药物化学构效关系
药物化学构效关系
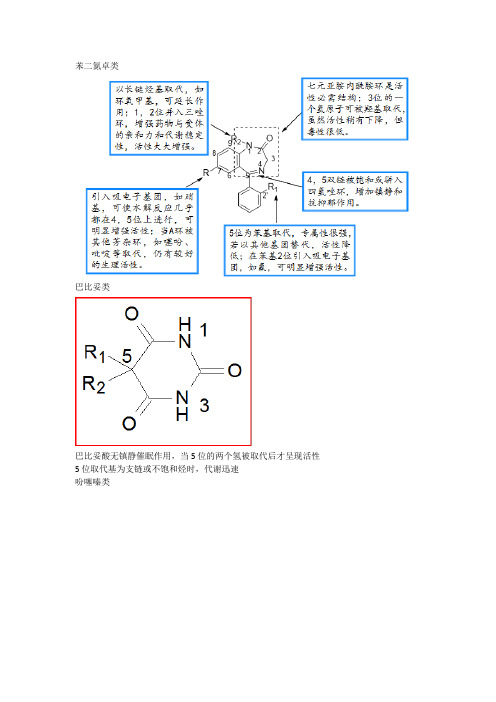
苯二氮卓类巴比妥类巴比妥酸无镇静催眠作用,当5位的两个氢被取代后才呈现活性5位取代基为支链或不饱和烃时,代谢迅速吩噻嗪类吗啡胆碱酯类M受体合成M受体拮抗剂R1和R2部分为较大基团,通过疏水性力或范德华力与M受体结合,阻碍乙酰胆碱与受体的接近和结合。
R3可以是H,OH,CH2OH或CONH2。
由于R3为OH或CH2OH时,可通过形成氢键使与受体结合增强,比R3为H时抗胆碱活性强,所以大多数M受体强效拮抗剂的R3为OH。
X一般是酯键-COO-,非活性必需。
氨基部分通常为季铵盐或叔胺结构。
R4、R5通常以甲基、乙基或异丙基等较小的烷基为好。
N上取代基也可形成杂环。
环取代基到氨基氮原子之间的距离,以n=2为最好,碳链长度一般在2~4个碳原子之间,再延长碳链则活性降低或消失。
局部麻醉药亲脂性部分可为芳烃、芳杂环,以苯环作用较强。
苯环上邻对位给电子取代基如氨基、烷氧基有利于增加活性;而吸电基会使活性下降。
中间部分-决定药物稳定性作用时间:-CH2CO->-CONH->-COS->-COO-作用强度:-COS->-COO-> -CH2CO-> -CONH-通常以n = 2-3碳原子为最好在苯环和羰基之间插入-CH2-,-O-,破坏了共轭体系,活性下降;插入-CH=CH-,则保持活性。
亲水性部分可为仲胺和叔胺,或脂环胺如吡咯烷、哌啶、吗啉等,以叔胺最为常见。
不可以是伯胺,不稳定而且毒性大。
青霉素喹诺酮吡啶酮酸的A 环是抗菌作用必需的基本药效基团,变化较小。
其中3位COOH和4位C=O 与DAN螺旋酶和拓扑异构酶Ⅳ结合,为抗菌活性不可缺少的部分。
B环可作较大改变,可以是并合的苯环(X=CH,Y=CH)、吡啶环(X=N,Y=CH)、嘧环(X=N,Y=N)等。
1位N上若为脂肪烃基取代时,以乙基或与乙基体积相似的乙烯基、氟乙基抗菌活性最好;若为脂环烃取代时其抗菌作用最好的取代基为环丙基、而且其抗菌活性大于乙基衍生物。
药物化学结构和药效的关系

例:
资料仅供参考,不当之处,请联系改正。
2.6 药物的电子云密度分布对药效的影响
如果药物分子中的电荷分布正好和其特定 受体相适应,药物与受体通过形成离子键、偶 极-偶极相互作用、范德华力、氢键等分子间引 力相互吸引,就容易形成复合物,而具有较高 活性。
资料仅供参考,不当之处,请联系改正。
下例为苯甲酸酯类局麻药分子与受体通过形成 离子键,偶极-偶极相互作用,范德华力相互作 用形成复合物的模型。
资料仅供参考,不当之处,请联系改正。
(2)增加药物分子的位阻:
抵抗青霉素酶得水解
资料仅供参考,不当之处,请联系改正。
(3)电性的影响:
资料仅供参考,不当之处,请联系改正。
2.卤素对药物生物活性的影响
强吸电子基,影响电荷分布
3.羟基、醚键对药物生物活性的影响
-OH增强与受体的结合力(氢键),增加水溶性,改变生物活性 -O-有利于定向排布,易于通过生物膜
资料仅供参考,不当之处,请联系改正。
药物的化学结构与生物活性(药效)间 的关系,通常称为构效关系(Structureactivity relationships, SAR),是药物化 学研究的主要内容之一。
资料仅供参考,不当之处,请联系改正。
本章内容
药物作用机制 受体学说 影响药物产生作用的主要因素 药物结构的官能团对药效的影响 药物的理化性质对药效的影响 药物的电子云密度分布对药效的影响 药物的立体结构对药效的影响
4.磺酸基、羧基与酯对药物生物活性的影响
-SO3H、-COOH使水溶性、解离度增大,不易通过生物膜, 生物活性减弱;
-COOR使脂溶性增大,生物活性增大
5.酰胺基与胺基对药物生物活性的影响
第一章药物化学结构与构效关系

第一章药物化学结构与构效关系药物化学结构与构效关系是研究药物分子结构与其生物活性之间关系的重要学科之一、了解药物分子的结构特征以及其与生物活性之间的相互作用对于新药的研发和优化具有重要意义。
本章将从药物分子的结构与活性关系的概念、分子结构对活性的影响、构象与构效关系等方面进行论述。
药物分子的结构与活性关系是以药物分子的结构特征为基础,通过对药物分子的结构与活性进行定性和定量的研究,从而提高药物的活性、选择性和毒性。
药物分子的结构特征包括分子量、电荷分布、功能团、立体构型等。
药物分子的活性受多种因素的影响,例如药物分子与靶点的相互作用方式、药物分子的生物转化、药物分子的分布与代谢等。
因此,通过研究药物分子的结构与活性关系,可以揭示药物分子与靶点之间的相互作用机制,为药物设计提供理论依据和指导。
药物分子的结构对活性的影响可以从药物内部结构、药物分子与靶点相互作用等角度进行分析。
药物内部结构中,有机化合物的骨架结构、取代基的位置和类型等对于药物分子的活性具有重要影响。
骨架结构的特定形状可以影响药物分子在靶点上的识别和结合,在保持药物分子与靶点相互作用的基础上,通过改变骨架结构来提高药物的活性和选择性。
取代基的位置和类型也可以影响药物分子的立体构型和电荷分布,从而影响药物与靶点之间的相互作用。
药物分子与靶点相互作用是药物发挥生物活性的基础,通过研究药物与靶点之间的相互作用方式,可以揭示药物分子活性的机制,并为药物的设计和优化提供指导。
药物分子的构象与构效关系是研究药物分子构象特征与其生物活性之间的关系。
药物分子的构象是指药物分子在空间中的排列方式,包括键角、键长、手性等方面的信息。
药物分子的构象特征对于药物的活性和选择性具有重要影响。
例如,药物分子的手性结构可以影响药物分子与靶点的亲和力和选择性,手性药物分子的活性常常和其对映异构体(对映体)的结构密切相关。
此外,药物分子的构象特征还可以通过分子模拟等方法进行预测和研究,为药物的设计和优化提供指导。
药物化学构效关系

药物化学构效关系1.局部麻醉药的构效关系:①亲脂性部分:可变范围较大,可为芳环或芳杂环,但以苯环的作用较强,是局麻药物的必需部位。
当酯类药物苯环的邻位或对位引入给电子集团,如氨基、烷氧基时,局麻作用均较未取代得苯甲酸衍生物强;对氨基苯甲酸酯类苯环的邻位上若再有其他取代基如氯、氨基、烷氧基时,由于位阻作用而延长了酯的水解,因此活性增强,作用时间延长。
②中间连接部分:由羰基部分和烷基部分共同组成。
羰基部分与麻醉药持效时间及作用强度有关,作用持续时间为:酮﹥酰胺﹥硫代酯﹥酯;麻醉作用强度:硫代酯﹥酯﹥酮﹥酰胺。
烷基部分碳原子数以2~3个为好,当烷基部分为—CH2CH2CH2—时,麻醉作用最强。
③亲水性部分:大多数为叔胺,易形成可溶性的盐类。
氮原子上取代基的碳原子总和以3~5时作用最强,也可为酯环胺,其中以哌啶的作用最强。
2. 苯二氮卓类药物的构效关系:① 1、2位拼入三氮唑环,使代谢稳定性增加,提高与受体的亲和力,活性显著增加;② 3位引入手性碳,分子构想更稳定,对受体亲和力增强;③ 4、5位引入恶唑环,增强稳定性;④7位有吸电子取代基时,药物活性明显增强,且吸电子性越强,活性增加越明显,NO2>Br>CF3>Cl;⑤ 5位苯环的2’位引入体积较小的吸电子基团如F、Cl,可使活性增强。
①镇静作用的强度和起效快慢,与药物的理化性质有关。
【酸性解离常数pKa】巴比妥酸和5位取代的巴比妥类有较强的酸性,在生理pH=7.4几乎全都电离成离子状态,不易透过血脑屏障,无镇静催眠作用;5,5-二取代的巴比妥类,酸性减弱,生理pH条件下不易电离,易进入脑中发挥作用,显效快,作用强。
【脂水分配系数】5位无取代基时,分子有一定极性,亲脂性强,不易透过血脑屏障,无镇静催眠作用;5位取代基碳原子总数在7~8之间作用最强,若亲脂性过强,作用下降甚至出现惊厥。
药物有最适当的的脂溶性,有利于药物透过细胞膜和血脑屏障,起效快,作用强。
药物化学构效关系

局部麻醉药构sheng效关系1.分类芳酸酯类、酰胺类、氨基醚类、氨基酮类、其他类2.构效关系亲酯部分中间链亲水部分⑴亲脂部分:芳烃或芳杂环,这一部分修饰对理化性质变化大,但苯环作用较强。
苯环上引入给电子取代基,麻醉作用增强,而吸电子取代基则作用减弱。
⑵中间部分:此部分决定药物稳定性,和局麻作用持续时间有关⑶亲水部分:常为仲胺和叔胺,仲胺刺激性较大;烃基链3~4个碳原子作用最强,杂环以哌啶环作用最强巴比妥类药构效关系(1)、分子中5位上应有两个取代基。
(2)、5位上的两个取代基的总碳数以4—8为最好(3)、5位上的两个取代基的总碳数以4—8为最好. (4)、在酰亚胺氮原于上引入甲基,可降低酸性和增加脂溶性。
(5)、将C2上的氧原子以硫原子代替,则脂溶性增加,起效快,作用时间短。
苯二氮卓类药物的构效关系(1)1,3-二氢-5-苯基-2H-1,4-苯二氮卓-2-酮是此类药物基本结构;(2)环A7位引入吸电子取代基活性增加(3)环B为七元亚胺-内酰胺结构是产生药理作用的必要结构(4)5位苯环上的取代基时产生药效的重要结构之一,(5)1,2位的酰胺键和4,5位的亚胺键在酸性条件下易水解开环.吩噻嗪类药构效关系R1 部分必须由三个成直链的碳原子组成,若为支链,与多巴胺受体B 部分立体上不匹配,抗精神病活性明显下降,抗组胺作用增强。
顺式吩噻嗪类药物与多巴胺的优势构象能部分重叠,活性高(当侧链与氯取代的苯环同侧时,成为顺式构象)。
丁酰苯类药物的构效关系(1)丁酰苯基为必需的基本骨架(2)侧链末端连一碱性叔胺(3)苯环的对位一般具有氟取代(4)侧链湠基于碱基之间以三个碳原子最好镇痛药的一般特征(1)分子中具有一个平坦的芳香结构(2)有一个碱性中心能在生理PH条件下大部分电离为阳离子(3)含有哌啶或类似于哌啶的空间结构吗啡的构效关系(半合成类镇痛药)叔胺是镇痛活性的关键基团,氮原子引入不同的取代基可使μ 受体激动剂转变为拮抗剂。
药物的化学结构与药效

第二章药物的化学结构与药效的关系本章以药物的化学结构为主线,重点介绍药物产生药效的决定因素、药物的构效关系、药物的结构与性质,药物的化学结构修饰和新药的开发途径及方法。
第一节药物化学结构的改造药物的化学结构与药效的关系(构效关系)是药物化学和分子药理学长期以来所探讨的问题。
由分子生物学、分子药理学、量子有机化学和受体学说等学科的进一步发展,促使药物构效关系的深入研究和发展一、生物电子等排原理在药物结构改造和构效关系的研究中,把具有外层电子相同的原子和原子团称为电子等排体,在生物领域里表现为生物电子等排,已被广泛用于药物结构的优化研究中。
所以把凡具有相似的物理性质和化学性质,又能产生相似生物活性的基团或分子都称为生物电子等排体。
利用药物基本结构的可变部分,以生物电子等排体的相互替换,对药物进行结构的改造,以提高药物的疗效,降低药物的毒副作用的理论称为药物的生物电子等排原理。
生物电子等排原理中常见的生物电子等排体可分为经典生物电子等排体和非经典生物电子等排体两大类。
(一)经典生物电子等排体1.一价原子和基团如F、Cl、OH、-NH2、-CH3等都有7个外层电子。
2.二价原子和基团如O、S、—NH—、—CH2—等都有6个外层电子。
3.三价原子和基团如—CH=、—N=等都有5个外层电子。
4.四价基团如=C=、=N+=、=P+=等都有四个外层电子。
这些电子等排体常以等价交换形式相互替换。
如普鲁卡因(3-1)酯键上的氧以NH取代,替换成普鲁卡因胺(3-2),二者都有局部麻醉作用和抗心律失常作用,但在作用的强弱和稳定性方面有差别。
(3-2)(3-1)O NHCH 2CH 2N(C 2H 5)2O C H 2N CH 2CH 2N(C 2H 5)2OCH 2N(二)非经典生物电子等排体常见可相互替代的非经典生物电子等排体,如—CH =、—S —、—O —、—NH —、—CH 2—在药物结构中可以通过基团的倒转、极性相似基团的替换、范德华半径相似原子的替换、开链成环和分子相近似等进行电子等排体的相互替换,找到疗效更高,毒性更小的新药。
构效关系指药物的化学结构与生物活性之间的关系

构效关系指药物的化学结构与生物活性之间的关系新药研发是创新药物研发的基础,关键在于理解药物的构效关系,揭示药物的化学结构与生物活性之间的关系。
构效关系是生物活性化学和医药物理学领域最重要的研究内容之一,研究其实质是研究药物的“结构定义活性”问题,即探索化学结构对活性的影响,寻找有效的药物研发策略。
构效关系是以药物的化学结构与生物活性之间的关系为基础的研究,也可以称为构效学或构物活性关系学。
它是研究药物结构与活性之间关系的学科,是药物开发、药效学研究和药代动力学研究的基础。
其中,药效学研究是以“活性定义结构”为基础,研究药物含量,主要追求药物的药效。
药物开发是以“无形定义活性”为基础,研究药物的结晶度,追求药物的质量控制。
药代动力学研究是以药物的“动力学定义活性”为基础,追求药物的药代动力学性质。
构效关系的研究包括对药物的有效性和毒性的研究,以及对药物的毒副作用的研究。
在药物的有效性和毒性方面,主要是研究药物的化学结构与药物的活性之间的关系,以探索和开发药物的有效结构和活性。
在药物的毒副作用方面,则是研究药物的化学结构与其副作用之间的关系,以探索和开发药物的低毒、高活性结构。
构效关系开发的重要性是不言而喻的。
通过对药物的结构和性质进行深入研究,有助于开发新型药物,提高药物的疗效,并降低药物毒副作用的发生率,从而丰富药物资源,为临床治疗提供有效的技术支持,满足人们的医疗需求。
构效关系的研究主要包括药物结构分析、体外实验、药效学模型建立和药物活性预测等内容。
首先是在不同实验条件下研究药物的性质,以揭示药物的活性和毒副作用;其次是建立药效学模型,以揭示药物结构与功能之间的关系;最后,利用计算机模拟药物的结构,以预测它的活性及其作用机制。
综上所述,构效关系可以说是药物学的基础理论之一,它的研究包括药物的有效性和毒性的研究,以及药物的毒副作用的研究。
该领域的研究主要侧重于研究药物的“结构定义活性”问题,以及药物化学结构与生物活性之间的关系,旨在开发有效的药物研发策略,丰富药物资源,为临床治疗提供有效的技术支持。
药物化学构效关系

巴比妥类药物巴比妥类药物属于结构非特异性药物.结构非特异性药物:药物的生物活性与药物的化学结构关系不大,与理化性质有关.结构特异性药:药物的作用依赖于药物分子的特异化学结构及空间相互排列.巴比妥类药物的作用强弱和起效时间的快慢与药物的解离常数,PKa,脂水分配系数有密切关系.解离常数:药物以分子的形式透过生物膜,以离子的形式产生作用.油水分配系数:药物既可以在体液中转运,又可以透过血脑屏障到达作用部位.该类药物5位上有两个取代基才有活性,当两个取代基的碳原子总数在4到8之间时,分配系数适中,活性最好.当碳原子总数超过8时,产生作用过强,易产生惊厥作用.结构中酰亚胺上的N原子上有甲基取代时可降低酸性和增加脂溶性,起效快.将C-2位的O用S替代时.脂溶性增加,易透过血脑屏障,起效快.巴比妥类药物的体内代谢过程与药物的代谢时间有关.二.苯二氮卓类药物(地西泮)A环为活性所必须.B环可以被其他芳杂环取代,仍保留其活性.1位一般为N-CH3.-CH3可在代谢中脱掉,但仍保留其活性1.2位可骈入杂环(三唑仑:稳定性增加活性增加)3位一个H原子可被-OH取代活性稍微降低,但毒性很低.4,5为双键被饱和.活性降低,并入恶唑环增加镇静和抗抑郁作用.5位-苯基的2位引入吸电子基(F,Cl,Br.....)活性增强.7位引入吸电子基,活性明显增强NO2>CF3>Br>Cl三.芳基丙酸类药物(布洛芬)苯环与羧基之间间距一个或一个以上的碳原子.羧基的a位又一个-CH3,限制了羧基的自由旋转,使其保持在适合与受体或酶结合的构像,以增加抗炎镇痛作用.由于羧基a位的-CH3的引入,使其产生了不对称中心,通常是S-构型的活性高于R-构型,在体内手性异构体之间可以相互转换,通常是无活性的R-构型转换为有活性的S-构型.芳环上可以引入另一个疏水基如,环己基,烯丙氧基等.在苯环羧基的间位引入一个吸电子基如F.Cl等.抗炎活性好.四.胆碱受体激动剂(氯贝胆碱)的构效关系1,季氨基.(1)带正电荷的氮原子是活性所必须.若以As,Se取代活性降低.(2)氮原子上以甲基取代为好,若以较大基团取代如乙基则活性降低,若为三个乙基则变为抗胆碱活性.2,乙酰氧基.(1)当乙酰或丁酰基等取代时活性降低,(2)乙酰基上的氢被芳环或较大基团取代时变为抗胆碱活性.(3)酯基的快速水解是乙酰胆碱作用时间短暂和不稳定的因素,因此用不易水解的基团取代乙酰基可以增加稳定性和作用时间.如用氨甲酰基取代乙酰基,由于氮上孤对电子的参与,羰基碳的亲电性较乙酰基为低,不易水解.3,亚乙基桥.(1)亚乙基桥的长度对活性有关键影响,两个碳为最好.随着碳链的延长,活性逐渐降低.(2)季氨氮原子的a位有甲基取代,整体活性降低,但N样作用大于M样作用.(3季氨氮原子的B位有甲基取代,可阻止胆碱酯酶的作用,延长作用时间,M 样作用于乙酰胆碱相当,N样作用大大减弱,成为选择性的M受体激动剂.肾上腺受体激动剂的构效关系苯乙胺的基本结构是活性所必须,碳链的延长或缩短均使作用减弱.苯环上的酚羟基可显著增强拟肾上腺素作用,尤其以3,4位最为明显,但作用时间短暂.以其他环状结构取代苯环,外周作用仍被保留,中枢兴奋作用降低.N上的取代基对a和B受体效应的相对强弱有显著影响.取代基从甲基到叔丁基,a受体效应减弱.B受体效应增强,且对B2受体的选择性提高.B-碳上通常连有羟基.其绝对构型以R-构型为活性体.局麻药的构效关系.(图自己想)邻对位给电子基取代,有利于两性离子的形成,活性增强.若有吸电子存在则活性下降.可以为芳环,芳杂环,此部分的修饰对活性的影响较大,活性顺序为苯环>吡咯>噻吩>呋喃通常以2-3个碳原子为最好有仲胺,叔胺或吡咯烷,哌啶.吗啉等,以叔胺最为常见.在苯环和羧基之间插入-CH2,-O-等基团,破坏了两性离子的形成.活性降低.若连入可以形成共轭的基团,如-CH2=CH2-等.活性可保持不变.酰胺也可形成两性离子.此部分决定药物的稳定性,按作用时间顺序羰基+-O->....+-S->....+-NH->....+-CH2-;按作用强度顺序:-S->-O->-CH2->-NH-B受体阻滞剂的构效关系(图自己想).苯乙醇胺类和芳氧丙醇胺类可以是苯,萘,杂环,稠环和脂肪性的不饱和杂环.可以有甲基,氯,硝基,甲氧基等取代基,在2,4和2,3,6位取代时活性最佳.用S,-CH2,-NCH3取代时,活性降低.S-构型异构体活性增加,R-构型异构体活性降低或消失.R-构型异构体活性增加,S-构型异构体活性降低或消失以叔丁基和异丙基取代时最好,甲基上的氢原子数小于3或N-N双取代时,活性降低.由于B 受体阻滞剂的结构组成自由度很大,所以其溶解度也有较大差异,这与其副作用和体位消除的位置有关.亲脂性-肝代谢-速率较快.亲水性-肾消除二氢吡啶类药物的构效关系(硝苯地平)二氢吡啶环为活性所必须.若变为吡啶环或六氢吡啶环则活性消失,环上的NH不被取代时,活性保持最佳.2,6位取代基为低级烷烃3,5位的羧酸脂为活性所必须,若变为乙酰基或氰基则活性降低,若变为硝基则激活钙通道. 3,5位羧酸脂不同,C4为手性中心,酯基的大小对活性影响不大,但不对称脂则影响作用部位. C4若为手性碳,具有立体选择性4位取代基与活性的关系:苯基或取代苯基>环烷基>甲基>H苯环上邻间位上有吸电子基取代活性较佳,对位取代时活性降低H2受体拮抗剂的构效关系(雷尼替丁).H2受体拮抗剂的结构有三部分组成:碱性的芳杂环结构,易曲饶的四原子链和平面极性基团.碱性的芳杂环和碱性的基团取代的芳杂环是活性所必须,咪唑环作为质子转移的机制,被异噻唑,恶唑置换后碱性下降,活性也随之降低.被亲脂性的芳杂环(苯环)取代时活性降低.被碱性基团取代,呋喃,噻唑置换后,是良好的H2受体拮抗剂.平面极性基团.是具有胍,脒基样的结构.在生理PH条件下,电离程度低的极性基团作为脒脲基团与受体形成一个以上的氢键保持活性.易曲饶的链或芳杂环:链长为4个原子.链长与其拮抗性有关.自由旋转受限使其活性下降,中间连接的链可被钢性环所取代.青霉素的构效关系(1)6位侧链的酰胺基团主要决定其抗菌谱,改变其极性,使其易于透过细胞膜,可扩大其抗菌谱.例如,在芳环乙酰胺基的a位引入极性-NH2,-COOH,-SO2等亲水性基团,扩大抗菌谱.增加亲水性.有利于对格兰阴性菌的抑菌作用,并能增强对青霉素结合蛋白的亲和力.在分子中的适当位置引入立体位阻基团.如在侧链引入立体位阻较大的基团和在6位引入甲氧基和甲酰胺基,因其立体位阻的效应降低了钝化酶的结构适应性,保护B-内酰胺环不被B-内酰胺酶进攻.因而得到耐酶的抗生素.青霉素的噻唑环上的羧基是基本活性基团,虽可被硫代酸和酰胺取代但活性降低.若羧基被还原为醇则失去抗菌活性.对于羧基可利用前药原理制成脂,改进口服吸收和药物代谢动力学性质.青霉烷酸分子中三个手性碳的构型对其活性至关重要.但青霉素的噻唑环上的两个甲基不是活性的必要因素.半合成头孢菌素的构效关系在7位侧链引入亲脂性的基团,如苯基,环稀基,噻吩和含氮的杂环.可增强抗菌活性,扩大其抗菌谱.同时改变3位取代基,引入杂环,可改进口服吸收分布也可扩大其抗菌谱.在7位酰胺的a位引入亲水性的-SO3H,-NH2,-COOH,等极性基团.可扩大抗菌谱同时改变3位取代基,引入-Cl,CH3,和含氮的杂环,可增强口服吸收扩大抗菌谱.带有7B为顺势”氨噻肟”的侧链可提高对B-内酰胺酶的稳定性,扩大抗菌谱.这主要是由于引入肟后,甲氧基占据了靠近B-内酰胺环的位置.阻止了酶分子对B-内酰胺环的靠近,因而使药物有耐酶,广谱的性质.5位的S用生物电子等排体O,CH2等取代,分别称为氧头孢菌素和碳头孢菌素,活性不降低.3位取代基的改造,如乙酰氧基可被甲基,氯等取代可扩大抗菌谱并且改变药物在体内的吸收分布和药物的渗透性的药物代谢动力学性质.2,3位的双键移位失活.2位-COOH可制成前药增加口服吸收.喹诺酮类药物的构效关系.A环(吡啶酮酸部分)是抗菌作用所必须的基本药效基团.其中3位的羧基和4位的酮基是与靶酶的结合位点,是抗菌活性必不可少的部分.B环部分可做较大改变,可并入苯环.吡啶环和嘧啶环等.N1位置取代基对抗菌活性的贡献较大,若由烃基,环烃基取代活性增加,尤以乙基,氟乙基和环丙基取代活性最佳.2位引入取代基活性减弱或消失,可能是由于空间位阻阻止了与受体的结合.3位的羧基和4位的酮基是抗菌活性中不可缺少的部分.被其他基团取代活性消失,与铁铝钙络合产生副作用.5位氨基取代活性增加.其他基团取代活性均降低.6位取代基对活性的影响很重要.活性取代顺序位:F>Cl>CN>=CH2>=H.引入F可比H的抗菌活性增强30倍.因为F代物与DNA螺旋酶亲和力增强2-17倍.对细菌细胞壁的穿透力增强1-70倍.7位引入取代基增强活性的顺序为:哌嗪基>二甲氨基>甲基>卤素>氢.其中以哌嗪基取代活性最好.8位以F,Cl,-OCH3,等取代活性增强,但以F取代光毒性也增强.若1,8位间成环,产生的光化学异构体之间也有较大差异.。
第四章 构效关系-1

沙利度胺 S-异构体 R-异构体
强致畸 无
心脏毒性:毒性基团或分子本身,抑制hERG受体—引起心 律失常,导致心脏猝死。
F N N N OCH3 阿司咪唑,1999年停止使用 H N
第五节
基团变化对活性的影响
基团变换的总原则: 改变药效团特征,直接影响活性
N-氧化 物、N- 羟胺、 胺类及 在体内 可以转 化成胺 的化合 物
烷基硫 酸酯或 磺酸酯 及β-卤 代硫醚 类
β-内酯 及醌类
可生成 阳碳离 子或自 由基的 某些含 卤素的 烷烃及 含卤素 的芳烃 和硝基 芳烃
2. 经代谢诱导生成的毒性基团
HO O Michael reaction HO HO H3C HO OH O O R1 N N R2 N O O R O 自由基反应 O O O Michael reaction H2C O Michael reaction O
1. 药效团与优势结构
药效团是不连续的散在性的基团或片断, 分子骨架 具有连续的结构特征,没有适合的骨架支撑,药效 团无法准确具现。 优势结构(privileged structure): 反复出现在 作用于多种受体的配体结构中的片断或骨架。
优势结构与药效团的恰当配臵,是研制创新药物特 别是模拟创新药物(follow-on drug)的策略基 础。
7. 醚基和硫醚基
醚基的键角与C-C-C相似
氧原子上有未偶电子对和较强电负性,可以形成 氢键,使分子增加极性 氧原子的亲水性和碳原子的亲脂性,使醚类化合 物在脂-水界面处定向排布。
H3CO
H3C N N H 奥美拉唑 H2 S C O
OCH3 CH 3 N
药物化学结构与药效的关系

化学结构相似的药物,能与同一受体结合,引起相似 作用(激动药,拟似药)或相反的作用(拮抗药,阻断药).
例:
乙酰胆碱
(神经递质)
氨甲酰胆碱
(拟胆碱药)
D=药物;R=受体;DR=药物-受体复合物 E=药理效应;
药物-受体复合物的键合方式包括:共价键、 氢键、离子键、离子-偶极和偶极-偶极作用、 范德华力等。
5. 受体激动药与受体拮抗药
根据药物与受体结合后所产生效应的不同,将药 物分为受体激动药与受体拮抗药
激动药(agonist):对受体既有亲和力又有内在 活性的药物,它们与受体结合并激活受体产生效 应。
2.2 受体学说
1. 受体的概念
受体(Receptor,R)是指对生物活性物质具有 识别能力,并选择性与之结合,传递信息,引起 特定效应的生物大分子。
受体存在于细胞内,具有一定坚固性的三维结 构. 各种药物的受体是不相同的, 但是它们可能 都具有:
(1) 一个高度折叠的近似球状的肽链; (2) 有一个空穴,此空穴至少部分被多肽区域 所 包围.
2.1 药物的作用机制:
药物的作用机制(mechanism of drug action)是研究药物如何与机体不 同靶细胞结合,又如何发挥作用的。
一.药物的作用机制简介:
1、理化作用 2、参与或干扰细胞代谢 3、影响酶的活性 4、影响生理物质的合成、释放与转运 5、影响离子通道 6、影响核酸代谢 7、影响免疫机制 8、作用于受体
2.7 药物的立体结构对药效的影响
1.官能团间的距离对药效的影响
第二章 药物的构效关系 药物化学 课件

第二章 药物的构效关系
第四节 药物其它特性对药效的影响
二、电子云密度对药效的影响
各种元素的原子核对其核外电子的吸引力各不相同而显示 电负性的差异。由电负性不同的原子组成的化合物分子就存在 电子密度分布不均匀状态。药物分子的电子密度分布如果和酶 蛋白分子的电荷分布恰好相反,则有利于相互作用而结合,形 成复合物。
化学工业出版社
第二章 药物的构效关系
第一节 药物的基本结构和药效的关系
药物作用过程的三个阶段
过程分类 发生过程 研究目的
药剂相
药物的释放
优化处方和 给药途径
药物动力学
药效相
吸收、分布和消除 药物-受体在靶 (代谢及排泄) 组织的相互作用
优化生物利用度
优化所需的 生物效应
化学工业出版社
化学工业出版社
P=CO/CW
化学工业出版社
第二章 药物的构效关系
第二节 药物的理化性质和药效的关系
二、药物的解离度对药效的影响 多数药物为弱酸、弱碱及其盐类,体液中部分解离,
以离子型和非离子型(分子型)同时存在。药物常以分子型 通过生物膜,在膜内的水介质中解离成离子型,再起作用。 因此药物需有适宜的解离度。
胃肠道各部分的pH不同,不同pKa药物在胃肠道各部分 的吸收情况也就有差异。
化学工业出版社
第二章 药物的构效关系
第一节 药物的基本结构和药效的关系
三、药物的特异结构与非特异结构 (一)结构非特异性药物
药物活性主要取决于药物分子的各种理化性质,与化学结 构的关系不大。临床应用的非特异性药物较少,主要有全身吸 入麻醉药,酚类和长链季铵盐的杀菌药以及巴比妥的催眠药等。 (二)结构特异性药物
药物化学构效关系(第二版尤启冬主编)

药物化学构效关系(第二版尤启冬主编)主要药物的构效关系应用抗肿瘤作用机理:1、药物在体内能形成缺电子活泼中间体(碳正离子)或其他具有活泼的亲电性基团的化合物,进而与肿瘤细胞的生物大分子(DNA,RNA,酶)中富电子基团(氨基,巯基,羟基等)发生共价结合,使其丧失活性,致肿瘤细胞死亡。
2、属细胞毒类药物,在抑制和毒害增生活跃的肿瘤细胞的同时,对其它增生较快的细胞产生抑制。
如骨髓细胞、肠上皮细胞、毛发细胞和生殖细胞等。
副作用大:影响造血功能和机体免疫功能,恶心、呕吐、骨髓抑制、脱发等。
氮芥类药物脂肪氮芥:氮原子的碱性比较强,在游离状态和生理PH(7.4)时,易和β位的氯原子作用生成高度活泼的亚乙基亚胺离子,为亲电性的强烷化剂,极易与细胞成分的,亲核中心发生烷基化反应。
脂肪族氮芥:烷化历程是双分子亲核取代反应(SN2),反应速率取决于烷化剂和亲核中心的浓度。
脂肪氮芥属强烷化剂,对肿瘤细胞的杀伤能力也较大,抗肿瘤谱较广;但选择性比较差,毒性也较大。
芳香族氮芥:氮原子与苯环共轭,减弱了碱性,碳正离子中间体,单分子的亲核取代反应。
氮芥类药物及大多数烷化剂主要是通过和,DNA上鸟嘌呤或胞嘧啶碱基发生烷基化,产生DNA链内、链间交联或DNA蛋白质交联而抑制,DNA的合成,阻止细胞分裂。
β-内酰胺类抗生素的化学结构特点:1分子内有一个四元的β-内酰胺环,除了单环β-内酰胺外,该四元环通过N原子和邻近的第三碳原子与另一个五元环或六元环相稠合。
2除单环β-内酰胺外,与β-内酰胺环稠合的环上都有一个羧基。
3所有β-内酰胺类抗生素的β-内酰胺环羰基α-碳都有一个酰胺基侧链。
4β-内酰胺环为一个平面结构,但两稠环不共平面β-内酰胺类药物可抑制粘肽转肽酶的活性和青霉素结合蛋白青霉素构效关系(1)6位的侧链酰胺基团决定其抗菌谱。
改变其极性,使之易于透过细胞膜可以扩大抗菌谱。
例如,在芳环乙酰氨基的α位上引入-NH2、-COOH、和-SO3H等亲水性基团,可以扩大抗菌谱,增强亲水性有利于对革兰阴性菌的抑制作用并能增强对青霉素结合蛋白的亲和力。
药物的构效关系

药物的构效关系药物的构效关系(Structure-activity relationship, SAR)是指药物的结构与其生物活性之间的关系。
通过研究不同化合物的结构特征和生物活性数据,可以揭示药物分子的作用机制,指导药物设计和优化,提高研发效率和成功率。
药物的构效关系研究对于药物化学、药理学和药代动力学等领域都有重要的意义。
以下是一些常见的构效关系的参考内容:1. 功能团对药效的影响:研究表明,药物分子中的特定功能团如羟基、酰胺、酯等,可以影响药物的生物活性。
例如,对于抗菌药物,羟基和酰胺基团通常与细菌靶标结合,从而发挥药效。
2. 结构类似性对药效的影响:药物分子的结构类似性对于药效也有重要的影响。
通常来说,结构相似的化合物可能具有相似的生物活性。
因此,通过对已知药物结构进行改良和优化,可以获得具有更高活性和选择性的新化合物。
3. 空间构型对药效的影响:药物分子的空间结构对于其与靶标的相互作用和选择性也起着重要作用。
例如,药物分子的立体异构体可能具有不同的生物活性。
研究不同空间构型的药效差异,有助于设计和优化具有更好活性和选择性的药物。
4. 电子结构对药效的影响:电子结构指的是药物分子中原子和键的电荷分布和云密度。
电子结构的差异可以影响药物分子与靶标的相互作用和药效。
例如,芳香环的电子密度与药物的溶解度、生物利用度和靶标的亲和性有关。
5. 氢键和离子键对药效的影响:氢键和离子键是药物分子与靶标相互作用的常见方式。
氢键的强度和方向性可以影响分子的亲和性和选择性。
离子键的形成可以改变药物分子的溶解度和稳定性。
6. 毒性与构效关系:药物的构效关系研究中还要考虑药物的毒性和副作用。
通过研究药物结构与毒性之间的关系,可以优化药物的安全性和耐受性,减少不良反应。
总的来说,药物的构效关系研究可以从多个角度考察药物分子的结构与生物活性之间的关系。
通过深入理解药物分子的作用机制,可以为药物设计和优化提供重要的理论指导。
药学综合考研之药物化学构效关系总结

药学综合考研之药物化学构效关系总结一、概述药物化学构效关系,即药物化学结构与生物活性之间的关系,是药学领域的重要研究方向之一。
在药学综合考研中,药物化学构效关系的学习和理解对于理解药物作用机制、药物设计与优化、新药研发等方面具有至关重要的意义。
药物化学构效关系研究主要关注药物分子结构与其生物活性之间的相互影响和关联。
通过系统研究药物化学结构的变化如何影响其生物活性,我们可以更好地理解药物作用的本质,为新药的设计和研发提供理论基础和实践指导。
药物化学构效关系不仅涉及到化学结构的知识,还需要深入理解生物学、生理学、病理学等领域的知识,是一个多学科交叉的领域。
随着现代科学技术的发展,尤其是计算机技术和生物技术的不断进步,药物化学构效关系的研究方法也在不断发展和完善。
从传统的合成、提取、筛选等实验方法,到现代的计算机模拟、大数据分析等高科技手段,药物化学构效关系的研究正在逐步深入。
对药物化学构效关系的考研复习者来说,不仅需要掌握基础的理论知识,还需要具备跨学科的综合能力,以适应这个领域的研究和发展。
药物化学构效关系是药学研究的重要基础,对于指导新药设计、优化药物作用机制等方面具有重要意义。
本文旨在对药学综合考研中的药物化学构效关系进行总结,以期为考研学生提供系统的学习资料和复习指导。
1. 简述药物化学构效关系的重要性。
药物化学构效关系,作为药物设计与研发领域中的核心原理,具有极其重要的地位。
其重要性主要体现在以下几个方面:药物化学构效关系是药物研发的基础。
药物的疗效与其化学结构之间有着密切的联系,通过对药物分子结构的深入研究,可以预测和优化药物的生物活性,从而有针对性地设计合成新药物。
构效关系研究有助于提高药物研发的效率。
随着现代医药产业的飞速发展,药物研发已经进入了一个竞争激烈的时代,如何快速、高效地发现和优化具有优良药效的药物成为了一个重要的挑战。
而药物化学构效关系的研究,可以指导科研人员快速筛选出具有潜力的药物分子,从而大大提高药物研发的效率。
药物化学药物的化学结构与药效的关系

CH3
利多卡因
达克罗宁
普鲁卡因
H N
H
δ
CO
Oδ
CH2CH2
C 2H 5 H
N
C 2H5
V
V
V
D
E
O
C 2H5
N O
CO O
CH2CH2
N C 2H5
无局麻作用
O
O
N .HCl
H2N
普鲁卡因的局麻作用似与分子极化有平行关系:
◆供e基甲氧基、乙氧基、二甲氨基取代-NH2, ED50减小 ◆吸e基硝基取代-NH2,ED50增大 ◆在苯环和碳基间嵌入乙撑基, 共轭效应被阻, ED50增大 ◆在苯环和碳基间嵌入乙烯基, 共轭效应不变, ED50不变
N-甲 酰 溶 肉 瘤 素
H
ClCH2CH2
N
Np O
C lC H 2C H 2
N
HO
尿嘧啶氮芥
ClCH2CH2
O
环磷酰胺
二、结构改造
结构变化带来新的物理性质,也改 变了分子化学反应性,可导致药物在细 胞与组织中分布的改变,进而改变对酶 及受体作用部位的结合,改变对这些部 位的反应速率及排泄方式。
四价
=C= =N+= =P+= =As+= =Sb+=
环 内 等 价 -CH =CH - -S- -O - -NH -
a. 一 价 原 子 或 基 团 的 取 代
H2N
S O2NHCONHC4H9 丁 磺 酰 脲
H3C
S O2NHCONHC4H9 甲 磺 丁 脲
氯磺丁脲
Cl
S O2NHCONHC4H9
延长半衰期
减低毒性
b. 二 价 原 子 或 基 团 的 交 换
药物化学构效关系(Pharmacochemical structure-activity relationship)

药物化学构效关系(Pharmacochemical structure-activityrelationship)barbituratesBarbiturates are structural nonspecific drugs.Structure nonspecific drugs: the biological activity of the drug is not related to the chemical structure of the drug.Structural specific drugs: the effects of drugs depend on the specific chemical structure and spatial alignment of drug molecules.The function of barbiturates and the fast and slow time of the drug are closely related to the dissociation constant, PKa, and the distribution coefficient of lipid water.Dissociation constant: the drug ACTS as an ion in the form of molecules through the biofilm.Oil and water distribution coefficient: the drug can be transported both in body fluids and through the blood-brain barrier.The category 5 is active, there are two substituents on the total number of carbon atoms when two substituent between 4 to 8, the distribution coefficient is moderate, active best. When the total number of carbon atoms in more than eight, effect is strong, easy to produce convulsion. The structure of the N atoms on the methyl on the imide replaced can reduce acidity andincrease the fat soluble, work fast. Use of C - 2 O S alternative. Increase lipid solubility and easy through the blood brain barrier, it works fast.The metabolism of barbiturates is associated with the metabolic time of the drug.2. Benzodiazepines (diazepam)A ring is necessary for the activity.B ring can be replaced by other aromatic rings, and it retains its activity.One is generally n-ch3. - CH3 can be removed in metabolism, but retains its activity1.2 bits can be added to the heterocyclic ring (triazolam: increased stability and increased activity)Three of the H atoms can be reduced slightly by -oh, but very low toxicity.4, 5 for the double bond to be saturated. The activity decreases, and incorporation into oxazole to increase sedation and antidepressant effect.The 2 bits of 5 - phenyl are introduced into the electron base (F, Cl, Br...). Increased activity.7 bits were introduced into the electron base, and the activity significantly enhanced NO2 > CF3 > Br > Br > ClThree. Aryl propionic acid (ibuprofen)One or more carbon atoms are spaced between a benzene ring and a carboxyl group.The carboxyl group of a is another - CH3, which limits the free rotation of carboxyl group, keeping it fit for the binding of the receptor or enzyme to increase the anti - inflammatory analgesic effect.Because of carboxyl a bit - the introduction of CH3, make its produce the asymmetric center, is usually a S - configuration activity is higher than R - configuration, can mutual transformation between chiral isomers in the body, is normally inactive R - S - configuration into active configuration.Another hydrophobic base, cyclohexyl and allyl oxide, can be introduced in the aromatic ring.The interposition of benzene ring carboxyl group is introduced into a suction electron base such as F.C l, etc.Anti-inflammation activity is good.Iv. The constitutive effect of cholinergic receptor agonists (choline choline)Positively charged amino 1, quarter. (1) the nitrogen atom is must be active. If the As, Se replace activity. (2) methyl instead As well on the nitrogen atoms, if replace with larger groups such As ethyl activity decreased, if three ethyl into choline active resistance.2 acetoxyl group. (1) when the acetyl or butyl acyl replace activity, (2) on the acetyl hydrogen is replaced by aromatic ring or larger groups to fight choline activity. (3) the rapid hydrolysis of ester base for acetylcholine effect time short and unstable factors, so it not easy to replace acetyl groups can increase the stability of the hydrolysis and action time. Such as acetyl ammonia formyl replaced, because in the nitrogen lone pair electrons, carbonyl carbon is electrophilic acetyl is low, not easy to hydrolysis.3, ethylidene bridge. (1) ethylidene has key influence on activity of the length of the bridge, two carbon for the best. With the extension of carbon chain, the activity is gradually reduced. (2) the quaternary ammonia nitrogen atom of a bit have replaced by methyl, overall activity decreased, but lack of the sample N greater than M. (season 3 of ammonia nitrogen atom B have replaced by methyl, can prevent the effect of cholinesterase, extend the action time, effect on the acetylcholine M sample, sample N effect greatly abate, be selective M receptor agonist.The constitutive effect of adrenal receptor agonistThe basic structure of phenylethylamine is the activity and the elongation or shortening of the carbon chain reduces the effect.Phenol hydroxyl groups in benzene ring significantly enhanced the role of pseudo adrenalin, especially in the 3 or 4 digits, but the effect time was brief.The peripheral function was retained and the central excitation decreased.N substituent on the relative strength of a and B receptor effect have a significant impact. The alternative to base from methyl tert-butyl, a receptor effect weakened. B receptor effect increase, and the selective increase of B2 receptor.B - carbon usually has hydroxyl group. Its absolute configuration is R - configurational.The constitutive relationship of anesthetics.The adjacent position gives the electron base substitution, which is beneficial to the formation of the amphoteric ion and the activity is enhanced. If there is an electron absorption, the activity decreases.It can be aromatic ring, aromatic heterocyclic, the modification of this part has a great influence on the activity, the active sequence is the benzene ring > pyrrol > furanIt's usually best to have 2 or 3 carbonsThere are secondary amine, tertiary amine or pyrrolidine, piperidine, etc., and tertiary amine is the most common.Insert - CH2, between benzene ring and carboxyl - O - groups, such as damage to the formation of zwitterionic. Activity. If connected to conjugate groups can be formed, such as - CH2 =CH2 -. Activity can remain the same. Amide also can form the amphoteric ion.In this part, the stability of the drug is determined, and the time sequence of carbonyl + -o->... + - S - >... + - NH - >... + - CH2 -; The order of strength: -s-> - o->-ch2 - > - NHThe constitutive relationship of beta blockers (figure of figure).Phenylethanolamine and aromatic propanolamineIt can be benzene, naphthalene, heterocyclic, dense ring, and fatty unsaturated heterocyclic. It can be substituted for methyl, chlorine, nitro, methoxide, etc., with the best activity in 2, 4 and 2, 3 and 6 positions.When substituted with S, CH2, -nch3, the activity decreases.The activity of s-configurational isomers increased, and the activity of r-configurational isomers decreased or disappeared.The activity of r-configurational isomers increased, and the activity of s-configurational isomers decreased or disappearedTertiary butyl and isopropyl instead at best, the hydrogen atoms on the methyl for less than 3 or N - N double replaced, activity decreased. Due to the structure of great B receptor blockers, so its solubility also have bigger difference, which related to its side effects and the location of the positionto eliminate. Pro fatty liver metabolism - rate faster. The hydrophilic - renal eliminationStructure-effect relationship of dihydropyridine drugs (nifedipine)The activity of dihydropyridine ring is necessary for the activity. If the activity of pyridine ring or hexahydropyridine ring is lost, the NH in the ring is not replaced, and the activity remains the best.2, 6 substituents are low - grade alkanesThe 3, 5 carboxylic acid lipid is necessary for the activity, and if it becomes acetyl or cyano, the activity decreases, and if it becomes nitro, the calcium channel can be activated.The 3, 5 carboxylic acid is different, the C4 is chiral center, the size of the ester group has little influence on the activity, but the asymmetric fat affects the part.C4 is stereoscopic for chiral carbonThe relationship between the four substituents and the activity: phenyl or substituted phenyl > ring alkyl > methyl > HIn the adjacent Spaces of the benzene ring, there is a better activity of the electron base substitutionThe constitutive effect of H2 receptor antagonist (ranitidine).The structure of the H2 receptor antagonist consists of three parts: the basic aromatic ring structure, and the four atomic chain and the polar group.Alkaline aromatic heterocyclic and alkaline groups substituted aromatic heterocyclic are necessary to active, imidazole ring as the proton transfer mechanism, by different thiazole, pbo replacement after alkaline, activity also decreases. By lipophilic aromatic heterocyclic (benzene) replaced activity. Was replaced by the alkaline groups, furan, thiazole, after displacement is good H2 receptor antagonist.Planar polar groups. It is the structure of guanidine and amidine. In the physiological PH condition, the polar groups with low ionization degree can form more than one hydrogen bond with the receptor forming one of the hydrogen bonds.The chain length is 4 atoms long. The chain length is related to antagonism. Free rotation and restriction make its activity fall, and the chain of intermediate connection can be replaced by the steel ring.The constitutive effect of penicillin(1) six side chain of amide groups mainly decided its antibacterial spectrum, change its polarity, makes it easy to through the cell membrane, can enlarge its antibacterial spectrum. For example, in a place of aromatic ring ethyl amide group introduced polarity - NH2, - COOH, - SO2 hydrophilic groups, such as antibacterial spectrum. Increase thehydrophilicity. Is advantageous to the bacteriostatic action of bacterium of negative of glen, and can enhance the affinity of binding protein to penicillin.The position of the three-dimensional resistance group is introduced in the appropriate position of the molecule.As in the side chain is introduced into the three-dimensional steric larger groups and introduced in six methoxyl and formamide, because of its three-dimensional steric effect reduces the passivation enzyme structure adaptability, protect B - lactam ring from B - lactamase attack. So get resistance to antibiotics of enzyme.Penicillin thiazole ring of carboxyl is basic reactive group, although can be replaced by sulfur acid and amide activity. If the reduction of carboxyl of alcohol, antimicrobial activity was lost. For fat carboxyl made available before the medicine principle, improve the oral absorption and pharmacokinetic properties.The configuration of three chiral carbons in penicillium is essential for its activity, but the two methyl groups on the thiazole ring of penicillin are not necessary for the activity.The constitutive effect of semi-synthetic cephalosporinIn seven side chain introduction of lipophilic groups, such as phenyl, dilute base, thiophene and nitrogen heterocyclic. Can be enhanced antibacterial activity, expand its antibacterial spectrum. Change 3 substituent at the same time, theintroduction of heterocyclic, can improve the oral absorption distribution also can expand its antibacterial spectrum.A position of 7 amides was introduced into the hydrophilic - SO3H, -nh2, -cooh, isopolar group. The antibacterial spectrum could be expanded to change 3 substituents, and the introduction of -cl, CH3, and nitrogen-containing heterocyclic could enhance oral absorption and expand the antibacterial spectrum.With 7 B as the side chain of ammonia oxime "conveniently" can improve the stability of B - lactamase, expand the antibacterial spectrum. This is mainly due to the introduction of oxime, methoxy occupy the position near the B - lactam ring. Prevents the enzyme molecules of B - lactam ring close to, so that drug resistant enzymes, the nature of broad spectrum.5 S with biological electronic body O, CH2, etc, respectively called cephalosporins cephalosporin and carbon, oxygen activity is not reduced. 3 substituent of transformation, such as acetoxyl group can be methyl, chloride to replace you can expand the antibacterial spectrum and change the absorption of drug in the body distribution and pharmacokinetic properties of the permeability of drugs.2-3 double - bond shift inactivation. 2 - COOH can be made into premedication to increase oral absorption.The constitutive effect of quinolone drugs.A ring (pyridine keto acid) is the basic therapeutic effectsof antibacterial action necessary to group. One of the three carboxyl and four ketone group is with its target enzyme binding sites, is A crucial part of the antimicrobial activity.The B ring part can be changed greatly, and can be incorporated into the benzene ring, pyridine ring and pyrimidine ring.The substitution of the N1 position substituents for the antibacterial activity was greater, and if the hydroxyl group was replaced by the hydroxyl group, the activity was better than that of ethyl, fluoroethyl and cyclopropyl.Two substituents were introduced to reduce or disappear the substituents, possibly due to space resistance blocking the binding of receptors.The carboxyl group and the 4-digit ketones are indispensable part of antibacterial activity.Five amino substituted activity increased. Other groups replaced the activity.Six substituent effect on the activity is very important. Active replace order: F > Cl > > CN = CH2 > = h. introduced F than H antibacterial activity of 30 times. Because the F generation and DNA spiral enzyme enhanced affinity 2-17 times. The penetration enhancement 1-70 times of bacterial cell walls.The order of 7 substituents to be substituted for base enhancement is piperazine > dimethylamine > methylamine > hydrogen, which is the best substitute for piperazine.The 8 bits were replaced by F, Cl and -och3, but they were also enhanced by the substitution of F instead of the phototoxicity. If 1, 8 rings, the photochemical isomers produced by the other were also significantly different.。
构效关系的名词解释

构效关系的名词解释
构效关系的名词解释是:药物或其他生理活性物质的化学结构与其生理活性之间的关系,是药物化学的主要研究内容之一。
狭义的构效关系研究的对象是药物,广义的构效关系研究的对象则是一切具有生理活性的化学物质,包括药物、农药、化学毒剂等。
最早期的构效关系研究以直观的方式定性推测生理活性物质结构与活性的关系,进而推测靶酶活性位点的结构和设计新的活性物质结构,随着信息技术的发展,以计算机为辅助工具的定量构效关系成为构效关系研究的主要方向,定量构效关系也成为合理药物设计的重要方法之一。
药物的构效关系:
例如拟胆碱药的化学结构与乙酰胆碱相似,都有季胺或叔胺基团,都能与胆碱受体结合,形成具有活性的复合物,因而表现出相似的作用。
又如,磺胺药与对氨基苯甲酸化学结构相似,因而能与对氨基苯甲酸竞争二氢叶酸合成酶而影响细菌叶酸的代谢。
在具有基本结构的任何一类药物中,药理作用的类型是由它们的基本结构决定的,而它们的药理作用的相对强度,是由基本结构上各个取代基团的性质决定的,如磺胺类药物。
但也有化学结构相似而作用相拮抗的情况,如磺胺与对氨基苯甲酸、氨丙啉与硫胺等。
同时,也有化学结构不同而药理作用相似的情况,如麻黄碱与茶碱。
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表本人。
本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。
如发现本站有涉嫌抄袭侵权/违法违规的内容请联系客服!。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
苯乙醇胺类和芳氧丙醇胺类
可以是苯,萘,杂环,稠环和脂肪性的不饱和杂环.可以有甲基,氯,硝基,甲氧基等取代基,在2,4和2,3,6位取代时活性最佳.
用S,-CH2,-NCH3取代时,活性降低.
S-构型异构体活性增加,R-构型异构体活性降低或消失.
R-构型异构体活性增加,S-构型异构体活性降低或消失
B-碳上通常连有羟基.其绝对构型以R-构型为活性体.
局麻药的构效关系.(图自己想)
邻对位给电子基取代,有利于两性离子的形成,活性增强.若有吸电子存在则活性下降.
可以为芳环,芳杂环,此部分的修饰对活性的影响较大,活性顺序为苯环>吡咯>噻吩>呋喃
通常以2-3个碳原子为最好
有仲胺,叔胺或吡咯烷,哌啶.吗啉等,以叔胺最为常见.
青霉烷酸分子中三个手性碳的构型对其活性至关重要.但青霉素的噻唑环上的两个甲基不是活性的必要因素.
半合成头孢菌素的构效关系
在7位侧链引入亲脂性的基团,如苯基,环稀基,噻吩和含氮的杂环.可增强抗菌活性,扩大其抗菌谱.同时改变3位取代基,引入杂环,可改进口服吸收分布也可扩大其抗菌谱.
在7位酰胺的a位引入亲水性的-SO3H,-NH2,-COOH,等极性基团.可扩大抗菌谱同时改变3位取代基,引入-Cl,CH3,和含氮的杂环,可增强口服吸收扩大抗菌谱.
油水分配系数:药物既可以在体液中转运,又可以透过血脑屏障到达作用部位.
该类药物5位上有两个取代基才有活性,当两个取代基的碳原子总数在4到8之间时,分配系数适中,活性最好.当碳原子总数超过8时,产生作用过强,易产生惊厥作用.结构中酰亚胺上的N原子上有甲基取代时可降低酸性和增加脂溶性,起效快.将C-2位的O用S替代时.脂溶性增加,易透过血脑屏障,起效快.
2,6位取代基为则活性降低,若变为硝基则激活钙通道.
3,5位羧酸脂不同,C4为手性中心,酯基的大小对活性影响不大,但不对称脂则影响作用部位.
C4若为手性碳,具有立体选择性
4位取代基与活性的关系:苯基或取代苯基>环烷基>甲基>H
苯环上邻间位上有吸电子基取代活性较佳,对位取代时活性降低
巴比妥类药物
巴比妥类药物属于结构非特异性药物.
结构非特异性药物:药物的生物活性与药物的化学结构关系不大,与理化性质有关.
结构特异性药:药物的作用依赖于药物分子的特异化学结构及空间相互排列.
巴比妥类药物的作用强弱和起效时间的快慢与药物的解离常数,PKa,脂水分配系数有密切关系.
解离常数:药物以分子的形式透过生物膜,以离子的形式产生作用.
平面极性基团.是具有胍,脒基样的结构.在生理PH条件下,电离程度低的极性基团作为脒脲基团与受体形成一个以上的氢键保持活性.
易曲饶的链或芳杂环:链长为4个原子.链长与其拮抗性有关.自由旋转受限使其活性下降,中间连接的链可被钢性环所取代.
青霉素的构效关系
(1)6位侧链的酰胺基团主要决定其抗菌谱,改变其极性,使其易于透过细胞膜,可扩大其抗菌谱.例如,在芳环乙酰胺基的a位引入极性-NH2,-COOH,-SO2等亲水性基团,扩大抗菌谱.增加亲水性.有利于对格兰阴性菌的抑菌作用,并能增强对青霉素结合蛋白的亲和力.
在苯环和羧基之间插入-CH2,-O-等基团,破坏了两性离子的形成.活性降低.若连入可以形成共轭的基团,如-CH2=CH2-等.活性可保持不变.酰胺也可形成两性离子.
此部分决定药物的稳定性,按作用时间顺序羰基+-O->....+-S->....+-NH->....+-CH2-;按作用强度顺序:-S->-O->-CH2->-NH-
3,亚乙基桥.(1)亚乙基桥的长度对活性有关键影响,两个碳为最好.随着碳链的延长,活性逐渐降低.(2)季氨氮原子的a位有甲基取代,整体活性降低,但N样作用大于M样作用.(3季氨氮原子的B位有甲基取代,可阻止胆碱酯酶的作用,延长作用时间,M 样作用于乙酰胆碱相当,N样作用大大减弱,成为选择性的M受体激动剂.
巴比妥类药物的体内代谢过程与药物的代谢时间有关.
二.苯二氮卓类药物(地西泮)
A环为活性所必须.B环可以被其他芳杂环取代,仍保留其活性.
1位一般为N-CH3.-CH3可在代谢中脱掉,但仍保留其活性
1.2位可骈入杂环(三唑仑:稳定性增加活性增加)
3位一个H原子可被-OH取代活性稍微降低,但毒性很低.
2位引入取代基活性减弱或消失,可能是由于空间位阻阻止了与受体的结合.
3位的羧基和4位的酮基是抗菌活性中不可缺少的部分.被其他基团取代活性消失,与铁铝钙络合产生副作用.
5位氨基取代活性增加.其他基团取代活性均降低.
6位取代基对活性的影响很重要.活性取代顺序位:F>Cl>CN>=CH2>=H.引入F可比H的抗菌活性增强30倍.因为F代物与DNA螺旋酶亲和力增强2-17倍.对细菌细胞壁的穿透力增强1-70倍.
以叔丁基和异丙基取代时最好,甲基上的氢原子数小于3或N-N双取代时,活性降低.由于B受体阻滞剂的结构组成自由度很大,所以其溶解度也有较大差异,这与其副作用和体位消除的位置有关.亲脂性-肝代谢-速率较快.亲水性-肾消除
二氢吡啶类药物的构效关系(硝苯地平)
二氢吡啶环为活性所必须.若变为吡啶环或六氢吡啶环则活性消失,环上的NH不被取代时,活性保持最佳.
4,5为双键被饱和.活性降低,并入恶唑环增加镇静和抗抑郁作用.
5位-苯基的2位引入吸电子基(F,Cl,Br.....)活性增强.
7位引入吸电子基,活性明显增强NO2>CF3>Br>Cl
三.芳基丙酸类药物(布洛芬)
苯环与羧基之间间距一个或一个以上的碳原子.
羧基的a位又一个-CH3,限制了羧基的自由旋转,使其保持在适合与受体或酶结合的构像,以增加抗炎镇痛作用.
带有7B为顺势”氨噻肟”的侧链可提高对B-内酰胺酶的稳定性,扩大抗菌谱.这主要是由于引入肟后,甲氧基占据了靠近B-内酰胺环的位置.阻止了酶分子对B-内酰胺环的靠近,因而使药物有耐酶,广谱的性质.
5位的S用生物电子等排体O,CH2等取代,分别称为氧头孢菌素和碳头孢菌素,活性不降低.3位取代基的改造,如乙酰氧基可被甲基,氯等取代可扩大抗菌谱并且改变药物在体内的吸收分布和药物的渗透性的药物代谢动力学性质.
7位引入取代基增强活性的顺序为:哌嗪基>二甲氨基>甲基>卤素>氢.其中以哌嗪基取代活性最好.
8位以F,Cl,-OCH3,等取代活性增强,但以F取代光毒性也增强.若1,8位间成环,产生的光化学异构体之间也有较大差异.
在分子中的适当位置引入立体位阻基团.如在侧链引入立体位阻较大的基团和在6位引入甲氧基和甲酰胺基,因其立体位阻的效应降低了钝化酶的结构适应性,保护B-内酰胺环不被B-内酰胺酶进攻.因而得到耐酶的抗生素.
青霉素的噻唑环上的羧基是基本活性基团,虽可被硫代酸和酰胺取代但活性降低.若羧基被还原为醇则失去抗菌活性.对于羧基可利用前药原理制成脂,改进口服吸收和药物代谢动力学性质.
H2受体拮抗剂的构效关系(雷尼替丁).
H2受体拮抗剂的结构有三部分组成:碱性的芳杂环结构,易曲饶的四原子链和平面极性基团.
碱性的芳杂环和碱性的基团取代的芳杂环是活性所必须,咪唑环作为质子转移的机制,被异噻唑,恶唑置换后碱性下降,活性也随之降低.被亲脂性的芳杂环(苯环)取代时活性降低.被碱性基团取代,呋喃,噻唑置换后,是良好的H2受体拮抗剂.
由于羧基a位的-CH3的引入,使其产生了不对称中心,通常是S-构型的活性高于R-构型,在体内手性异构体之间可以相互转换,通常是无活性的R-构型转换为有活性的S-构型.
芳环上可以引入另一个疏水基如,环己基,烯丙氧基等.
在苯环羧基的间位引入一个吸电子基如F.Cl等.抗炎活性好.
四.胆碱受体激动剂(氯贝胆碱)的构效关系
肾上腺受体激动剂的构效关系
苯乙胺的基本结构是活性所必须,碳链的延长或缩短均使作用减弱.
苯环上的酚羟基可显著增强拟肾上腺素作用,尤其以3,4位最为明显,但作用时间短暂.
以其他环状结构取代苯环,外周作用仍被保留,中枢兴奋作用降低.
N上的取代基对a和B受体效应的相对强弱有显著影响.取代基从甲基到叔丁基,a受体效应减弱.B受体效应增强,且对B2受体的选择性提高.
1,季氨基.(1)带正电荷的氮原子是活性所必须.若以As,Se取代活性降低.(2)氮原子上以甲基取代为好,若以较大基团取代如乙基则活性降低,若为三个乙基则变为抗胆碱活性.
2,乙酰氧基.(1)当乙酰或丁酰基等取代时活性降低,(2)乙酰基上的氢被芳环或较大基团取代时变为抗胆碱活性.(3)酯基的快速水解是乙酰胆碱作用时间短暂和不稳定的因素,因此用不易水解的基团取代乙酰基可以增加稳定性和作用时间.如用氨甲酰基取代乙酰基,由于氮上孤对电子的参与,羰基碳的亲电性较乙酰基为低,不易水解.
2,3位的双键移位失活.2位-COOH可制成前药增加口服吸收.
喹诺酮类药物的构效关系.
A环(吡啶酮酸部分)是抗菌作用所必须的基本药效基团.其中3位的羧基和4位的酮基是与靶酶的结合位点,是抗菌活性必不可少的部分.
B环部分可做较大改变,可并入苯环.吡啶环和嘧啶环等.
N1位置取代基对抗菌活性的贡献较大,若由烃基,环烃基取代活性增加,尤以乙基,氟乙基和环丙基取代活性最佳.