配合物的化学键理论
第二章 配合物的化学键理论

第二章配合物的化学键理论配合物的化学键理论:中心离子与配位体之间的化学键。
第一节配合物的静电理论静电理论的基本观点:认为中心离子是带正电荷的粒子,而配位体是带负电荷的粒子,配合物中的配位键是由单纯的静电相互吸引而形成的,形成配合物的结合能有两项:1 配体与中心离子的吸引能2 配体与配体之间的排斥能U结合=U吸引+U排斥有一些现象不能很好解释:配体一定时,半径相近的金属离子与之形成的配合物的稳定性应相近。
如Na b半径为0.95, Cu+半径为0.96形成的配合物的稳定性却相差很大改进的静电理论:静电极化理论,即中心离子和配位体在静电的作用下,相互会产生变形,中心离子的正电荷可吸引配体的电子,而配体的负电荷则排斥中心离子的电子。
1 自己变形两方面的作用2 使对方变形Na b , K+ , Mg2+ , Ca2+等离子,具有8电子结构,极化弱,因而与配体配位时,变形小Cu2+,Ag+ , Zn2+ , Cd2+等离子,具有18电子结构,极化强,与配体配位时,变形大规则:中心离子具有较强的极化作用或变形性强,与变形性强的配位体配位时,形成的配合物稳定静电理论的评价:提出较早,对早期的配合物的化学键理论有贡献。
不足之处:1 不能解释象Ni(CO)4这样的配合物2 不能解释配合物的磁性和光谱第二节价键理论Sidywich 的配键理论主要的价键理论 Pauling 的电价和共价配位理论 Taube 的内轨和外轨理论主要介绍的价键理论的内容:1 Pauling 提出的杂化轨道理论 2 Taube 提出的内轨和外轨理论 IIIB IVB VB VIB VIIB VB IB IIB Sc Ti V Cr Mn Fe Co Ni Cu Zn 3d 1 4S 2 3d 2 4S 2 3d 3 4S 2 3d 5 4S 1 3d 5 4S 2 3d 6 4S 2 3d 7 4S 2 3d 8 4S 2 3d 94S 13d 10 4S 2Pauling 杂化轨道理论,用于处理配合物的形成其基本假设:中心离子,主要指过渡金属离子的价电子层中能量相近的(n-1)d, ns, np 或nd 轨道杂化后,形成能量等同的杂化轨道,接受配体的孤对电子而形成配合物,Pauling 称这种成键方式形成的为共价配合物。
第9章 9.7配合物的化学键理论
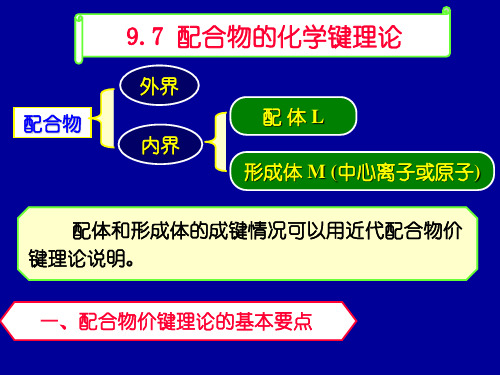
F-, H2O, OH-
常形成外轨型配离子 ( 因配位原子F, O电负性高)
配 CN-, CO
常形成内轨型配离子
体
(C的电负性较低,易给出孤对电子)
NH3, Cl-
内, 外轨型配离子均可形成 ( 由中心离子决定)
4、内轨型和外轨型配合物的稳定性和磁性
1)稳定性(解离程度)
一般来说 , 内轨型配 离子比外轨型配离子稳 定,解离程度小。
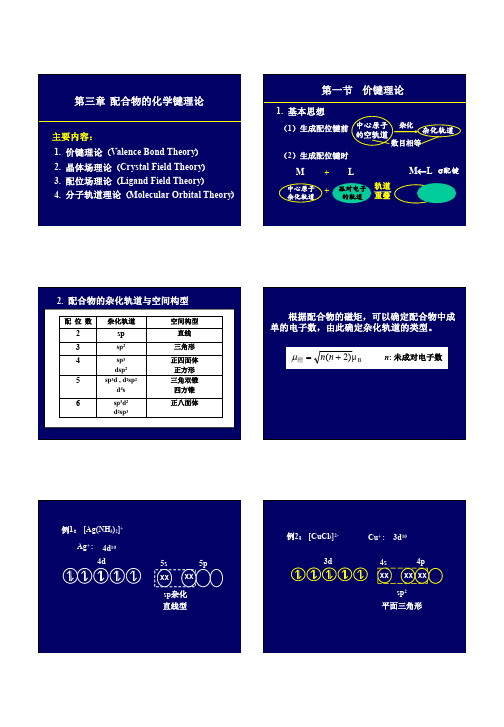
常见的轨道杂化类型与配合物几何构型的对应关系
杂化类型 配位数 几何构型
实例
spp ssp2 spp33 dspp22 dspp33 sp33dd22 d22sp33
2
直线型
[Ag(NH3)2]+,[Cu(NH3)2]+
3 平面正三角形
[ CuCl3]-
4
正四面体
[Ni(NH3)4]2+,[Zn(NH3)4]2+
4
正方形
[Cu(NH3)4]2+,[Ni(CN)4]2-
5
三角双锥
[Fe(CO)5]
6
正八面体
[FeF6]3-,[Co(NH3)6]2+
6
正八面体
[Fe(CN)6]3-,[Co(NH3)6]3+
sp, sp2, sp3, sp3d2杂化 形成外轨型配合物
dsp2,dsp3,d2sp3杂化 形成内轨型配合物
正八面体
配体 中心离子
三角双锥
[Zn(NH3)4]2+是正四面体
Zn2+
Fe3+
[FeF6]3-是正八面体
Ni2+
[Ni(CN)4]2-是正方形
Ni2+
F e
配合物的化学键理论

dxy
dyz
2/5Δt
dxz
t2
3/5Δt
Δt=(4/9)Δ0 e
dx2-y2
dz2
由于正四面体场中的e轨道和t轨道都 不象正八面体场中那样直接指向配体,所 以它们受到配体的排斥作用没有在正八面 体场中受到的排斥作用大。 根据计算,在配体及中心离子都相同,并 且配体与中心离子的距离和八面体相等的 条件下:
e 0.58e e - 3 3 1.26 r r r
2
2
2
当n=6, q=2时, 配离子的生成能为:
2e 1.66e e - 6 6 2.04 r r r
2
2
2
表2-1 配离子生成能(-e2/r) q n 1 2 3 4(四面体) 4(平面正方形) 5 6 7 8 1 1.00 1.50 1.25 0.32 0.16 2 2.00 3.50 4.26 4.32 4.16 3.10 2.04 3 3.00 5.50 7.26 8.32 8.16 8.10 8.04 6.02 4.24 4 4.00 7.50 10.26 12.32 12.Байду номын сангаас6 13.10 14.04 13.02 12.24
eg t2g eg
t2g
1. 成对能(P)
定义:当一个轨道中已有一个电子时,它对第二个 电子的引入有其排斥作用。因此需要一定的能量克 服这种排斥才能引入第二个电子与之成对,所需要 的能量称为成对能。 2.高自旋配合物和低自旋配合物
当Δ<P时,Δ较小为 弱场配体,形成高 自旋配合物。 当Δ>P时,Δ较大为 强场配体,形成低 自旋配合物。
dxy轨道其 极大值方 向与x轴和 y轴成45°, 故其能量 低于dx2-y2 轨道
配位化学-配合物的化学键理论
3d
4s
4p
xx xx xx
sp2 平面三角形
1
2017-9-12
例3: [Co(NCS)4]23d
Co2+ 3d7
4s
4p
xx xx xx xx
sp3 四面体
例4:[Ni(CN)4]2- Ni2+: 3d8
3d
4s
4p
电子归并, 杂化
xx xx xx xx dsp2
平面正方形
问题:什么情况下,内层d电子归并?
d. 价键理论不能解释配合物的颜色及吸收光谱。
e. 对非经典配合物无法解释。
3
2017-9-12
第二节 晶体场理论 1929年由Bethe提出
基本思想:
50年代以后得到发展
中心原子 静电作用
配体
①
具有电子结构
无电子结构 静电场
的离子
② 在配体静电场作用下,中心原子原来简并的5个d轨道能
级发生分裂,分裂能量的大小与空间构型及配体、中心原子 的性质有关。
2017-9-12
第三章 配合物的化学键理论
主要内容: 1. 价键理论(Valence Bond Theory) 2. 晶体场理论(Crystal Field Theory) 3. 配位场理论(Ligand Field Theory) 4. 分子轨道理论(Molecular Orbital Theory)
的排斥作用相对较小,能量降低。 dxz
eg (dx2—y2、dz2)
o
d
自由金属离子 球形场
t2g (dxy、dyz、dxz)
八面体场
d轨道在八面体场中的分裂
o:(1)由电子光谱得到;(2)由量子力学微扰理
第四章 配位键和配位化合物第二节 配合物的化学键理论

中心离子的氧化数相同,随半径增大,d电子离核越远,受晶体场 的影响越大,分裂能越大。如
[CrCl6]3- △○=162.7kJ.mol-1 [MoCl6]3- △○=229.7kJ.mol-1
2023/2/19
20
型
数
dxy
dyz
dxz
Dx2-y2
dz2
△
正八面体 6 -4.00 -4.00 -4.00 6.00 6.00 10.00
正四面体 4 1.78 1.78 1.78 -2.67 -2.67 4.45
平面正方 4 2.28 -5.14 -5.14 12.28 -4.28 17.42
直线
2 -6.28 1.14 1.14 -6.28 10.28 16.56
2023/2/19
9
●内轨型配合物
——定义 中心离子以部分次外层轨道((n-1)d轨道)与外层轨 道(ns、np轨道)杂化,再与配体成键 ——特点
•配体对中心离子影响大 •d轨道电子排布发生了变化,未成对电子数减小,磁性减小 •配位键稳定性强,键的共价性较强,水溶液中较难离解为简 单离子 ——示例 [Ni(CN)4]2-、[Fe(CN)6]3-、Fe(CO)5、[Cr(H2O)6]3+
4d
5s
5p
sp杂化
2023/2/19
3
(2)配位数为4的配合物 有两种构型。例,Ni2+
3d
4s
4p
●四面体构型 例,[Ni(NH3)4]2+。sp3杂化
3d
4s
4p
sp3杂化 ●平面正方形构型 例,[Ni(CN)4]2-,dsp2杂化,方向指向平面正 方形的四个顶点,Ni2+位于中心,4个CN-分占4个角顶
配合物的化学键理论

12
根据这个结构, 可以推测 Cu2+的配合物应当很容易地失去未配对的4p电子而迅速氧化为Cu3+, 但事实并非如 此。
因此, 价键理论有其局限性。它被配位场理论或分子轨道理论取代是必然的。
13
4.2 晶体场理论 1929年由Bethe提出, 30年代中期为Van Vleck等所发展, 及Puling的价键理论处于同一时代, 但当时并未引起
17
由于电子的总能量, 亦即各轨道总能量保持不变, eg能量的升高总值必然等于t2g轨道能量下降的总值, 这就是所谓的重 心守恒原理(原来简并的轨道在外电场作用下如果发生分裂, 则分裂后所有轨道的能量改变值的代数和为零)。
将eg和t2g这两组轨道间的能量差用△o或10Dq来表示, △o或10 Dq称为分裂能, 根据重心守恒原理, 则
的伸展较3d轨道远, 5d轨道在空间的伸展又比4d轨道远, 因而易受到配体场的强烈作用。
27
(4) 配体的本性 将一些常见配体按光谱实验测得的分裂能从小到大次序排列起来, 便得光谱化学序:
这个化学序代表了配位场的强度顺序。由此顺序可见:对同一金属离子, 造成△值最大的是CN-离子, 最小的是I -离子, 通常把CN-、NO2-等离子称作强场配体, I-、Br-、F-离子称为弱场配体。
优点:能够说明一些配合物的配位数、几何构型和稳定性。 缺点:将中心原子和配体都看作是没有内部结构的点电荷——离子键,不能说明配合物的磁学性质和光学性质。
3
➢价键理论 Sidgwick(1923)和Pauling(1928)提出了配位共价键模型,考虑了中心原子和配体的结构,能较好地说明许多配
合物的配位数、几何构型、磁性质和一些反应活性等问题。
显然, 配合物的配位数就是中心原子在成键时动用的空轨道数。
配合物的化学键理论

杂化
轨道 sp3d2 d2sp3
sp3
dsp2
配键 类型 外轨型 内轨型
外轨型
内轨型
Kf 1014
稳定性
<
1042
107. 96
1031. 3
<
磁性
Ni2+的d电子构型 杂化轨道 配键类型
未成对电子数 磁性
[Ni(NH3)4]2+ [Ni(CN)4]2 d8
sp3 外轨型
dsp2 内轨型
2 顺磁性
弱场配体
强场配体
——以上称为光谱化学序列
4. 电子成对能和配合物高、低自旋
电子在分裂后轨道上的分布遵循: 能量最低原理和洪特规则
如 Cr3+ d3
eg
E t2g
八面体场
d4d7构型的离子, d电子分布有高、低自旋两种方式。
如 Cr2+ d4
[Cr(H2O)6]2+
eg
△o t2g
[Cr(CN)6]4-
中心离子和配体之间以静电引力相互作用而形 成化学键。
中心离子的5个能量相同的d轨道受配体负电场 的排斥作用,发生能级分裂(有的轨道能量升 高,有的能量降低)。
2. 正八面体场中d轨道的能级分裂
无外电场作用下的d轨道 Edxy= Edxz= Edyz= Edx2-y2= Edz2
在带负电荷均匀球形场的作用下,d轨道能量 均升高相同值,能级不发生分裂。
请问: [Zn(NH3)4]2+、 [Ag(NH3)2]+呈现什么颜色?
中心离子d 轨道全空(d0)或全满(d10), 不能发生 d-d跃迁,其水合离子为无色。
解释配合物的稳定性
Eeg=+0.
配合物化学键理论

强场:o > P 弱场:o < P
d5 型
强场o > P
弱场o < P
(4) 影响CFSE的因素 ① d电子数目; ② 配位体的强弱; ③ 晶体场的类型
表1 过渡金属络离子的稳定化能(CFSE)
弱场CFSE/Dq
dn d0 离子 Ca2+,Sc3+ 正方型 0 正八面体 0 正四面 体 0 正方型 0
中心离子用外层(n-1)d,ns,np杂化轨道与电负性 较小的配位原子,如CN-、NO2-等形成内轨型配合 物。例如[Fe(CN)6]3-配离子,Fe采用d2sp3内轨型 杂化轨道,配合物的键能大,稳定,在水中不易 离解。
(3)内、外轨型配合物的测定---磁矩
由磁矩可判断内轨或外轨型配合物
s n—分子中未成对电子数
z
y
x
x
dz2
y z
dx2-y2
z
x
x
y
dxy
dxz
dyz
1.分裂能 (1)分裂能与配合物几何构型的关系
八面体型的配合物
在八面体型的配合物中,6个配位体分别占据八 面体的6个顶点,由此产生的静电场叫做八面体场。
(1)八面体场
八面体场中d轨道能级分裂
dz2 dx2-y2 eg 3 5 Δo =6Dq Δ o =10Dq 2 5 Δ o = 4Dq t2g dxy dxz dyz
[CrCl6]313600
[MoCl6]319200
分裂能与配位体的关系:光谱化学序列
[CoF6]3- [Co(H2O)6]3+ [Co(NH3)6]3+ o/cm-1 13000 18600 22900 [Co(CN)6]334000
第二章 2.3 配合物的成键理论

计算d 6(高自旋)、d 6 (低自旋)和
Solution
d 3、d 8 四种组态的CFSE。
d3:
CFSE = [3×(-0.4△0)] = -1.2△0 d8:
CFSE = 6×(-0.4△0)+2×0.6△0 = -1.2△0 d6(高自旋):
CFSE = 4×(-0.4△0)+2×0.6△0= - 0.4△0 d6(低自旋):
[Fe(CO)5] [FeF6]3[Fe(CN)6]3[Fe(CN)6]4[Fe(H2O)6]2+ [MnCl4]2[Mn(CN)6]4[Cr(NH3)6]3+
sp
sp sp3 dsp2 sp3 sp3 dsp3 sp3d2 d2sp3 d2sp3 sp3d2 sp3 d2sp3 d2sp3
2.3.2 晶体场理论
(2)由化学反应快慢确定电子构型 高自旋构型为活性配合物,化学反应速率常数大。
Question
八面体Co(Ⅱ)配合物的磁矩为 4.0 μB,试推断其电子组态。
Solution
Co( Ⅱ ) 配 合 物 可 能 有 两 种 组 态 : t2g5eg2(3 个 未成对电子,高自旋)和 t2g6eg1 (1个未成对电 子,低自旋),相应的自旋磁矩分别为3.87和
( [FeF6]4-中Fe 2+有4个不成对电子)
sp3d2杂化
[Fe(CN)6 ]34 -
Inner orbital complexes
内轨配合物 配位原子的电负性较小,如氰
基(CN-,以C配位),氮(NO2-, 以N配位),较易给出孤电子对, 对中心离子的影响较大,使电子层结构发生变化,(n-1)d 轨
排布原则 : ● 能量最低原理 ● Hund规则 ● Pauli不相容原理
第4讲 配合物的化学键理论

第4讲 配合物的化学键理论
3-1配合物的化学键理论简介 配合物的化学键理论,主要研究中心原子和配体之 间结合力的本性;
并用来说明配合物的物理和化学性质:如配位数、 几何构型、磁学性质、光学性质、热力学稳定性、动 力学反应性等。
静电理论
价键理论
晶体场理论
分子轨道理论
配位场理论
洛阳师范学院
Co原子(3d74s2)
洛阳师范学院
3. 反馈π键
在配合物形成过程中,中心原子(或离子)与配
体形成σ键时,如果中心原子的某些d轨道(如 dxy、 dyz、dzx) 有孤对电子,而配体有空的π分子轨道(如 CO有空的π *轨道)或空的p或d轨道,而且两者的对称 性又匹配时,则中心原子可以反过来将其孤对d电子 给予配体形成所谓“反馈π键”。
洛阳师范学院
配体场理论
配位场理论是晶体场理论的发展,分别取其晶体 场理论和分子轨道理论的优点结合而成。对中心离 子与配体静电作用部分用晶体场理论来处理,而共 价作用部分用分子轨道理论来处理。 遵循成键三原则:能量近似、最大重叠和对称性匹 配原则。 在理论上比晶体场理论等方法更为严谨,所得的 结果常用来补充晶体场理论的不足。
② 成键特征:(n-1)d 轨道参与杂化,形成内轨配键, 配合物属内轨型,低自旋态。
③ 规律:中心离子 sp3d2 与d2sp3杂化,配离子的空 间构型均为正八面体形。
洛阳师范学院
(二)判断配合物的成键类型
1、中心原子的结构不发生变化,仅使用其外层 的空轨道与配体结合,形成的配合物称为外轨型配 合物(高自旋配合物)。
sp杂化 5p
H3N NH3
结果: [Ag(NH3)2]+形成前后, 中心原子的d电子排 布没有变化 ,络合物是直线型(μ = 0)。
配合物中的化学键理论.
例:(见例4、例6、)
8
②、内轨型配合物: A、定义: 指形成配合物时,中心离子提供外层 ( ns,np ) 和 次 外 层 空 轨 道 (n - 1)d 进 行 杂 化而与配体结合所形成的配合物。 B、特点: a、提供外层(ns, np)和次外层空轨道 (n-1)d进行杂化成键。 b、杂化类型为:dsp2和d2sp3杂化。 c、配合物有较少(或没有)未成对电子数。
10
例 : [Co(NH3)6]2+ 为 外 轨 型 , 则 [Co(NH3)6]3+为内轨型。 D、内轨型配离子的稳定性大于外轨型配离 子。
原因:由于次外层轨道能级比最外层的 低。
11
内容小结:
①:杂化——构型——类型
n M用以杂 杂化 空间构型 类型 化的轨道 轨道
示例
2 ns、np sp 直线型
外层轨道
成键类型: 外轨配键
内轨配键
配合物的类型: 外轨型
内轨型
成单电子状态: 高自旋
低自旋
空间构型
正四面体
平面正方形
5
规律:中心离子dsp2 杂化,配离子的空间构 型为平面正方形。
类似有:[Cu(NH3)4]2+、[Pt(NH3)4]2+等。
③、配位数为6的配离子 也有两种成键方式
A、以 SP3d2 杂化轨道成键: 例:
Ag(NH3)2+
4 ns、np sp3 正四面体 外轨型 Ni(NH3)42+
(n-1)d、 dsp2 平面四方 内轨型 Ni(CN)42ns、np
6 ns、np、 sp3d 正八面体 外轨型
nd
2
无机化学-配位化学基础-配合物的化学键理论

解得: Et2 = + 1.78 Dq Ee = - 2.67 Dq
dxy ,dxz 和 dyz 轨道(即t 轨道) d x2-y2和 d z2轨道(即e 轨道)
( 3 ) 正方形场
sq = 17.42 Dq
四面体、八面体或正方形场中,中心金属离子5个d 轨道的能级分裂
t = 4.45 Dq
sq = 17.42 Dq
中心离子
电荷↑,半径↑, △ ↑
同一几何构型配合物的 △ : 八面体场△o
第二过渡系列中心离子 > 第一过渡系列(40 - 50%)
第三过渡系列中心离子 > 第二过渡系列(20 - 25%)
正八面体配合物ML6的△o (cm-1)
1 cm-1 = 1.23977 10-4 eV = 1.19 10-2 kJ.mol-1
电荷迁移跃迁: KMnO4 , K2CrO4 , HgO 等
(中心离子为d 0 或d 10的化合物)
互相极化 e(荷移跃迁) Mn+ ——— O2- ———→ Mn+
h
→ 显示互补色
E
hν e
O2-
1951年,几位化学家用CFT解释了 [Ti(H2O)6]3+的吸收光谱,应用于配合物,迅 速发展。
9.3.2.1 要点
1. 静电模型:配合物中Mn+ - L纯粹是静电作用,均
为点电荷,L是阴离子成偶极分子.
2. d 轨道能量分裂:
中心离子的d 轨道的能量在非球形对称的配位体形成
的晶体场中都升高,且发生分裂,分离能为 △ :
d4 – d7 构型中心离子在 八面体强场和弱场中d电子的排布
弱 场 ( △o < P )
d4
第3章 配合物的化学键理论

Mn2+ < Co2+ Ni2+ < V2+ < Fe3+ < Cr3+ < Co3+ < Mo3+ < Rh3+ < Ir3+ < Pt4+
3. 晶体场理论
3. 晶体场理论
(3)配体的性质和光谱化学序
(A)同一金属、不同配位原子对的影响 I < Br < Cl < S < F < O < N < C
MXL5:拉长 / 缩短八面体
3. 晶体场理论
3. 晶体场理论
3.2 晶体场分裂能( )及其影响因素
晶体场分裂能( ):d轨道能量分裂后,最高能量d轨道与最低能量 d轨道之间的能量差。相当于1个电子从能量最低d轨道跃迁至能量最高d 轨道所需吸收的能量。
影响因素:
(1)晶体场类型
八面体场、四面体场、平面正方形场· · · · · ·
[Co(NH3)6]3+
o = 23000 cm-1
(C)中心金属离子半径:半径越大, 越大。 中心离子半径越大,d轨道离核越远,易在配体场作用下改变能量, 增加。 同族元素, 随中心离子轨道主量子数的增加而增加: 3d4d, 增加约40%50%; [Co(NH3)6]3+ [Rh(NH3)6]3+ 4d5d, 增加约20%25%
原子半径减小 电负性减小
(B)光谱化学序列 (spectrochemical series) 弱场 I-<Br-<S2-<SCN-<Cl-<NO3-<F-<(NH2)2CO<OH- ~
CH3COO- ~ HCOO-<C2O42-<H2O<NCS-<gly-<CH3CN<edta4<py < NH3<en<NH2OH<bpy<Phen<NO2-<PPh3<CN-<CO 强场
第三章 配合物化学键理论

1 d 轨道在晶体场中的分裂
d 轨道在八面体场中的能级分裂
•dxy、dxz 、 dyz 、 dx2-y2 、 dz2在球形 对称场中,受到的作用相等,为简 并轨道;
•若有一个配合物ML6,M处于八面 体场oh中,由于L沿着x、y、z轴方向 接近中心离子, dxy、dxz 、 dyz 正好 插入配位体L的空隙中间,受静电排 斥相对较小,能量较低、而dx2-y2 、 dz2正好正对着配位体L,受静电排 斥相对较大,能量较高。
(σ-π的协同效应)
当配位体给出电子对与中心元素形成 键时,如果中心元素的
某些d 轨道(如dxy、dyz、dxz)有孤电子对,而配位体有空的p分子轨
道(如CO中有空的 p*轨道) 或空的 p或 d 轨道,而两者的对称性又匹 配时,则中心元素的孤对 d 电子也可以反过来给予配位体形成所谓
的“反馈 p 键”,它可用下式简示:
⑴ 正八面体配离子中d轨道的能级分裂 在过渡金属的自由离子中,五个d轨道的空间取向虽 然不同,但他们的能量却是相同的,是五个简并轨道,设 其能量为E。如果中心离子处在球形对称的电场中,由于 负电场在各个方向的斥力相同,五个d轨道能量升高的程 度也相同,因此,五个d轨道的能量虽都有升高,但并不 发生分裂,设其能量E=0 Dq。
d 轨道在四面体场中的能级分裂
在球型场中 在四面体场中
组轨道的能量与八面体场中正好相反。其能量差用 符号△T表示: △T = E(t2g) - E(eg)
d 轨道在平面正方形场中的能级分裂
在球型场中 在平面四边形场中
dx2–y2
dx2–y2
dz2
Δ
0
eg
dxy dxy dxz dyz dxz
2/3Δ dz20 dyz1/12Δ 0
配合物的化学键理论

配合物的化学键理论The Chemical Bond Theories of Complexes配合物的化学键理论处理中心原子(或离子)与配体之间的键合本质问题,用以阐明中心原子的配位数、配位化合物的立体结构以及配合物的热力学性质、动力学性质、光谱性质和磁性质等。
几十年来,提出来的化学键理论有: 静电理论(EST) Electrostatic Theory 价键理论(VBT) Valence Bond Theory 晶体场理论(CFT) Crystal Field Theory分子轨道理论(MOT) Molecular Orbital Theory 角重叠模型(AOM) Angular Overlap Model在这一节中,我们讲授配合物的价键理论和晶体场理论。
分子轨道理论和角重叠模型在后续课程中学习。
一、价键理论(Valence Bond Theory )L .Pauling 等人在二十世纪30年代初提出了杂化轨道理论,首先用此理论来处理配合物的形成、配合物的几何构型、配合物的磁性等问题,建立了配合物的价键理论,在配合物的化学键理论的领域内占统治地位达二十多年之久。
1.价键理论的基本内容:(1) 配合物的中心体M 与配体L 之间的结合,一般是靠配体单方面提供孤对电子对与M 共用,形成配键M ←∶L ,这种键的本质是共价性质的,称为σ配键。
(2) 形成配位键的必要条件是:配体L 至少含有一对孤对电子对,而中心体M必须有空的价轨道。
(3) 在形成配合物(或配离子)时,中心体所提供的空轨道(s 、p ,d 、s 、p 或s 、p 、d)必须首先进行杂化,形成能量相同的与配位原子数目相等的新的杂化轨道。
2.实例:(1) 主族元素配合物 Be 4O(CH 3COO)6:每个Be 原子都采取sp 3杂化-4BF :B 原子为sp 3杂化,正四面体构型 -36AlF :-3][ Al 3+周围共有12个价电子 Al 3+采取sp 3d 2杂化 (2) 过渡元素配合物a .(n - 1)d 10电子构型中心体+243)Zn(NH sp 3杂化 正四面体-3HgI sp 2杂化 平面三角形b .(n - 1)d 8电子构型中心体F Al F F F FF+243])[Ni(NH sp 3杂化 正四面体 -24]Ni(CN)[ dsp 2杂化 平面四方-24PtCl dsp 2杂化 平面四方c .(n - 1)d x (x <8)电子构型中心体-36Fe(CN) d 2sp 3杂化 正八面体+363])[Co(NH d 2sp 3杂化 正八面体 +263])[Co(NH sp 3d 2杂化 正八面体-36FeF sp 3d 2杂化 正八面体3.讨论:(1) 配合物中的中心体可以使用两种杂化形式来形成共价键:一种杂化形式为(n - 1)d 、n s 、n p 杂化,称为内轨型杂化。
配合物的化学键理论

K[PtCl5(NH3)] 五氯一氨合铂(Ⅳ)酸钾
2020/6/15
再如:
[Zn(OH)(H2O)3]NO3 硝酸一羟基三水合锌(Ⅱ)
[Co(NH3)5 (H2O)]Cl3 氯化五氨一水合钴(Ⅲ)
[Fe(CO)5]
五羰(基)合铁
[Co(NO2)3 (NH3)3] 三硝基•三氨合钴(Ⅲ)
[Ca(EDTA)]2- 乙二胺四乙酸根合钙(Ⅱ)配离子
2020/6/15
试试看: [CoCl(NH3)(en)2]SO4 命名: 硫酸 一氯 一氨 二(乙二胺)合钴(Ⅲ)
内界:
[CoCl(NH3)(en)2]2+
外界:
SO42-
中心原子: Co3+
配位体: Cl- NH3 en 配位原子: Cl N
配位数:
6
2020/6/15
配合物的分类
螯合物: 一个中心原子与多齿配体成键形成具有
H3N OH
反式 trans-
HO NH3
草酸 草酸
H3N O Pt
H3N O
Pt
H3N OH 不能反应
O C
CO
两种不同的二氯二氨合铂异构体具有不同的化学性质, 顺2式020/具6/15有抗癌活性,而反式则没有。
第二节 配合物的化学键理论
一.价键理论 (一)理论要点
1.中心原子与配体之间通过配位键形成配离子。 配位键:特殊的共价键。 一方提供共用电子,另一方提供空轨道。 如 NH4+等
向进攻配离子时,dz2、dx2-y2轨道和配位体处于迎头相 碰的状态,这些轨道受负电荷配体的静电排斥较大, 因而能量升高。而dxy dxz dyz不处于迎头相碰的状态, 因而能量降低。
第6讲 配合物的化学键理论-分子轨道理论

洛阳师范学院
在八面体弱场中,稳定化能的次序为:d0< d1 < d2 < d3 > d4 > d5 < d6 < d7 < d8 > d9 > d10,一般地,稳 定化能大的配合物应该比较稳定;
但配合物的稳定性与中心离子的d电子数有关, 通常随d电子数↗,稳定性↗,其顺序为:d0 < d1 < d2 < d3 < d4 < d5 < d6 < d7 < d8 < d9 < d10; 把两者联系起来,可解释过渡金属配合物的稳定 次序:d0< d1 < d2 < d3 >or< d4 > d5 < d6 < d7 < d8 >or< d9 < d10。
g↔u
但 g↔g u↔u
宇称定则
☻ 自旋和宇称允许时, 很大(一般在103-105之间)。 ☻ 自旋允许, 宇称禁阻时, 较小(一般在10-102之间)。 ☻ 自旋和宇称都禁阻, 极小(一般在10-2-10之间)。 ☻ 配合物的荷移跃迁(CT), 极大, 在105以上。
洛阳师范学院
E 0 h h c
0: 10000~30000 cm-1
可见光波数:14000~25000 cm-1。 所以 d-d 跃迁一般落在可见紫外光范 围,因此,过渡金属配合物绝大多数 是有颜色的。
洛阳师范学院
● 关于d-d 跃迁吸收强度 拉波特(Laporte)选择定则: S=0 自旋定则
洛阳师范学院
例如:Cu2+(d9)八面体构型时:
有两种排布: 第一种
屏蔽作用
第二种
第三章配合物的化学键理论

d x2-y2
d
dz2
dxy
dxy , dyz , dxz dyz , dxz
八面体场中Ni2+ (d8)的电子排布 C不FS发E生= 0畸变(t2g6eg2)
无论采用哪一种几何畸变, 都会 引起能级的进一步分裂, 消除简并, 其 中一个能级降低, 从而获得额外的稳 定化能(左图为第一种情况的能级图)。
根据△G=△H -T△S=-RTlnK, 配合物的稳定性将 由△G决定, 由于各种配合物的△S相差不大, 所以主要决 定于△H, 显然, △H值越负, 则MLm愈稳定。
设m=6、4……时, 上述配合反应的△H值为 △H正八面体=6△bH(M-L)-CFSE正八面体 △H正四面体=4△bH(M-L)-CFSE正四面体 △H正方形 =4△bH(M-L)-CFSE正方形 ……
思考题:
组态为d1-10的离子在八面体对称场中 有哪些可能的电子排布?
eg d
t2g
㈢ 自旋交叉(Spin Crossover)
高自旋(HS) T⇌/h低自旋(LS)
例:配合物[Fe(phen)2(NCS)2]
NN
N F e N CS
N N CS
粉红色
H S (t2g4eg2)
白色
具有这种性质的物质 在一种持久外场的微 扰下 ,就能发生一种 稳定态向另一种稳定 态的转变 ,从而达到 信息储存和开关的作用。
L S (t2g6eg0)
T
FeII(phen)2(NCS)2的变温磁化率图
2-5 晶体场稳定化能(CFSE) •晶体场稳定化能(CFSE)
定义:由于d轨道的分裂所造成的体系总 能量的降低,即稳定性的增加
eg
6 Dq
t2g -4 Dq
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
μ= 0
n=0
05.12.2020
教学ppt
7
[NiCl4]2-的空间构型为四面体,μ=2.83B.M.
28Ni:3d84s2
μ= 2.83
n=2
05.12.2020
教学ppt
8
6配位的配合物绝大多数是八面体构型,形成体可
能采取d2sp3或sp3d2杂化轨道成键。
26Fe:3d64s2
eg t2g eg t2g d7
eg
eg
t2g
t2g
d 教学ppt 7
eg
o < P
t2g 弱场
强场低自旋
弱场高自旋
23
4. 晶体场稳定化能与Jahn-Teller 效应
在八面体场和四面体场中d轨道的分裂情况不同,且 Δ值也不同
05.12.2020
教学ppt
17
2. 配合物的颜色
所吸收光子的频率与 分裂能大小有关
颜色的深浅与跃迁电 子数目有关
05.12.2020
教学ppt
18
3.d电子的排布与配合物的磁性
电子成对能(p):当中心离子的一个d轨 道中已有一个电子,另一个电子继续进入 与之配对时,必须克服电子间的相互排斥 作用,所需之能量叫做电子成对能 如果o < p (弱场), 高自旋,磁矩较大 如果o > p(强场), 低自旋,磁矩较小
[Fe(H2O)6]3+ [Fe(H2O)6]2+
o /cm-1 13700
10400
[CrCl6]3-
[MoCl6]3-
o /cm-1 13600
19200
电荷Z增大, o增大 主量子数n增大, o增大
05.12.2020
教学ppt
15
(2)配位体的影响:光谱化学序列
[CoF6]3- [Co(H2O)6]3+ [Co(NH3)6]3+ [Co(CN)6]3-
教学ppt
2
思考题2
利用光谱化学序列和磁矩数据确定下列配合 物的配体哪些是强场配体,哪些是弱场配体? 并确定d电子的排布及未成对电子数。
[Co(NO2)6]3- = 0 B.M. [Fe(NH3)6]2+ = 5.2 B.M.
[Fe(CN)6]3[FeF6]3-
05.12.2020
教学ppt
3
思考题3
晶体场理论要点
在配合物中,中心离子M处于带负电荷的配体L形成 的静电场中,二者完全靠静电作用结合在一起
晶体场对M的d 电子产生排斥作用,使M的d 轨道发 生能级分裂,有些d轨道能量升高,有些则降低
在空间构型不同的配合物中,配体形成不同的晶体 场,对中心离子d轨道的影响也不相同
05.12.2020
教学ppt
已知Co3+的p=17800cm-1,Co3+与下列配体
形成配合物的∆为:
∆/cm-1
F13000
H2O 18600
NH3 23000
试回答:
(1)Co3+的d电子在这些配合物中的排布情况
以及这些配合物的类型和磁矩。
(2)计算这些配合物的晶体场稳定化能。
05.12.2020
教学ppt
4
3.1 价键理论(VBT)
05.12.2020
教学ppt
21
八面体场中d电子的排布
eg
eg
t2g
t2g
d1
d2
eg t2g
d3
eg t2g d10
05.12.2020
eg t2g
d9
教学ppt
eg t2g d8
22
八面体场中d电子的排布
o > P 强场
ห้องสมุดไป่ตู้
d4 eg t2g
d5 eg
t2g
d6 eg
t2g
o < P 弱场
o > P 强场
价键理论要点
中心原子M和配体L间的结合是由M提供空轨 道,L提供孤电子对形成配位键
M提供的空轨道必须进行杂化,杂化轨道的 类型决定配离子的空间构型和稳定性
05.12.2020
教学ppt
5
杂化轨道的类型与空间构型
配位数 杂化轨道
2
sp
3
sp2
sp3 4
dsp2
dsp3 5
d2sp2
d2sp3 6
sp3d2
12
八面体场对d轨道的作用
05.12.2020
教学ppt
13
八面体场中d轨道能级分裂
能
o
量
球形场
自由离子
05.12.2020
八面体场
教学ppt
14
1.影响分裂能o的因素(中心离子,配位体,晶体场)
(1)中心离子:
[Cr(H2O)6]3+ [Cr(H2O)6]2+
o /cm-1 17600
14000
第三章 配合物的化学键理论
3.1 价键理论和杂化原子轨道 3.2 晶体场理论 3.3 分子轨道理论
05.12.2020
教学ppt
1
思考题1
已知: [Co(H2O)6]2+ = 4.3 B.M. [Co(EDTA)]- = 0 B.M.
指出分子构型、中心离子的价层电子排布和杂化方式
05.12.2020
例:[Fe(CN)6]3μ= 2.4B.M.
内轨型配合物
低自旋
例:[FeF6]3-
μ= 5.90B.M.
外轨型配合物
高自旋
05.12.2020
教学ppt
9
思考题1
已知: [Co(H2O)6]2+ = 4.3 B.M. [Co(EDTA)]- = 0 B.M.
指出分子构型、中心离子的价层电子排布和杂化方式
05.12.2020
教学ppt
19
某些过渡金属离子的电子成对能与晶体场分裂能
05.12.2020
教学ppt
20
以d 4为例
o < P ------弱场
eg
t2g
排布规律:
o > P ------强场
eg t2g
弱场( o <P )中,电子优先占据不同的轨道; 强场( o >P )中,电子最后占据eg轨道。
05.12.2020
空间构型
直线 平面三角形 四面体 平面正方形 三角双锥 四方锥 八面体 八面体
教学ppt
例子
[Ag(NH3)2]+ HgI3[Zn(NH3)4 ]2+ [Ni(CN)4]2[Fe(CO)5] [VO(acac)2] [Fe(CN)6]3[FeF6]3-
6
[Ni(CN)4]2-的空间构型为平面正方形,μ= 0
05.12.2020
教学ppt
10
价键理论的优缺点
很好地解释了配合物的空间构型、磁性,直 观明了
无法解释配合物的颜色(吸收光谱) 无法解释配合物的稳定性随Mn+的d电子数目
的多少而变化 Fe3+的外轨配合物动用了高能量的4d轨道似乎
不大可能
05.12.2020
教学ppt
11
3.2 晶体场理论(CFT)
o /cm-1 13000
18600
22900
34000
各种配体对同一M产生的晶体场分裂能的值由小到大的顺序
I-<Br-<Cl-,SCN-<F-<OH-<C2O42<H2O<NCS-
<edta<NH3<en<bipy<phen<SO32-<NO2<CO, CN-
光谱化学序列
教学ppt
(3)晶体场类型的影响