Selumetinib plus docetaxel for KRAS-mutant
蔓洛舒

每日500毫克的分量 所提供的花青苷,相等於
7杯蔓越莓果汁!
透過Bio-Shield®輸送系統, 達到高身體吸收利用率
• Bio-Shield® 將蔓越莓活性成分 包覆於保護層中,以免它們遭 到胃酸破壞或失去活性
• 將活性成分輸送至吸收力最佳的腸道 部位
• 在12-16小時內,控制式地將活性成 分釋放,以達到最高的身體吸收利用 率及效益
Nn Cran-Max® Plus
善用兩種富含強效的專利 成分,對抗念珠菌侵害及 尿道感染:
500mg Cran-Max®
(已獲臨床驗證的蔓越莓萃取物)
200mg UTIROSE™
(取自洛神花的天然萃取物)
只需每天2顆
爲什麽 Cran-Max® 比其他蔓越莓萃
取物更優越?
取自全顆蔓越莓果實 的專利萃取物
(資料來源:美國國家腎臟基金會和美國國立衛生研究院)Biblioteka 增加患上酵母菌感染風險 的因素
•飲食不當 •抗生素 •長期精神壓力及焦慮
不安 •曝露環境毒素中 •糖尿病 •懷孕 •避孕丸及類固醇之類
的藥物 •免疫機能衰弱
念珠菌導致
發胖!
“念珠菌會對阻礙進行 減重目標造成障礙。。。 人體新陳代謝機能會嚴 重衰退,而身體脂肪卻 無法減去,導致無法成
•濃縮洛神花(Hibiscus sabdariffa)萃取物
•含有 •>40% 有機酸 •>5% sambubiosides •>45% 酚類化合物
證實具高身體 吸收利用率
•在尿液中發現完整的 UTIROSE™ 活性成分
•這表示 UTIROSE™ 能通 過腸道及腎臟的障礙, 發揮直接的抗菌功效, 以對付能引發尿道感染 的細菌及念珠菌
初级癌症疗法后使用的含硼蛋白酶体抑制剂[发明专利]
![初级癌症疗法后使用的含硼蛋白酶体抑制剂[发明专利]](https://img.taocdn.com/s3/m/1b1214e47cd184254b3535ea.png)
专利名称:初级癌症疗法后使用的含硼蛋白酶体抑制剂
专利类型:发明专利
发明人:A·惠,R·拉波特卡,N·古普塔,K·文卡塔克里斯南,G·吕申请号:CN201580029362.6
申请日:20150519
公开号:CN106659761A
公开日:
20170510
专利内容由知识产权出版社提供
摘要:本公开涉及包括式(I)的蛋白酶体抑制剂或其药学上可接受的盐的方法或给药方案,所述方法或给药方案用于治疗癌症或预防癌症复发或进展,其中环A、Z和Z如本文所定义。
申请人:千年药物公司
地址:美国马萨诸塞
国籍:US
代理机构:中国国际贸易促进委员会专利商标事务所
代理人:袁志明
更多信息请下载全文后查看。
吉西他滨及顺铂经动脉、静脉注射后血浆、组织药物浓度的变化

吉西他滨及顺铂经动脉、静脉注射后血浆、组织药物浓度的变化目的分析不同化療药物通过动脉及静脉途径注射后血浆及组织内药物浓度的变化情况。
方法40只带瘤裸大鼠,随机分为8组,其中4组为动脉组,另4组为静脉组,带瘤裸大鼠分别经动脉及静脉注射吉西他滨及顺铂。
于注射后5、10、20、40、80、120、360、720 min采血液标本,注射后10、40、120、720 min取组织标本,以高效液相色谱法测定血浆及肿瘤组织中吉西他滨浓度,ICP-MS法测定血浆及肿瘤组织中的铂含量,计算药代动力学参数。
结果经动脉及静脉注射两种药物后,血浆及肿瘤组织中的药物浓度出现规律性变化,其变化过程均可用两室模型来描述。
动脉注射两组药物的药代动力学参数与静脉注射的药代动力学参数不同,动脉组注射药物后,血浆药物峰浓度[吉西他滨:(20.84±10.11)μg/mL,顺铂:(15.13±7.12)μg/mL]均低于静脉组[吉西他滨:(28.96±7.02)μg/mL,顺铂:(21.64±9.72)μg/mL],靶组织内药物峰浓度[吉西他滨:(20.18±9.43)μg/mL,顺铂:(6.98±0.31)μg/mL]均高于静脉组[吉西他滨:(18.19±10.30)μg/mL,顺铂:(3.04±0.11)μg/mL],靶组织内药物曲线下面积[吉西他滨:(2641±411)μg/(min·mL),顺铂:(6025±870)μg/(min·mL)]均明显高于静脉组[吉西他滨:(1663±568)μg/(min·mL),顺铂:(1780±883)μg/(min·mL)],差异均有统计学意义(P < 0.05或P < 0.01)。
结论动脉注射吉西他滨和顺铂较静脉注射有不同程度的优势,这种优势与药物的药理特性有关。
FDA批准治疗葡萄膜黑色素瘤药品selumetinib

selumetinib是一种口服、强效、选择性MEK抑制剂,在I期和II期临床试验中,已被证明作为单药疗法或与标准化疗方案相结合的组合疗法,对多种实体瘤表现出疗效,这些数据支持了selumetinib在MEK依赖性肿瘤中的开发潜力。
MEK是MAPK通路的一部分,该通路在癌症中常常被激活,同时在许多不同实体瘤中升高,包括携带KRAS突变的肿瘤,该突变存在于20%的人类肿瘤,以及20%-30%的NSCLC肿瘤。
如、处方药物、非处方药物、处方药物、药物配置、药物注射以及各类非住院患者的药物分发、咨询、配送等各项服务。如今国内患者可以依托科技,实现远程的病历交互,由美国医生根据患者病情开具电子处方,以正规渠道在美国药房购买到处方药。
目前,阿斯利康正在开展一项III期临床研究,评估selumetinib联合化疗用于转移性葡萄膜黑色素瘤的一线治疗,该研究的数据预计在今年晚些时候获得。除了葡萄膜黑色素瘤,阿斯利康也正在III期临床调查selumetinib治疗KRAS突变阳性肺癌和甲状腺癌的治疗,同时也在一项II期研究中调查selumetinib用于I型神经纤维瘤病(neurofibromatosis,NF)儿科患者的治疗。
产品名称:Selumetinib
化学名:司美替尼
2015年4月17日讯/生物谷BIOON/ --英国制药巨头阿斯利康(AZN)收获FDA的孤儿药地位。 口服小分子抑制剂selumetinib收获了治疗葡萄膜黑色素瘤(uveal melanoma)的孤儿药地位。
葡萄膜黑色素瘤是一种罕见、恶性程度极高的肿瘤,癌细胞在眼组织中形成,一旦扩散出眼组织,没有有效的治疗选择。该病是成人中最常见的原发性恶性眼内肿瘤,约占所有黑色素瘤病例的5%。
Plin2在肺癌中的表达及临床意义

2040Chin J Lab Diagn,December»2020,Vol 24,No. 12文章编号:1007 — 4287(2020)12 — 2040 — 02Plin2在肺癌中的表达及临床意义王晶莹ia,刘洋2,刘启迪ib*(1.吉林大学中日联谊医院a.检验科;b.呼吸内科,吉林长春130033;2.吉林大学第二临床医院放射线科)全球每年的新增肺癌数量高达180万,其中一 半以上死于该病[1]。
流行病学对肺癌的15年生存 率调查结果显示,不同地区不同疾病进展阶段的患者的生存率从4%到17%不等[2]。
而较为常规的治 疗手段是手术切除[3],此外,肺癌的5年生存率也基 于不同的病理分区,呈现出较大的差异[4]。
因此发 现可以用于早期诊断的肿瘤标志物对于改善患者的 预后是十分有意义的。
紫苏脂素2(Pl i n2)是一种与细胞内脂滴(LDs)代谢有关的蛋白质。
然而,对其表达的调控会影响多种代谢和年龄相关疾病的严重程度,如脂肪肝、胰 岛素抵抗和2型糖尿病(T2D)、心血管疾病、动脉粥 样硬化、肌萎缩和癌症[5’6],研究显示该蛋白可能在这些病理状态中起作用。
本研究目的在于寻找肺癌 的差异表达蛋白,探索其作为肿瘤标志物的意义。
1材料与方法1.1研究对象收集手术中切取的新鲜肿瘤组织、正常组织,置于无菌容器内,P B S冲洗,液氮速冻。
1.2方法通过生物信息数据库T C G A检索,初 步确认P l i n2的表达情况,免疫印迹进一步观察其在肺癌组织及癌旁组织中的差异表达情况.免疫组 化染色,探讨P l i n2的表达意义。
1.3主要试剂 N,N’_亚甲双丙烯酰胺,蛋白定量 试剂盒,甘氨酸,无水乙醇,蛋白抽提试剂盒,丙烯酰 胺,D A B试剂盒,Tris-base,二甲苯。
1.4统计学处理数据采用卡方检验,用SPSS12.0 软件分析,P<〇. 05定义为有统计学意义。
2结果2. 1TCGA数据库检索差异表达基因Plin2通过在线检索T C G A数据库,搜索肺癌中的差 异表达基因,发现P l i n2基因在肺癌组织中的转录水平显著低于癌旁组织。
Development and Validation of a Liquid Chromatogra

J. Chem. Chem. Eng. 5 (2011) 1-6.Development and Validation of a LiquidChromatography–Tandem Mass Spectrometry Method for Determination of Artemisinin in Rat PlasmaElhassan Gamal1,2, Yuen Kah1, Wong Jiawoei1, Chitneni Mallikarjun1,3, Al-Dahli Samer1, Khan Jiyauddin1 and Javed Qureshi31. School of Pharmaceutical Sciences, Universiti Sains Malaysia, Minden 11800, Penang, Malaysia2. Local Pharmaceutical Manufacturing Department, General Pharmacy Directorate, MOH, 11111, Khartoum-Sudan3. School of Pharmacy and Health Sciences, International Medical University, 5700, Kula Lumpur, MalaysiaReceived: September 03, 2010 / Accepted: October 11, 2010 / Published: January 10, 2011.Abstract: Artemisinin is a potent anti-malarial drug isolated from traditional Chinese medicinal herb, Artemisia annua. The objective of this study was to develop and validate a sensitive and specific LC-MS/MS method for the determination of artemisinin in rat plasma using amlodipine as Internal Standard. The method consist of a simple liquid-liquid extraction with methyl tertiary butyl ether (MTBE) with subsequent evaporation of the supernatant to dryness followed by the analysis of the reconstituted sample by LC-MS/MS with a Z-spray atmospheric pressure ionization (API) interface in the positive ion-multiple reaction monitoring mode to monitor precursor→product ions of m/z 282.70→m/z 209.0 for artemisinin and m/z 408.9→m/z 237.0 for amlodipine respectively. The method was linear (0.999) over the concentration range of 7.8–2000 ng/mL in rat plasma. The intra and inter-day accuracy were measured to be within 94-104.2% and precision (CV) were all less than 5%. The extraction recovery means for internal standard and all the artemisinin concentrations used were between 82-85%.Key words: Artemisinin, LC-MS/MS, amlodipine, plasma, accuracy and precision.1. IntroductionArtemsinin is the name given to the active principle of qinghaosu, an extract of the Chinese medicinal plant qinghaosu or green Artemisia (Artemisinin annua L.) which has been used for many years centuries in Chinese traditional medicine for treatment of fever and malaria [1]. In 1972, Chinese researchers isolated artemisinin from Artemisia annua L. sweet wormwood) and its structure was elucidate in 1979 as show in Fig. 1.The determination of artemisinin and its derivatives in biological matrices have previously been characterized using several analytical techniques suchCorresponding author: Gamal Osman Elhassan Ph.D., research field: pharmaceutical technology. E-mail: ******************.as LC, HPLC, GC-MS etc [3-8]. However, some of these methods suffer from few drawbacks. In particulars, interference with endogenous constituents in the plasma at the absorption wave length of the derivatized compounds may render these techniques unsatisfactory and few of them lacked the required sensitivity to be used for measurement of drugFig. 1 The chemical structure of artemisinin [2].ll Rights Reserved.Development and Validation of a Liquid Chromatography–Tandem Mass Spectrometry Method forDetermination of Artemisinin in Rat Plasma2concentration in blood sample obtained from clinical investigation [9].To increase the specificity and sensitivity of HPLC-UV method, some workers combined it with a mass spectrometry (MS) and the total system is described as LC-MS technique [10, 11]. The development of LC-tandem mass spectrometry (LC-MS/MS) has made a more specific and sensitive analysis of artemisinin and its derivatives possible [12, 13]. The objective of this study was to develop a sensitive and specific LC-MS/MS method for the determination of artemisinin in rat plasma by simple liquid-liquid extraction procedure.2. Materials and Methods2.1 MaterialsArtemisinin was purchased from Kunming Pharmaceutical Corporation (Kunming, China). Amlodipine was obtained from Sigma Chemical (Louis, USA). Acetonitrile (ACN), formic acid and methyl tertiary butyl ether (MTBE) were purchased from J.T Baker (USA).3. Methods3.1 Instrumentation and ConditionsThe instrumentation comprised of Quattro-micro tandem mass spectrometer with Z-spray atomospheric pressure ionization (API) source (Micromass, Manchester, UK) using electrospray ionization (ESI) operated at positive mode. Chromatography was performed on an Alliance 2,695 separation module (Waters, M.A, USA). The delivery system consisted of an autosampler and a column heater. The chromatographic separation was obtained using an X Terra MS C8 encapped (5 μm) (150 × 2.1 mm) analytical column (Water, USA).3.2 Sample PreparationA 250 μL aliquot of plasma was pipetted into a screw-capped culture tube, followed by 100 μL of internal standard solution (50 ng/mL). To each tube, 5 mL (MTBE) extraction solvent was then added and the mixture was vortexed for 2.5 minutes followed by centrifuging for 15 minutes at 3,500 rpm. The upper layer was transferred to a reactive vial and dried under nitrogen flow at 40 °C. The residue was then reconstituted with 250 μL of mobile phase and 20 μL was injected into the LC-MS/MS system.3.3 Assay ValidationCalibration curve at a concentration range of 7.8–2,000 ng/mL were constructed by spiking blank human plasma with a known amount of artemisinin. Plasma sample spiked with artemisinin at these concentrations 7.8, 62.5, 250, 2,000 ng/mL were used to determine the within and between-day accuracy and precision. For within-day accuracy and precision, replicates analysis (n = 6) for each concentration were performed in a single day. For between-day evaluation, analysis was carried out with a single sample of each concentration daily over 6 days, with calibration curve constructed on each day of analysis. The extraction recovery of artemisinin was estimated by comparing the peak height obtained after extraction of the samples from plasma with that of aqueous artemisinin solution of the corresponding concentration.4. Results and DiscussionBoth electrospray (TIS) and atmospheric pressure chemical ionisation (APCI) methods have been reported previously for the quantification of artemisinin derivatives in biological fluids [11, 12, 14-16]. According to the previously reported methods TIS was found to be superior to APCI for the quantification of artesunate and dihydroartemisinin (DHA) mainly because of improved linearity [16]. Therefore in this method electrospray ionization was used. When artemisinin and amlodipine were injected directly into the mass spectrometer along with mobile phase in the positive mode, the protonated molecules of artemisinin and amlodipine were set as precursorll Rights Reserved.Development and Validation of a Liquid Chromatography–Tandem Mass Spectrometry Method forDetermination of Artemisinin in Rat Plasma3(a)(b)Fig. 2 (a) Positive-ionization electrospray mass spectra of precursor ion for artemisinin; (b) Positive-ionization electrospray mass spectra of product ion for artemisinin.ions with m/z of 282.7 and 408.7, respectively. The product ion that gave the highest intensity was m/z of 209.0 for artemisinin and 237.7 for amlodipine. Fig 2(a) shows the spectra precursor ion, 2(b) production for artemisinin.Artemisinin and amlodipine have retention time of approximately 6.9 and 1.65 minutes, respectively (Fig.3). The peak was well resolved and free from interference from endogenous compounds in rat plasma (Fig. 4).ll Rights Reserved.Development and Validation of a Liquid Chromatography–Tandem Mass Spectrometry Method forDetermination of Artemisinin in Rat Plasma4Fig. 3 Plasma spiked with 500 ng/ml artemisinin and amlodipine 50 ng/mL.Fig. 4 Chromatograms for analysis of artemisinin in plasma (Rat blank plasma).Calibration curve was linear over the entire range of calibration curves with a mean correlation coefficient greater than 0.9995 (Fig. 5).The limit of quantification (LOQ) of the assay method was 7.8 ng/mL being the lowest concentration used to construct the calibration curve whereas the limit of detection (LOD) was 3.9 ng/mL at a signal to noise ratio of 3. The validation data demonstrated a good precision, accuracy and recovery. The extraction recovery means for internal standard and all artemisinin concentrations used were 75-85% (Table 1). The within-day and between-day accuracy and precision values are given in Table 2.Neither artemisinin nor the internal standard producedll Rights Reserved.Development and Validation of a Liquid Chromatography–Tandem Mass Spectrometry Method forDetermination of Artemisinin in Rat Plasma5Fig. 5 Mean calibration curve of artemisinin (ng/mL).Table 1 Extraction recovery.Concentration (ng/mL) Mean recovery (%) CV (%)7.81 75.081.5062.50 82.161.94250.00 82.03 2.072000.00 85.23 1.48Table 2 Within-day and between-day precision andaccuracy.Added (ng/mL)Within-day Between-day Accuracy (%) C.V (%) Accuracy (%) C.V (%)7.81 96.00 4.60 104.11 2.30 62.50 98.10 1.60 94.10 2.20 250.00 98.10 1.50 98.10 1.60 2000.00 96.10 2.50 97.10 1.80any detectable carry-over after three injections of upper limit of quantification. Blank rat plasma showed no interference with artemisinin. Interfering signals from blank plasma contributed less than 20% of the artemisinin signal at LOQ. There was no interference of artemisinin on the internal standard or vice versa. A small enhancement for artemisinin and the internal standard could be detected when references in neat injection solvent were compared with references in extracted blank biological matrix. The normalized matrix effects (artemisinin/internal standard) were close to 1 with a low variation in accordance with international guidelines. Post-column infusion experiments confirmed the absence of regions with severe matrix effects (i.e., no sharp drops or increases in the response) for blank human plasma extracted with the developed method.Xing et al. used artmether as an internal standard for the analysis of artemisinin [17]while for the analysis of artemisinin derivatives; artemisinin was used as internal standard [14]. In the present study amlodipine was found to be suitable because it could be separated chromatographically, ionized and fragmented under the conditions that optimized the intensity of artemisinin peak (Fig. 3).The analysis of artemisinin and its derivatives with mass spectrometry are most often performed with a different mode of ionization. Xing et al. used ESI inletin the positive ion-multiple reaction monitoring mode which relatively producing a higher sensitivity than in the SIM mode. Therefore, the mass spectrometry was operated at positive ion-MRM mode.4. ConclusionThe LC-MS/MS method described in this work is suitable for the determination of artemisinin in plasma. The assay procedure is simple with a relatively shortll Rights Reserved.Development and Validation of a Liquid Chromatography–Tandem Mass Spectrometry Method forDetermination of Artemisinin in Rat Plasma6retention time allowing sufficient sample to beprocessed to be applied to pharmacokinetic and bioavailability studies of artemisinin. The accuracy and precision of the assay method, as well as the recovery of extraction procedure were found to be satisfactory.References[1] D.L. Klayman, Qinghasou (Artemisinin): An antimalaria drug from China, Science 228 (1985) 1049-1055.[2] X.D. Luo, C.C. Shen, The chemistry, pharmacology andclinical applications of Qinghaosu (artemisinin) and it’sderivatives, Med. Res. Rev. 7 (1987) 29-52.[3] K.T. Batty, M. Ashton, K.F. Llett, G . Edwards, T.M. Davis,Selective high-performance liquid chromatography ofartesunate and α-and β-dihydroartemisinin in patients withfalciparum malaria, J. Chromatog. B 677 (2-3) (1996)345-350.[4] J. Karbwang, K. Na-Bangchang, P. Molunto, V . Banmairuroi, Determination of artemisinin and its majormetabolite, dihydroartemisinin, in plasma usinghigh-performance liquid chromatography withelectrochemical detector, J. Chromatog. B 7 (1-2) (1997)259-265.[5] K.L. Chan, K.H. Yuen, H. Takayanki, S. Jinandasa, K.K. Peh, Polymorphism of artemisinin from Artemisia annua,Phytochemistry 46 (7) (1997) 1209-1214.[6] G .Q. Li, T.O. Peggins, L.L. Fleckenstein, K. Masonic,M.H. Heiffles, T.G . Brewer, The pharmacokinetics andbiovailability of dihydroartemisinin, arteether, artemether,artesunic acid and artelinic acid in rats, J. Pharm.Pharmacol 5 (1998) 173-182.[7] B.A. Avery, K.K. Venkatesh, M.A. Avery, Rapid determination of artemisinin and related analogues usinghigh-perfomance liquid chromatography and anevaporative light scattering detector, J. Chromat. B 730 (1)(1999) 71-80.[8] S.S. Mohamed, S.A. Khalid, S.A. Ward, T.S.M. Wan,H.P.O. Tang, M. Zheng, R.K. Haynes, G . Edwards,Simultaneous determination of artemether and its majormetabolite dihydroartemisinin in plasma by gaschromatography-mass spectrometry-selected ionmonitoring, J. Chromat. B 731(1999) 251-260.[9] K.T. Batty, M. Ashton, K.F. Llett, G . Edward, T.M. Davis,The pharmacokinetics of artemisinin (ART) and artesunate (ARTS) in healthy volunteers, Am J. Trop Med. Hyg. 58(2) (1998) 125-126.[10] C. Souppart, N. Gouducheau, N. Sandenan, F. Richard,Development and validation of a high-performance liquid chromatography-mass spectrometry assay for the determination of artemisinin and its metabolite dihydraartemisinin in human plasma, J. Chromat. B 774(2002) 195-203.[11] H. Naik, D.J. Murry, L.E. Kirsch, L. Fleckenstein,Development and validation of high-performance liquid chromatography-mass spectroscopy assay for determination of artesunate and dihydrroartemisinin in human plasma, J. Chromat. B 816 (1-2) (2005) 233-242. [12] J. Xing, H. Yan, S. Zhang, G . Ren, Y . Gao, A high-performance liquid chromatography/tandem mass spectrometry method for the determination of artemisinin in rat plasma, Rapid Commun in Mass Spectro. 20 (9) (2006) 1463-1468. [13] J. Xing, H.X. Yan, R.L. Wang, L.F. Zhang, S.Q. Zhang,Liquid chromatography-tandem mass spectrometry assay for the quantitation of β-dihydroartemisinin in rat plasma, J. Chromat. B 852 (1-2) (2007) 202-207. [14] M. Rajanikanth, K.P. Madhusudanan, R.C. Gupta, An HPLC-MS method for simultaneous estimation of alpha, beta-arteether and its metabolite dihydroartemisinin, in rat plasma for application to pharmacokinetic study, J Biomed. Chromat. 17 (7) (2003) 440-446. [15] Y . Gu, Q. Li, M.V . Elendez, P. Weina, Comparison of HPLC with electrochemical detection and LC–MS/for the separation and validation of artesunate and dihydroartemisinin in animal and human plasma, J. Chromatogr B 867 (2008) 213-218. [16] W. Hanpithakpong, B. Kamanikom, A.M. Dondorp, P.Singhasivanon, N.J. White, N.P. Day, N. Lindegardh, A liquid chromatographic-tandem mass spectrometric method for determination of artesunate and its metabolite dihydroartemisinin in human plasma, J. Chromatogr. B 876 (2008) 61-68. [17] Y . Xing, H. Yan, S. Zhang, G . Ren, Y . Gao, A high-performance liquid chromatography/tandem mass spectrometry method for the determination of artemisinin rat plasma, Rapid Communication in Mass Spectrometry 20 (9) (2006) 1463-1468.ll Rights Reserved.。
激酶抑制剂(inase inhibitors)列表
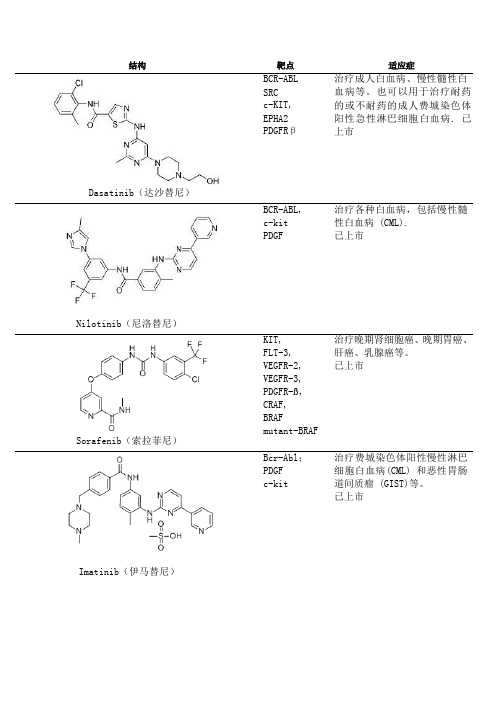
期临床试验;治疗淋巴瘤已经
完成1期临床研究
BI6727(Volasertib) GSK461364 ON01910 HMN-214
PLK1
该化合物已完成一期临床试 验,而对于膀胱癌的治疗已进 入二期临床试验阶段
PLK1
对于实体瘤的治疗已完成一期 临床试验
PLK CDK 等
对胰腺癌的治疗已进入三 期 临床试验阶段。
治疗费城染色体阳性慢性淋巴 细胞白血病(CML) 和恶性胃肠 道间质瘤 (GIST)等。 已上市
Gefitinib(吉非替尼) Erlotinib(埃罗替尼) Sunitinib(苏尼替尼) Lapatinib(拉帕替尼)
Vemurafenib
EGFR
用于铂类或多紫杉醇类药物化 疗失败后的局部晚期或转移性 非小细胞肺 癌患者 的 持续治 疗。已上市
CDK Aurora
JNJ-7706621是新型有效的广 谱CDK和Aurora激酶抑制剂;作 用于CDK1和2具有高度有效性。
CDK
治疗淋巴瘤已经完成二期临床
研究;治疗成人实体瘤已经完
成一期临床研究。
UCN-01 BI2536
PLK
治疗实体瘤、急性粒细胞白血
病、非小细胞肺癌、胰腺癌、
小细胞癌、前列腺癌已完成二
Everolimus(依维莫司) Cabozantinib(XL-184)
VEGFR2 RET
广谱络氨酸激酶抑制剂。用于 治疗转移性甲状腺髓样癌。 已上市
mTOR
用于治疗肾细胞癌;也用于治 疗乳腺癌、淋巴瘤、风湿性关 节炎以及多发性骨髓瘤。 已 上市
Temsirolimus(西罗莫司)
Rapamycin (Sirolimus, AY22989, NSC226080)
通过给予生长激素释放化合物或其拮抗剂来治疗肿瘤[发明专利]
![通过给予生长激素释放化合物或其拮抗剂来治疗肿瘤[发明专利]](https://img.taocdn.com/s3/m/d2c5ef80011ca300a7c390e5.png)
专利名称:通过给予生长激素释放化合物或其拮抗剂来治疗肿瘤
专利类型:发明专利
发明人:G·姆西奥利,M·帕波提,E·吉戈,R·戴根吉
申请号:CN200410045995.X
申请日:19991111
公开号:CN1594358A
公开日:
20050316
专利内容由知识产权出版社提供
摘要:治疗哺乳动物体内肿瘤的方法,所述的方法包括给予有效剂量的生长激素释放肽或其拮抗剂以降低或抑制肿瘤发生细胞在哺乳动物体内的增殖。
特别地,所治疗的肿瘤为肺肿瘤、乳腺肿瘤、甲状腺肿瘤或胰腺肿瘤。
优选的化合物是一些含有甲基色氨酸和赖氨酸单位的肽类。
申请人:赞塔里斯股份公司
地址:联邦德国法兰克福
国籍:DE
代理机构:中国国际贸易促进委员会专利商标事务所
代理人:陈轶兰
更多信息请下载全文后查看。
儿童急性髓系白血病的遗传基因异常及其意义

中国小儿血液与肿瘤杂志 2021年 6月第 26卷第 3期 JChinaPediatrBloodCancer,June2021,Vol26,No.3
·131·
2 异常基因在 MRD监测中的作用 21 融 合 基 因 RUNX1RUNX1T1/t(8;21)和 CBFBMYH11/inv(16)/t(16;16)均可用 于 治 疗 后 的 MRD监测 。 [1,811] 诱导或巩固结束后,RTqPCR 检测其转录产物 mRNA拷贝数(ABL做内参),较初 诊时降低 >3个 log者预后良好。究竟在诱导后还 是巩固后降低 >3个 log较有预后意义,视不同方案 而定。如果诱导结束后降低不足 3个 log,在巩固期 间继续降低,直到治疗完全结束后 <初诊时的 4个 log者预后仍好,提示连续监测的重要性。但如果将 RUNX1RUNX1T1和 CBFBMYH11分开统计,可 能 有不同 的 结 果,因 此 需 后 续 更 多 的 研 究[10]。关 于 KMT2A(MLL)基 因 重 排,在 AML 只 有 MLLT3 KMT2A/t(9;11)有详细研究[4],结果显示诱导缓解 后 RTqPCR <10-3和后续监测仍 <10-3复发率低。 BCRABL融合基因在 AML发生率低,用于 MRD监 测的意义尚未有足够的临床验证 。 [11] 以上研究结 果提示在用融合基因检测 MRD方面,连续监测的 重要性,治疗结束后仍低度阳性不影响预后。 22 突变基因 不是所有与 AML相关的突变基因 都适用于 MRD监 测 。 [1,45,1011] 造 血 干 细 胞 基 因 突 变后,获得竞争优势导致克隆造血,但仍有多系分化 和成熟的造血功能不属于白血病细胞,只有在继发 其他突变后才出现分化阻滞和无限增殖,发展为白 血病。随年龄增长干细胞的突变率增加,因此克隆 造血主要见于老年人,儿童少见。较常见的克隆造 血突变基因是编码表观遗传修饰因子的基因 DNMT3A、TET2、IDH、ASXL1,以 及 RNA 剪 接 子 和 Cohesins蛋 白 的 基 因 等。胚 系 突 变 基 因 RUNX1、 GATA2、CEBPA、DDX41和 ANKRD26也 不 适 用 于 MRD检测。与治疗前比较,缓解后突变基因的表达 水平仍没有明显下降,需考虑是克隆造血突变或胚 系突变基因。胚系突变基因可通过检测胚系组织 / 细胞(例如口腔黏膜等)DNA发现。
Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease trans

Reproduction numbers and sub-threshold endemicequilibria for compartmental models of disease transmissionP.van den Driesschea,1,James Watmough b,*,2aDepartment of Mathematics and Statistics,University of Victoria,Victoria,BC,Canada V8W 3P4b Department of Mathematics and Statistics,University of New Brunswick,Fredericton,NB,Canada E3B 5A3Received 26April 2001;received in revised form 27June 2001;accepted 27June 2001Dedicated to the memory of John JacquezAbstractA precise definition of the basic reproduction number,R 0,is presented for a general compartmental disease transmission model based on a system of ordinary differential equations.It is shown that,if R 0<1,then the disease free equilibrium is locally asymptotically stable;whereas if R 0>1,then it is unstable.Thus,R 0is a threshold parameter for the model.An analysis of the local centre manifold yields a simple criterion for the existence and stability of super-and sub-threshold endemic equilibria for R 0near one.This criterion,together with the definition of R 0,is illustrated by treatment,multigroup,staged progression,multistrain and vector–host models and can be applied to more complex models.The results are significant for disease control.Ó2002Elsevier Science Inc.All rights reserved.Keywords:Basic reproduction number;Sub-threshold equilibrium;Disease transmission model;Disease control1.IntroductionOne of the most important concerns about any infectious disease is its ability to invade a population.Many epidemiological models have a disease free equilibrium (DFE)at whichtheMathematical Biosciences 180(2002)29–48/locate/mbs*Corresponding author.Tel.:+1-5064587323;fax:+1-5064534705.E-mail addresses:pvdd@math.uvic.ca (P.van den Driessche),watmough@unb.ca (J.Watmough).URL:http://www.math.unb.ca/$watmough.1Research supported in part by an NSERC Research Grant,the University of Victoria Committee on faculty research and travel and MITACS.2Research supported by an NSERC Postdoctoral Fellowship tenured at the University of Victoria.0025-5564/02/$-see front matter Ó2002Elsevier Science Inc.All rights reserved.PII:S0025-5564(02)00108-630P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–48population remains in the absence of disease.These models usually have a threshold parameter, known as the basic reproduction number,R0,such that if R0<1,then the DFE is locally as-ymptotically stable,and the disease cannot invade the population,but if R0>1,then the DFE is unstable and invasion is always possible(see the survey paper by Hethcote[1]).Diekmann et al.[2]define R0as the spectral radius of the next generation matrix.We write down in detail a general compartmental disease transmission model suited to heterogeneous populations that can be modelled by a system of ordinary differential equations.We derive an expression for the next generation matrix for this model and examine the threshold R0¼1in detail.The model is suited to a heterogeneous population in which the vital and epidemiological parameters for an individual may depend on such factors as the stage of the disease,spatial position,age or behaviour.However,we assume that the population can be broken into homo-geneous subpopulations,or compartments,such that individuals in a given compartment are indistinguishable from one another.That is,the parameters may vary from compartment to compartment,but are identical for all individuals within a given compartment.We also assume that the parameters do not depend on the length of time an individual has spent in a compart-ment.The model is based on a system of ordinary equations describing the evolution of the number of individuals in each compartment.In addition to showing that R0is a threshold parameter for the local stability of the DFE, we apply centre manifold theory to determine the existence and stability of endemic equilib-ria near the threshold.We show that some models may have unstable endemic equilibria near the DFE for R0<1.This suggests that even though the DFE is locally stable,the disease may persist.The model is developed in Section2.The basic reproduction number is defined and shown to bea threshold parameter in Section3,and the definition is illustrated by several examples in Section4.The analysis of the centre manifold is presented in Section5.The epidemiological ramifications of the results are presented in Section6.2.A general compartmental epidemic model for a heterogeneous populationConsider a heterogeneous population whose individuals are distinguishable by age,behaviour, spatial position and/or stage of disease,but can be grouped into n homogeneous compartments.A general epidemic model for such a population is developed in this section.Let x¼ðx1;...;x nÞt, with each x i P0,be the number of individuals in each compartment.For clarity we sort the compartments so that thefirst m compartments correspond to infected individuals.The distinc-tion between infected and uninfected compartments must be determined from the epidemiological interpretation of the model and cannot be deduced from the structure of the equations alone,as we shall discuss below.It is plausible that more than one interpretation is possible for some models.A simple epidemic model illustrating this is given in Section4.1.The basic reproduction number can not be determined from the structure of the mathematical model alone,but depends on the definition of infected and uninfected compartments.We define X s to be the set of all disease free states.That isX s¼f x P0j x i¼0;i¼1;...;m g:In order to compute R0,it is important to distinguish new infections from all other changes inpopulation.Let F iðxÞbe the rate of appearance of new infections in compartment i,Vþi ðxÞbe therate of transfer of individuals into compartment i by all other means,and VÀi ðxÞbe the rate oftransfer of individuals out of compartment i.It is assumed that each function is continuously differentiable at least twice in each variable.The disease transmission model consists of non-negative initial conditions together with the following system of equations:_x i¼f iðxÞ¼F iðxÞÀV iðxÞ;i¼1;...;n;ð1Þwhere V i¼VÀi ÀVþiand the functions satisfy assumptions(A1)–(A5)described below.Sinceeach function represents a directed transfer of individuals,they are all non-negative.Thus,(A1)if x P0,then F i;Vþi ;VÀiP0for i¼1;...;n.If a compartment is empty,then there can be no transfer of individuals out of the compartment by death,infection,nor any other means.Thus,(A2)if x i¼0then VÀi ¼0.In particular,if x2X s then VÀi¼0for i¼1;...;m.Consider the disease transmission model given by(1)with f iðxÞ,i¼1;...;n,satisfying con-ditions(A1)and(A2).If x i¼0,then f iðxÞP0and hence,the non-negative cone(x i P0, i¼1;...;n)is forward invariant.By Theorems1.1.8and1.1.9of Wiggins[3,p.37]for each non-negative initial condition there is a unique,non-negative solution.The next condition arises from the simple fact that the incidence of infection for uninfected compartments is zero.(A3)F i¼0if i>m.To ensure that the disease free subspace is invariant,we assume that if the population is free of disease then the population will remain free of disease.That is,there is no(density independent) immigration of infectives.This condition is stated as follows:(A4)if x2X s then F iðxÞ¼0and VþiðxÞ¼0for i¼1;...;m.The remaining condition is based on the derivatives of f near a DFE.For our purposes,we define a DFE of(1)to be a(locally asymptotically)stable equilibrium solution of the disease free model,i.e.,(1)restricted to X s.Note that we need not assume that the model has a unique DFE. Consider a population near the DFE x0.If the population remains near the DFE(i.e.,if the introduction of a few infective individuals does not result in an epidemic)then the population will return to the DFE according to the linearized system_x¼Dfðx0ÞðxÀx0Þ;ð2Þwhere Dfðx0Þis the derivative½o f i=o x j evaluated at the DFE,x0(i.e.,the Jacobian matrix).Here, and in what follows,some derivatives are one sided,since x0is on the domain boundary.We restrict our attention to systems in which the DFE is stable in the absence of new infection.That is, (A5)If FðxÞis set to zero,then all eigenvalues of Dfðx0Þhave negative real parts.P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–4831The conditions listed above allow us to partition the matrix Df ðx 0Þas shown by the following lemma.Lemma 1.If x 0is a DFE of (1)and f i ðx Þsatisfies (A1)–(A5),then the derivatives D F ðx 0Þand D V ðx 0Þare partitioned asD F ðx 0Þ¼F 000 ;D V ðx 0Þ¼V 0J 3J 4;where F and V are the m Âm matrices defined byF ¼o F i o x j ðx 0Þ !and V ¼o V i o x jðx 0Þ !with 16i ;j 6m :Further ,F is non-negative ,V is a non-singular M-matrix and all eigenvalues of J 4have positive real part .Proof.Let x 02X s be a DFE.By (A3)and (A4),ðo F i =o x j Þðx 0Þ¼0if either i >m or j >m .Similarly,by (A2)and (A4),if x 2X s then V i ðx Þ¼0for i 6m .Hence,ðo V i =o x j Þðx 0Þ¼0for i 6m and j >m .This shows the stated partition and zero blocks.The non-negativity of F follows from (A1)and (A4).Let f e j g be the Euclidean basis vectors.That is,e j is the j th column of the n Ân identity matrix.Then,for j ¼1;...;m ,o V i o x jðx 0Þ¼lim h !0þV i ðx 0þhe j ÞÀV i ðx 0Þh :To show that V is a non-singular M-matrix,note that if x 0is a DFE,then by (A2)and (A4),V i ðx 0Þ¼0for i ¼1;...;m ,and if i ¼j ,then the i th component of x 0þhe j ¼0and V i ðx 0þhe j Þ60,by (A1)and (A2).Hence,o V i =o x j 0for i m and j ¼i and V has the Z sign pattern (see Appendix A).Additionally,by (A5),all eigenvalues of V have positive real parts.These two conditions imply that V is a non-singular M-matrix [4,p.135(G 20)].Condition (A5)also implies that the eigenvalues of J 4have positive real part.Ã3.The basic reproduction numberThe basic reproduction number,denoted R 0,is ‘the expected number of secondary cases produced,in a completely susceptible population,by a typical infective individual’[2];see also [5,p.17].If R 0<1,then on average an infected individual produces less than one new infected individual over the course of its infectious period,and the infection cannot grow.Conversely,if R 0>1,then each infected individual produces,on average,more than one new infection,and the disease can invade the population.For the case of a single infected compartment,R 0is simply the product of the infection rate and the mean duration of the infection.However,for more complicated models with several infected compartments this simple heuristic definition of R 0is32P.van den Driessche,J.Watmough /Mathematical Biosciences 180(2002)29–48insufficient.A more general basic reproduction number can be defined as the number of new infections produced by a typical infective individual in a population at a DFE.To determine the fate of a‘typical’infective individual introduced into the population,we consider the dynamics of the linearized system(2)with reinfection turned off.That is,the system _x¼ÀD Vðx0ÞðxÀx0Þ:ð3ÞBy(A5),the DFE is locally asymptotically stable in this system.Thus,(3)can be used to de-termine the fate of a small number of infected individuals introduced to a disease free population.Let wi ð0Þbe the number of infected individuals initially in compartment i and letwðtÞ¼w1ðtÞ;...;w mðtÞðÞt be the number of these initially infected individuals remaining in the infected compartments after t time units.That is the vector w is thefirst m components of x.The partitioning of D Vðx0Þimplies that wðtÞsatisfies w0ðtÞ¼ÀV wðtÞ,which has the unique solution wðtÞ¼eÀVt wð0Þ.By Lemma1,V is a non-singular M-matrix and is,therefore,invertible and all of its eigenvalues have positive real parts.Thus,integrating F wðtÞfrom zero to infinity gives the expected number of new infections produced by the initially infected individuals as the vector FVÀ1wð0Þ.Since F is non-negative and V is a non-singular M-matrix,VÀ1is non-negative[4,p.137 (N38)],as is FVÀ1.To interpret the entries of FVÀ1and develop a meaningful definition of R0,consider the fate of an infected individual introduced into compartment k of a disease free population.The(j;k)entry of VÀ1is the average length of time this individual spends in compartment j during its lifetime, assuming that the population remains near the DFE and barring reinfection.The(i;j)entry of F is the rate at which infected individuals in compartment j produce new infections in compartment i. Hence,the(i;k)entry of the product FVÀ1is the expected number of new infections in com-partment i produced by the infected individual originally introduced into compartment k.Fol-lowing Diekmann et al.[2],we call FVÀ1the next generation matrix for the model and set R0¼qðFVÀ1Þ;ð4Þwhere qðAÞdenotes the spectral radius of a matrix A.The DFE,x0,is locally asymptotically stable if all the eigenvalues of the matrix Dfðx0Þhave negative real parts and unstable if any eigenvalue of Dfðx0Þhas a positive real part.By Lemma1, the eigenvalues of Dfðx0Þcan be partitioned into two sets corresponding to the infected and uninfected compartments.These two sets are the eigenvalues of FÀV and those ofÀJ4.Again by Lemma1,the eigenvalues ofÀJ4all have negative real part,thus the stability of the DFE is determined by the eigenvalues of FÀV.The following theorem states that R0is a threshold parameter for the stability of the DFE.Theorem2.Consider the disease transmission model given by(1)with fðxÞsatisfying conditions (A1)–(A5).If x0is a DFE of the model,then x0is locally asymptotically stable if R0<1,but un-stable if R0>1,where R0is defined by(4).Proof.Let J1¼FÀV.Since V is a non-singular M-matrix and F is non-negative,ÀJ1¼VÀF has the Z sign pattern(see Appendix A).Thus,sðJ1Þ<0()ÀJ1is a non-singular M-matrix;P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–483334P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–48where sðJ1Þdenotes the maximum real part of all the eigenvalues of the matrix J1(the spectral abscissa of J1).Since FVÀ1is non-negative,ÀJ1VÀ1¼IÀFVÀ1also has the Z sign pattern.Ap-plying Lemma5of Appendix A,with H¼V and B¼ÀJ1¼VÀF,we have ÀJ1is a non-singular M-matrix()IÀFVÀ1is a non-singular M-matrix:Finally,since FVÀ1is non-negative,all eigenvalues of FVÀ1have magnitude less than or equal to qðFVÀ1Þ.Thus,IÀFVÀ1is a non-singular M-matrix;()qðFVÀ1Þ<1:Hence,sðJ1Þ<0if and only if R0<1.Similarly,it follows thatsðJ1Þ¼0()ÀJ1is a singular M-matrix;()IÀFVÀ1is a singular M-matrix;()qðFVÀ1Þ¼1:The second equivalence follows from Lemma6of Appendix A,with H¼V and K¼F.The remainder of the equivalences follow as with the non-singular case.Hence,sðJ1Þ¼0if and only if R0¼1.It follows that sðJ1Þ>0if and only if R0>1.ÃA similar result can be found in the recent book by Diekmann and Heesterbeek[6,Theorem6.13].This result is known for the special case in which J1is irreducible and V is a positive di-agonal matrix[7–10].The special case in which V has positive diagonal and negative subdiagonal elements is proven in Hyman et al.[11,Appendix B];however,our approach is much simpler(see Section4.3).4.Examples4.1.Treatment modelThe decomposition of fðxÞinto the components F and V is illustrated using a simple treat-ment model.The model is based on the tuberculosis model of Castillo-Chavez and Feng[12,Eq.(1.1)],but also includes treatment failure used in their more elaborate two-strain model[12,Eq.(2.1)].A similar tuberculosis model with two treated compartments is proposed by Blower et al.[13].The population is divided into four compartments,namely,individuals susceptible to tu-berculosis(S),exposed individuals(E),infectious individuals(I)and treated individuals(T).The dynamics are illustrated in Fig.1.Susceptible and treated individuals enter the exposed com-partment at rates b1I=N and b2I=N,respectively,where N¼EþIþSþT.Exposed individuals progress to the infectious compartment at the rate m.All newborns are susceptible,and all indi-viduals die at the rate d>0.Thus,the core of the model is an SEI model using standard inci-dence.The treatment rates are r1for exposed individuals and r2for infectious individuals. However,only a fraction q of the treatments of infectious individuals are successful.Unsuc-cessfully treated infectious individuals re-enter the exposed compartment(p¼1Àq).The diseasetransmission model consists of the following differential equations together with non-negative initial conditions:_E¼b1SI=Nþb2TI=NÀðdþmþr1ÞEþpr2I;ð5aÞ_I¼m EÀðdþr2ÞI;ð5bÞ_S¼bðNÞÀdSÀb1SI=N;ð5cÞ_T¼ÀdTþr1Eþqr2IÀb2TI=N:ð5dÞProgression from E to I and failure of treatment are not considered to be new infections,but rather the progression of an infected individual through the various compartments.Hence,F¼b1SI=Nþb2TI=NB B@1C CA and V¼ðdþmþr1ÞEÀpr2IÀm Eþðdþr2ÞIÀbðNÞþdSþb1SI=NdTÀr1EÀqr2Iþb2TI=NB B@1C CA:ð6ÞThe infected compartments are E and I,giving m¼2.An equilibrium solution with E¼I¼0has the form x0¼ð0;0;S0;0Þt,where S0is any positive solution of bðS0Þ¼dS0.This will be a DFE if and only if b0ðS0Þ<d.Without loss of generality,assume S0¼1is a DFE.Then,F¼0b100;V¼dþmþr1Àpr2Àm dþr2;givingVÀ1¼1ðdþmþr1Þðdþr2ÞÀm pr2dþr2pr2m dþmþr1and R0¼b1m=ððdþmþr1Þðdþr2ÞÀm pr2Þ.A heuristic derivation of the(2;1)entry of VÀ1and R0are as follows:a fraction h1¼m=ðdþmþr1Þof exposed individuals progress to compartment I,a fraction h2¼pr2=ðdþr2Þof infectious individuals re-enter compartment E.Hence,a fractionh1of exposed individuals pass through compartment I at least once,a fraction h21h2passthroughat least twice,and a fraction h k 1h k À12pass through at least k times,spending an average of s ¼1=ðd þr 2Þtime units in compartment I on each pass.Thus,an individual introduced into com-partment E spends,on average,s ðh 1þh 21h 2þÁÁÁÞ¼s h 1=ð1Àh 1h 2Þ¼m =ððd þm þr 1Þðd þr 2ÞÀm pr 2Þtime units in compartment I over its expected lifetime.Multiplying this by b 1gives R 0.The model without treatment (r 1¼r 2¼0)is an SEI model with R 0¼b 1m =ðd ðd þm ÞÞ.The interpretation of R 0for this case is simpler.Only a fraction m =ðd þm Þof exposed individuals progress from compartment E to compartment I ,and individuals entering compartment I spend,on average,1=d time units there.Although conditions (A1)–(A5)do not restrict the decomposition of f i ðx Þto a single choice for F i ,only one such choice is epidemiologically correct.Different choices for the function F lead to different values for the spectral radius of FV À1,as shown in Table 1.In column (a),treatment failure is considered to be a new infection and in column (b),both treatment failure and pro-gression to infectiousness are considered new infections.In each case the condition q ðFV À1Þ<1yields the same portion of parameter space.Thus,q ðFV À1Þis a threshold parameter in both cases.The difference between the numbers lies in the epidemiological interpretation rather than the mathematical analysis.For example,in column (a),the infection rate is b 1þpr 2and an exposed individual is expected to spend m =ððd þm þr 1Þðd þr 2ÞÞtime units in compartment I .However,this reasoning is biologically flawed since treatment failure does not give rise to a newly infected individual.Table 1Decomposition of f leading to alternative thresholds(a)(b)Fb 1SI =N þb 2TI =N þpr 2I 0000B B @1C C A b 1SI =N þb 2TI =N þpr 2I m E 000B B @1C C A Vðd þm þr 1ÞE Àm E þðd þr 2ÞI Àb ðN ÞþdS þb 1SI =N dT Àr 1E Àqr 2I þb 2TI =N 0B B @1C C A ðd þm þr 1ÞE ðd þr 2ÞI Àb ðN ÞþdS þb 1SI =N dT Àr 1E Àqr 2I þb 2TI =N 0B B @1C C A F0b 1þpr 200 0b 1þpr 2m 0 V d þm þr 10Àm d þr 2d þm þr 100d þr 2 q (FV À1)b 1m þpr 2mðd þm þr 1Þðd þr 2Þffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffib 1m þpr 2mðd þm þr 1Þðd þr 2Þs 36P.van den Driessche,J.Watmough /Mathematical Biosciences 180(2002)29–484.2.Multigroup modelIn the epidemiological literature,the term‘multigroup’usually refers to the division of a het-erogeneous population into several homogeneous groups based on individual behaviour(e.g., [14]).Each group is then subdivided into epidemiological compartments.The majority of mul-tigroup models in the literature are used for sexually transmitted diseases,such as HIV/AIDS or gonorrhea,where behaviour is an important factor in the probability of contracting the disease [7,8,14,15].As an example,we use an m-group SIRS-vaccination model of Hethcote[7,14]with a generalized incidence term.The sample model includes several SI multigroup models of HIV/ AIDS as special cases[8,15].The model equations are as follows:_I i ¼X mj¼1b ijðxÞS i I jÀðd iþc iþ iÞI i;ð7aÞ_S i ¼ð1Àp iÞb iÀðd iþh iÞS iþr i R iÀX mj¼1b ijðxÞS i I j;ð7bÞ_Ri¼p i b iþc i I iþh i S iÀðd iþr iÞR i;ð7cÞfor i¼1;...;m,where x¼ðI1;...;I m;S1;...;S m;R1;...;R mÞt.Susceptible and removed individu-als die at the rate d i>0,whereas infected individuals die at the faster rate d iþ i.Infected in-dividuals recover with temporary immunity from re-infection at the rate c i,and immunity lasts an expected1=r i time units.All newborns are susceptible,and a constant fraction b i are born into each group.A fraction p i of newborns are vaccinated at birth.Thereafter,susceptible individuals are vaccinated at the rate h i.The incidence,b ijðxÞdepends on individual behaviour,which determines the amount of mixing between the different groups(see,e.g.,Jacquez et al.[16]). The DFE for this model isx0¼ð0;...;0;S01;...;S0m;R01;...;R0mÞt;whereS0 i ¼b i d ið1Àp iÞþr iðÞd iðd iþh iþr iÞ;R0 i ¼b iðh iþd i p iÞd iðd iþh iþr iÞ:Linearizing(7a)about x¼x0givesF¼S0i b ijðx0ÞÂÃandV¼½ðd iþc iþ iÞd ij ;where d ij is one if i¼j,but zero otherwise.Thus,FVÀ1¼S0i b ijðx0Þ=ðd iÂþc iþ iÞÃ:P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–4837For the special case with b ij separable,that is,b ijðxÞ¼a iðxÞk jðxÞ,F has rank one,and the basic reproduction number isR0¼X mi¼1S0ia iðx0Þk iðx0Þd iþc iþ i:ð8ÞThat is,the basic reproduction number of the disease is the sum of the‘reproduction numbers’for each group.4.3.Staged progression modelThe staged progression model[11,Section3and Appendix B]has a single uninfected com-partment,and infected individuals progress through several stages of the disease with changing infectivity.The model is applicable to many diseases,particularly HIV/AIDS,where transmission probabilities vary as the viral load in an infected individual changes.The model equations are as follows(see Fig.2):_I 1¼X mÀ1k¼1b k SI k=NÀðm1þd1ÞI1;ð9aÞ_Ii¼m iÀ1I iÀ1Àðm iþd iÞI i;i¼2;...;mÀ1;ð9bÞ_Im¼m mÀ1I mÀ1Àd m I m;ð9cÞ_S¼bÀbSÀX mÀ1k¼1b k SI k=N:ð9dÞThe model assumes standard incidence,death rates d i>0in each infectious stage,and thefinal stage has a zero infectivity due to morbidity.Infected individuals spend,on average,1=m i time units in stage i.The unique DFE has I i¼0,i¼1;...;m and S¼1.For simplicity,define m m¼0. Then F¼½F ij and V¼½V ij ,whereF ij¼b j i¼1;j6mÀ1;0otherwise;&ð10ÞV ij¼m iþd i j¼i;Àm j i¼1þj;0otherwise:8<:ð11ÞLet a ij be the(i;j)entry of VÀ1.Thena ij¼0i<j;1=ðm iþd iÞi¼j;Q iÀ1k¼jm kQ ik¼jðm kþd kÞj<i:8>>><>>>:ð12ÞThus,R0¼b1m1þd1þb2m1ðm1þd1Þðm2þd2Þþb3m1m2ðm1þd1Þðm2þd2Þðm3þd3ÞþÁÁÁþb mÀ1m1...m mÀ2ðm1þd1Þ...ðm mÀ1þd mÀ1Þ:ð13ÞThe i th term in R0represents the number of new infections produced by a typical individual during the time it spends in the i th infectious stage.More specifically,m iÀ1=ðm iÀ1þd iÀ1Þis the fraction of individuals reaching stage iÀ1that progress to stage i,and1=ðm iþd iÞis the average time an individual entering stage i spends in stage i.Hence,the i th term in R0is the product of the infectivity of individuals in stage i,the fraction of initially infected individuals surviving at least to stage i,and the average infectious period of an individual in stage i.4.4.Multistrain modelThe recent emergence of resistant viral and bacterial strains,and the effect of treatment on their proliferation is becoming increasingly important[12,13].One framework for studying such sys-tems is the multistrain model shown in Fig.3,which is a caricature of the more detailed treatment model of Castillo-Chavez and Feng[12,Section2]for tuberculosis and the coupled two-strain vector–host model of Feng and Velasco-Hern a ndez[17]for Dengue fever.The model has only a single susceptible compartment,but has two infectious compartments corresponding to the two infectious agents.Each strain is modelled as a simple SIS system.However,strain one may ‘super-infect’an individual infected with strain two,giving rise to a new infection incompartment。
参丹散结胶囊联合XELOX方案化学治疗晚期胃癌患者的疗效研究

・
ll 4】・
・
中 医中药 ・
参丹散结胶囊联合 X E L O X方案化学治疗 晚期 胃癌患者 的疗效研 究
刘 娟 高 峻 杨牡 丹
胃癌在全世界很多 国家的发病率都很高 ,中 国每年都有
较其他 国家更多的新发 胃癌病例 ,且 经常到晚期才 诊断 , 失
部分 缓解 , 疾病控制 率( d i s e a s e c o n t r o l r a t e , D C R) : 完全缓解+ 部分缓解+ 稳定 。 1 . 3 . 2 毒副反应情况 :参照美 国国立癌症研究所通过的常规 化学治疗毒性评定标准( N C T . C T C ) 监测 2 组 治疗 期间毒副反
的安 全 性 。
足综合 征等的发生率 明显 升高 , 迫使部分患 者不能耐受全 身 化学治疗 。因此 , 除积极治疗原发病外 , 还应 降低化学治疗药 物 的毒 副作 用及 提高机体免疫 功能 ,使患 者完成化学治疗 ,
1 . 3 . 3 细胞免疫功能 的测定 : 2 组 患者治疗前 后 ( 采血 2次 ,
统计学意义。 治疗后 , 试验组 N K细胞 活性 、 T细胞亚群 C D 3 + ,
2组患 者均采用 X E L O X化学治疗 方案 :奥 沙利铂 1 3 0
C D 4 圾 C D 4  ̄ / C D 8 + 的比值升高 , C D 8 + 活性降低 , 同组治疗前后 差异具 有统计学 意义( £ 分别 为 2 . 8 9 、 2 . 6 0 、 2 . 1 7 、 2 . 3 2 、 3 . 0 4 , P <
性3 6例 , 女性 2 4例 ; 年龄 4 2 ~ 7 O岁 , 平均 ( 5 4 . 2 + 2 . 1 ) 岁。2 组
澳沙利铂联合ELF方案治疗晚期胃癌

ቤተ መጻሕፍቲ ባይዱ
医刊 20 年 9月第 3 卷第 1 期 08 5 7
C ie o r a o r c cl dc e e .0 8 V 1 5 N .7 — ns J u n l—P at a Me in p 2 0 , o 3 , o 1 h e f i i S .
续静 脉 泵 入 2 , 1 2h 第 ~3天 , 3周 重 复 , 少 完成 3个 周 期 判 断 疗 效 。结 果 本 组 3 每 至 2例 , 全 缓 解 ( R) 完 C 2例 ( . % ) 62 、
部分缓解 P 5例 (6 9 ) 无 变化 ( C)2例 (7 5 、 R1 4.% 、 N 1 3 、 %) 进展 ( D) P 2例 ( . % ) P 6 3 。K S评 分提 高 2 0分 以上 者 4 . % 06
1 对 象 与 方 法
的患者 出现 了骨 髓 抑制 。但 r度 和 Ⅱ度 的较 多 , Ⅲ度 以上偏
少, 用粒细胞集落刺激 因子治疗后 , 全部 患者完成 4周 期化疗 。 有 胃肠 道反应 患者经 应用 昂丹 司琼治 疗后均 不影 响进食 。本 组 患者 出现 神经毒性较 多 , 以感 觉神经 毒性 为主 , 神经 毒性 均 为可逆 性。本组 患者未见肝 、 肾功能损 害和心 电图异常。 表 1 澳沙利铂联 合 E F方案化疗 的毒 副反应 [ ( ) L 例 % ]
受 , 经 济 代 价 患者 可承 受 , 得 临 床 推 广 应 用 。 且 值
【 关键词 】 澳沙利铂 ;L E F方案 ; 晚期 胃癌
胃癌是 我国恶性肿瘤发病率和死亡率 最高 的肿 瘤之一 , 且 因缺 乏特异 性早期 表现 ,0 的患者确 诊时 已属晚期 , 4% 化疗 仍 是 晚期 胃癌 的主要治疗 手段 。近年 来 随着新 药在 胃癌治 疗 中
前列腺癌新辅助化疗进展

前列腺癌新辅助化疗进展发布时间:2021-09-07T05:44:11.471Z 来源:《科学与技术》2021年5月第13期作者:梁鹏陈飞飞田志崇胡俊超[导读] 根治性前列腺切除术(RP)是前列腺癌患者的重要治疗手段,梁鹏陈飞飞田志崇胡俊超华北理工大学附属医院河北省唐山市 063000摘要:根治性前列腺切除术(RP)是前列腺癌患者的重要治疗手段,但是前列腺患者术前分期困难重重,为了使得更多病人能在RP中获益,新辅助治疗的地位也在越来越高。
其中,以多西他赛为基础的新辅助化疗手段值得我们关注。
关键词:前列腺癌;根治性前列腺切除术(RP);新辅助治疗;新辅助化疗;多西他赛前列腺癌是最常被诊断出的癌症之一,由于与发达国家相比,我国早期发现和筛查相关的诊断活动较差,故前列腺癌并不是我们诊断最常见的癌症类型,但可能包括逐步实施前列腺特异性抗原筛查和改进的活检技术又或日益西化的生活方式的影响,前列腺癌在我国的发病率及病死率正逐步上升【1】。
总体而言,高危和局部进展期的前列腺癌是威胁患者生命的主要原因,这部分患者在初诊时比例可达20%-30%【2】。
前列腺癌根治性切除术是低危局限性前列腺癌患者的最佳治疗方式,随着近些年来的研究,指南逐渐将手术适应症放宽到中-低危前列腺癌,甚至于已有研究者在转移性前列腺癌中实施包括手术在内的综合性治疗。
但随之而来的,是手术失败、放疗失败、生化复发率高等问题【3,4】,故许多学者开始对前列腺癌的新辅助治疗进行了探索。
前列腺癌新辅助包括辅助内分泌治疗(neoad-juvanthormonaltherapy,NHT)、新辅助化疗(neoadjuvantchemotherapy,NCT)以及两者的结合,其中NHT研究占了大多数。
然而,目前欧洲泌尿外科学会的指南不推荐在根治性切除术前进行新辅助内分泌治疗(证据等级别strong),美国国家综合癌症网络也不建议患者接受除临床试验以外的新辅助内分泌治疗。
头痛宁胶囊联合阿司匹林肠溶片治疗

志,2019,41(12):881-890.[9]张萍,艾斌.实体瘤免疫治疗疗效评价标准[J].国际肿瘤学杂志,2016,43(11):848-851.[10]纪晓娜,李钧,宋伟.表皮生长因子受体G719A 、L833V 、TP53联合突变非小细胞肺癌一例[J].国际肿瘤学杂志,2020,47(4):254-256.[11]刘旭阳,郑静.阿帕替尼联合多西他赛二线化疗治疗晚期非鳞非小细胞肺癌的疗效观察[J].实用心脑肺血管病杂志,2018,26(6):104-107.[12]ARRIETA O,BARR ÓN F,RAM ÍREZ-TIRADO LA,et al.Efficacyand safety of pembrolizumab plus docetaxel vs docetaxel alone in pa-tients with previously treated advanced non-small cell lung cancer:The PROLUNG phase 2randomized clinical trial [J].JAMA Oncol,2020,6(6):856-864.[13]郑波,刘君.安罗替尼联合多西他赛与多西他赛单药治疗晚期非小细胞肺癌的疗效观察[J].中国肿瘤临床与康复,2020,27(5):549-551.[14]徐芹,鲍瑜,亢晓彬.多西他赛联合阿帕替尼二线治疗进展期非小细胞肺癌的临床效果[J].中国医药导报,2020,17(23):95-98.[15]LI Z,LIU Z,WU Y ,et al.Efficacy and safety of apatinib alone or apa-tinib plus paclitaxel/docetaxel versus paclitaxel/docetaxel in the treat-ment of advanced non-small cell lung cancer:A meta-analysis [J].Thorac Cancer,2021,12(21):2838-2848.[16]GE L,ZHOU L.Apatinib sensitizes chemoresistant NSCLC cells todoxetaxel via regulating autophagy and enhances the therapeutic effi-cacy in advanced and refractory/recurrent NSCLC [J].Mol Med Rep,2020,22(5):3935-3943.[17]张明晖,孙雅丽,杨艳,等.甲磺酸阿帕替尼联合埃克替尼治疗埃克替尼耐药的非小细胞肺癌的效果分析[J].癌症进展,2019,17(7):798-800,807.[18]王莉,赵莹,张辉,等.多种血清肿瘤标志物联合检测诊断非小细胞肺癌的价值分析[J].河北医药,2019,41(17):2594-2597.[19]刘辉楠,蔡忠仁,单桂芹,等.不同药物治疗晚期非小细胞肺癌的近期疗效及对血清肿瘤标志物的影响[J].癌症进展,2020,18(3):282-285.[20]尚可,胡若洋,胡继德.阿帕替尼联合多西他赛二线以上治疗对晚期非小细胞肺癌患者CEA 、SCC 及CA125的影响[J].实用癌症杂志,2019,34(7):1172-1175.[21]丁婷婷,芮晓艳,范洪峰,等.阿帕替尼联合多西他赛对多线治疗晚期非小细胞肺癌患者血清肿瘤标志物、免疫功能及生活质量的影响[J].现代生物医学进展,2021,21(6):1160-1164.(收稿日期:2022-3-21)头痛宁胶囊联合阿司匹林肠溶片治疗偏头痛的效果及对患者血管内皮功能和脑血流动力学的影响雷杰喻1,段旺旺2,李慧1,赵文娟3,康乐乐41.延安大学附属医院心脑血管病医院药剂科,陕西延安716000;2.延安大学附属医院临床药学科,陕西延安716000;3.陕西省康复医院药剂科,陕西西安710065;4.延安大学附属医院心脑血管病医院神经内科,陕西延安716000【摘要】目的观察头痛宁胶囊联合阿司匹林肠溶片治疗偏头痛的效果,并探讨其对患者血管内皮功能和脑血流动力学的影响。
替雷利珠单抗联合紫杉醇加铂类一线治疗肺鳞癌的效果

①川北医学院 四川 南充 637000②广元市中心医院通信作者:石平替雷利珠单抗联合紫杉醇加铂类一线治疗肺鳞癌的效果杜淑兰①② 石平② 张再军②【摘要】 目的:探讨替雷利珠单抗联合紫杉醇加铂类一线治疗肺鳞癌的效果。
方法:选取2020年5月—2021年5月广元市中心医院收治的76例肺鳞癌患者作为研究对象,采用随机数表法将其分为对照组和试验组,各38例。
对照组行紫杉醇+顺铂治疗,试验组行紫杉醇+顺铂+替雷利珠单抗治疗。
比较两组肿瘤标志物、T 淋巴细胞亚群水平变化情况,观察两组治疗效果及药物不良反应发生情况。
结果:治疗后,试验组鳞状上皮细胞癌抗原(SCC)、组织多肽抗原(TPA)、神经元特异性烯醇化酶(NSE)、细胞角蛋白21-1片段(CYFRA21-1)、基质金属蛋白酶-9(MMP-9)、组织金属蛋白酶抑制剂-1(TIMP-1)水平均显著低于对照组,差异有统计学意义(P <0.05)。
治疗后,试验组CD3+、CD4+、CD4+/CD8+水平均高于对照组,CD8+水平低于对照组,差异有统计学意义(P <0.05)。
试验组疾病控制率为89.47%,高于对照组的71.05%,差异有统计学意义(P <0.05)。
两组各项不良反应发生率比较,差异无统计学意义(P >0.05)。
结论:替雷利珠单抗联合紫杉醇加铂类一线治疗肺鳞癌患者,可改善患者免疫功能,增强抗肿瘤效果,降低肿瘤标志物水平,且不会增加不良反应发生率。
【关键词】 肺鳞癌 替雷利珠单抗 紫杉醇 顺铂 doi:10.14033/ki.cfmr.2023.24.004 文献标识码 A 文章编号 1674-6805(2023)24-0015-05 Efficacy of Tislelizumab Combined with Paclitaxel and Platinum in First-line Treatment of Lung Squamous Cell Carcinoma/DU Shulan, SHI Ping, ZHANG Zaijun. //Chinese and Foreign Medical Research, 2023, 21(24): 15-19 [Abstract] Objective: To explore the efficacy of Tislelizumab combined with Paclitaxel and Platinum in the first-line treatment of lung squamous cell carcinoma. Method: A total of 76 patients with lung squamous cell carcinoma admitted to Guangyuan Central Hospital from May 2020 to May 2021 were selected as the research objects, and they were divided into the control group and the experimental group by random number table method, with 38 cases in each group. The control group was treated with Paclitaxel+Cisplatin, and the experimental group was treated with Paclitaxel+Cisplatin+Tislelizumab. The levels of tumor markers and T lymphocyte subsets in the two groups were compared, and the treatment effects and adverse drug reaction of the two groups were observed. Result: After treatment, the levels of squamous cell carcinoma antigen (SCC), tissue peptide antigen (TPA), neuron specific enolase (NSE), cytokeratin 21-1 fragment (CYFRA21-1), matrix metalloproteinase-9 (MMP-9), tissue metalloproteinase-1 (TIMP-1) in the experimental group were significantly lower than those in the control group, the differences were statistically significant (P <0.05). After treatment, the levels of CD3+, CD4+, CD4+/CD8+ in the experimental group were higher than those in the control group, while the levels of CD8+ was lower than that in the control group, the differences were statistically significant (P <0.05). The disease control rate in the experimental group was 89.47%, which was higher than 71.05% in the control group, the difference was statistically significant (P <0.05). There were no statistically significant differences in the incidence of adverse reactions between the two groups (P >0.05). Conclusion: Tislelizumab combined with Paclitaxel and Platinum in first-line treatment of lung squamous cell carcinoma patients can improve their immune function, enhance anti-tumor effects, reduce tumor marker levels, and do not increase the incidence of adverse reactions. [Key words] Lung squamous cell carcinoma Tislelizumab Paclitaxel Cisplatin First-author's address: North Sichuan Medical College, Nanchong 637000, China 肺癌是全球致死率最高的恶性肿瘤,也是全球最主要的一种公共卫生疾病。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Selumetinib plus docetaxel for KRAS-mutant advancednon-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 studyPasi A Jänne, Alice T Shaw, José Rodrigues Pereira, Gaëlle Jeannin, Johan Vansteenkiste, Carlos Barrios, Fabio Andre Franke, Lynda Grinsted, Victoria Zazulina, Paul Smith, Ian Smith, Lucio CrinòSummaryBackground No targeted therapies are available for KRAS-mutant non-small-cell lung cancer (NSCLC). Selumetinib is an inhibitor of MEK1/MEK2, downstream of KRAS, with preclinical evidence of synergistic activity with docetaxel in KRAS-mutant cancers. We did a prospective, randomised, phase 2 trial to assess selumetinib plus docetaxel in previously treated patients with advanced KRAS-mutant NSCLC.Methods Eligible patients were older than 18 years of age; had histologically or cytologically confi rmed stage IIIB–IV KRAS-mutant NSCLC; had failed fi rst-line therapy for advanced NSCLC; had WHO performance status of 0–1; had not received previous therapy with either a MEK inhibitor or docetaxel; and had adequate bone marrow, renal, and liver function. Patients were randomly assigned (in a 1:1 ratio) to either oral selumetinib (75 mg twice daily in a 21 day cycle) or placebo; all patients received intravenous docetaxel (75 mg/m² on day 1 of a 21 day cycle). Randomisation was done with an interactive voice response system and investigators, patients, data analysts, and the trial sponsor were masked to treatment assignment. The primary endpoint was overall survival, analysed for all patients with confi rmed KRAS mutations. This study is registered with , number NCT00890825.Findings Between April 20, 2009, and June 30, 2010, we randomly assigned 44 patients to receive selumetinib and docetaxel (selumetinib group) and 43 to receive placebo and docetaxel (placebo group). Of these, one patient in the selumetinib group and three in the placebo group were excluded from effi cacy analyses because their tumours were not confi rmed to be KRAS-mutation positive. Median overall survival was 9∙4 months (6∙8–13∙6) in the selumetinib group and 5∙2 months (95% CI 3∙8–non-calculable) in the placebo group (hazard ratio [HR] for death 0∙80, 80% CI 0∙56–1∙14; one-sided p=0∙21). Median progression-free survival was 5∙3 months (4∙6–6∙4) in the selumetinib group and 2∙1 months (95% CI 1∙4–3∙7) in the placebo group (HR for progression 0∙58, 80% CI 0∙42–0∙79; one-sided p=0∙014). 16 (37%) patients in the selumetinib group and none in the placebo group had an objective response (p<0∙0001). Adverse events of grade 3 or higher occurred in 36 (82%) patients in the selumetinib group and 28 (67%) patients in the placebo group. The most common grade 3–4 adverse events were neutropenia (29 [67%] of 43 patients in the selumetinib group vs 23 [55%] of 42 patients in the placebo group), febrile neutropenia (eight [18%] of 44 patients in the selumetinib group vs none in the placebo group), dyspnoea (one [2%] of 44 patients in the selumetinib group vs fi ve [12%] of 42 in the placebo group), and asthenia (four [9%] of 44 patients in the selumetinib group vs none in the placebo group).Interpretation Selumetinib plus docetaxel has promising effi cacy, albeit with a higher number of adverse events than with docetaxel alone, in previously treated advanced KRAS-mutant NSCLC. These fi ndings warrant further clinical investigation of selumetinib plus docetaxel in KRAS-mutant NSCLC.Funding AstraZeneca.IntroductionLung cancer is one of the leading causes of cancer mortality worldwide,1 with 5 year survival of less than 15% across all stages of disease in Europe.2 I n unselected patients with advanced non-small-cell lung cancer (NSCLC), there have been few improvements in outcomes for patients after chemotherapy.3 By contrast, kinase inhibitors have been particularly effective for subsets of patients with NSCLC defined by molecular oncogenic alterations such as EGFR muta t ions (gefi tinib and erlotinib)4,5 and ALK rearrangements (crizotinib).6KRAS is the most frequently mutated oncogene in NSCLC, with mutations detected in about 20% of tumours in white people. KRAS mutations also predict for absence of benefit from EGFR tyrosine kinase inhibitors,7 are a negative prognostic factor for survival,8 and may be associated with a reduced benefi t from adjuvant chemotherapy.9Although KRAS mutations were identified in lung cancer nearly 20 years ago,10 no approved therapies exist for KRAS-mutant NSCLC and few trials have specifi cally addressed this population of patients. Up to now, direct inhibition of KRAS has proven clinically challenging. Farnesyl transferase inhibitors, designed to specifi cally inhibit RAS proteins, showed little clinical effi cacy.11 Eff orts to inhibit mutant KRAS in NSCLC have thereforeLancet Oncol 2013; 14: 38–47Published OnlineNovember 28, 2012 /10.1016/S1470-2045(12)70489-8See Comment page 3 Lowe Center for ThoracicOncology and the Belfer Institute for Applied CancerScience, Dana-Farber CancerInstitute, Boston, MA, USA (P A Jänne MD);Massachusetts General Hospital Cancer Center,Boston, MA, USA(A T Shaw MD);InstitutoBrasileiro de CancerologiaTorácica, Sao Paulo, Brazil (J Rodrigues Pereira MD);Service de Pneumologie, Hôpital Gabriel Montpied, Clermont-Ferrand, France (G Jeannin MD); Respiratory Oncology Unit and Trial Unit, Department ofPneumology, UniversityHospital Gasthuisberg,Leuven, Belgium (Prof J Vansteenkiste MD);PUCRS School of Medicine,Centro de Pesquisa em Oncologia, Porto Alegre, Brazil(C Barrios MD);CACON,Hospital de Caridade de Ijuí,Ijuí, Brazil (F A Franke MD); AstraZeneca UK, Macclesfi eld,UK (L Grinsted MSc, V Zazulina MD, P Smith PhD,I Smith MD);and Medical Oncology Division, Hospital S Maria della Misericordia, Perugia, Italy (Prof L Crinò MD)Correspondence to: Dr Pasi A Jänne, Lowe Center for Thoracic Oncology, Dana-FarberCancer Institute, Boston,MA 02215, USApjanne@targeted eff ector proteins downstream in the RAS-RAF-MEK-ERK (MAPK) signalling pathway.The MAPK pathway converges at the MEK1/MEK2 (also known as MAP2K1 and MAP2K2) kinases, for which the only known substrates are the ERK1/ERK2 (also known as MAPK3 and MAPK1) kinases. MEK inhib-ition would therefore be expected to block ERK signal-ling irrespective of the upstream stimulus. Selumetinib (AZD6244, ARRY-142886; AstraZeneca, Alderley Park, Cheshire, UK) is an orally available, potent, selective, non-ATP competitive inhibitor of MEK1/MEK2 kinases.12 Preclinical data from KRAS-mutant NSCLC tumour xenografts showed that selumetinib signifi cantly sup-pressed tumour growth.13 Early phase clinical studies of selumetinib monotherapy showed target inhibition14 and tumour responses.15 However, in a phase 2 study, selumetinib monotherapy showed little clinical activity in an unselected pretreated population with NSCLC.16 Results of additional preclinical in-vivo studies have shown that the combination of selumetinib and docetaxel leads to greater tumour-growth inhibition or regression, and apoptosis,17,18 and this eff ect is synergistic.13 A phase 1 study of selumetinib plus docetaxel showed a man-ageable tolerability profi le in advanced solid tumours.19 The aim of this phase 2 trial was to assess the effi cacy and safety of selumetinib in combination with docetaxel as second-line treatment for patients with KRAS-mutant advanced NSCLC.MethodsStudy design and patientsWe did this double-blind, placebo-controlled, randomised phase 2 study in 67 sites across 12 countries. We included patients aged 18 years or older with histologically or cytologically confirmed, locally advanced or metastatic, KRAS-mutant NSCLC (stage IIIB–IV), after failure of fi rst-line chemotherapy. KRAS-mutation status was confi rmed before randomisation by an AstraZeneca-appointed cen-tral laboratory with Amplification Refractory Mutation System analysis,20 or by an AstraZeneca-agreed local la-boratory with agreed methods, or an accredited com-mercial laboratory with proprietary-accredited methods. No subsequent cross validation of mutational testing was done. Additional inclusion criteria were WHO perform-ance status 0–1, and adequate bone marrow, renal, and liver function. Patients were ineligible if they had received more than one chemotherapy, any previous treatment with a MEK inhibitor or docetaxel-containing regimen, or had mixed small cell and NSCLC histology. Patients with brain metastases or spinal-cord compression were eligible if they had received therapy and had been stable without steroids and anticonvulsants for at least 1 month.All patients provided written informed consent and the study was done in accordance with the Declaration of Helsinki. The protocol was approved by the institutional review board at each study site and complied with local country regulations.Randomisation and maskingEligible patients were randomly assigned, in a1:1 ratio (block size of four), to receive docetaxel incombination with either selumetinib (selumetinibgroup) or placebo (placebo group). Treatment-groupassignment was done by means of an interactive voiceresponse system at a central location. The voice responsesystem allocated randomisation numbers and medicationpack codes. Patients and study team were masked to thetreatment assigned; selu m etinib and placebo capsuleswere identical in appear a nce and presented in the samepackaging to ensure blinding of the medication.ProceduresPatients received intravenous docetaxel 75 mg/m² onday 1 of every 21 day cycle and were expected to receiveup to six cycles. Investigators were allowed to reduce thenumber of cycles if severe toxic eff ects developed, or toadminister more than six cycles when such treatmentdid not contravene local practice. I nvestigators wereadvised to consider administration of granulocyteFigure 1: Trial profi le30 patients in the selumetinib group completed the study, 13 were still receiving treatment at the time of data cutoff , and one discontinued treatment voluntarily. 29 patients in the placebo group completed the study and 14 were still receiving treatment at the time of data cutoff .colony-stimulating factor after occurrence of either febrile neutropenia or grade 4 neutropenia (according to the National Cancer I nstitute Common Terminology Criteria for Adverse Events [CTCAE], version 321), lasting for more than 7 days. Granulocyte colony-stimulating factor was administered in line with local practice and ASCO guidelines.22Patients received either selumetinib hydrogen sulphate capsules 75 mg twice a day or matched placebo until disease progression or unacceptable toxic eff ects occurred. The dose reduction algorithm in the study allowed for three steps: 75 mg once a day (fi rst dose reduction), 50 mg twice a day (dose adjustment), and 50 mg once a day (fi nal dose reduction). Once reduced or adjusted, the dose was not allowed to return to the previous level. Treatment with selumetinib or placebo was withheld if an adverse event of grade 3 or higher or an intolerable adverse event, irrespective of grade, was recorded despite optimum supportive care for all adverse events deemed at least partly related to treatment.After disease progression, patients could receive any subsequent therapy at the discretion of the treating physician. No crossover was allowed.The primary endpoint was overall survival, defi ned as the time from randomisation until the date of death from any cause. Secondary endpoints were progression-free survival (time from randomisation to docu-mentation of disease progression or death), proportion of patients with an objective response (ie, a complete or partial response), duration of response, change in tumour size, 6 month progression-free survival, and safety. Assessments of prevalence, severity, and change over time of NSCLC-specifi c symptoms were exploratory endpoints.Tumour response was based on investigator assess-ment of target and non-target lesions using CT or MRI at baseline, week 6, week 12, and then every 12 weeks,relative to date of randomisation. Tumour measure-ments according to Response Evaluation Criteria in Solid Tumors (RECI ST version 1.0) were used to estab l ish progression-free survival, the proportion of patients with an objective response, duration of response, and change in tumour size at 12 weeks. Confi rmation of response was done at the next scheduled RECIST assessment (no less than 4 weeks) after the date the criteria for response were fi rst met.We assessed safety throughout the study, from in-formed consent until 30 days after study treatment was discontinued. We graded adverse events according to the National Cancer Institute CTCAE (version 3).21Patient-reported outcomes were assessed with a Lung Cancer Specifi c Symptom Questionnaire (LSSQ), which includes the Lung Cancer Subscale (LCS). The LSSQ consists of questions GP1, GP2, GP4, and GF1 from the Functional Assessment of Cancer Therapy-Lung plus the Additional Concerns section, excluding questions B5, Q3, and L5.23 Patients completed questionnaires on day 1 and every 3 weeks until discontinuation from study treatment. Statistical analysisThe primary objective of the trial was to assess effi cacy in the selumetinib plus docetaxel group. The protocol was amended on Nov 1, 2010 (after enrolment was complete and before analysis), to change the primary endpoint from progression-free survival to overall survival; the sample size was not changed. This change was undertaken to allow decisions to be made based on data for overall survival without breaking the study blind on the earlier endpoint of progression-free survival, therefore maintaining trial integrity, and was agreed by local regulatory authorities and ethics committees.We planned overall survival analysis for after about 58 events, assuming a hazard ratio (HR) of 0∙57, giving roughly 80% power to show a statistically signifi cant diff erence for overall survival, with a one-sided 10% sig-nifi cance level. About 80 patients were to be randomised. We analysed efficacy data according to randomised treatment, excluding patients who were randomised withFigure 2: Kaplan-Meier estimates of overall and progression-free survival(A) Overall survival. (B) Progression-free survival. Symbols show censored observations.an unconfi rmed KRAS-mutation test. We analysed endpoints of progression-free survival and overall sur-vival with a Cox proportional hazards model, allowing for the eff ect of treatment and including terms for WHO performance status, sex, histology, and smoking status. All patients who received at least one dose of selumetinib or placebo were included in the safety population. Analysis of the LCS score was done post hoc once the study results were unmasked. Improvement rate is the proportion of patients with improvement; improvement was defined as an increase of at least 3 points from baseline in LCS score (0–28); worsening was defi ned as at least a 3 point decrease. No change was defi ned as a score within 2 points. We analysed time-to-deterioration with a Cox proportional hazards model, allowing for the effect of treatment and including terms for sex, WHO performance status, histology, and smoking status. Improvement rate was analysed with a logistic regression model, allowing for the eff ect of treatment and including terms for sex, WHO performance status, and smoking status. We summarised total LSSQ scores by treatment. Absolute values and changes from baseline of total LSSQ scores were summarised by visit (excluding screening). Compliance was also summarised by visit and treatment. We did data analysis with SAS (version 8.1). 95% CI values for median time-to-event data were calculated with the method proposed by Brookmeyer and Crowley24 with S-Plus software (version 8.1.1). This study is registered with , number NCT00890825. Role of the funding sourceThe trial was funded by the study sponsor and designed by the principal investigator (PAJ) and the sponsor. The sponsor was responsible for data collection, analysed the data, and had a role in data interpretation. The authors vouch for the completeness and accuracy of the data and the data analyses. This report was written by the lead author, with editorial assistance funded by the sponsor, and was reviewed and approved for publication by all coauthors and the sponsor. The corresponding author had full access to all the data in the study and had fi nal responsibility for the decision to submit the publication. ResultsWe recruited patients between April 20, 2009, and June 30, 2010. 422 patients were screened; 299 (71%) were excluded because their KRAS-mutation status was negative or unknown. 87 (21%) patients were random i sed: 44 to the selumetinib group and 43 to the placebo group (fi gure 1). One patient was randomised to the placebo group and received doxetaxel but died before receiving placebo, thus 86 patients were included in the safety analysis. Four patients—one in the selumetinib group and three in the placebo group—were excluded from effi cacy analyses because their tumours were not confi rmed to be KRAS-mutation positive. Baseline characteristics were generally well balanced between the two groups (tables 1, 2, 3). We recorded slight imbalances between groups in the number of patients with locally advanced disease, previous response to therapy, previous radio t herapy, and previous treatment with vinca alkaloids (tables 1, 2).Median treatment duration was 117 days (range 7–435) for selumetinib and 68 days (7–463) for placebo. Median number of docetaxel cycles was fi ve (range one to nine) in the selumetinib group and four (one to 12) in the placebo group. Relative dose intensity of docetaxel was 86% (SD 18∙2) in the selumetinib group and 91% (17∙9) in the placebo group. 16 patients (36%) had a dose reduction and 24 patients(55%) had a dose interruption of selumetinib (table 4). Four patients (10%) had a dose interruption of placebo; there were no dose reductions of placebo (table 4).At the time of data cutoff(on May 1, 2011), 56 of 83 patients available for effi cacy analyses had died—29 (67%) patients in the selumetinib group and 27 (68%) patients in the placebo group. The survival status was known for 82 of 83 patients; median follow-up was 7∙2 months (95% CI 3∙5–10∙7). Median overall survival was 287 days (9∙4 months; 95% CI 6∙8–13∙6) in the selumetinib group and 157 days (5∙2 months; 3∙8–not calculable [NC]) in the placebo group; hazards were non-proportional (HR for death with selumetinib, 0∙80, 80% CI 0∙56–1∙14; one-sided p=0∙21; fi gure 2A). Overall survival was much the same when analysed with all patients, irrespective of con fi rmation of KRAS-mutation status (HR for death with selumetinib 0∙81, 80% CI 0∙58–1∙15; one-sided p=0∙22).35 (81%) patients in the selumetinib group and36 (90%) in the placebo group had progressed or died at the time of data cutoff .Median progression-free survival was 162 days (5∙3 months, 95% CI 4∙6–6∙4) inthe selu m etinib group and 63 days (2∙1 months, 1∙4–3∙7) in the placebo group (HR for progression with selumetinib 0∙58, 80% C I 0∙42–0∙79; one-sided p=0∙014, fi gure 2B). Most patients had progression of existing lesions (data not shown). 6-month progression-free survival was 37·1% (95%CI 22·3–51·9)in the selumetinib group and 15·8% (95%CI 4·2–27·4)in the placebo group (HR 0·54, 80% CI0·37–0·78; one-sided p=0·016).No patient had a complete response. 16 patients in the selumetinib group had a partial response (11 confi rmed, fi ve unconfirmed); thus, 16 (37%) of patients had an objective response (table 5). By contrast, no patient in the placebo group had an objective response (two-sided p<0∙0001, table 5). One of the two non-evaluable patients in the placebo group was deemed non-assessable because of unmeasurable non-target lesions. However, according to RECIST 1.1 criteria, this patientwould have had a partial response. Median duration ofresponse in the selu m etinib group was 182 days(6·0 months, 95% C I 3·8–non-calculable). Meandecrease in tumour size is shown in table 6 and awaterfall plot of change in tumour size at week 12 isshown in fi gure 3. Post-study use of anticancer therapywas much the same in the selumetinib (44% [19 of 43])and placebo (55% [22 of 40]) groups. A breakdown ofpost-discontinuation treatments can be found in theappendix. No crossover was allowed.The proportion of serious adverse events was higherin the selumetinib group than in the placebo group(table 7). The most frequent serious adverse eventswere febrile neutropenia (selumetinib group 14% [sixof 44]; placebo group 0), pneumonia (selumetinib 9%[four of 44]; placebo 0), neutropenia (selumetinib 7%See Online for appendixFigure 3: Change in tumour size at 12 weeks(A) Selumetinib plus docetaxel group. (B) Placebo plus docetaxel group. Week 12 tumour size was defi ned as the assessment within 7 days (before or after) of the scheduled assessment, or imputed with linear regression for patients with follow-up data showing a decrease but no assessment in window or, for those with follow-up data showing an increase, we imputed a 20% increase when progression (including death) occurred before the visit.[three of 44]; placebo 7% [three of 42]), and respiratory failure (selumetinib 7% [three of 44]; placebo group 5% [two of 42]).The number of adverse events leading to death was low in both groups (selumetinib group: respiratory failure n=3, pneumonia n=1; placebo group: respiratory failure n=1, pneumonitis n=1, lower respiratory tract infection n=1, cardiac arrest n=1). None were deemed by the investigator to be related to study treatment.Grade 3–4 neutropenia, febrile neutropenia, and asthenia were more common in the selumetinib group than in the placebo group; by contrast, grade 3–4 dys-pnoea was more common in the placebo group than the selumetinib group (table 8). The most frequent adverse events of any grade in the selumetinib group were neutropenia, diarrhoea, nausea, vomiting, peripheral oedema, rashes, and stomatitis, all of which occurred more frequently in the selumetinib group than in the placebo group (table 8). Dyspnoea, exertional dyspnoea, musculoskeletal chest pain, arthralgia, fatigue, and myalgia all occurred more frequently in the placebo group than in the selumetinib group (table 8).Febrile neutropenia or neutropenic infection and pyr-exia were more common in the selumetinib group than in the placebo group. Febrile neutropenia or neu t ropenic infections, together with respiratory infections, respiratory disorders, and other isolated adverse events contributed to the greater incidence of admission to hospital of patients in the selumetinib plus docetaxel group (table 7). Selumetinib seemed to increase the severity of neutropenia associated with docetaxel—18 (42%) patients had grade 4 reduction in neutrophils from baseline grade 0 in the selumetinib group, compared with ten (24%) of those in the placebo group; this outcome led to increased use of granulocyte-colony stimulating factor in the selumetinib plus docetaxel group (32% [14 of 44] vs 17% [seven of 42]). Discontinuation of docetaxel because of adverse events was much the same between groups (table 7).Overall compliance for completion of the LSSQ was 86% (37 of 43) for the selumetinib group and 85% (34 of 40) for the placebo group. I n post-hoc exploratory analyses, the proportion of patients who had an improvement in LCS score was greater for the selumetinib group (44%) than the placebo group (24%; odds ratio 2∙50; 80% CI 1∙34–4∙77; one-sided p=0∙029). Median time-to-deterioration of LCS score was 186 days (6∙1 months, 95% C I 2∙8–non-calculable) in the selumetinib group and 49 days (1∙6 months, 1∙4–3∙5) in the placebo group (HR for deterioration 0∙33, 80% CI 0∙22–0∙49; one-sided p=0∙0002; fi gure 4). DiscussionIn this phase 2 trial, we found that although the addition of the MEK1/MEK2 inhibitor selumetinib to docetaxel as second-line treatment did not signifi cantly improve overall survival for patients with KRAS-mutant NSCLC, the combination resulted in clinically meaningful and statistically signifi cant improvements in progression-free survival, responses, and patient-reported outcomes.To our knowledge, this is the first prospective study to assess patients with KRAS-mutant NSCLC (panel). Retrospective studies suggest that patients with NSCLC with KRAS-mutant tumours obtain less clinical benefi t from chemotherapy compared with the general NSCLC population.9,25 Our prospective study seems to support this notion, because patients who received docetaxel alone had fewer responses and reduced progression-free and overall survival compared with previous phase 3 trials of second-line docetaxel in a general NSCLC population.26,27 The mechanisms for the particularly poor effi cacy of docetaxel in KRAS-mutant NSCLC are unknown and warrant further investigation. Notably, overall survival and the proportion of patients with an objective response in the control group of our study were much the same as those in a retrospective subgroup analysis of patients with NSCLC with KRAS-mutant tumours receiving docetaxel (n=27).28The improved outcomes with selumetinib plus docetaxel were associated with an increase in adverse events and serious adverse events relative to placebo and docetaxel. The most common safety findings were as expected on the basis of monotherapy tolerability profi les of selumetinib and docetaxel.16,26,27 We noted a higher occurrence of adverse events including diarrhoea, vomiting, stomatitis, and dry skin with selumetinib plus docetaxel. Furthermore, the combination was also asso-ciated with greater neutropenic effects than would be expected with docetaxel alone. The increased toxic eff ects of the combination are thus a potential issue, but we anticipate this problem will be reduced with appropriateguidance and symptomatic management, such as the use of growth factor support to decrease the incidence of neutropenia and, subsequently, the occurrence of admission to hospital and infections. Although we recorded more admissions to hospital and dose alterations (interruption and reduction) with the combination of selumetinib and docetaxel than with placebo and docetaxel, these events did not result in a greater number of discontinuations because of adverse events or deaths.n addition to increased p roportions of responses and improved progression-free survival, we also noted a clinically meaningful improvement in disease-related symptoms. In post-hoc analyses, more patients had im-provements in lung cancer symptoms with selumetinib plus docetaxel than with placebo and docetaxel, and occurrence of dyspnoea (all grades) was lower in the selumetinib group. These benefi ts might be attributable to the cytoreductive eff ects of the treatment.The combination of the two drugs in our study seems to have a synergistic eff ect compared with monotherapy activity.16,27 This fi nding is consistent with preclinical data for selumetinib,13 but contrasts with most previous studies in NSCLC, in which addition of a targeted agent to chemotherapy has not resulted in improved effi cacy. For example, studies of vandetanib plus chemotherapy as second-line treatment in unselected patients showed little or no benefi t.29 Additional studies are therefore needed to understand why the combination studied here displays synergistic activity and whether this fi nding will also be recorded with other agents targeting MAPK signalling or other chemotherapies.Additional factors warrant further investigation to define a subpopulation of KRAS-mutant NSCLC in which the combination of selumetinib and docetaxel leads to improved effi cacy. Diff erences in effi cacy might arise depending on the type of KRAS mutation present.I n our study most mutations were found in codon 12 (table 3) but the sample size is too small to determine whether any of the specific codon 12 mutations or codon 13 mutations affect the effi cacy of selumetinib plus docetaxel. The therapeutic eff ect of specifi c KRAS mutations should be assessed in future clinical trials. Other factors, such as concurrent tumour suppressor loss, including P53 and LKB1,17 and dosing schedule, could also modulate the activity of the combination, thus, additional dosing and scheduling might need to be assessed. I n-vivo mechanistic drug sequencing studies have shown that administration of selumetinib after docetaxel, rather than before, induced more apoptosis.18 This fi nding could have important clinical implications for any dosing schedule of this combination.Potential limitations of our study include the small sample size and absence of independent confi rmation of progression-free survival and tumour response. The study design was felt to be appropriate for a phase 2 proof-of-concept trial and was suffi ciently powered toFigure 4: Kaplan-Meier estimate of time-to-deterioration of the lung cancer subscale Symbols show censored observations.。