AFLPProtocol
AFLP分子标记的发展及应用

AFLP分子标记的发展及应用分子标记是指能反映生物个体或种群间基因组中某种差异特征的DNA片段,直接体现基因组DNA 间的差异。
在过去30多年,DNA 分子标记得到了迅速发展。
扩增片段长度多态性(Amplified Fragment Length Polymorphism, AFLP) 技术为第二代分子标记技术,是在随机扩增多态性(Random Amplified Polymorphic DNA, RAPD) 和限制性片段长度多态性(Restriction Fragment Length Polymorphism, RFLP) 技术上发展起来的DNA 多态性检测技术,同时拥有RFLP的高重复性和RAPD简便快捷的特点。
同其他以PCR 为基础的标记技术相比,AFLP 技术能同时检测到大量的位点和多态性标记,具有覆盖面广、高保真性、高效性、高分辨率、DNA用量少、事先勿需知道序列任何信息、可在全基因组产生标记、标记的分离遵循孟德尔遗传规律等优点,自1995年由Zabeau和Vos创建以来,已在动物、植物和微生物的分子系统学研究中得到应用,具有广阔的发展前景。
1 AFLP 标记的基本原理、操作流程及技术关键1.1 AFLP 标记的原理AFLP 标记的原理是对基因组总DNA 酶切后经PCR 进行选择性扩增。
先将基因组DNA 用两种限制性内切酶酶切成大小不等的片段,并与含有粘性末端的人工接头相连,形成酶切位点不同的限制性酶切片段,然后用与接头和位点相匹配的引物进行预扩增,预扩增产物作为进一步PCR 扩增的模板,再选用特异引物进行选择性扩增,扩增后的产物经聚丙烯酰胺凝胶电泳将特异的限制性片段分离。
由于不同来源DNA 的酶切片段存在差异,因而产生了扩增产物的多态性。
1.2 操作流程AFLP 标记的具体操作流程为: ①DNA 提取和质量检测;②双酶切和酶切片段连接;③酶切连接片段的预扩增;④选择性扩增;⑤扩增产物在聚丙烯酰胺变性凝胶上电泳; ⑥将电泳后的凝胶进行显影检测及数据分析。
AFLP分子标记的发展及应用

收稿 日期:0 0一 l 1 2 1 O -5 作者简介 : 静( 99 ) 女, 刘 1 一 , 硕士, 师。E— a :u r9 1 @13 CI 7 讲 m i l j ̄ 7 2 6 .O l ii n
京林业大学学报 , 0 0 2 ( ) 7 — 3 20 ,2 6 :9 8 .
[] 李 6
1 AL F P标记 的基本 原 理 、 操作 流程 及 技
术 关键
1 1 A IP 记的原 理 . FJ 标
AL F P标记 的原理是对基 因组 总 D A 酶切 N
后 经 P R进行选 择性扩 增 。先将 基 因组 D A用 c N 两 种限制 性 内切 酶 酶 切成 大 小 不 等 的 片段 , 与 并
不断改进和优化 , 由此衍生出多种相关技术 , 并 使其 在遗传多样 性、 种质 鉴定 、 遗传图谱构 建 、 因定位 等研 基 究 中得到广泛的应用。本文 即对 A L FP的原理、 衍生技术及其应用等进行了介绍 。
关键词 : 分子标记技术 ; F P 应用 AL ; 中图分类号 : 53 Q 0 文献标识号 : A 文章编号 :0 1 44 (00 0 - 0 0 0 10 - 9 2 2 1 ) 5 0 1 - 5
为第 二代分子标 记技术 , 是在 随机 扩增 多态性 ( a dm mpie oy op i D A,R P R n o A l dP l rh N i f m c A D)和
限制 性 片 段 长 度 多 态 性 ( etco r m n R sii Fa et r tn g
nt Pl o hs R L )技术上发展起来的 gI o m r i I y p m, F P D A多态性检测技术 , N 同时拥有 R L F P的高重复 性和 R P A D简便快捷的特点。同其他 以 P R为 C 基础 的标 记 技 术 相 比 , F P 技术 能 同 时 检 测 到 AL 大量 的位 点 和多态性 标记 , 具有覆 盖面广 、 高保 真 性、 高效性、 高分辨率、 N D A用量少、 事先勿需 知 道序列任何信息、 可在全基因组产生标记、 标记的 分离遵循孟德 尔遗传 规律等优点 , 19 自 95年 由 Zba V s aeu和 ou 创建以来 , 已在动物、 植物和微生
AFLP 技术的基本原理
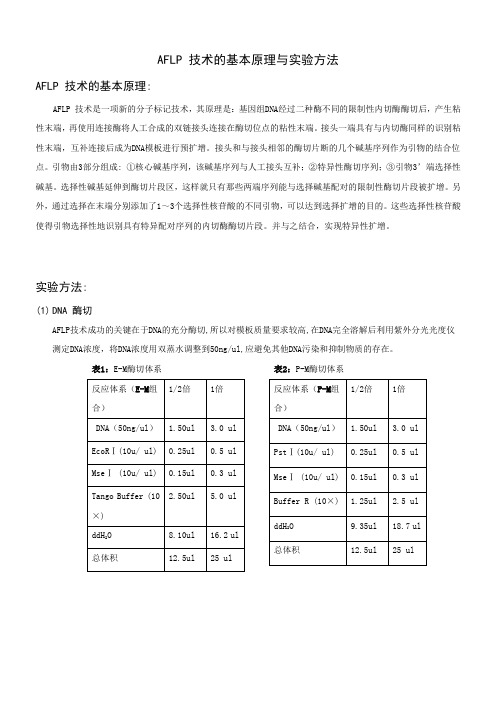
AFLP 技术的基本原理与实验方法AFLP 技术的基本原理:AFLP 技术是一项新的分子标记技术,其原理是:基因组DNA经过二种酶不同的限制性内切酶酶切后,产生粘性末端,再使用连接酶将人工合成的双链接头连接在酶切位点的粘性末端。
接头一端具有与内切酶同样的识别粘性末端,互补连接后成为DNA模板进行预扩增。
接头和与接头相邻的酶切片断的几个碱基序列作为引物的结合位点。
引物由3部分组成: ①核心碱基序列,该碱基序列与人工接头互补;②特异性酶切序列;③引物3’端选择性碱基。
选择性碱基延伸到酶切片段区,这样就只有那些两端序列能与选择碱基配对的限制性酶切片段被扩增。
另外,通过选择在末端分别添加了1~3个选择性核苷酸的不同引物,可以达到选择扩增的目的。
这些选择性核苷酸使得引物选择性地识别具有特异配对序列的内切酶酶切片段。
并与之结合,实现特异性扩增。
实验方法:(1)DNA 酶切AFLP技术成功的关键在于DNA的充分酶切,所以对模板质量要求较高,在DNA完全溶解后利用紫外分光光度仪测定DNA浓度,将DNA浓度用双蒸水调整到50ng/ul,应避免其他DNA污染和抑制物质的存在。
表1:E-M酶切体系表2:P-M酶切体系表3:P-T酶切体系表4:E-T酶切体系表5:M-S酶切体系表6:T-S酶切体系※先将模板吸入PCR板中,再将内切酶、Buffer、双蒸水配制为Mix(因内切酶用量很少或者因少数枪头质量问题,每次吸取内切酶时一定要注意观察吸取是否足量),将配制好的Mix 加入到模板中,最后在配好的反应体系中加入矿物油覆盖离心,放入PCR仪或水浴锅中37℃条件下5-6小时,然后立即转入65℃条件下1小时完全酶切。
一般做1/2倍体系即可。
目前本实验室拥有的内切酶有:EcoRⅠ;MseⅠ;PstⅠ;TaqⅠ;SacⅠ,以上内切酶均为Fermentas 公司生产。
(2)连接表8:连接体系※在连接的过程中不同的内切酶都有其对应的接头(表7),不同的酶切组合就用相应的接头组合。
nature原文

Genetic diversity of Chilean and Brazilian Alstroemeria species assessedby AFLP analysisTAE-HO HAN*,MARJO DE JEU,HERMAN VAN ECK&EVERT JACOBSEN Laboratory of Plant Breeding,The Graduate School of Experimental Plant Sciences,Wageningen University,PO Box386,NL-6700AJ Wageningen,The NetherlandsOne to three accessions of22Alstroemeria species,an interspeci®c hybrid(A.aurea´A.inodora), and single accessions of Bomarea salsilla and Leontochir ovallei were evaluated using the AFLP-marker technique to estimate the genetic diversity within the genus Alstroemeria.Three primer combinations generated716markers and discriminated all Alstroemeria species.The dendrogram inferred from the AFLP®ngerprints supported the conjecture of the generic separation of the Chilean and Brazilian Alstroemeria species.The principal co-ordinate plot showed the separate allocation of the A.ligtu group and the allocation of A.aurea,which has a wide range of geographical distribution and genetic variation,in the middle of other Alstroemeria species.The genetic distances,based on AFLP markers,determined the genomic contribution of the parents to the interspeci®c hybrid.Keywords:Alstroemeriaceae,Bomarea,classi®cation,Inca lily,Leontochir,Monocotyledonae.IntroductionThe genus Alstroemeria includes approximately60 described species of rhizomatous,herbaceous plants, with Chile and Brazil as the main centres of diversity (Uphof,1952;Bayer,1987;Aker&Healy,1990).The Chilean and Brazilian Alstroemeria are recognized as representatives of di erent branches of the genus.The family of Alstroemeriaceae,to which Alstroemeria belongs,includes several related genera,such as Bomarea Mirbel,the monotype Leontochir ovallei Phil. and Schickendantzia Pax(Dahlgren&Cli ord,1982; Hutchinson,1973).The species classi®cation in Alstroemeria is based on an evaluation of morphological traits of the¯ower, stem,leaf,fruit and rhizome(Bayer,1987).The avail-able biosystematic information on Alstroemeria species is restricted to the Chilean species,as described in the monograph of Bayer(1987).Little is known about the classi®cation of the Brazilian species(Meerow& Tombolato,1996).Furthermore,morphology-based identi®cation is rather di cult because morphological characteristics can vary considerably in di erent envi-ronmental conditions(Bayer,1987).The immense genetic variation present in the genus Alstroemeria o ers many opportunities for the improve-ment and renewal of cultivars.Therefore,identi®cation of genetic relationships at the species level could be very useful for breeding in supporting the selection of crossing combinations from large sets of parental genotypes,thus broadening the genetic basis of breeding programmes(Frei et al.,1986).The species used in the study reported here are commonly used in the breeding programme of Alstroemeria for cut¯owers and pot plants.Molecular techniques have become increasingly sig-ni®cant for biosystematic studies(Soltis et al.,1992). RAPD markers were used for the identi®cation of genetic relationships between Alstroemeria species and cultivars(Anastassopoulos&Keil,1996;Dubouzet et al.,1997;Picton&Hughes,1997).In recent years a novel PCR-based marker technique,AFLP(Vos et al., 1995),has been developed and used for genetic studies in numerous plants including lettuce(Hill et al.,1996), lentil(Sharma et al.,1996),bean(Tohme et al.,1996), tea(Paul et al.,1997),barley(Schut et al.,1997),and wild potato species(Kardolus et al.,1998).These studies indicated that AFLP is highly applicable for molecular discrimination at the species level.The technique has also been optimized for use in species such as*Correspondence:Tae-Ho Han,Laboratory of Plant Breeding,Wageningen University,PO Box386,NL-6700AJ Wageningen,The Netherlands.Tel.:31317483597;Fax:31317483457;E-mail:tae-ho.han@users.pv.wau.nlHeredity84(2000)564±569Received21June1999,accepted15November1999564Ó2000The Genetical Society of Great Britain.Alstroemeria spp.,which are characterized by a large genome size(2C-value:37±79pg)(Han et al.,1999). In this study,we produced AFLP®ngerprints of 22Alstroemeria species,one interspeci®c hybrid (A.aurea´A.inodora)and the distantly related species Bomarea salsilla and Leontochir ovallei,and we analysed their genetic relationships.The interspeci®c hybrid was included in our study in order to investigate the possibility of identifying the parental genotypes. Materials and methodsPlant materialSeeds and plants of22Alstroemeria species were obtained from botanical gardens and commercial breeders.The collection has been maintained for many years in the greenhouse of Unifarm at the Wageningen Agricultural University.When available,three acces-sions were selected for each Alstroemeria species,and both B.salsilla and L.ovallei were chosen as outgroups. One interspeci®c hybrid(A.aurea´A.inodora)was obtained from earlier research(Buitendijk et al.,1995) (Table1).All accessions were identi®ed according to their morphological traits(Uphof,1952;Bayer,1987).AFLP protocolGenomic DNA was isolated from young leaves of greenhouse-grown plants using the cetyltrimethy-lammonium bromide(CTAB)method according to Rogers&Bendich(1988).The AFLP technique followed the method of Vos et al.(1995)with modi®-cations of selective bases of pre-and®nal ampli®cationsTable1Accessions and origin of Alstroemeria species for AFLP analysisCode Plant material Accession Distribution/altitudeàChilean speciesC1 A.andina Phil.IX-2Chile26°±31°S.L.,2900±3700m(1)C2 A.angustifolia Herb.ssp.angustifolia AN1S,AN2S,AN7K Chile,33°S.L.,<1000m(1)C3 A.aurea Grah.A001,A002,A003Chile,36°±42°/47°S.L.,200±1800m(1) C4 A.diluta Bayer AD2W,AD4K,AD5K Chile,29°±31°S.L.,0±100m(1)C5 A.exserens Meyen AO2S,AO5S,AO7Z Chile,34°±36°S.L.,1500±2100m(1)C6 A.garaventae Bayer AH6Z,AH8K Chile,33°S.L.,2000m(1)C7 A.gayana Phil.XIII-2Chile29°±32°S.L.,0±200m(1)C8 A.haemantha Ruiz and Pav.J091±1.J091±4Chile,33°±35°S.L.,0±1800m(1)C9 A.hookeri Lodd.ssp.c umminghiana AQ5S,AQ6Z,AQ7Z Chile,32°±34°S.L.,0±500m(1)C10 A.hookeri Lodd.ssp.hookeri AP2S,AP3S,AP8K Chile,35°±37°S.L.,0±300m(1)C11 A.ligtu L.ssp.incarnata AJ7S,AJ12K Chile,35°S.L.,1100±1400m(1)C12 A.ligtu L.ssp.ligtu AL4S,AL6K,AL11K Chile,33°±38°S.L.,0±800m(1)C13 A.ligtu L.ssp.s imsii AM6K,AM7K,K101±1Chile,33°±35°S.L.,0±1800m(1)C14 A.magni®ca Herb.ssp.magni®ca Q001±4,Q001±5,Q007Chile,29°±32°S.L.,0±200m(1)C15 A.modesta Phil.AK2W,AK3W Chile29°±31°S.L.,200±1500m(1)C16 A.pallida Grah.AG4Z,AG7K,AG8K Chile33°±34°S.L.,1500±2800m(1)C17A.pelegrina L.AR4S,C057±1,C100±1Chile,32°±33°S.L.,0±50m(1)C18 A.pulchra Sims.ssp.pulchra AB3W,AB7S,AB8S Chile,32°±34°S.L.,0±1000m(1)C19 A.umbellata Meyen AU2Z Chile,33°±34°S.L.,2000±3000m(1) Brazilian speciesB1 A.brasiliensis Sprengel BA1K,BA2K,R001±1,Central Brazil(2)R001±2B2A.inodora Herb.P002,P004±6,P008±3Central and Southern Brazil(2)B3 A.pstittacina(D)Lehm.D031,D032,D92±02±1Northern Brazil(2)B4 A.pstittacina(Z)Lehm.93Z390±2,93Z390±4,Northern Brazil(2)96Z390±6O1Bomarea salsilla Mirbel.M121Central and Southern South America(3) O2Leontochir ovallei Phil.U001Central Chile(4)Interspeci®c hybridF1A1P2±2(A001´P002)-2Buitendijk et al.(1995)Codes from accessions of species maintained at the Laboratory of Plant Breeding,Wageningen University and Research centre.àLiterature source:(1)Bayer,1987;(2)Aker&Healy,1990;(3)Hutchinson1959;(4)Wilkin(1997).EVALUATION OF THE CHILEAN AND BRAZILIAN ALSTROEMERIA SPP.565ÓThe Genetical Society of Great Britain,Heredity,84,564±569.(Han et al.,1999).To assess interspeci®c variation, autoradiograms comprising the AFLP®ngerprints of a mixture of three accessions per species were analysed by pooling5l L of the®nal selective ampli®cation products according to Mhameed et al.(1997).The low level of variation between individual samples showed that pool-ing accessions was justi®ed.Three primer combinations (E+ACCA/M+CATG,E+ACCT/M+CATC and E+AGCC/M+CACC)were selected from a test of96primer combinations,and these produced272, 211and233bands,respectively(Table2).The choice of the primers used in the study was based upon the visual clarity of banding patterns generated and a preferably low®ngerprint complexity.The complexity of the banding pattern is a major limiting factor for scoring AFLP®ngerprints of large-size genomes.Data analysisPositions of unequivocally visible and polymorphic AFLP markers were transformed into a binary matrix, with`1'for the presence,and`0'for the absence of a band at a particular position.The genetic distance(GD) between species was based on pair-wise comparisons and calculated according to the equation:GD xy 1) [2N xy/(N x+N y)],where N x and N y are the numbers of fragments to individuals x and y,respectively,and N xy is the number of fragments shared by both(Nei&Li, 1979).Genetic distances were computed by the software package TREECON(v.1.3b)(Van De Peer&De Wachter, 1993).The dendrogram of the22Alstroemeria species, the interspeci®c hybrid,Bomarea and Leontochir was generated based on the GD matrix by using cluster analysis,the UPGMA(unweighted pair group method using arithmetic averages)method with1000bootstraps (Sneath&Sokal,1973;Felsenstein,1985)(Fig.1). Principal co-ordinate analysis was performed to access interspecies relationships based on the Nei&Li(1979) coe cient[2N xy/(N x+N y)]using the NTSYS-PC pro-gram(Rohlf,1989).Results and discussionThe average genetic distance among species excluding Bomarea,Leontochir,the interspeci®c hybrid and A.umbellata was0.65GD(a table showing the genetic distances between all the species studied is available from the authors on request).Alstroemeria umbellata was excluded because the accessions used were found to be highly related and possibly wrongly classi®ed as di erent from A.pelegrina.The average GD among accessions within a species was0.32GD(data not shown).In addition,the average GD between Brazilian species(GD:0.27)and between Chilean species(GD: 0.33)was not signi®cantly di erent.Buitendijk&Ramanna(1996)suggested that the Chilean and Brazilian species form distinct lineages.The genetic diversi®cation of Alstroemeria species as detected by the AFLP technique revealed three main clusters with99%bootstrap values:the Chilean species,the Brazilian species and the outgroup(Fig.1).This®nding would support an early divergence of these groups and is consistent with the occurrence of interspeci®c cross-ing barriers between the Chilean and Brazilian species (De Jeu&Jacobsen,1995;Lu&Bridgen,1997).The variance of the®rst three principal co-ordinates accounted for34.9%of the total variation,di erentia-ted e ectively among the species and re¯ected the main clustering of the dendrogram.From the principal co-ordinate plot,four groups were clearly demarcated:Table2Sequences of adaptors and primers usedEco RI adaptor5¢-CTCGTAGACTGCGTACC-3¢3¢-CTGACGCATGGTTAA-5¢Mse I adaptor5¢-GACGATGAGTCCTGAG-3¢3¢-TACTCAGGACTCAT-5¢Eco RI+0primer E005¢-GACTGCGTACCAATTC-3¢Eco RI+2primers E+AC5¢-GACTGCGTACCAATTCAC-3¢E+AG5¢-GACTGCGTACCAATTCAG-3¢Eco RI+4primers E+ACCA5¢-GACTGCGTACCAATTCACCA-3¢E+ACCT5¢-GACTGCGTACCAATTCACCT-3¢E+AGCC5¢-GACTGCGTACCAATTCAGCC-3¢Mse I+0primer M005¢-GATGAGTCCTGAGTAA-3¢Mse I+2primers M+CA5¢-GATGAGTCCTGAGTAACA-3¢M+CT5¢-GATGAGTCCTGAGTAACT-3¢Mse I+4primers M+CACC5¢-GATGAGTCCTGAGTAACACC-3¢M+CTAC5¢-GATGAGTCCTGAGTAACTAC-3¢M+CTAG5¢-GATGAGTCCTGAGTAACTAG-3¢566T.-H.HAN ET AL.ÓThe Genetical Society of Great Britain,Heredity,84,564±569.(i)the Brazilian group;(ii)the Chilean group;(iii)the A.ligtu group;and (iv)the outgroup (Fig.2).The Brazilian species (A.brasiliensis , A.psittacina and A.inodora )were consistently assigned to one cluster with 98%bootstrap values,whereas the Chilean species were rather weakly clustered with 62%bootstrap values containing several subgroups within the Chilean group (Figs 1and 2).The dispersion of the Chilean species on the principal co-ordinate plot re¯ected a wider geneticvariation than the Brazilian species.However,the narrow variation of the Brazilian species might be caused by the limited number of species investigated.Buitendijk &Ramanna (1996)described the similar-ities between C-banding patterns of A.inodora and A.psittacina ;in our study these species clustered strongly,reinforcing this ®nding (Fig.1).The similarity between A.psittacina and A.inodora was also revealed by allozyme analysis (Meerow &Tombolato,1996)and by a study using species-speci®c repetitive probes (De Jeu et al.,1995).These ®ndings are also supported by the fact that A.inodora and A.psittacina are easily crossed (De Jeu &Jacobsen,1995).In addition,the Chilean species A.aurea was posi-tioned between three subgroups (Fig.2).The unique position of A.aurea ,and the observation that this species has a wide geographical spread,suggest that other Chilean species may have evolved from A.aurea ecotypes.Alstroemeria aurea is indeed a widespread inhabitant in the regions with higher rainfall at the more southern latitudes between 33and 47°S in Chile (Bayer,1987;Buitendijk &Ramanna,1996).It is not found in Brazil,although A.aurea plants are found on both sides of the Andes mountains in Argentina,supporting the possibility that A.aurea ecotypes were also the ancestors of the Brazilian species (A.F.C.Tombolato,personal communication).Alstroemeria pelegrina and A.umbellata were assigned as sister species with a GD of 0.26showing a remarkable genetic similarity (data available on request).The species we coded under the name A.umbellata actually seemed to be an A.pelegrina species that did not ¯ower for many years.Alstroemeria haemantha was assigned to a group together with A.ligtu ssp.ligtu ,A.ligtussp.Fig.1Dendrogram of 22Alstroemeria species,Bomarea salsilla and Leontochir ovallei resulting from a UPGMA cluster analysis based on Nei's genetic distances obtained from 716AFLP bands.The bootstrap analysis was conducted using TREECON (v.1.3b)with 1000bootstrap subsamples of the data matrix.Percent-age values for those branches occurring in at least 60%of the bootstrap topologies areshown.Fig.2Relationships among 22Alstroemeria species,the F 1hybrid,Bomarea salsilla and Leontochir ovallei by principal co-ordinate analysis using Nei and Li coe cients.The three principal co-ordinates accounted for 34.9%of the totalvariation.PC1,PC2and PC3:®rst,second and third principal co-ordinates.See Table 1for species names.EVALUATION OF THE CHILEAN AND BRAZILIAN ALSTROEMERIA SPP.567ÓThe Genetical Society of Great Britain,Heredity ,84,564±569.incarnata and A.ligtu ssp.simsii(Figs1and2)(Aker& Healy,1990;Ishikawa et al.,1997).Bayer(1987) suggested the synonymous name of A.ligtu ssp.ligtu for A.haemantha Ruiz and Pavon.Our results support this hypothesis.Alstroemeria exserens was positioned between the Chilean group and the A.ligtu group (Fig.2).Alstroemeria andina and A.angustifolia ssp. angustifolia,and A.hookeri ssp.cumminghiana and A.hookeri ssp.hookeri were clustered together with 95%and93%bootstrap values,respectively.The interspeci®c hybrid(A1P2±2)was included in our study in order to investigate the possibility of the identi®cation of the parental genotypes.The F1hybrid A1P2±2showed a0.45-GD value with A.inodora and 0.59GD value with A.aurea showing genomic contri-bution of both parents(data available on request).It indicated the feasibility of the AFLP technique as a tool for the identi®cation of parental genotypes (Sharma et al.,1996;Marsan et al.,1998).Bomarea and Leontochir showed the mean GD value of0.83as the outgroup,thus showing large genetic distances within the Alstroemeriaceae family.In conclusion,the genetic variation and the genetic relationships among Alstroemeria species were e ciently rationalized by using AFLP markers for the character-ization of germplasm resources.In general,the topolo-gies of the dendrogram and the principal co-ordinate analysis of our study were in agreement with Bayer's views(Bayer,1987)on the classi®cation of the Als-troemeria species.Furthermore,this technique might be useful for the identi®cation of parental genotypes in interspeci®c hybrids.AcknowledgementThe authors would like to thank Anja G.J.Kuipers and Jaap B.Buntjer for critical reading of the manuscript and for helpful comments.ReferencesAKER,S.AND HEALY,W.1990.The phytogeography of the genus Alstroemeria.Herbertia,46,76±87. ANASTASSOPOULOS,E.AND KEIL,M.1996.Assessment of natural and induced genetic variation in Alstroemeria using random ampli®ed polymorphic DNA(RAPD)markers.Euphytica, 90,235±244.BAYER,E.1987.Die Gattung Alstroemeria in Chile.Mitt.Bot. Staatsamml.MuÈnchen,24,1±362.BUITENDIJK,J.H.AND RAMANNA,M.S.1996.Giemsa C-banded karyotypes of eight species of Alstroemeria L.and some of their hybrids.Ann.Bot.,78,449±457. BUITENDIJK,J.H.,PINSONNEAUX,N.A.C.,VAN DONK,M.S.AND LAMMEREN, A. A.M.1995.Embryo rescue by half-ovuleculture for the production of interspeci®c hybrids in Alstroemeria.Sci.Hortic.,64,65±75.DAHLGREN,R.M.T.AND CLIFFORD,H.T.1982.Monocotyledons.A Comparative Study.Academic Press,London.DE JEU,M.J.AND JACOBSEN, E.1995.Early postfertilization ovule culture in Alstroemeria L.and barriers to interspeci®c hybridization.Euphytica,86,15±23.DE JEU,M.J.,LASSCHUIT,J.,CHEVALIER,F.AND VISSER,R.G.F. 1995.Hybrid detection in Alstroemeria by use of species-speci®c repetitive probes.Acta Hortic.,420,62±64. DUBOUZET,J.G.,MURATA,N.AND SHINODA,K.1997.RAPD analysis of genetic relationships among Alstroemeria L. cultivars.Sci.Hortic.,68,181±189. FELSENSTEIN,J.1985.Con®dence limits on phylogenies:an approach using the bootstrap.Evolution,39,783±791. FREI,O.M.,STUBER,C.W.AND GOODMAN,e of allozymes as genetic markers for predicting performance in maize single cross hybrids.Crop Sci.,26,37±42.HAN,T.H.,VAN ECK,H.J.,DE JEU,M.J.AND JACOBSEN,E.1999. Optimization of AFLP®ngerprinting of organisms with a large genome size:a study on Alstroemeria spp.Theor.Appl. Genet.,98,465±471.HILL,M.,WITSENBOER,H.,ZABEAU,M.,VOS,P.,KESSELI,R.AND MICHELMORE,R.1996.PCR-based®ngerprinting using AFLPs as a tool for studying genetic relationships in Lactuca spp.Theor.Appl.Genet.,93,1202±1210. HUTCHINSON,J.1973.The Families of Flowering Plants. Clarendon Press,Oxford.ISHIKAWA,T.,TAKAYAMA,T.,ISHIZAKA,H.,ISHIKAWA,K.AND MII,M.1997.Production of interspeci®c hybrids between Alstroemeria ligtu L.hybrid and A.pelegrina L.var.rosea by ovule culture.Breed.Sci.,47,15±20. KARDOLUS,J.P.,VAN ECK,H.J.AND VAN DEN BERG,R.G.1998. The potential of AFLPs in biosystematics:a®rst application in Solanum taxonomy.Pl.Syst.Evol.,210,87±103.LU,C.AND BRIDGEN,M.P.1997.Chromosome doubling and fertility study of Alstroemeria aurea´A.caryophyllaea. Euphytica,94,75±81.MARSAN,P.A.,CASTIGLIONI,P.,FUSARI, F.,KUIPER,M.AND MOTTO,M.1998.Genetic diversity and its relationship to hybrid performance in maize as revealed by RFLP and AFLP markers.Theor.Appl.Genet.,96,219±227. MEEROW,A.W.AND TOMBOLATO,A.F.C.1996.The Alstroemeria of Itatiaia.Herbertia,51,14±21.MHAMEED,S.,SHARON,D.,KAUFMAN,D.,LAHAV,E.,HILLEL,J., DEGANI,C.AND LAVI,U.1997.Genetic relationships within avocado(Persea americana Mill.)cultivars and between Persea species.Theor.Appl.Genet.,94,279±286.NEI,M.AND LI,W.H.1979.Mathematical model for studying genetic variation in terms of restriction endonucleases.Proc. Natl.Acad.Sci.U.S.A.,76,5269±5273.PAUL,S.,WACHIRA, F.N.,POWELL,W.AND WAUGH,R.1997. Diversity and genetic di erentiation among populations of Indian and Kenyan tea(Camellia sinensis(L.)O.Kuntze) revealed by AFLP markers.Theor.Appl.Genet.,94,255±263. PICTON, D.D.AND HUGHES,H.G.1997.Characterization of Alstroemeria species using Random Ampli®ed Polymorphic DNA(RAPD)analysis.HortScience,32,482,Abstract:323.568T.-H.HAN ET AL.ÓThe Genetical Society of Great Britain,Heredity,84,564±569.ROGERS,S.O.AND BENDICH,A.J.1988.Extraction of DNA from plant tissues.Plant Mol.Biol.Manual,6,1±10. ROHLF, F.J.1989.NTSYS-Pc Numerical Taxonomy and Multivariate Analysis System,version1.80.Exeter Publica-tions,New York,NY.SCHUT,J.W.,QI,X.AND STAM,P.1997.Association between relationship measures based on AFLP markers,pedigree data and morphological traits in barley.Theor.Appl.Genet., 95,1161±1168.SHARMA,S.K.,KNOX,M.R.AND ELLIS,T.H.1996.AFLP analysis of the diversity and phylogeny of Lens and its comparison with RAPD analysis.Theor.Appl.Genet.,93, 751±758.SNEATH,P.H.A.AND SOKAL,R.R.1973.Numerical Taxonomy. W.H.Freeman,San Francisco,CA.SOLTIS,P.S.,SOLTIS, D.E.AND DOYLE,J.J.1992.Molecular Systematics of Plants.Chapman&Hall,New York,NY. TOHME,J.,GONZALEZ,D.O.,BEEBE,S.AND DUQUE,M.C.1996. AFLP analysis of gene pool of a wild bean core collection. Crop Sci.,36,1375±1384.UPHOF,J.C.T.1952.A review of the genus Alstroemeria.Plant Life,8,37±53.VAN DE PEER,Y.AND DE WACHTER,R.1993.TREECON:a software package for the construction and drawing of evolutionary put.Applic.Biosci.,9,177±182.VOS,P.,HOGERS,R.,BLEEKER,M.,REIJANS,M.,VAN DE LEE,T., HORNES,M.ET AL.1995.AFLP:a new technique for DNA ®ngerprinting.Nucl.Acids Res.,23,4407±4414. WILKIN,P.1997.Leontochir ovallei Alstroemeriaceae.Curtis's Bot.Magazine,14,7±12.EVALUATION OF THE CHILEAN AND BRAZILIAN ALSTROEMERIA SPP.569ÓThe Genetical Society of Great Britain,Heredity,84,564±569.。
cDNA-AFLP实验流程v2.0

不同发育期油菜种子cDNA-AFLP实验流程Ver 2.0一、总RNA提取1. 采集花后不同日期的油菜种子2. 用具RNase灭活处理(玻璃器皿:160℃烘烤2h;塑料制品:DEPC水浸泡过夜,高压灭菌)3. 液氮下研磨样品,分装100mg/管,-80℃保存4. 每100mg样品加入1ml Trizol,混匀,室温温育5min5. 加入200μl氯仿,剧烈震荡6. 4℃,12000rpm离心15min,上层水相转移至新管7. 加入0.5倍体积异丙醇,室温静置10min8. 4℃,12000rpm离心15min,弃上清,加入200μl 75%乙醇,重悬洗涤沉淀2次9. 4℃,12000rpm离心15min,弃上清,吸尽乙醇,超净台内吹干(不要干透)10. 加入50μl水溶解,-80℃保存。
1%琼脂糖凝胶电泳检测提取结果注:若Trizol对种子RNA提取效果不佳,考虑改用CTAB提取二、RNA结合磁珠1.取20ul总RNA(约2ug),65℃温育10min2.加入1ul 生物素化oligo(dT)引物(700ug/ul),1.1ul 20×SSC,轻轻混匀至完全冷却至室温3.重悬磁珠,磁力架收集,弃上清。
4.用300ul 0.5×SSC重悬磁珠,磁力架收集,弃上清。
洗三次5.将清洗后的磁珠重悬于100ul 0.5×SSC中6.取10ul磁珠,置于管中,加入结合oligo(dT)引物的RNA,室温温育10min,保持磁珠悬浮7.磁力架收集磁珠,100ul 0.1×SSC清洗四次三、cDNA合成1. cDNA第一链合成:按下表加样,42℃温育2h磁珠--- biotinylated oligo(dT) primer (700μg/μl)1μl H2O 24μl 5× First Strand Buffer 8μl0.1M DTT 4μleach 10mM dNTP mix 2μl 2.5mM Superscript II (200U/μl) 1μl M-MLV RTase, RNase H-Final V olume 40μl 或20ulT10E0.1重悬磁珠,H2O 4ul5× First Strand Buffer:(-20℃)250mM Tris-HCl (pH 8.3)15mM MgCl2375mM KCl2. 磁力架收集磁珠,仔细吸弃上清,用5×第二链buffer平衡磁珠,磁力架收集,弃上清3. cDNA第二链合成:按下表加样,12℃温育1h,22℃温育1h磁珠--- 5× Second Strand Buffer 20μl 10×buffer:10μl10mM dNTP mix 2μl0.1M DTT 4μlE.Coli DNA ligase (10U/μl) 1μl 10UDNA Polymerase I (10U/μl) 3μl 30U RNase H (5U/μl) 1μl 5U H2O 69μl Final V olume 100μl或20ulT10E0.1重悬磁珠,H2O 49ul5× Second Strand Buffer: (-20℃)7.0)(pH100mMTris-HCl20mM MgCl2450mMKCl750μM NAD+50mM (NH4)2SO44. 磁力架收集磁珠,仔细吸弃上清,用1×RL-Buffer清洗磁珠,磁力架收集(或20ulT10E0.1清洗)四、Bst YI 酶切1. 向磁珠加入10U BstYI (1μl,10U/μl),4μl 10×RL-Buffer,35μl H2O,共40μl,混匀。
AFLP分子标记技术及其应用2

涪陵师范学院生命科学系 主讲人:江 波
AFLP分子标记技术及其应用
? 一、什么叫AFLP ? ? 二、AFLP 的基本原理是什么? ? 三、AFLP 的实验过程怎样? ? 四、AFLP 的应用研究如何? ? 五、对AFLP 的评价
我们都知道世界是多姿多彩的,可以说是 五彩斑斓,花样百出,无奇不有!从生物
?AFLP (Amplified Fragment Length Polymorphism,
扩增片断长度多态性) ? 事实上,还有很多分子标记术像SSR、ISSR、
SRAP(相关序列扩增多态性)另外,还有现在号称 为第三代分子标记的SNP( 单核苷酸多态性) 等
AFLP 的基本原理
? 基因组DNA经两种限制性内切酶酶切,形成分子 量大小不等的随机限制性酶切片段,将特定的人 工合成的短的双链接头连在这些片段的两端,形 成一个带接头的特异片段,通过接头序列和PCR 引物3ˊ端选择性碱基的识别,对特异性片段进行 预扩增和选择性扩增。最后只有那些两端序列能 与选择性碱基配对的限制性酶切片段才能被扩增; 最后将选择性扩增产物在高分辨率的变性聚丙烯 酰胺凝胶上电泳,寻找多态性扩增片段。
叫做扩增片断长度多 h.所得数据可在实验室之间交
态性。
流和比较
分子标记的历史:
? 第一代分子标记技术 RFLP (Restriction Fragment Length Polymorphism,限制性片段长度多态性)
? 第二代分子标记技术 RAPD(Random Amplified Polymorphic DNA,随机扩增多态性DNA )
a.多态性高;
b.共显性遗传,在二倍体的生 物中能区分纯合与杂合状态;
AFLP分子标记及其应用

基因组DNA 的双酶切与接头连接
每个取4μ L基因组DNA,用PstⅠ 和MseⅠ 进行双酶切,酶切片段与PstⅠ和MseⅠ接 头连接。
AFLP扩增反应
预扩增:采用不含选择性碱基(+0)的 预扩增引物,每个样品反应总体积为 25 μ L,包括模板DNA (酶切连接产物 1:10 稀释)2 μ L ,Pre-ampmix 1 μ L dNTPs1 μ L 、10×PCR buffer 2.5 μ L 、 DNA 聚合酶0.5 μ L 、ddH2O 18 μ L , 反应条件为94 ℃ 30 s,56℃ 30 s, 72 ℃80 s, 30个循环。72 ℃延伸5 min。
EcoR I
Mse I
(限制性内切酶)
EcoR I接头
Mse I接头
(连接)
EcoR I引物+A
Mse I引物+C
(预扩增)
EcoR I引物+AAC
Mse I引物+CAA
(选择扩增)
(电泳检测)
4种分子标记比较
(1)DNA需要量少,检测效 率高,理论上可产生无限多 的AFLP标记。
(2)多态性高。AFLP分析可以通 过改变限制性内切酶和选择性碱基 的种类与数目,来调节扩增的条带 ,具有较强的多态分辨能力。 AFLP特点
凝胶电泳
荧光标记的PCR 扩增产物用4%琼脂糖凝胶 电泳检测,再向2 μ L的扩增产物中加2 μ L 上样缓冲液(10%蓝色葡聚糖),同时加1μL GENEMARK500 荧光DNA 梯度标准品, 标准品分子量大小50~500 nt, 梯度为25 nt。 90℃变性2min,取1 μ L 通过ABI377 测序 仪走4%含尿素的变性聚丙烯酰胺,从 Sequence Analysis上进行AFLP指纹图谱 的分析
AFLP的原理及其应用

基础理论 Basic researchA FLP的原理及其应用王 斌 翁曼丽(中国科学院遗传研究所 100101)提 要:AFLP是检测DNA多态性的一种新的分子标记技术。
对其起源、基本原理、技术程序和应用范围及前景进行了介绍和描述。
关键词:分子标记技术 AFLP 原理 应用1 分子标记技术的快速发展在过去10a中,分子标记技术得到了突飞猛进的发展,至今已有10余种分子标记技术相继出现,并在各个研究领域得到了应用。
其中在植物分子生物学领域中应用最广泛的是RF LP和RA PD。
R FL P 的结果稳定可靠,重复性好,特别适应于建立连锁图,例如水稻RF LP连锁图的建立〔1,2〕。
RF L P比较作图进一步揭示了在主要粮食作物中,R FL P标记在染色体上的排列具有类似的顺序〔3,4〕。
因此R FL P 自出现至今虽然已有10多a了,但它仍是当今应用最广泛的一种分子标记。
然而,R FL P必须经过滤膜转移和So uthern杂交,费时、费力、周期长。
另外, RF L P对DN A多态性检出的灵敏度不高,RF L P连锁图上还有很多大的空间区,这限制了它的进一步发展。
PCR对分子标记技术的发展产生了巨大的推动作用,迄今所用的分子标记技术尽管可以分为多种类型,其实除了以传统的Souther n杂交为基础的RF L P外,其它各类分子标记都涉及P CR。
R AP D就是以随机引物为模板通过P CR扩增进行DN A多态性研究的,与RF LP相比,它较便宜,方便易行,非常灵敏,DN A用量少,而且不需要同位素,安全性好。
继RF L P之后,RA PD是应用最广泛的,特别是在寻找与目的基因连锁的分子标记方面,近年来报道了大量的与各种目的基因连锁的R AP D标记。
近来西红柿P to基因和水稻Xa21基因的成功分离就是首先找到了与目的基因紧密连锁的RA PD标记,然后通过M BC(M ap based clo ning)方法克隆了目的基因〔5,6〕。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Amplified Fragment Length Polymorphism (AFLP) ProtocolIntroduction taken fromhttp://biologi.uio.no/FellesAvdelinger/DNA_KAFFE/Kaffe_Resources/AFLP.html;Protocol modified from Vos et al. (1995) NAR 23:4407-4414;Written by Scott R. Santos, last modified March 20, 2000 The AFLP technique, originally known as selective restriction fragment amplification (SRFA) (Zabeau and Vos 1993), produces highly complex DNA profiles by arbitrary amplification of restriction fragments ligated to double-stranded adaptors with hemi-specific primers harboring adaptor-complementary 5' termini (Vos et al. 1995). The technique has been widely used in the construction of genetic maps containing high densities of DNA marker loci. The AFLP protocol amplifies restriction fragments obtained by endonuclease digestion of target DNA using "universal" AFLP primers complementary to the restriction site and adapter sequence. However, not all restriction fragments are amplified because AFLP primers also contain selective nucleotides at the 3' termini that extend into the amplified restriction fragments. These arbitrary terminal sequences result in the amplification of only a small subset of possible restriction fragments. The number of amplified fragments (generally kept around 50-100) can therefore be "tailored" by extending the number of arbitrary nucleotides added to the primer termini. Alternatively, the use of endonuclease combinations that vary in their restriction frequency can also be used to tune the number of amplicons. Generally, the abundant restriction fragments produced from complex genomes require AFLP primers with longer selective regions. Conversely, analysis of small genomes require only few arbitrary nucleotides added at the primer 3' termini. The resulting AFLP fingerprints are usually a rich source of DNA polymorphisms that can be used in mapping and general fingerprinting endeavors.The AFLP protocol can be divided into the following steps: (1) DNA digestion with two different restriction enzymes (generally a rare and a frequent cutter), (2) ligation of double-stranded adapters to the ends of the restriction fragments, (3) optional DNA pre-amplification of ligated product directed by primers complementary to adapter and restriction site sequences, and (4) DNA amplification of subsets of restriction fragments using selective AFLP primers and labeling of amplified products. Amplification of very small "genomes" (plasmids, cosmids, BACs) requires primers with no selective nucleotides. AFLP fingerprinting of bacteria and fungi generally requires primers with 2 selective bases. Complex genomes require the use of more than 2 selective bases in one or both primers. In the case of complex genomes it is recommended to carry the amplification in two consecutive steps (preamplification and selective amplification) to increase specificity and the amount of initial template. The AFLP fragments are usually detected by labeling one of the two AFLP primers. For example, radioactively labeled primers can be obtained by phosphorylating the 5' ends with g-33P-ATP and polynucleotide kinase or by the use of dye labeled primers. Do not label the two primers if the generation of doublets resulting from the different mobility of complementary strands in sequencing gels wants to be avoided. Finally, the labelled reaction products are separated by electrophoresis using denaturing polyacrylamide gels and exposed to X-rayfilms to visualize the AFLP fingerprints (if radioactive) or by running out the products on an automated DNA sequencer (i.e. the Li-Cor).CONSTRUCTION OF AFLP ADAPTORS1) Order all of the oligonucleotides listed at the end of this protocol or, if you have a different combination of oligos that you will be using, order those. This protocol is designed to use the Mse I/Eco RI combination. Dilute the oligos to 100 µM concentrations O.in ddH22) Recipe for making the adaptors:Mse I adaptors (50 µM [conc]) Eco RI adaptors (5µΜ [conc]) 100 µL 100 µM Mse I.1 10 µL 100 µM Eco RI.1100 µL 100 µM Mse I.2 10 µL 100 µM Eco RI.22 µL 1 M Tris-HCl (pH 8.0) 2 µL 1 M Tris-HCl (pH 8.0)2 µL 5 M NaCl 2 µL 5 M NaCl0.4 µL 0.5 M EDTA 0.4 µL 0.5 M EDTAO175.6 µL ddH2Briefly vortex. Final volume for both adaptor mixtures should be approximately 200 µL.3) You will need to heat the mixtures to 95°C and slowly cool them to room temperature so that the two oligos in the mixture will hydrogen bond and form the adaptors. I find the easiest way to do this is by using a thermocycler. Aliquot the above mixtures into 50 µL volumes in thin wall PCR tubes. Place in a thermocycler and run the following program.Step 1: 95°C 2 minutesStep 2: 95°C decreasing to 25°C by 1°C increments per 1 minute intervals Step 3: END4) Store the aliquots at -20°C.DNA DIGESTION WITH TWO DIFFERENT RESTRICTION ENZYMES5) Make up 10 mL AFLP Digestion/Ligation (DL) Buffer (recipe is at the end of this protocol). This can be made up ahead of time and stored in at -20°C.6) Genomic DNA can also be prepared ahead of time. You should have approximately 100-250 ng of DNA/sample that you want to generate AFLPs for. The DNA should be of good quality and clean. Phenol-chloroformed isolated DNA works well. Resuspend the DNA in the lowest volume of liquid possible to keep the concentration high.7) Determine the volume of liquid required to obtain approximately 100-250 ng of DNA/sample. Aliquot the DNA into a 1.5 mL eppie. If some samples have very concentrated DNA (small volumes) while others have dilute DNA (larger volumes), dilute the concentrated samples so that all samples have the sample volume of liquid (this makes the calculations easier). Record the volume value.8) Mix the following reagents together into a master mix.Recipe for digestion (40 µL total volume/sample):Reagent amount needed/sampleEco RI enzyme 5 U1Mse I enzyme 5 U110X AFLP DL Buffer 4.4 µLddH2O up to 40 µL21 = Different companies pack their restriction enzymes at different unit concentrations so volumes will differ.2 = Remember that you want 40 µL total volume/digest, which includes the genomic DNA that you will be adding. Be sure to subtract the volume value of genomic DNAfrom the ddH2O volume value. That’s why its easier if all samples have the samevolume!!9) Vortex the master mix briefly, centrifuge briefly and aliquot the required volume of master mix to each eppie of genomic DNA so that the total final volume is 40 µL. Mix well by pipetting up and down several times.10) Place in 37°C water bath for 1 hour.LIGATION OF DOUBLE-STRANDED ADAPTERS TO THE ENDS OF THERESTRICTION FRAGMENTS11) Near the end of the 1 hour 37°C water bath incubation, make up the following ligation master mix.Recipe for ligation (10 µL total volume/sample):Reagent amount needed/sampleT4 ligase 1 U310X AFLP DL Buffer 1.1 µL5 µM Eco RI adaptors 1.0 µL50 µM Mse I adaptors 1.0 µL10 mM ATP 1.0 µLO up to 10 µLddH23 = Different companies pack their ligase at different unit concentrations so volumes will differ.12) Aliquot 10 µL into each 1.5 mL eppie that contains the digestion reaction. Mix well by pipetting up and down several times. Total volume of each tube should now be 50 µL. Place into 37°C water bath for 3 hours (the total time duration of digestion/ligation should be at least 4 hours).13) Following incubation, dilute digestion/ligation reaction with approximately 450 µL ddHO (1:9 dilution), vortex and place in -20°C or proceed to next step.2OPTIONAL DNA PRESELECTION OF LIGATED PRODUCT DIRECTED BY PRIMERS COMPLEMENTARY TO ADAPTER AND RESTRICTION SITESEQUENCESIf your organism contains a simple or small genome, you may want to skip this step. This step is mainly for organisms with large complex genomes and is designed to reduce background smears in the final DNA fingerprint and to provide almost unlimited amount of template. If these things are important to you, you should consider this step. 14) Aliquot 5.0 µL of diluted digestion/ligation reaction into appropriately labeled 0.5 mL thin-walled PCR tubes.15) Set up the following PCR amplification master mix.Recipe for preselective (PS)-AFLP amplification:Reagent amount needed/sample10X 1.5 mM MgCl2 PCR Buffer 2.0 µL10 mM dNTPs 0.4 µL2.75 µM Eco RI primer 2.0 µL2.75 µM Mse I primer 2.0 µLUB Taq polymerase 0.25 µLddH2O 8.35 µL16) Vortex briefly, centrifuge and aliquot 15.0 µL master mix/PCR sample (20 µL total volume/sample). Mix well by pipetting up and down several times.17) Place in thermocycler and run the following program (this program is for an MJ PTC-100 thermocycler; you may have to modify the time intervals if you have a different thermocycler):Step 1: 72°C 2 minStep 2: 94°C 30 secStep 3: 56°C 1 minStep 4: 72°C 1 minStep 5: Goto Step 2 20X*Step 6: 72°C 2 minStep 7: 60°C 15 minStep 8: Hold 4°C (you only need to do this if the machine is running o/n)Step 9: END* if the sample had less than 25 ng total genomic DNA added, you may have to increase the number of cycles to increase yield.18) Once the PCR is done (approximately 2 hr), run out 5 µL PCR product in a 2% agarose gel to confirm amplification next to 5 µL of the dilution used as template. If amplification has occurred (evident by a smear when the two lanes are compared), diluteremaining PCR product 1:9 with ddH2O, vortex and place in -20°C or proceed to next step. If PCR was weak, increase the number of cycles (see above).SELECTIVE DNA AMPLIFICATION OF SUBSETS OF RESTRICTION FRAGMENTS USING AFLP PRIMERS AND LABELLING OF AMPLIFIEDPRODUCTS19) Aliquot 5.0 µL of diluted PS-AFLP reaction into appropriately labeled 0.5 mL thin-walled PCR tubes.20) Set up the following PCR amplification master mix.Recipe for selective AFLP amplification:Reagent amount needed/sample10X 1.5 mM MgCl2 PCR Buffer 2.0 µL10 mM dNTPs 0.4 µL0.46 µM Eco RIAF labeled primer 2.0 µL2.75 µM Mse IAF primer 2.0 µL4UB Taq polymerase 0.25 µLddH2O 8.35 µL4 = this is the selective primer. Depending on your organism, you may have to change the selective nucleotides at this primer’s (or both this primer and the Eco RIAF primer’s) 3’ end. The only way to know is to empirically test and tailor the primers to your organism.21) Vortex briefly, centrifuge and aliquot 15.0 µL master mix/PCR sample (20 µL total volume/sample). Mix well by pipetting up and down several times.22) Place in thermocycler and run the following program (this program is for an MJ PTC-100 thermocycler; you may have to modify the time intervals if you have a different thermocycler):Step 1: 94°C 30 secStep 2: 65°C 30 sec decrease by 0.7°C/cycleStep 3: 72°C 1 minStep 4: Goto Step 1 12XStep 5: 94°C 30 secStep 6: 56°C 30 secStep 7: 72°C 1 minStep 8: Goto Step 5 23XStep 9: 60°C 30 minStep 10: Hold 4°C (you only need to do this if the machine is running o/n)Step 11: END23) Add 20 µL of formamide loading buffer (LB). If your samples still look overloaded(evident by “blob”-like bands, dilute PCR with 10 µL ddH2O and then add 30 µL LB.AFLP PAGE USING Li-COR AUTOMATED SEQUENCER24) Prepare a 6.5% acrylamide gel for running out your labeled samples I have used Li-Cor’s KB Plus gel matrix with great success. You may also try the following recipe for a 7% 19:1 acrylamide/bis-acrylamide gel.Reagent amount needed 40% stock 19:1 acrylamide/bis-acrylamide 5.25 mL10X AFLP PAGE Running Buffer 1.5 mLUrea 13.5 g25) Dissolve and mix above reagents well, measure in graduate cylinder and top off to 30 mL with ddH2O.26) Clean glass plates and assemble gel rig according to Li-Cor directions. If using a 32 well-square tooth comb (recommended by Li-Cor) or any other square tooth comb, apply bind silane to both plates before putting the rig together. γ-methacryloxypropyltrimethoxysilane (bind silane) working stock is made by mixing 50 µL bind silane dissolved in 10 mL 100% EtOH. Add 100 µL of the working stock with 100 µL 10% acetic acid in 1.5 mL eppie, vortex well, apply to both plates where comb will be mounted. Let dry for 3 minutes, DO NOT wipe area with alcohol after applying.27) When ready to pour gel, add 225 µL freshly-made 10% APS (0.1 g APS/1 mLddH2O) and 22.5 µL TEMED to the gel matrix solution. ONCE THESE REAGENTSARE ADDED, POUR THE GEL QUICKLY USING A 60 CC SYRINGE SINCE IT WILL START TO POLYMERIZE!!!!28) Let the gel polymerize for at least 1.5 hours before using.29) Prepare the gel by washing off excess acrylamide, pull the 32 well-square tooth comb (recommended by Li-Cor) and pre-run the gel for approximately 8 minutes (see Li-Cor manual if you have questions on how to do this). The setting should be set to the following:1500 V40 mA40 W45°C30) Denature samples for 3-4 minutes at 94° C and place immediately on ice. Load samples and begin electrophoresis.AFLP Data Sheet Date:Determine the volume of liquid required to obtain approximately 0.5 µg of DNA/sample. Aliquot the DNA into a 1.5 mL eppie. Dilute concentrated samples so that all have the sample volume of liquid (this makes the calculations easier). Record the volume value.Notes for DNA samples and concentrations:I. DIGESTION W/ ECO RI AND MSE I.Reagent Amount needed/sample Amount used/sample # Samples Total10X Buffer 4.4 µLEco RI1 5 UMse I1 5 UGenomic DNA 0.5 µgddH2O2up to 40 µL1 = Different companies pack their restriction enzymes at different unit concentrations so volumes will differ.2 = Remember that you want 40 µL total volume/digest, which includes the genomic DNA that you will be adding. Be sure to subtract the volume value of genomic DNA from the ddH2O volume value. That’s why its easier if all samples have the same volume!!II. Add master mix to eppies with DNA so that total volume is 40 µL.III. Incubate @ 37°C for approximately 1 hour.IV. Digestion/ligation with Eco RI, Mse I and T4 ligase.Reagent Amount needed/sample Amount used/sample # Samples Total10X Buffer 1.1 µLT4 ligase3 1 UEco RI adaptors 1 µL 5 µMMse I adaptors 1 µL 50 µMATP 1 µL 10 µMddH2O up to 10 µL3 = Different companies pack their ligase at different unit concentrations so volumes will differ. You may have to adjust water volume/sample.V. Add 10 µL of above master mix to each tube (tubes will now have 50 µL volume).VI. Incubate @ 37°C for approximately 3 hour (4 hour total incubation). Place @ -20°C.AFLP Reagent Recipes and Oligonucleotide Sequences10X AFLP digestion/ligation (DL) Buffer (10 mL)(initial concentrations in parenthesis after reagent)0.121 g Tris-base (100 mM)0.2145 g MgAc (100 mM)0.4907 g KAc (500 mM)0.077 g DTT (50 mM)pH to 7.5 with acetic acidadd 100 µL of 10 mg/mL BSA (100 ng/µL)Obring up to 10 mL with ddH2Eco RI-adaptor Structure5’-CTCGTAGACTGCGTACC OLIGO #1CATCTGACGCATGGTTAA-5’ OLIGO #2Mse I-adaptor Structure5’-GACGATGAGTCCTGAG OLIGO #3TACTCAGGACTCAT-5’ OLIGO #4AFLP PrimersAFLP primers consist of three parts; 1) core sequence = corresponds to the adaptors; 2) enzyme-specific = cleavage recognition sequence to the enzymes being used, and; 3) selective sequence = selects which fragments will be amplified. The enzyme-specific and selective sequences can be customized for different enzymes and amplifications, respectively. Below is the basic structure of the Eco RI and Mse I primers. N = any nucleotide.Core Enzyme-specific SelectiveEco RI 5’-GACTGCGTACC AATTC NNN-3’Mse I 5’-GATGAGTCCTGAG TAA NNN-3’For the preselective (PS)-AFLP amplification, one Eco RI and one Mse I primer containing a single selective nucleotide on each are required. For the selective AFLP amplification, at least one Eco RI and one Mse I primer containing a two to three selective nucleotides on each are required. You may want to order several different primers with different selective nucleotides and determine which ones give you the best results for your organism.10X PAGE Running Buffer (1000 mL)(initial concentrations in parenthesis after reagent) 10X TBEUse at 1X strength for running buffer and gel matrix 40% Stock 19:1 acrylamide/bis-acrylamide19 g acrylamide1 g bis-acrylamidebring up to 50 mL with ddH2Oshelf life is one month at 4°C。