05为差异有统计学意义㊂2结果2.1 H C C组织中S N H G17的表达水平及与病人生存情况的相关性利用G E P I A数据库T C G A数据集分析发现, H C C中S N H G17表达量明显增高(P<0.001),且与H C C的分级密切相关,与低级别H C C(S t a g e Ⅰ/Ⅱ)相比较,高级别H C C(S t a g eⅢ/Ⅴ)组织中S N H G17表达明显升高(P<0.01)㊂见图1A㊁B㊂S N H G17表达水平也与H C C病人的O S和D F S高度相关,与S N H G17表达水平较高的病人相比较, S N H G17表达较低的病人的O S和D F S时间延长(P<0.05)㊂见图1C㊁D㊂同样地,与来自G E O的R N A-s e q数据集G S E36376㊁G S E14520中的正常组织相比,H C C组织中S N H G17的表达明显上调(P<0.01)㊂见图1E㊁F㊂2.2 G E O数据集中S N H G17靶基因的功能富集分析通过l n C A R平台分析G E O的R N A-s e q数据集G S E14520,得到与S N H G17相关靶基因共200个(r>0.50或<-0.50)并构建基因相关性网络㊂见图2A㊂利用M e t a s c a p e平台分析200个相关性靶基因,G O生物过程分析发现基因主要集中在核蛋白的复合体生成㊁m R N A的翻译以及核蛋白的组装㊂见图2B㊂K E G G信号通路富集分析发现,排名第一的是与细胞周期相关的通路㊂见图2C㊂对G E O的R N A-s e q数据集G S E36376㊁G S E14520相关靶基因分析取交集找到36个共同靶基因,其中C D K4以促进H C C细胞的G1/S转变调控细胞周期的信号通路[10]㊂同时我们用G E P I A的P e r a r s o n 相关性分析进行验证,结果显示在T C G A数据集中C D K4与S N H G17的表达呈高度相关(P<0.01)㊂见图2D㊂852青岛大学学报(医学版)59卷A:H C C组织中S N H G17表达量明显增高(G E P I A数据库,P<0.001);B:S N H G17与H C C组织的分级密切相关(P<0.01);C㊁D: S N H G17的表达与病人的O S和D F S相关(P<0.05);E㊁F:H C C组织中S N H G17的表达明显上调(数据来源于G S E36376㊁G S E14520,P< 0.001)㊂图1S N H G17在H C C中表达以及对生存的影响A:S N H G17基因相关性网络构建(G S E14520,R>0.50或<-0.50);B:S N H G17相关基因的G O分析;C:S N H G17相关基因的K E G G分析;D:C D K4与S N H G17的表达高度相关㊂图2S N H G17相关基因富集分析以及靶基因C D K4的筛选6期刘建东,等.基于生物信息学方法构建肝细胞癌S N H G 17-m i R N A -C D K 4预后预测模型8532.3 H C C 中C D K 4表达与生存时间关系利用U A L C A N 数据库T C G A 数据集分析发现,H C C 中的C D K 4表达明显增高(P <0.001)㊂见图3A ㊂C D K 4表达与人种㊁性别㊁年龄㊁体质量无关(P >0.05)㊂见图3B ~E ㊂与高级别和高分期的病人相比,部分低级别和低分期病人C D K 4的表达量降低(P <0.05)㊂见图3F ㊁G ㊂C D K 4水平也与H C C 病人的O S 和D F S 高度相关,与C D K 4表达水平较高的病人相比,C D K 4表达较低的病人的O S 和D F S 延长(P <0.001)㊂见图3H ㊁I㊂A :H C C 中C D K 4表达明显增高(U A L C A N 数据库,P <0.001);B ~E :H C C 中C D K 4的表达水平与人种㊁性别㊁年龄㊁体质量均无相关性(U A L C A N 数据库,P >0.05);F ㊁G :C D K 4的表达与H C C 的分期㊁分级密切相关(P <0.005);H ㊁I :C D K 4的表达与H C C 病人的O S 和D F S 相关(G E P I A 数据库,P <0.01)㊂图3 T C G A 数据集中C D K 4在H C C 中表达以及对生存的影响2.4 S N H G 17-m i R N A -C D K 4调控模型中m i R N A的筛选在多种肿瘤中,S N H G 17可以作为c e R N A 发挥 分子海绵 作用,吸附m i R N A ,调控下游靶基因的表达㊂我们通过S t a r B a s e ㊁m i R B D 预测H C C 中与S N H G 17和C D K 4结合的m i R N A 并取交集,发现h s a -m i R -335-5p ㊁h s a -m i R -520a -5p㊁h s a -m i R -525-5p ㊁h s a -m i R -124-3p ㊁h s a -m i R -506-3p 可以同时结合S N H G 17和C D K 4㊂见图4㊂3 讨 论S NH G 家族通过多种分子调控机制参与H C C的发生发展过程[11]㊂例如,S N H G 1通过吸附m i R-图4 S N H G 17-m i R N A -C D K 4调控模型中m i R N A 的筛选377-3p 促进H CC 进展和转移[12];S N H G 3可直接结合m i R -128从而促进C D 151表达,激活上皮间充质转变相关通路的表达,促进H C C 的侵袭和转移[13];S N H G 5高表达解除了m i R -26a -5p 对糖原合854青岛大学学报(医学版)59卷成酶激酶3β的抑制作用,从而促进了H C C的发生[14];S N H G17在多种肿瘤中表达上调并促进癌症进展[15-16]㊂然而,S N H G17在H C C中的功能和机制的相关研究较少㊂本研究利用生物信息学技术分析显示,S N H G17在H C C组织中的表达显著高于癌旁正常肝组织,并且S N H G17高表达与H C C病人较短的O S和D F S显著相关,提示S N H G17可能在H C C中发挥癌基因功能,调控H C C的发生发展㊂对G S E14520数据集进行进一步分析结果表明,与S N H G17相关的基因高度富集在细胞周期相关通路上,在T C G A数据集以及不同的G E O数据集中同时发现C D K4的表达水平与S N H G17显著相关㊂C D K4蛋白是一种由C D K4基因编码的广泛表达的丝氨酸/苏氨酸激酶㊂在有丝分裂原刺激下,细胞周期蛋白D(C y c l i n D)与激活的C D K4组成C D K4-C y c l i n D复合物,对于细胞周期G1/S期转换至关重要,在癌细胞周期调控中发挥重要的作用㊂例如,复制蛋白A1作为潜在的癌基因通过C D K4/ C y c l i n D途径促进肿瘤增殖,从而在H C C中发挥关键作用[17]㊂本研究系统分析了C D K4在T C G A H C C数据集中的表达水平,并分析了其与肿瘤分期分级及病人种族㊁年龄㊁性别㊁体质量的相关性㊂结果显示,C D K4是H C C独立预后因素,在H C C的发生发展中起重要作用㊂本文生信分析结果显示,H C C组织S N H G17-m i R N A-C D K4预后预测模型的构建,为S N H G17在H C C中的机制研究提供了重要的参考㊂然而,由于本研究样本量较少,因此该结果需进一步通过大样本研究证实㊂[参考文献][1]E L-S E R A G HB,R U D O L P H KL.H e p a t o c e l l u l a r c a r c i n o m a:e p i d e m i o l o g y a n dm o l e c u l a r c a r c i n o g e n e s i s[J].G a s t r o e n t e r o l o-g y,2007,132(7):2557-2576.[2]F O R N E R A,R E I G M,B R U I XJ.H e p a t o c e l l u l a rc a r c i n o m a[J].L a n c e t,2018,391(10127):1301-1314.[3]J E MA L A,B R A Y F,C E N T E R M M,e ta l.G l o b a l c a n c e rs t a t i s t i c s[J].2011,61(2):69-90.[4]J A C O B R,Z A N D E RS,G U T S C H N E R T.T h ed a r ks i d eo ft h ee p i t r a n s c r i p t o m e:c h e m i c a lm o d i f i c a t i o n s i nl o n g n o n-c o-d i n g R N A s[J].I n te r n a t i o n a lJ o u r n a lof M o l e c u l a rS c i e n c e s,2017,18(11):2387.[5]D U FP,H O U Q.S N H G17d r i v e sm a l i g n a n t b e h a v i o r s i na s-t r o c y t o m ab y t a r g e t i n g m i R-876-5p/E R L I N2a x i s[J].B M CC a n c e r,2020,20(1):839.[6]L I UJ,Z HA N Y,WA N GJF,e t a l.l n c R N A-S N H G17p r o-m o t e s c o l o n a d e n o c a r c i n o m a p r o g r e s s i o n a n d s e r v e s a s a s p o n g e f o rm i R-375t o r e g u l a t eC B X3e x p r e s s i o n[J].A m e r i c a n J o u r n a l o fT r a n s l a t i o n a lR e s e a r c h,2020,12(9):5283-5295.[7]L I U Y,L IQ S,T A N G D X,e ta l.S N H G17p r o m o t e s t h ep r o l i f e r a t i o na n d m i g r a t i o no fc o l o r e c t a l a d e n o c a r c i n o m ac e l l sb y m o d u l a t i n g C X C L12-m e d i a t e da n g i o g e n e s i s[J].C a nc e rC e l lI n t e r n a t i o n a l,2020,20(1):566.[8]Q I N Y,S U N W,WA N GZ H,e t a l.L o n g n o n-c o d i n g s m a l ln u c l e o l a rR N Ah o s t g e n e s(S N H G s)i ne n d o c r i n e-r e l a t e dc a n-c e r s[J].O n c o T a r g e t s a n dT h e r a p y,2020,13:7699-7717.[9]Y A N G H,J I A N GZ,WA N GS,e t a l.L o n g n o n-c o d i n g s m a l ln u c l e o l a rR N A h o s t g e n e si n d i g e s t i v ec a n c e r s[J].C a n c e r M e d i c i n e,2019,8(18):7693-7704.[10]C H E NL,WA N GX,C H E N G H H,e t a l.C y c l i nYb i n d s a n da c t i v a t e sC D K4t o p r o m o t e t h eG1/S p h a s e t r a n s i t i o n i nh e p a-t o c e l l u l a r c a r c i n o m a c e l l s v i aR b s i g n a l i n g[J].B i o c h e m i c a l a n dB i o p h y s i c a lR e s e a r c hC o mm u n i c a t i o n s,2020,533(4):1162-1169.[11]H A NS W,Y A N G X,Q IQ,e t a l.C a n s m a l l n u c l e o l a rR N Ab e an o v e lm o l ec u l a r t a r g e t f o rh e p a t o c e l l u l a rc a r c i n o m a[J]?G e n e,2020,733:144384.[12]Q U A,Y A N G Q.L n c R N AS N H G1p r o m o t e s c e l l p r o g r e s s i o na n dm e t a s t a s i s v i a s p o n g i n g m i R-377-3p i nh e p a t o c e l l u l a r c a r-c i n o m a[J].N e o p l a s m a,2020,67(3):557-566.[13]Z H A N GPF,WA N G F,WUJ,e t a l.L n c R N AS N H G3i n-d u ce sE M Ta n ds o r af e n i br e s i s t a n c eb y m o d u l a t i ng th e mi R-128/C D151p a t h w a y i nh e p a t o c e l l u l a r c a r c i n o m a[J].J o u r n a l o fC e l l u l a rP h y s i o l o g y,2019,234(3):2788-2794.[14]L IY R,G U O D,Z H A O Y,e ta l.L o n g n o n-c o d i n g R N AS N H G5p r o m o t e s h u m a nh e p a t o c e l l u l a r c a r c i n o m a p r o g r e s s i o nb y r e g u l a t i n g m i R-26a-5p/G S K3βs i g n a l p a t h w a y[J].C e l lD e a t h&D i s e a s e,2018,9(9):888.[15]P A N X F,G U O Z H,C H E N Y Y,e ta l.S T A T3-i n d u c e dl n c R N AS N H G17e x e r t so n c o g e n i ce f f e c t so no v a r i a nc a n c e r t h r o u g h r e g u l a t i n g C D K6[J].M o l e c u l a r T h e r a p y N u c l e i cA c i d s,2020,22:38-49.[16]Z HA N G Y T,Y A N G G Y.L n c R N A S N H G17a c t sa sac e R N Ao fm i R-324-3p t oc o n t r i b u t e t h e p r o g r e s s i o no f o s t e o-s a r c o m a[J].J o u r n a l o fB i o l o g i c a lR e g u l a t o r s a n dH o m e o s t a t i cA g e n t s,2020,34(4):1529-1533.[17]WA N GJC,Y A N GT,C H E N H,e t a l.O n c o g e n eR P A1p r o-m o t e s p r o l i f e r a t i o n o f h e p a t o c e l l u l a r c a r c i n o m a v i aC D K4/C y c-l i n-D p a t h w a y[J].B i o c h e m i c a l a n dB i o p h y s i c a l R e s e a r c hC o m-m u n i c a t i o n s,2018,498(3):424-430.(本文编辑牛兆山)。
基于TCGA数据库胃癌LincRNA生物标志物筛选及生物学功能分析

基于TCGA数据库胃癌LincRNA生物标志物筛选及生物学功能分析摘要:目的:预测与胃癌相关的LincRNA,及与其相关的信号通路,并筛选出特定的LincRNA作为胃癌预后潜在的生物标志物。
方法:第一步,从TCGA(The Cancer Genome Atlas,TCGA)数据库中下载375例胃腺癌组织和32例癌旁组织的RNA-seq数据(HTseq-RNA Counts)及胃癌患者相关临床信息,并分别提取相关的LincRNA和mRNA。
第二步,使用R语言/Bioconductor的edgeR包分别筛选出差异表达的LincRNA、mRNA。
第三步,使用加权基因共表达网络分析(WGCNA)方法对数据进行加权,得到与胃癌临床特征相关性最大的模块及相对应的临床特征。
第四步,通过CytoHubba 软件寻找关键基因,得到与胃癌发生相关性最大的关键LincRNA。
第五步,对所得到的关键基因LincRNA进行靶基因预测及生物学功能分析,研究与胃癌发生、转移有关的LincRNA与mRNA的相关性,并使用Cytoscape构建LincRNA与mRNA的共表达网络。
第六步,将得到的胃癌关键LincRNA使用Kaplan-Meier软件进行生存分析,以研究LincRNA与胃癌患者的总体存活之间的关联。
结果:1.此研究筛选出2200个差异表达的LincRNA以及4623个差异表达的mRNA;2.得到与肿瘤分级相关性最大的MEbrown模块、MEturquoise模块(模块中为基因群);3.筛选出LincRNA相关性最大模块中的5个关键基因(Hub基因),分别是BARX1-AS1、CARMN、FENDRR、GAS1RR、LINC01354;4.得到胃癌发病可能相关的MAPK Signaling Pathway、CELL CYCLE Signaling Pathway、P53 Signaling Pathway、P13K-AKT Signaling Pathway、WNT Signaling Pathway、TGF-BETA Signaling Pathway、ADHERENS JUNCTION Signaling Pathway七条主要的KEGG生物信号通路;5.构建LincRNA 与mRNA共表达网络;6.BARX1-AS1、CARMN、FENDRR、GAS1RR、LINC01354高表达组生存率均较低表达组低。
植物中lncRNAs的研究进展

植物中lncRNAs的研究进展王艳芳;苏婉玉;张琳;曹绍玉;吕霞;张应华;许俊强【摘要】With the rapid development of high-throughput sequencing technology,more and more lncRNAs (Long non-coding RNA) which have biological functions were found in plants,lncRNAs are important non-coding RNA longer than 200 nucleotides.The researches on the function and mechanism of lncRNAs were more deeply in animals so far,not only in epigenetic level,transcription and post-transcription,but also playing important roles in wide ranges of biological processes such as genomic imprinting,chromatin remodeling,transcriptional pared to animals,some studies have to do with lncRNAs in plants,but the research of lncRNAs in plants is a little backward due to the lack of effective technologies and methods.Therefore,based on the latest advances on lncRNA recently,we reviewed the origin,structuralcharacteristics,classification,molecular mechanism and function,which providing a guide for further study of function and molecular mechanism of lncRNAs in plants.%随着高通量测序技术的飞速发展,越来越多的lncRNAs在植物中被发现,并具有生物学功能.lncRNAs是长度大于200个核苷酸且不具备编码蛋白质功能的一类RNA分子.目前lncRNAs在动物中的功能、作用机制等方面的研究较深入,lncRNAs不仅在表观遗传水平、转录水平、转录后水平上调控基因的表达,也参与诸多生命过程调控如基因组印迹、染色体重塑、转录激活等.在植物中虽有关于lncRNAs的研究,但由于缺乏有效的技术方法而相对滞后.该文就近几年来国内外关于植物lncRNAs的来源、结构特征、分类、功能等方面的研究进展进行综述,为进一步理解和研究植物中lncRNAs的功能和分子机制提供参考.【期刊名称】《西北植物学报》【年(卷),期】2018(038)003【总页数】7页(P582-588)【关键词】lncRNA;来源;结构特征;分类;植物【作者】王艳芳;苏婉玉;张琳;曹绍玉;吕霞;张应华;许俊强【作者单位】云南农业大学园林园艺学院,昆明650201;云南农业大学园林园艺学院,昆明650201;云南农业大学资源与环境学院,昆明650201;云南农业大学园林园艺学院,昆明650201;云南农业大学云南省滇台特色农业产业化工程研究中心,昆明650201;云南农业大学云南省滇台特色农业产业化工程研究中心,昆明650201;云南农业大学云南省滇台特色农业产业化工程研究中心,昆明650201【正文语种】中文【中图分类】Q753;Q789RNA是DNA经转录、翻译成蛋白质的纽带和桥梁,在生物体内扮演着十分重要的作用。
lncRNA与miRNA相互调控作用及其与肿瘤的关系

coding RNA, lncRNA ) 是一类转录本长度大于 200 bp 的非编码 摘要 长链非编码 RNA ( long nonRNA, 可作为人类基因组中一类重要的调控分子通过多种方式发挥其生物学功能 . 近年来的研究表 lncRNA 也可以作为一种竞争性内源性 RNA ( competing endogenous RNA,ceRNA) 与 miRNA 相 明, 互作用, 参与靶基因的表达调控, 并在肿瘤的发生发展中发挥重要 的 作 用. 本 综 述 在 简 要 介 绍 lncRNA 功能研究现状和主要研究方法的基础上 , 进一步分析了 lncRNA 与 miRNA 之间的互相调控 关系及其在肿瘤发生发展中的作用 , 以便为后续的研究提供新的思路 . 关键词 长链非编码 RNA ( lncRNA) ; microRNA; 研究方法; 肿瘤 中图分类号 Q1 ; Q3 ; R74 ; Q75
Smart-seq 2还是10x Genomics Chromium

单细胞转录组:Smart-seq2还是10x Genomics Chromium?应用单细胞测序技术研究科学问题越来越普遍,当下应用最火热的是10x Genomics公司的Chromium解决方案(以下简称10X)。
但是基于2017年Christoph Ziegenhain et al[1]对6种单细胞转录组技术及2019Broad研究所的团队对7种单细胞RNA测序方法进行了比较[2],某些特殊或者少量细胞样本的单细胞转录组研究中,Smart-seq2技术还是一项研究利器。
Smart-seq2与10x技术的区别与应用的科研场景有哪些呢?北京大学张泽民团队在2019年发表的预印文章“Direct Comparative Analysis of10x Genomics Chromium and Smart-seq2”中给予了答案。
那么今天小编再次对Smart-seq2与10x Genomics Chromium的技术原理和应用进行阐明。
Smart-Seq简介Smart-Seq(Switching mechanism at5'end of the RNA transcript)于2012年发表[3],2013年发表了其改进技术的应用Smart-Seq2[4],2014年Smart-Seq2protocol发表[5]。
Smart-Seq2对原始的Smart-Seq实验流程进行了多项改进优化,它不再需要纯化步骤,可大大提高产量,最重要的改进是下面两项:•TSO3'端最后一个鸟苷酸替换为锁核酸LNA(locked nucleic acid)。
LNA单体的热稳定性增强,其退火温度增强非模板cDNA的3'延伸能力。
•甜菜碱(一种具有两个重要作用的甲基供体:它会增加蛋白质的热稳定性,并通过破坏DNA螺旋来降低甚至消除了DNA热融变对碱基对组成的依赖性)与较高的MgCl2浓度结合使用。
解决某些RNA形成二级结构(例如发夹或环)由于空间位阻,可能导致酶终止链延长的问题。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
RNA Extraction Preparation for Next-Generation SequencingTotal RNA was extracted using TRK-1001 total RNA purification kit (LC Science, Houston, TX) following the manufacturer's procedure. The total RNA quantity and purity were analysis of Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA) with RIN number >7.0. Approximately 5 ug of total RNA representing a specific adipose type was used to deplete ribosomal RNA according to the manufacturer’s instructions of the Ribo-Zero™ rRNA Removal kit (Epidemiology version) (Epicentre, an Illumina company, Madison, WI). Following purification, the poly(A)- or poly(A)+ RNA fractions is fragmented into small pieces using divalent cations under elevated temperature. Then the cleaved RNA fragments were reverse-transcribed to construct cDNA library using the dUTP method as described [1], the average insert size for the paired-end libraries was 300 bp (±50 bp). RNA libraries were then sequenced on the Illumina HiSeq 2000 or 2500 platform using 125 bp paired-end reads.RNA-Sequencing Data AnalysisAll RNA-seq data were aligned to hg19 using TopHat[2]v2.0.9 with default parameters. The mapped reads were assembled using Cufflinks[3] v2.11. All multiple assembled transcript files (GTF format) were then merged to produce a unique set of transcriptomes using the Cuffmerge utility provided by the Cufflinks package. Cuffdiff v2.11 was used for all differential expression analyses with Gencode v21.0 annotation. Transcript abundances were estimated by Cufflinks in Fragments Per Kilobase per Million mapped reads (FPKM) for pairedend reads or Reads Per Kilobase per Million mapped reads (RPKM) for single-end reads[4]. In all differentialexpression tests, a gene was considered significant if the q value was less than 0.05 (Cuffdiff Default).lncRNA DiscoveryWe filtered the assembled novel transcripts 2 cell lines to obtain putative lncRNAs. Firstly, identical and overlapping transcripts were merged to remove redundancy. Then, transcripts overlapping with known exons of genes were removed. Only transcripts with length>200nt were retained. To identify potential known lncRNA transcripts, we compare the merged transcriptome with lncRNA and protein-coding genes in public databases including GENCODE[5], NONCODE[6], RefSeq [7], and Ensembl[8]to eliminate potential known lncRNA transcripts with further filtering of length>200 nucleotides.In order to obtain a reliable dataset of putative lncRNAs, single exon models were filtered out. Next, we removed transcripts that were likely to be assembly artifacts or PCR run-on fragments according to class code annotated by Cuffmerage. Among the different classes, only those a nnotated by ‘‘u’’, ‘‘i’’and ‘‘x’’ were retained, which repres ent novel intergenic, intronic and cis-antisense transcripts, respectively. But here, most analyses were focused on intergenic, intronic and cis antisense lncRNAs. Extremely low gene expression is generally considered to be transcriptional noise [9]. On average, 85% of the initial reads could be aligned to the hg19 assembly of the human genome sequence. Transcripts with RPKM/FPKM under lower bound of single tail 85% confidence interval (<0.3) for all expression values were removed.Lastly, we calculated the protein-coding capacity of novel transcripts using CPC (:) which incorporates the sequence features into a support vectormachine to assess the protein-coding potential of each transcript. The proportion of coding transcripts miss-classified as non-coding RNAs by CPC was previously shown to be marginal [10,11], suggesting CPC is a robust approach for distinguishing coding from noncoding RNAs. Then those putative transcripts with CPC score<-1 were retained as candidate lncRNAs for the further analysis.Putative lncRNA ClassificationThe assembled putative lncRNAs were divided into three categories: (1) lncRNAs without any overlaps with any genes (RefSeq or Ensembl) were classified as intergenic overlap lncRNAs (intergenic lncRNAs); (2) lncRNAs that were entirely contained within intron of any protein-coding genes in either sense or antisense orientation were classified as intronic overlap lncRNAs (intronic lncRNAs); (3) lncRNAs with exonic overlaps with any exons of RefSeq transcript on the opposite strand were classified as cis-antisense overlap lncRNAs (cis-antisense lncRNAs).Gene Function Enrichment AnalysisDAVID (:/) [14] was used to perform gene function enrichment analysis based on GO (:/) [12], KEGG (:genome.jp/kegg/) [13] annotation by submitting closest gene lists for putative intergenic lncRNAs, host genes for putative embryonic brain intronic lncRNAs and overlapping genes for putative embryonic brain cis-antisense lncRNAs, respectively. Only putative intergenic lncRNAs with distance to closest genes <500 kb were kept for this analysis, which would eliminate longdistance irrelevant genes. Furthermore, putative intronic lncRNAs embedded in long introns (<100 kb) of known genes were also discarded to avoid the bias of large introns. Functional terms with Benjamini-Hochberg adjusted p-values<0.05 were considered to be significantly。