【医药】如何控制基因毒性杂质
基因毒性杂质的评估与控制

6. Istituto Superiore di Sanita (Italy) 7. Prous Institute (Spain) 8. Swedish Toxicology Science Research Center(Sweden)
1.临床使用剂量增加 2.用药时长显著增加 3.病情严重或危及生命的病患状态下采用较高可接 受摄入量,变为不那么严重的病患情况后,原有的 杂质可接受摄入量不再适当 4.使用新的给药途径 5.扩大使用患者群包括孕妇和(或)小儿
◆新的杂质被确知属于第一类或第二类诱变性杂
质
THREE
基因毒性杂质的评估
一、评估内容
五、杂质残留风险评估考量
基毒杂质清除因子计算表
代码
GTI
到API 步骤
3 2 1
反应活 性
1-100 X X
溶解性
1-10(100) X X
挥发性
1-10 X X
电离性
5 X X
消除因 子/步
XXX
总消除因 子
清除 原理
Xx,xxx,xxx
参考文献: Teasdale A, Elder D, Chang S-J, et al. Risk assessment of genotoxic impurities in new chemical entities: strategies to demonstrate control[J]. Org Process Res Dev, 2013, 17: 221-230.
FDA
2008 年签发《原料药和成品药中遗传 毒性和致癌性杂质,推荐方法》
内容和EMA指南基本一致,主要包括: ◆ 原料药和制剂中的基因毒性杂质生 成的预防办法
欧盟关于基因毒性杂质问题的解答

欧盟医药管理局(EMA)发布了《基因毒性杂质限度指引》问答。
目的是对《基因毒性杂质限制指引》(EMEA/CHMP/QWP/251344/2006)进行统一与说明,共有9个问答,具体内容如下:问题1:该指引并不要求对已批准销售的产品进行基因毒性杂质再评估,除非有一个特定的“重要原因”(cause-for-concern)。
请问什么是“重要原因”?回答:如果原料药的生产过程基本上没有改变,就不需要对基因毒性杂质进行重新评价。
但是,如果新知识表明有新原因时,例如几年前发现的甲磺酸盐药物可能形成甲磺酸烷基的基因毒性杂质,这需要进行基因毒性杂质的再评估,包括EP药典中收载的所有甲磺酸盐类产品,并出示“生产声明”。
问题2:该指引指出:即使按决策树程序其水平低于毒理学关注阈值(threshold of toxicological concern,TTC),也要尽可能地减少已知或未知的诱变杂质(mutagenic impurity)。
如果已知其诱变杂质的水平低于TTC(TTC是一个非常保守的值),为什么要还进一步减少呢?实际上这还涉及定量限在1ppm 左右的分析方法,可以这样做但可能没结果,这是否有必要呢?回答:如果一个诱变杂质的水平低于毒理学关注阈值(相当于临床剂量≤1.5微克/天),就没有必要这样做。
除非它具有一个高度关注的风险结构:如N -亚硝基,黄曲霉毒素类和氧化偶氮物就需要这样做。
问题3:该指引规定:“当一个潜在的杂质包含“警示结构”(structural alerts)时,应考虑用细菌突变试验对其杂质进行的基因毒性分析”。
i)如果一个杂质能诱发“警示结构”,该杂质的致突变试验(Ames)结果为阴性时,是否就足以得出结论:该化合物不属于关注的遗传毒性杂质?是否还需要进一步的确认研究?ii)“警示结构”不存在就足以说明不属于关注杂质呢?iii)假设某杂质属于“警报结构”,但只要加以控制确保其杂质水平低于TTC,不进行常规检测是否可以接受?回答:i)是的。
药物中基因毒性杂质分析方法的研究

药物中基因毒性杂质分析方法的研究2山东辰龙药业有限公司 272300摘要:遗传性的毒素会破坏DNA,使其具有致癌性,具有很大的危险性。
因为它的结构性较强,所以在服用药物时,会有吸收此类杂质的危险。
在一些国家,对有毒物质的限制已成为一种主要的药品进入市场。
本文介绍了遗传毒性杂质的基本概念、相关标准、部分杂质的检测限度,为检测基因毒性杂质提供了理论基础,保证了患者的使用安全。
关键词:药物;基因毒性;杂质检测方法引言药品的安全,并不是由其本身的毒性决定的,而是由其含有的杂质决定的。
有机杂质可引起遗传变异、染色体断裂、重组等。
因其来源广泛、有毒,已严重危害人类健康。
现有的方法已不能满足对微量基因毒性物质的检测需求,因此,如何对其进行高效的分析具有重要的现实意义。
本文介绍了近年来在检测方法、检测极限等方面的研究进展。
为药品中的基因毒性物质的检测与控制提供了基础和基础,确保了用药的安全性。
一基因毒性杂质研究现状1.1基因毒性杂质来源基因毒性杂质是一种常见的药物。
原料、中间体、副产物、助剂、残留剂、贮存不当等都会引起基因毒性。
目前,基因毒性杂质普遍存在,对人体健康构成极大威胁。
因此,要对其进行严格的科学检验,并对其进行定量检测。
Duane和Ambavaram提出,利用评估决策树来决定生产中含有或预期的有害物质的生产工艺。
Duane介绍了在加工过程中,如何使用评价决策树对遗传毒素的影响,从而为制药企业在生产过程中,不能识别出有毒物质的来源,提供了一个明确的思路。
1.2法规对基因毒性杂质的要求以及国内外药典规定现状为避免遗传毒素对患者的伤害,全球的监管机构一直在更新和改进遗传毒素。
美国食品及药物管理局和欧洲食品药品监督管理局的指南均推荐采用与“毒理学关注阈值”相关的限制标准,以控制基因毒性杂质进入制药公司。
TTC是指确定可被接受的化学物质的摄入量。
如果终生服药,TTC估算出每天可吸收的基因毒性杂质不能超过1.5μg。
遗传毒性杂质在医药工业中的来源与控制路径

遗传毒性杂质在医药工业中的来源与控制路径摘要:制药企业生产出的药品如果存在遗传毒性杂质,使得药品带有可遗传的毒性,会对人类健康造成严重威胁。
近年来,药品中遗传毒性杂质问题已成为了药品监管机构重点关注的问题之一。
本文将简要概括遗传毒性杂质的属性和含义,详细分析遗传毒性杂质的具体来源,并在最后提出如何控制生产药品中遗传毒性杂质的具体途径。
关键词:遗传毒性杂质;医药工业;来源;控制路径在制药环节中,很多药品通过合成或者天然产物结构修饰制成。
相关制药企业为了在复杂的合成过程中尽可能提高生产效率,而使用大剂量的化学试剂。
这种化学试剂过量会使反应继续发生,进而发生副反应,产生副产物最后仍然储存在药品中售卖。
这样的药品中含有大量不明杂质,可能会影响人类的身体健康。
药品监管局了解到这一问题后,开始聚焦遗传毒性杂质在药品中的含量这一指标,这一问题也成了各位专家的研究重点。
一、遗传毒性杂质的属性和含义首先,我们要明确遗传毒性是指物理或化学的某些因素与生物体内的DNA等遗传物质相结合,进而发生作用并最终表现为毒性。
遗传物质进入人体后,会刺激和加快基因突变或者使人体细胞发生癌变,会对人体健康造成不利影响。
因此,遗传毒性杂质本身具有致突变性和致癌性两种基本属性,容易使得生物体发生基因突变或者发生致癌现象,这种突发性大多情况下是无法及时反应或者预测的。
二、遗传毒性杂质的来源遗传毒性杂质主要来源于药品生产过程中。
药品生产过程涉及到的原料或产物有很多,都从属于化学试剂。
例如反应物、催化剂、副产物等等。
根据研究,遗传毒性杂质的遗传毒性机制是嘧啶和嘌呤碱的N原子、O原子以及磷酸二甲酯骨架,在特殊情况下进入DNA找到碱基的亲核中心,破坏连接的键,进而使得整条DNA双链断裂。
遗传毒性杂质的常见来源包括试剂、副反应的生成物和有机溶剂三种方式。
(一)试剂含有遗传毒性杂质的试剂包括硼酸、芳香胺类、烷基卤化物、环氧乙烷、肼试剂、氮氧化物等。
环氧乙烷自身带有环,而DNA中心受到环的张力会与该物质发生亲核反应,进而产生大量遗传毒性杂质。
基因毒性杂质控制

1
1
基因毒性杂质的定义
2
ICH M7介绍
3
基因毒性杂质的控制
4
当前需要关注的一些方面
2020/3/24
2
杂质按照毒性分为一般杂质和毒性杂质。基因毒性杂质 是一类可与DNA反应,造成DNA损伤,在很低水平下即可诱发
基因突变,并可能致癌的杂质。
PGIs(potentially genotoxic impurities 潜在基因毒性的杂质) GIs( genotoxic impurities 基因毒性杂质)
需控杂质与已建立可接受限度的中间体物化特性相似,清除方式相似, 且残留更低。
案例4:高反应活性基因毒性物质(二氯亚砜)。
根据反应活性不可能残留。
上述案例详见M7 第25页:
2020/3/24
26
附 关于杂质限度的界定方法
在满足以下1或多种条件视为得到界定(制剂同原料药)
(1) The observed level and proposed acceptance criterion for the impurity do not exceed the level observed in the reference listed drug product.
➢ 方法2:起始原料或中间体标准或中控过程中控制在可接受限度以下;
➢ 方法3:起始原料或中间体标准或中控过程中控制在可接受限度以上;
明确杂质的去向及清除过程; 根据实验室研究,成品残留在可接受限度的30%以下(推荐加标试验); 必要时有中试或商业化批数据支持。
➢ 方法4:充分理解工艺参数和影响杂质残留因素,有足够信心保证原 料药中残留在可接受限度以下,不需检测(不定入任何标准)。
基因毒性杂质的控制

原料药质量研究部2008~2010年度基因毒性杂质限 度验证列表:
序号 1 2 3 4 5 6 7 基因毒性杂质名称 保护基溴化物 邻氨基甲苯 间氨基甲苯 对氨基甲苯 邻硝基甲苯 叠氮酸 叠氮酸 杂质限度 15ppm 7.5ppm 7.5ppm 7.5ppm 7.5ppm 10ppm 10ppm 产品名称 氯沙坦钾 托拉塞米 托拉塞米 托拉塞米 托拉塞米 坎地沙坦酯 缬沙坦
——基因毒性杂质卤代烃的风险评估
有数据表明氯乙烷、氯甲烷为基因毒性杂质, 因此有理由怀疑其他低分子卤代烃类也有类 似的作用。在生产中应该对其进行相应的控 制。
——基因毒性杂质卤代烃的风险评估
在氨基物盐酸盐使用醇类溶剂精制的时候, 基本都会产生卤代烃。 产生的条件和温度、水分、浓度、时间等有 关系。 对于控制低级卤代烃的方法可以参考控制甲 磺酸酯的相关建议。
是否检查了起始物料,如甲磺酸盐(苯磺酸 盐,对甲苯磺酸盐,羟乙基磺酸),中的烷 基磺酸酯或芳基磺酸酯杂质(如甲磺酸中的 EMS 和MMS)及相应的酰氯?是否有这些 杂质的适宜标准和验证过的方法? 当被磺酸酯或相关物质所污染了的磺酸作为 起始物料用于药物活性成分时,是否能保证 药物活性成分中潜在基因毒性杂质不超过其 TTC值?应当要考虑各种烷基或芳基取代磺 酸酯杂质的累加风险。
缬沙坦 缬沙坦 西酞普兰氢溴酸盐 西酞普兰氢溴酸盐 西酞普兰氢溴酸盐 西酞普兰氢溴酸盐 盐酸吡格列酮 盐酸吡格列酮 盐酸吡格列酮
原料药质量研究部2008~2010年度基因毒性杂质限 度验证列表:
18 19 20 酞嗪二酮 联苯溴化物 二甲海因 0.04% 10ppm 0.05% RP-氯沙坦钾 RP-氯沙坦钾 RP-氯沙坦钾
不能进行阀值鉴定的基因毒性杂质的可接受剂量
ema基因毒杂质控制指导原则

ema基因毒杂质控制指导原则EMA基因毒杂质控制指导原则随着基因工程技术的发展,基因毒杂质成为了EMA制造过程中一个重要而复杂的问题。
基因毒杂质是指在基因工程制造过程中产生的潜在有害的物质,其可阻碍产品的质量和安全性。
为了有效控制基因毒杂质,EMA制定了一套基因毒杂质控制指导原则。
本文将对此进行详细阐述。
一、建立完善的产品质量控制体系首要任务是建立和完善完整的产品质量控制体系,确保产品符合EMA要求的标准和规范。
这包括建立实验室测试方法,测试产品中的毒性物质含量,并确保测试结果的可靠性和准确性。
此外,在产品制造过程中,要对关键步骤进行严格的控制和监测,确保产品质量的稳定性和一致性。
二、有效控制原材料的选择和管理EMA强调原材料的选择和管理对于基因毒杂质控制的重要性。
制造过程中使用的原材料必须来自可靠的供应商,并经过严格的筛选和审核,确保其质量的稳定性和安全性。
EMA要求企业建立原材料管理系统,对采购的原材料进行检验和验证,及时发现和处理不合格的原材料。
三、确保生产设备的清洁和消毒生产设备的清洁和消毒是控制基因毒杂质的关键环节。
EMA要求企业建立标准的清洁和消毒程序,确保生产设备的表面和内部的清洁度。
清洁和消毒程序应当包括适当的清洗剂和消毒剂的选择和使用,以及正确的清洗和消毒方法。
此外,生产设备应定期进行验证和验证,以确保其清洁和消毒效果符合要求。
四、实施适当的监察和审查程序为了确保基因毒杂质控制的有效性,企业应实施适当的监察和审查程序。
这包括对生产现场的定期和不定期监察,对关键环节的抽样和检验,以确保产品的质量和安全性。
此外,应建立完善的记录和文档管理系统,记录并跟踪基因毒杂质控制的各项活动和结果。
五、持续改进和故障分析EMA鼓励企业进行持续改进和故障分析,不断提高基因毒杂质控制的能力和水平。
企业应建立故障分析和纠正措施程序,对发生的问题进行追踪和分析,并采取相应的纠正措施,防止类似问题再次发生。
药物研发中基因毒性杂质的控制策略与方法探索进展

药物研发中基因毒性杂质的控制策略与方法探索进展摘要:本文简介了基因毒素杂质的概念,对药物制作过程中的潜在杂质进行简单的概述,并重点讨论的“避免-控制-清除”策略与方法。
关键词:药物研发;基因毒性杂质;控制策略;方法一、毒素门事件在制药行业中的影响2017年6月,在欧洲的药物检测中发现一些抗艾滋病药物中含有大量的基因毒素杂质,制药商决定召回所有的药物[1]。
2018年7月,花海制药的高血压药中被检测出含有微量的基因毒性杂质,全球市场被召回。
2018年8月印度制药公司检测出基因毒性杂质,相关的14批药物被公司召回。
毒素门事件不但给患者们带来了巨大的隐患,对企业也是一种巨大的经济损失,这件事情给整个制药行业带来了一个警告。
二、基因毒性杂质的基本概念基因毒素是一种能够直接将DNA破坏或者引起身体内大量基因突变的物质。
它能够使人类身体内的染色体断裂、插入或修饰复制中的DNA和使细胞发生突变。
三、可能具有基因毒性的警示结构图1展示出了一部分基因毒性的警示结构官能团,这些官能团都能够与DNA发生反应。
虽然这些官能团还不够详细,但是可以为基因毒素评估做出基本的评估。
还有很多其他的基因毒性官能团,它们在当前的一些商业软件中例如DERKK、Mcase等便能够做出基本的评估[2]。
由于基因毒性化合物例如芳香胺等作为原料制作药品时能够很大概率对药品成分带来基因毒性杂质污染,所以对GTI的检查、控制和预防一定要非常的严格。
四、基因毒性的控制策略与方法制药的过程中使用“避免”-“控制”-“清除”的策略(表4、表5)来控制基因毒性,这样能够发挥最大的能力减少药品原料中的基因毒性杂质[3]。
美国有的制药企业便规定必须按照如下规定来制作药品:(1)GTI或者PGI生成后保证至少还有四步才能得到最终产物,而且在每一步都要判断分析是否会清除PGI。
(2)当需要测试耐热性时,需要在分离纯化前添加足够的GTI或者PGI类似保证检验产品的纯度。
遗传毒性杂质控制指导原则

遗传毒性杂质控制指导原则遗传毒性杂质控制指导原则用于指导药物遗传毒性杂质的危害评估、分类、定性和限值制定,以控制药物中遗传毒性杂质潜在的致癌风险。
为药品标准制修订,上市药品安全性再评价提供参考。
一、总则遗传毒性(Genotoxcity)是指遗传物质中任何有害变化引起的毒性,而不考虑诱发该变化的机制,又称为基因毒性。
遗传毒性杂质(Genotoxic Impurities,GTIs)是指能引起遗传毒性的杂质,包括致突变性杂质和其它类型的无致突变性杂质。
其主要来源于原料药的生产过程,如起始原料、反应物、催化剂、试剂、溶剂、中间体、副产物、降解产物等。
致突变性杂质(Mutagenic Impurities)指在较低水平时也有可能直接引起DNA损伤,导致DNA突变,从而可能引发癌症的遗传毒性杂质。
本指导原则主要关注致突变机制的遗传毒性杂质,非致突变机制的遗传毒性杂质在杂质水平的剂量下,一般可忽略其致癌风险。
药品生产、药品标准提高及上市药品再评价过程中发现杂质后,可按本指导原则进行风险评估,确定其是否为遗传毒性杂质,尤其是致突变性杂质。
如果一个杂质被鉴定为具有潜在的致癌风险,应制定相应的限值。
在制订可忽略致癌风险的杂质限值时,应进一步分析生产工艺,兼顾安全性和质量风险管理成本两方面的因素,综合考虑制定合适的限值。
本指导原则包括危害评估方法、可接受摄入量计算方法和限值制定方法。
本指导原则中描述的对杂质潜在致突变性的评估方法不适用于以下类型的原料药和制剂:生物/生物技术制品、肽类、寡核苷酸、放射性药物、发酵产品、中药和动物或植物来源的粗制品。
也不适用于已上市药物中使用的辅料、调味剂、着色剂和香料,以及与药物包材相关的可浸出物。
本指导原则中对杂质潜在致突变性的评估方法不适用于用于晚期癌症适应症的原料药和制剂,以及用于其它适应症但本身在治疗剂量下就具有遗传毒性,且预计可能与癌症风险增加有关的原料药。
在这些情况下,致突变性杂质不会显著增加原料药的致癌风险。
基因毒性杂质控制

3
TTC限度或以下;或进行细菌致 含警示结构的物质,与API结构 突变性试验。 无关,无致突变性数据 如果无致突变性=第5类; 如果有致突变性=第2类。
所含警示结构与活性成份(API) 相同,或与已证实无基因毒性 按一般杂质控制 原料药结构相关化合物的警示 结构相同 无警示结构,或有充分的数据 证明其警示结构无致突变性 按一般杂质控制
d、成品基于TTC的可接受限度为50ppm。
结论: 起始物料Y中杂质B控制限度0.1%可接受,不需提供中试及商业化 批
数据。
案例3:关于结构相似的基因毒性杂质的控制(芳硝基位置异构体 杂质);
需控杂质与已建立可接受限度的中间体物化特性相似,清除方式相似, 且残留更低。
案例4:高反应活性基因毒性物质(二氯亚砜)。
(1)工艺杂质控制 方法1:原料பைடு நூலகம்质量标准中控制在可接受限度以下
至少连续6批中试或连续3批放大检出低于限度30%以下,可周期性检验。
方法2:起始原料或中间体标准或中控过程中控制在可接受限度以下; 方法3:起始原料或中间体标准或中控过程中控制在可接受限度以上;
明确杂质的去向及清除过程; 根据实验室研究,成品残留在可接受限度的30%以下(推荐加标试验); 必要时有中试或商业化批数据支持。
4、基因毒性杂质的限度
(3)非终生暴露(Less than lifetime,LTL)的控制 采用Staged TTC Approach
单个基因毒性杂质可接受摄入量
治疗期 日摄入量 (μg/day)
≤1月 120
>1-12月 20
>1-10年 10
>10年至 终生 1.5
间隔给药按实际给药天数计,例:某药治疗期2年,每周给药一次,2
基因毒杂质控制策略案例

基因毒杂质控制策略案例基因毒杂质(Genotoxic Impurities)控制策略是药物开发和制造过程中的重要环节,旨在确保药物产品中基因毒性杂质的控制和限制。
以下是一个基因毒杂质控制策略的案例示例:
1. 风险评估:首先,对药物候选化合物进行综合的基因毒性风险评估。
评估包括利用体外基因毒性测试(如Ames 试验)和计算毒性预测模型,对化合物进行筛选和分类。
2. 导入限值:基于风险评估结果,制定适当的基因毒杂质导入限值。
此限值应与国际指南(如ICH M7指南)和适用的监管要求相一致。
3. 合成和纯化策略:在药物合成和纯化过程中,采取特定的操作条件和工艺控制,包括选择合成路线、溶剂使用、温度控制、反应时间和条件等,以最小化基因毒杂质的产生和残留。
4. 检测和分析:开发和验证适当的分析方法,用于检测和定量基因毒杂质的存在。
常见的分析技术包括高效液相色谱(HPLC)、质谱法(如LC-MS/MS)、核磁共振(NMR)
等。
5. 清洁验证:使用适当的清洁验证方法和程序,确保生产设备和工艺的清洁性,在不同批次之间避免交叉污染和残留。
6. 临床监控:在临床阶段,对药物进行基因毒杂质的监控和评估,以确保在实际使用中的毒性风险得到控制。
这只是一个基本的基因毒杂质控制策略案例,具体策略会因药物特性、制造过程和监管要求等因素而有所不同。
在实际应用中,需要根据具体情况制定并执行适合的控制策略,并与相关的监管机构保持合作与沟通。
(完整版)基因毒性杂质的评估与控制

杂质 来源
降解杂质
加速试验、长期试验、光降 解、强制降解试验
环境污染
三、基因毒性杂质识别
工艺杂质
实际杂质
潜在杂质
API中实际观察到>ICH Q3A 报告限(0.05%)工艺杂质
1)合成API过程:起始物料,中间体,化学试剂 2)风险评估可能带入API中的,存在于起始物料,中间体中已识别 的杂质,以及合理机理预测产生的副产物(对于工艺早期杂质携带 入API的风险可忽略,但要提供基于风险论证的表明哪步后应该评估 杂质的潜在突变性) 3)对后工艺引入的起始物料,评价起始物料合成最后一步的潜在基 因毒性杂质
2018年
华海药业生产的缬沙坦原料药中含有微量基因毒性杂质N,N-二甲 基亚硝胺(NDMA),缬沙坦及其相关制剂从欧洲、美国和中国市 场被召回。
2018年
印度Torrent制药生产的缬沙坦片剂中也检测出含有NDMA,该公司 也从美国市场上自愿召回了14批相关药品。
二、相关概念
何为基因毒性杂质?
基因毒性杂质,也称遗传毒性杂质,通常指较低水平可直接造成DNA损伤,进而导致DNA突变, 因此可能引发癌症的DNA反应性物质。
EMA
2006年颁布《基因毒性杂质限度指南》
2010年发布《遗传毒性杂质限度指导 原则问答》 ◆为限制新活性物质中的基因毒性杂 质提供了解决问题的框架和具体方案。
◆新药必须进行基因毒性杂质分析
◆对于现有药品,不强制进行基因毒 性杂质分析评估
◆对已上市产品进行化学合成变更或 仿制药上市前,需对合成路线、过程 控制、杂质概括评价并与现有产品对 比,以确定未引入新的或更高水平的 基因毒性杂质
降解杂质
长 期 稳 定 性 试 验 , API 中 观 察 到>ICH Q3A报告限降解产物
基因毒性杂质控制相关文件及指南介绍
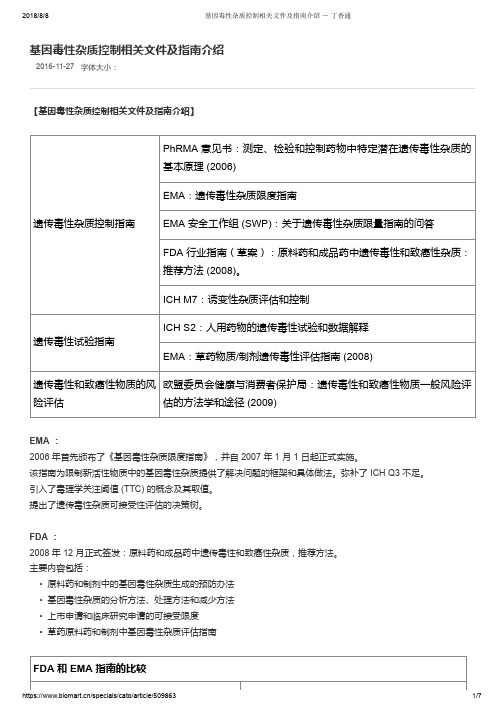
2016-11-27字体大小:基因毒性杂质控制相关文件及指南介绍【基因毒性杂质控制相关文件及指南介绍】遗传毒性杂质控制指南PhRMA 意见书:测定、检验和控制药物中特定潜在遗传毒性杂质的基本原理 (2006)EMA:遗传毒性杂质限度指南EMA 安全工作组 (SWP):关于遗传毒性杂质限量指南的问答FDA 行业指南(草案):原料药和成品药中遗传毒性和致癌性杂质:推荐方法 (2008)。
ICH M7:诱变性杂质评估和控制遗传毒性试验指南ICH S2:人用药物的遗传毒性试验和数据解释EMA:草药物质/制剂遗传毒性评估指南 (2008)遗传毒性和致癌性物质的风险评估欧盟委员会健康与消费者保护局:遗传毒性和致癌性物质一般风险评估的方法学和途径 (2009)EMA :2006 年首先颁布了《基因毒性杂质限度指南》,并自 2007 年 1 月 1 日起正式实施。
该指南为限制新活性物质中的基因毒性杂质提供了解决问题的框架和具体做法。
弥补了 ICH Q3 不足。
引入了毒理学关注阈值 (TTC) 的概念及其取值。
提出了遗传毒性杂质可接受性评估的决策树。
FDA :2008 年 12 月正式签发:原料药和成品药中遗传毒性和致癌性杂质,推荐方法。
主要内容包括:• 原料药和制剂中的基因毒性杂质生成的预防办法• 基因毒性杂质的分析方法、处理方法和减少方法• 上市申请和临床研究申请的可接受限度• 草药原料药和制剂中基因毒性杂质评估指南FDA 和 EMA 指南的比较相似点不同点推荐的鉴定和认证潜在遗传性杂质的方法相同 推荐的处理遗传毒性和致癌性杂质的方法相同FDA 指南包含致癌性杂质TTC 设定为 1.5 μg/天指南允许的 14 天内用药的 TTC 水平为 120 μg , 而非仅针对单次用药临床试验中短期暴露的 TTC 更高FDA 指南不允许根据现售药品的短期暴露情况而 提高 TTC ICH M7【基因毒性杂质的控制策略】具有阳性致癌数据的诱变杂质(第1类)---计算可接受摄入量( AI ): • M7 Addendum 中列出的 15 种化合物中有 10 个为该计算方法计算 • Carcinogenicity Potency Database (CPDB )中列明了 1574 种致癌物质的 TD50 值毒理学关注门槛---TTC 法(第 2/3 类): • ICHM7 主要讨论的方法,主要针对第 2/3 类基因毒性杂质,比如低级磺酸酯类等。
基因毒性杂质控制

(4) The observed level and proposed acceptance criterion for the impurity do not exceed the level that has been adequately evaluated in toxicity studies. 杂质的检测水平及拟定限度不超过经过充分的毒理学研究的水平。
(2)特定限度控制策略
• 计算:已知致癌活性(如半数致癌剂量TD50),线性外推可接受限度,按十万分之 一风险计算,TD50/50000,据此计算限度(见第16页,note4,环氧乙烷);
• 参考:如有化学结构相似,且特定限度已确定的化合物,在提供结构相似合理性 及支持数据前提下,可参考计算限度;
• 计算:如前例,已知NOEL,计算杂质的PDE,结合每日最大用药剂量计算限度;
➢ 方法3:起始原料或中间体标准或中控过程中控制在可接受限度以上;
明确杂质的去向及清除过程; 根据实验室研究,成品残留在可接受限度的30%以下(推荐加标试验); 必要时有中试或商业化批数据支持。
➢ 方法4:充分理解工艺参数和影响杂质残留因素,有足够信心保证原 料药中残留在可接受限度以下,不需检测(不定入任何标准)。
结论:按一般杂质控制。
三、基因毒性杂质的分类
需充分检索和对比文献后,确定控制策略!
三、基因毒性杂质的控制
4、基因毒性杂质(分类1、2和3)的限度
(Risk characterization M7 第7页)
(1)基于TTC的控制
一般为长期用药(>10年)、杂质无致癌性数据(分类2和3); 杂质控制水平为1.5μg/日;
摘自:FDA Guidance for Industry ANDAs: Impurities in Drug Substances 第4页
药品研发杂质控制策略

药品研发杂质控制策略如下:
1.杂质分类:药品杂质通常分为有机杂质、无机杂质、残留溶剂。
2.控制方法:
•性状、晶型、熔点、比旋度、异构体、有关物质、残留溶剂、氯化物、硫酸盐、pH值、溶液颜色和澄清度、炽灼残渣、重金属、元素杂质、基因毒性杂质等。
•控制目标(即测什么)、控制方法(用什么方法测)和控制限度(控制标准如何制定)。
3.控制策略:
•源头控制:从源头上避免杂质的产生,例如提高生产工艺的稳定性和清洁性,减少副反应的发生等。
•过程控制:在生产过程中对杂质进行控制,例如通过加入合适的试剂来降低杂质含量,或者通过精制来去除杂质。
•终点控制:在生产结束时对产品进行检测,控制杂质的限度。
4.具体措施:
•对于有机杂质,应关注起始原料、中间体、副产物、降解产物等,这些杂质可能是已鉴定或未鉴定的,挥发性的或非挥发性的。
•对于无机杂质,应关注生产过程,如反应试剂、配位体、催化剂、元素杂质、无机盐等。
•对于残留溶剂,应关注原料药或辅料的生产中以及制剂制备过程中使用的有机溶剂。
基因毒杂质控制策略案例

基因毒杂质控制策略案例基因毒杂质是指在制造过程中因杂质或不纯净的原料导致的可引发基因突变的物质,对人体健康造成潜在的危害。
因此,对于制药和生物技术行业来说,对基因毒杂质的控制至关重要。
本文将以一家制药公司为例,介绍其基因毒杂质控制策略的具体实施情况。
案例分析该公司制定了一套完善的基因毒杂质控制策略,主要包括以下几个方面:1.原料管理公司对每一批原料进行严格的检验和筛选,确保不含基因毒杂质。
在采购原料时,公司要求供应商提供详尽的质量报告,并进行审核。
同时,公司自身也要加强对原料的检测和把关,确保符合标准。
2.生产过程控制在生产过程中,公司制定了严格的操作规程,包括对生产设备、工艺流程和操作人员的管理。
同时,公司进行了一系列的监测和控制措施,确保生产过程中不发生基因毒杂质的产生。
例如,在每个生产环节都要进行抽样检验,确保产品的质量符合标准。
3.环境监测公司还加强了对生产环境的监测,保持生产场所的清洁和良好的通风环境。
同时,对环境中可能产生的基因毒杂质进行了严格的控制,确保不对产品质量造成影响。
4.技术改进为了进一步提高基因毒杂质的控制水平,公司还进行了技术改进,引进了先进的生产设备和检测技术。
同时,加强了员工的培训和技术交流,提高了员工的意识和能力,确保总体生产过程的控制效果更加可靠。
结果与效益经过一系列的控制措施和持续的改进,该公司成功地实现了基因毒杂质的控制目标,产品的质量稳定可靠。
同时,公司还得到了一些额外的效益。
首先,公司获得了客户的信任和好评,提高了市场的竞争力。
其次,公司降低了因产品质量问题而可能引发的风险,保障了持续稳定的生产。
最后,公司还提高了自身的技术水平和管理水平,为未来的发展打下了良好的基础。
结论基因毒杂质的控制对于制药和生物技术行业来说至关重要。
针对不同的企业特点和生产特点,制定相应的基因毒杂质控制策略是至关重要的。
通过采取科学有效的控制措施和持续不断的改进,企业可以实现基因毒杂质的全面控制,提高产品质量,增强市场竞争力,为企业的可持续发展奠定坚实的基础。
基因毒性杂质及其警示结构

基因毒性杂质及其警示结构
据研究,化合物具有基因毒性的原因可能是因为其能够与DNA结合并导致DNA损伤,从而引起基因突变和细胞死亡。
此外,一些化合物也可能会干扰细胞的DNA修复机制,从而进一步加剧其基因毒性。
在药品生产过程中,如何控制基因毒性杂质的含量?
为了控制基因毒性杂质的含量,药品生产企业需要采取一系列措施,包括选择合适的原料和反应条件、对反应过程进行严格监控、采用有效的分离和纯化技术、以及对最终产品进行全面的质量控制等。
此外,药品生产企业还需要对基因毒性杂质进行全面的风险评估,并制定相应的控制策略,以确保药品的安全性和有效性。
最后,基因毒性杂质的控制不仅是药品生产企业的责任,也需要相关监管机构的支持和监督。
只有通过全社会的共同努力,才能保障药品的安全性和有效性,为人民群众的健康保驾护航。
___夫妇在19世纪70年代对化合物致癌机理进行了深入研究,并提出了“亲电理论”。
该理论指出,构成DNA的四个碱基中存在许多亲核位点,如嘧啶环和嘌呤上的N和O等,这些位点可以与亲电试剂反应,从而引起基因突变,是诱发癌症的重要原因。
___在19世纪80年代提出了致癌性的警示结构(SAs)的概念,这些结构含有的化合物与DNA发生作用的可能性较高,可能诱发癌症。
___的相关机构在2009年的报告中收录了三十余种基因毒性警示结构,这些结构的具体信息也在报告中列举了出来。
基因毒性杂质控制相关文件及指南介绍
2016-11-27字体大小:基因毒性杂质控制相关文件及指南介绍【基因毒性杂质控制相关文件及指南介绍】遗传毒性杂质控制指南PhRMA 意见书:测定、检验和控制药物中特定潜在遗传毒性杂质的基本原理 (2006)EMA:遗传毒性杂质限度指南EMA 安全工作组 (SWP):关于遗传毒性杂质限量指南的问答FDA 行业指南(草案):原料药和成品药中遗传毒性和致癌性杂质:推荐方法 (2008)。
ICH M7:诱变性杂质评估和控制遗传毒性试验指南ICH S2:人用药物的遗传毒性试验和数据解释EMA:草药物质/制剂遗传毒性评估指南 (2008)遗传毒性和致癌性物质的风险评估欧盟委员会健康与消费者保护局:遗传毒性和致癌性物质一般风险评估的方法学和途径 (2009)EMA :2006 年首先颁布了《基因毒性杂质限度指南》,并自 2007 年 1 月 1 日起正式实施。
该指南为限制新活性物质中的基因毒性杂质提供了解决问题的框架和具体做法。
弥补了 ICH Q3 不足。
引入了毒理学关注阈值 (TTC) 的概念及其取值。
提出了遗传毒性杂质可接受性评估的决策树。
FDA :2008 年 12 月正式签发:原料药和成品药中遗传毒性和致癌性杂质,推荐方法。
主要内容包括:• 原料药和制剂中的基因毒性杂质生成的预防办法• 基因毒性杂质的分析方法、处理方法和减少方法• 上市申请和临床研究申请的可接受限度• 草药原料药和制剂中基因毒性杂质评估指南FDA 和 EMA 指南的比较相似点不同点推荐的鉴定和认证潜在遗传性杂质的方法相同 推荐的处理遗传毒性和致癌性杂质的方法相同FDA 指南包含致癌性杂质TTC 设定为 1.5 μg/天指南允许的 14 天内用药的 TTC 水平为 120 μg , 而非仅针对单次用药临床试验中短期暴露的 TTC 更高FDA 指南不允许根据现售药品的短期暴露情况而 提高 TTC ICH M7【基因毒性杂质的控制策略】具有阳性致癌数据的诱变杂质(第1类)---计算可接受摄入量( AI ): • M7 Addendum 中列出的 15 种化合物中有 10 个为该计算方法计算 • Carcinogenicity Potency Database (CPDB )中列明了 1574 种致癌物质的 TD50 值毒理学关注门槛---TTC 法(第 2/3 类): • ICHM7 主要讨论的方法,主要针对第 2/3 类基因毒性杂质,比如低级磺酸酯类等。
遗传毒性杂质控制指导原则
遗传毒性杂质控制指导原则遗传毒性杂质控制指导原则用于指导药物遗传毒性杂质的危害评估、分类、定性和限值制定,以控制药物中遗传毒性杂质潜在的致癌风险。
为药品标准制修订,上市药品安全性再评价提供参考。
一、总则遗传毒性(Genotoxcity)是指遗传物质中任何有害变化引起的毒性,而不考虑诱发该变化的机制,又称为基因毒性。
遗传毒性杂质(Genotoxic Impurities,GTIs)是指能引起遗传毒性的杂质,包括致突变性杂质和其它类型的无致突变性杂质。
其主要来源于原料药的生产过程,如起始原料、反应物、催化剂、试剂、溶剂、中间体、副产物、降解产物等。
致突变性杂质(Mutagenic Impurities)指在较低水平时也有可能直接引起DNA损伤,导致DNA突变,从而可能引发癌症的遗传毒性杂质。
本指导原则主要关注致突变机制的遗传毒性杂质,非致突变机制的遗传毒性杂质在杂质水平的剂量下,一般可忽略其致癌风险。
药品生产、药品标准提高及上市药品再评价过程中发现杂质后,可按本指导原则进行风险评估,确定其是否为遗传毒性杂质,尤其是致突变性杂质。
如果一个杂质被鉴定为具有潜在的致癌风险,应制定相应的限值。
在制订可忽略致癌风险的杂质限值时,应进一步分析生产工艺,兼顾安全性和质量风险管理成本两方面的因素,综合考虑制定合适的限值。
本指导原则包括危害评估方法、可接受摄入量计算方法和限值制定方法。
本指导原则中描述的对杂质潜在致突变性的评估方法不适用于以下类型的原料药和制剂:生物/生物技术制品、肽类、寡核苷酸、放射性药物、发酵产品、中药和动物或植物来源的粗制品。
也不适用于已上市药物中使用的辅料、调味剂、着色剂和香料,以及与药物包材相关的可浸出物。
本指导原则中对杂质潜在致突变性的评估方法不适用于用于晚期癌症适应症的原料药和制剂,以及用于其它适应症但本身在治疗剂量下就具有遗传毒性,且预计可能与癌症风险增加有关的原料药。
在这些情况下,致突变性杂质不会显著增加原料药的致癌风险。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
01、何为基因毒性杂质
基因毒性杂质(或遗传毒性杂质,Genotoxic Impurity,GTI)是指能直接或间接损害DNA,引起DNA突变、染色体断裂、DNA重组及DNA 复制过程中共价键结合或插入,导致基因突变或癌症的物质(如卤代烷
烃、烷基磺酸酯类等)。
潜在基因毒性杂质(Potential Genotoxic Impurity ,PGI)结构中含有与基因毒性杂质反应活性相似的基团(如肼类、环氧化合物、N-亚硝胺类等),通常也作为基因毒性杂质来评估。
基因毒性杂质主要来源于原料药合成过程中的起始物料、中间体、试剂和反应副产物。
此外,药物在合成、储存或者制剂过程中也可能会降解产生基因毒性杂质。
除此之外,有些药物通过激活正常细胞而产生基因毒性物质导致突变,如化疗药物顺铂等。
02、何为基因毒性杂质“警示结构”
由于杂质结构的多样性,一般很难进行归类,因此,在缺乏安全性数据支持的情况下,法规和指导原则采用“警示结构”用来区分普通杂质和基因毒性杂质。
所谓“警示结构”,是指杂质中的特殊基团可能与遗传物质发生化学反应,诱导基因突变或者染色体断裂,因此具有潜在的致癌风险。
对于含有警示结构的杂质,应当进行(Q)SAR预测和体内外遗传毒性和致癌性研究,或者将杂质水平控制在毒理学关注阈值(TTC)之下。
但是含有警示结构并不能说明该杂质一定具有遗传毒性,而确认有遗传毒性的物质也不一定会产生致癌作用。
杂质自身性质和结构特点会对其毒性产生抑制或调节作用。
警示结构的重要性在于它提示了可能存在的遗传毒性和致癌性,为进一步的杂质安全性评价与控制指明方向。
(关于基因毒杂质警示结构的详细信息可参考欧盟发布的警示结构《Development of
structure alerts for the in vivo micronucleus assay in rodents》)。
03、基因毒性杂质严格控制的必要性
基因毒性杂质最主要的特点是在极低浓度时即可造成人体遗传物质的损伤,导致基因突变并促使肿瘤发生。
因其毒性很强,对药物的安全性造成强烈的威胁。
由于基因毒性杂质控制不合规,已造成上市药品强行被召回,给企业造成了巨大的损失。
例如:奈非那韦(甲磺酸盐)是一种用于HIV治疗的抗病毒药物,分别于1997年和1998年由FDA和EMA 批准后上市。
2007年6月,罗氏制药接到6名患者投诉DNA序列异常后,立即召回产品,EMEA暂停其上市销售。
在后期内部调查时发现原料药存储罐中有残留的乙醇未清除干净,与甲基磺酸反应形成甲磺酸乙酯。
之后罗氏对存储罐这一步工艺进行修正并在生产过程中增加对甲磺酸乙酯的控制,要求低于0.5ppm。
EMA重新评估检查后,2007年10月才恢复其上市销售。
因此,在该事件后,各国的法规机构ICH、FDA、EMEA等都对基因毒性杂质有了更明确的要求,越来越多的药企在新药研发过程中也重点关注基因毒性杂质的控制和检测。
04、常见GTI/PGI
图1. 常见GTI/PGI种类
磺酸酯
如合成工艺中使用了磺酸类试剂(如:对-甲苯磺酸、甲磺酸)和低级醇(甲醇、乙醇等),则有可能形成磺酸酯化合物,根据取代基的不同,磺酸酯可分为烷基磺酸酯和芳基磺酸酯。
这些物质能与DNA 发生烷基化反应,从而可能成为引发癌症的诱因,尤其是甲磺酸甲酯、甲磺酸乙酯及甲磺酸异丙酯已被证明具有基因毒性,同时其它磺酸酯类也存在潜在基因毒性。
卤代烷烃
卤代烷是在药物合成过程中经常使用到的烷基化试剂,含有一个或者多个卤原子的化合物,结构种类繁多,可以分为氟代烷、氯代烷、溴代烷和碘代烷,是基因毒性杂质中最为常见的一类。
卤代烷烃的反应活性较强,能直接与生物大分子,如DNA、RNA及蛋白质发生烷基化反应,从而导DNA的突变。
酰卤
酰卤类化合物中以酰氯最为常用,是化学合成中重要的酰化试剂。
尽管尚无明确的数据证明酰卤类化合物具有基因毒性和致癌性,但是这类化合物含有警示结构基团,是潜在基因毒性杂质,在药物中需要严格的分析与监测。
肼
肼类化合物是已知的基因毒性杂质,具有潜在的致癌性。
该类化合物性质活泼,主要通过代谢活化生成碳正离子和碳氧自由基等活性较强的中间体,这些中间体能够与DNA发生烷基化反应或者导致DNA的其他病变。
叠氮
叠氮类化合物也是常见的基因毒杂质之一,该类化合物能够抑制细胞色素氧化酶及多种酶的活性,并导致磷酸化及细胞呼吸的异常。
叠氮类化合物的主要急性毒作用是能够引起血管张力极度降低,因此要严格控制。
05、基因毒性杂质基本控制思路
国外制药企业为了产品的成功上市,减少基因毒性杂质相关风险,对于基因毒性杂质的控制基本形成了以下思路:
a. 对工艺路线中所涉及的基因毒性杂质来源进行评估,分析其合成路线,全面考虑起始物质、试剂、溶剂等可能带入的基因毒性物质;
b. 对评估出来的杂质根据是否存在致癌性或者是否存在警示结构进行分类;
c. 确认临床试验中短期用药的每日允许摄入量;
d. 根据杂质类型分类,论证药物基因毒性杂质残留限度,设定严谨或合理的阈值,并使用可靠的分析手段对基因毒性杂质进行针对性的分析检测。