A histochemical method for the demonstration of calcifying cartilage [PMIDJ3145130]
病理切片染色(Pathologicalsectionstaining)

病理切片染色(Pathological section staining)This routine pathological examination notice.1. the tissue specimens cut during operation should be fixed in fixed liquid immediately (operation room should be provided with specimen bottle and fixative). Usually 10% neutral marlin is used as fixative. The amount of fixative shall not be less than 5~10 times the volume of the specimen. The mouth of the specimen should be big enough to be removed after the specimen is fixed. The specimen is fixed to the bottle wall and the bottom of the bottle. The absorbent is made of absorbent cotton pad. The floating surface is covered with absorbent cotton. If it is an infectious specimen, it should be careful not to pollute the outside of the container.2. specimen containers, should be affixed with the patient's name or label for single chain. As a patient has several tissues at the same time, or the same tissue is taken out of different parts, different containers shall be packed and marked separately. Specimens of different patients should not be placed in the same container.Take 3. samples, pay attention to not use toothed forceps or clamp, do not squeeze, to avoid man-made deformation, affect the diagnosis. Don't open the specimen before delivery. Keep the prototype and submit it all. When it is necessary to open the case, it is better to invite the pathologist to the scene. Otherwise, the specimen should be described in detail before and after the pathological examination application. Check the organization should not be too small, so as not to affect the diagnosis.4. specimens such as whole organ or department, or larger, is not fixed, but should send pathology treatment (treatment field or long specimen, can refer to section third for testing).5. samples of various body fluids, punctures and cytology should be examined immediately. If not for inspection immediately, then the centrifugal precipitation will make uniform sediment smear from 2 to 3, in a timely manner into the fixed ether and 95% ethanol mixture or 95% ethanol, and then together with the fixed liquid, or in smear coated with glycerol. Vaginal discharge, nasopharynx or other secretions, puncture smear, should be made after the smear, then placed in the fixed liquid fixed, and then submit.6. when you submit a specimen, you should fill out the application form for pathological examination in detail. When the clinical department has special requirements, it should indicate or contact in the application form.To organize the dehydrated and paraffin.(1) matters needing attention in the work;1. the specimens are not adequately fixed and shall not enter the procedure of dewatering treatment.2. each l specimen shall be numbered in the course of the processing. Be careful to prevent the specimen from being mixed with each other. Small specimens are wrapped in a tissue paper to avoid dehydration.3. units with many specimens can be dehydrated in batches according to the nature, size and classification of specimens, so as to ensure the quality according to the characteristics of the tissues, and the dehydration and transparency of the conventional specimens should be carried out at room temperature.4., usually ethanol as tissue dehydrating agent, from low concentration to high concentration gradually and repeatedly replaced, tissue moisture content can be fully removed, transparent, wax dipping can be carried out smoothly, to ensure the quality of the slice is very important.5., tissue block in xylene time should not be too long, so that tissue transparent. If the transparency is not good for a long time, the reasons should be checked. If the dehydration is not enough, the dehydration should be repeated.6. often pay attention to the quality of reagents used to reduce the situation, according to the need for timely replacement of new liquid, the replacement of ethanol can be downgraded, repeated use. Reagents should be regularly filtered to keep clean and avoid mixing.7. the melting point of paraffin used for routine production should be about 56 DEG C. In warm regions or summers, paraffin with higher melting point is used, and in cold regions or winters, paraffin with lower melting point is better.8. stearic acid wax can be used as an intermediary agent betweenethanol and paraffin wax, and can make the tissue moderate hardness, easy to slice.9. ethanol, xylene and other flammable products, the use of 2m may not be open flame. The whole process of heating, dehydration and soaking of wax is to use a water temperature box or an open flame device, heating up in advance and no drying oven. Wax bath, and a special guardian.Storage is kept in a fire free room, bright and easy to take. The laboratory should be well ventilated,Fire extinguishers and sand boxes are available. Acetone, ethyl ether, chloroform, ethyl chloride, collodion and other inflammable and explosive goods should be handled according to the above principle.(two) general steps and timeSee table 31-2-l.Table 31-2-1 steps and time of dehydration, transparency and waxing of tissue blocks of different thicknessesThe processing time of different thickness tissue blocks was (min)2mm below2 ~ 4mm4 ~ 5mm80% ethanol45~6060~120120~24095% ethanol I45~6060~120120~24095% ethanol II45~6060~120120~240Anhydrous ethanol I 45~6060 ~ T20120~240Anhydrous ethanol II 45~6060~120120~240Xylene, I*, or15~30ThirtySixtyStearic acid wax (3:2) One hundred and twenty One hundred and eighty Two hundred and forty Xylene II or15~30ThirtySixtyStearic acid wax (2:3)SixtyNinetyOne hundred and twentyWax I**SixtySixty60~120WaxSixtyOne hundred and twenty120~240* when using stearic acid wax, no need for xylene transparent. * * conditional units can be dipped in wax to increase wax leaching effect and shorten time. Pressure gauges and safetybottles shall be used.(three) the use and maintenance of the automatic tissue processor1. automatic tissue processor has a wide range of products, its structure and performance vary, before use should be familiar with the instructions in the machine manual and precautions, according to the instructions in the use of procedures and procedures.2. procedures and times for organizational processing see table 31-2-2.Table 31-2-2 procedure and time of tissue processor processing biopsy tissue (min)Processor processing time (min)80% ethanol95% ethanol, I, II, IIIAnhydrous ethanol I, II, IIIXylene I, II *Wax I / or stearic acid - wax (3:2)Wax II or stearic acid - wax (2:3)Wax III / waxSixtyThe 90The 60The 6090 / 18090 / 12060 / 60* use stearic acid - wax without xylene.3. according to the actual working conditions, design the operation procedure, time and temperature of the dehydrating machine. After the machine is adjusted, do not change at will. The use of the process should always pay attention to the operation, the temperature and time control is accurate.4., the machine should be placed in dry, ventilated, smooth place, often do cleaning, according to the instructions to do the necessary maintenance. When there is a fault, please ask the maintenance personnel to repair it.By HE staining of paraffin sections.(1) matters needing attention in dyeing1., slice dewaxing should be thorough, when the room temperature is low, should pay attention to, in the section of xylene in a transparent shape, or into the ethanol, no white spots on the patch is better.2. special staining, histochemical reaction and immunohistochemical staining were performed to ensure the effect according to the requirements of each method. The tissue fixation for enzyme reaction should be strictly required. Where the organization with mercuric chloride were fixed, sliced by dewaxing, mercury removal treatment, can be dyed. The method is: after washing section 2min, immersed in 10min solution, 0.5% iodine washed, 0.5% sodium thiosulfate solution 5min, rinse thoroughly with water, after washing with distilled water after dyeing. During the process of mercury removal,Beware of slicing off.3., a variety of dyes, reagents are generally used chemically pure. The dyeing effect of various dyes is often different because of the different batch of production plants and batch numbers. Any new products should be dyed first. With the dye solution should be contained in the colored reagent bottle, bottle label should specify the name, content, preparation date, order is preserved in the dark in the cupboard, need to put the refrigerator. Where preparations are to be used, the dosage shall be appropriate.4., the time required for dyeing each step is often affectedby temperature, slice thickness and so on, which can be adjusted according to what is observed under the microscope.But for chemical reactions (especially enzymes), the time, temperature and other factors must be constant, and should be done on the photos.Should update the tinting dye significantly decreased, staining reaction is not normal, should check whether the failure or misuse of dye and reagent.5. during the dyeing process, do not dry the slices so as not to affect the cell morphology. After dyeing, the result of dyeing is checked by microscope, and the sample type, number and quantity are checked.(two) dyeing procedure1. xylene dewaxing (2 times) 5 to 10min.2. anhydrous ethanol, 95% ethanol, 80% ethanol, 70% ethanol, 1 to 2min, and then distilled water 3min (change several times).3. hematoxylin staining about 5rain (as the dye type and color change).4. washed; l% acid ethanol (hydrochloric acid, 1mI, 70% ethanol, 99m1), or 1% hydrochloric acid differentiation. Microscopic control.5. water immersion at least 15min to the nucleus is blue. Canalso be washed after a short time, soaked at 40 degrees of lukewarm water to make the nucleus blue. The microscope observation, if nuclear hyperchromatism or insufficient, should be differentiated or redye.6. after 1 to 2min, eosin 2, 95% times, absolute ethanol 2 times, each time about 1 ~ 2min.7. phenol xylene (1:3) 5min.8. xylene 2 times, each time 3 ~ 5min.9. with neutral gum or DPX to cover mounting, label.Probably the process is first drawn, dehydration, paraffin embedding, sectioning, staining, dewaxing, dehydration, transparent, and mounting process. The material should be determined according to the purpose of your experiment. There is a special instrument for dehydration and immersion. Other instruments are embedded in it. Then, the slicer is sliced. If you want to slice the area, it is recommended to embed it into several wax blocks. Ready to bake slice, while hot into xylene and graded alcohol dewaxing until the distilled water, then the section with hematoxylin dewaxing, hematoxylin staining according to the effect, usually 2-3 minutes, rinse water to remove excess hydrochloric acid alcohol acid hematoxylin, 15 seconds, alkalescent water blue, then dye eosin, then gradient alcohol dehydration, xylene transparent balata. Dewaxing and dehydration can not be public, need to dye the cylinder about 23.The staining cylinder is better in copper. "The tissue shrinks when it is perfused." do you mean the heart perfusion of the animal? Then you pay attention to the osmotic pressure and pH value of the PBS used in the lavage, right?.Experiment five pathological tissue sections were stainedExperiment five pathological tissue sections were stainedFirst, the purpose of the experimentAny section, without staining, can only see the outline of the cells and tissues under the microscope, which is far from meeting the purpose of observation and diagnosis. The dye staining is prepared into solution, the tissue sections were immersed in dye, after a certain time, the tissue and cell components into different colors have different refractive index, it is convenient to observe under microscope. Therefore, staining has important practical value in tissue morphology, pathology diagnosis and research work.Two 、 experimental principleDyeing is the process of combining the dye with the tissue cell. There are differences in the principle of using different staining methods. Hematoxylin and eosin staining for histological staining methods most commonly used. It is suitable for all kinds of components of tissue cells, which is convenient for comprehensive observation of tissue components, and can be applied to any fixed liquid material. Specimens stained with H.E are not susceptible to fading and can bepreserved for a long time.In the conventional hematoxylin and eosin staining, hematoxylin after oxidation into haematein acid dyes, and the formation of a blue color with a positive charge with haematein and aluminum, positively charged blue color and the negatively charged DNA acid by polar sorption of positive and negative charge to complete the combination the nuclei were stained blue and purple. Eosin is an acid red cytoplasm dye. Their staining may be accomplished by osmosis or diffusion, making the cytoplasm red.Three 、 main instruments and testing materialsA full set of HE staining reagent, dyeing cylinder (horizontal), spreading enamel disc, cylinder, glass, neutral gum, toothpicks, microscope.Four, experimental methods and stepsRoutine paraffin section H.E staining procedure(1) dewaxing to water;1. xylene I 5-15 minutes2. xylene II 5-15 minutes3. absolute ethanol 3 minutes4.95% alcohol for 3 minutes5.80% alcohol for 3 minutes6.70% alcohol for 3 minutes7. tap water washing(two) staining;1.Harris hematoxylin solution for 5-7 minutes2. tap water for 5 minutes3.1% hydrochloric acid solution differentiation 30 seconds4. tap water for 5 minutes5.1% ammonia returns blue for 10 seconds6. tap water for 15-20 minutes7.95% alcohol for 3 minutes8.1% Yi Hong alcohol solution 1-2 minutes(three) dehydration, transparency and sealingL.95% alcohol for 2 minutes2. anhydrous alcohol I 2 minutes3. anhydrous alcohol II 2 minutes4. xylene I 5 minutes5. xylene II 5 minutes6. neutral gum sealingResults: the nucleus was blue and the cytoplasm was red.Five, experimental notes1. any paraffin sections must be removed by xylene to be dyed. Paraffin sections require plates to be dried so that the tissue is firmly adhered to the slides. The quality of the tissue section dewaxing is related to the temperature and time of xylene, and the xylene should be replaced in a timely manner.2. H.E in the dyeing process, the key to success lies in its differentiation, if improper differentiation should part of the end to remove differentiation decolorization, or differentiation caused by lack of uneven dyeing, the complex also cannot get the contrasting colors, in addition to water washing time on the back of blue color and whether the organization has a certain the relationship between. Need to mention is that in addition to the success or failure of staining staining outside, tissue material too old or long-term fixed in formaldehyde organization, due to excessive acidification will affect the dyeing; or tissue fixation is improper, lack of fixed tissue autolysis were the fuzzy staining.3., after the Iraqi Red dyeing must be gradient alcohol dehydration, especially after the need for absolute ethanol, dehydration must be thorough, otherwise, affect the transparent.4., after dehydration, transparent by xylene in order to seal. Transparency should be paid attention to enough time to achieve good results. First, neutral gum can not drop too much or too little. Good sealing sections should be placed in the stalls on spools promptly placed in the incubator, 40-50 C baking 15 hours or so, is conducive to the preservation of the section.Six, the experimental results are dealt withEach member received 2-3 slices of good quality and described pathological changes.Seven, thinking1. the principle and significance of pathological tissue section staining2. what are the procedures of pathological tissue section dyeing, what problems should be paid attention to?。
P53和Ki-67在胃高级别上皮内瘤变、分化型早期胃癌组织中的诊断价值

· 23 ·
P53和 Ki一67在 胃高级别 上皮 内 瘤 变 、分化型早期 胃癌组织 中的诊 断价值
高福 平 ,魏 谨 ,马 平 ,薛 松 ,夏莉 花 ,孙 琼 ,李文 洁 ,赵 绮莲
(南京市高淳人 民医院病理科 ,江苏 南京 21 1300)
[Key words]P53;Ki-67;early gastric cancer;high,-grade intraepithelial neopasm;immunohistochemisitry
我 国 胃癌 年发 病 率 约 为 36/10万 ,年 死 亡 率 约 为 26/10万 ,位于我 国恶性肿瘤死亡率第 3位 。早期 胃癌 的 预后 良好 ,术后 5 a生 存 率 大 于 90% 。 内镜 下黏膜剥离术常被应用于早期 胃癌 的治疗 ,其能达到
作者简 介 :高福平 (1982一),男 ,硕士 ,副主任医师 ,主要 从事肿瘤病 理 诊 断工作 。E—mail:gaofuping2l000@qq.com
[摘要 ] 目的 探讨 P53和 Ki-67在 胃高级别 上皮 内瘤变 、分 化型早期 胃癌组 织 中的诊断价 值 。方 法 用 免疫组 化法检测 P53和 Ki-67在 34例正常 胃黏 膜 ,25例 胃低 级别上 皮 内瘤 变 ,34例 胃高 级别 上皮 内瘤 变 、 分 化型早期 胃癌组织 中的表达情况 。结 果 P53在 胃高级别上皮 内瘤变 、分化 型早 期 胃癌组 织 中阳性表达 率 为 82.35% ,明显 高 于正 常 胃黏膜 (0.oo%)及 胃低 级 别上 皮 内瘤 变 (8.0o%),差 异 均有 统 计 学意 义 (P均 <0.01)。Ki-67在 胃高级别上皮 内瘤变 、分化型早期 胃癌组 织 中阳性表 达率为 88.24% ,明显 高于正 常 胃黏膜 (8.82% )及 胃低级别上皮 内瘤变 (20.00% ),差 异均有统 计学 意义 (P均 <0.O1)。结论 P53和 Ki-67在 胃高级别上皮 内瘤变 、分化 型早期 胃癌组 织 中存在 高表 达 ,两者联 合检 测有 助于早 期 胃癌 的鉴 别 诊 断。 [关键 词 ]P53;Ki-67;早期 胃癌 ;胃高级别 上皮内瘤变 ;免疫组化 DOI:10.3969/j.issn.1673-5412.2018.01.008 [中图分类 号 ]R735.2;R730.4 [文 献标识码 ]A [文章编号 ]1673—5412(2o18)Ol一0023—03
非靶向代谢组学方法英语

非靶向代谢组学方法英语Non-targeted Metabolomics Methods in EnglishIntroductionNon-targeted metabolomics is an innovative approach in the field of metabolomics that aims to identify and quantify as many metabolites as possible in a given biological sample without any prior knowledge or bias towards specific metabolites. This method provides comprehensive insights into the global biochemical changes occurring in a biological system, such as a cell, tissue, or organism. In recent years, non-targeted metabolomics has gained immense popularity due to its ability to unravel intricate metabolic pathways and discover novel biomarkers for various diseases.Sample Collection and PreparationThe first step in non-targeted metabolomics is the collection and preparation of the biological sample. The choice of sample depends on the research question and can range from blood, urine, tissues, or even fecal samples. It is crucial to handle the samples with extreme care to avoid any degradation or contamination of metabolites. Sample preparation involves various techniques such as extraction, filtration, and derivatization, to enhance the stability and visibility of metabolites during subsequent analysis.Mass Spectrometry-Based AnalysisMass spectrometry (MS) is the key analytical technique used in non-targeted metabolomics. It detects and quantifies metabolites based on their mass-to-charge ratio (m/z) and abundance. Liquid chromatography-massspectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS) are commonly used platforms for metabolite analysis. LC-MS is suitable for hydrophilic compounds, while GC-MS is preferred for volatile and thermally stable metabolites.Data Acquisition and PreprocessingOnce the samples are analyzed using MS, the raw data obtained needs to be processed and converted into a format suitable for downstream analysis. This step involves data acquisition, which includes peak picking, alignment, and normalization. Peak picking identifies and quantifies metabolite peaks in the acquired spectra, while alignment corrects any potential retention time variations. Normalization ensures that all samples are comparably represented, eliminating any technical biases.Statistical Analysis and IdentificationStatistical analysis is a crucial step in non-targeted metabolomics, as it helps in identifying significant metabolites and detecting patterns within the dataset. Multivariate statistical techniques, such as principal component analysis (PCA) and partial least squares-discriminant analysis (PLS-DA), are commonly used to visualize and interpret the data. Additionally, metabolite identification is performed by matching the acquired mass spectra with metabolite databases, such as the Human Metabolome Database (HMDB) and the Kyoto Encyclopedia of Genes and Genomes (KEGG), using tools like MassBank, MetFrag, or Metlin.Metabolic Pathway AnalysisOne of the key strengths of non-targeted metabolomics is its ability to unravel complex metabolic pathways. Pathway analysis tools, such as MetaboAnalyst, MetaboMiner, and Ingenuity Pathway Analysis (IPA), are used to identify significantly altered pathways and discover potential biomarkers. These analyses provide crucial insights into the underlying biochemical mechanisms and aid in understanding the disease pathogenesis or physiological responses.Challenges and Future PerspectivesDespite its numerous advantages, non-targeted metabolomics faces several challenges. Metabolite identification remains a major bottleneck due to the limited coverage of metabolite databases and the lack of standardization in data reporting. Additionally, the high complexity and dynamic range of metabolomes make it difficult to detect low-abundance metabolites accurately. Nevertheless, advancements in analytical techniques, bioinformatics, and collaborative efforts are steadily overcoming these challenges and driving the field forward.In conclusion, non-targeted metabolomics plays a vital role in understanding the complex metabolic dynamics within biological systems. Through the use of advanced mass spectrometry techniques, data analysis tools, and metabolite identification strategies, this approach has the potential to uncover novel biomarkers and therapeutic targets for various diseases. With continued advancements, non-targeted metabolomics is poised to revolutionize personalized medicine and contribute significantly to the field of biomedical research.。
非酒精性脂肪性肝病代谢组学研究进展

机制尚未完全明确,1998 年Day 等[12]提出“二次打击”学说。 开。同时NAFLD 肝硬化患者与酒精性肝硬化患者也可有效区
随后Tilg 等[13 -14]提出“多重平行打击”理论,包括遗传因素、 分开(AUC =0. 83)。他们认为此方法可作为区分NAFLD 纤维
IR、氧化应激、脂毒性、慢性炎症、纤维化、免疫和肠道菌群等, 化程度及诊断的无创生物标志物,且可以显著减少对肝活检的
黄酯和13 - cisRA 呈正相关。他们在人类组织中首次检测到 验证;单不饱和TAG 的增加可能是NAFLD 和CHB 患者NASH
atRA 的活性代谢物4 - oxo - atRA,表明这种类维生素A 可能 的特异性标志物。
有助于人体类维生素A 的信号传导。肝脏维生素A 的稳态平 2. 3 代谢组学对NAFLD 药物作用与疗效研究的推动作用
录组学、蛋白质组学为代表的系统生物学技术提供了新的技术 展的新学科,代谢组学较为全面的展示了机体的代谢结果,为
与思路。区别于其他组学技术,以内源性小分子代谢物为研究 临床医学提供了新的技术和方法。
对象的代谢组学可以很好的揭示机体变化的最终代谢结果。因 2 非酒精性脂肪性肝病(NAFLD)
收 基 作DO稿 金 者I:日 项 简10期 目 介. 3:::912上 栾研6709)2海究雨/0j.中婷-is医1s(n1药.1-19大090006学1—;修-附)5回,属2女5日第6,.期七主20:人2要210民.2从00医4事-.院01慢42人7-性才1肝7培病养计的划基(础XX与20临19床- 通信作者:顼志兵,xzb6160@ 163. com
和遗传易感密切相关的代谢应激性肝损伤,包括非酒精性单纯 1 代谢组学概述
性肝脂肪变(NAFL)、非酒精性脂肪性肝炎(NASH)、肝硬化和 1. 1 代谢组学含义 代谢组学最初于1999 年由Nicholson
2020中国动态血压监测指南

摘要高血压是心脑血管疾病的重要危险因素。
动态血压监测已成为识别和诊断高血压、评估心脑血管疾病风险、评估降压疗效、指导个体化降压治疗不可或缺的检测手段。
本指南对2015年发表的《动态血压监测临床应用专家共识》进行了更新,详细介绍了动态血压计的选择与监测方法、动态血压监测的结果判定与临床应用、动态血压监测的适应证、特殊人群动态血压监测、社区动态血压监测应用以及动态血压监测临床应用展望,旨在指导临床实践中动态血压监测的应用。
关键词 动态血压监测;血压管理;指南;高血压2020 Chinese Hypertension League Guidelines on Ambulatory Blood Pressure MonitoringWriting Group of the 2020 Chinese Hypertension League Guidelines on Ambulatory Blood Pressure Monitoring.Corresponding Author: WANG Jiguang, Email: jiguangwang@2020中国动态血压监测指南中国高血压联盟《动态血压监测指南》委员会指南与共识AbstractHypertension is an important risk factor for cardiovascular and cerebrovascular diseases. Ambulatory blood pressure monitoring (ABPM) has become an indispensable technique for the detection of hypertension, risk assessment of cardiovascular and cerebrovascular diseases, therapeutic monitoring, and guidance of the individualized treatment. Based on the “2015 expert consensus on the clinical use of ambulatory blood pressure monitoring”, the current guideline updates recommendations on the major issues of ABPM, such as the device requirements and methodology, interpretation of the reported results, clinical indications, application on special populations, the utility in the community and future perspectives. It aims to guide the clinical application practice of ABPM in China.Key words ambulatory blood pressure monitoring; blood pressure management; guideline; hypertension(Chinese Circulation Journal, 2021, 36: 313.)高血压是心脑血管疾病的重要危险因素,与心脑血管疾病发病和死亡密切相关[1-3]。
超分子溶剂萃取

第42 卷第 5 期2023 年5 月Vol.42 No.5559~567分析测试学报FENXI CESHI XUEBAO(Journal of Instrumental Analysis)超分子溶剂萃取/超高效液相色谱-串联质谱法测定血浆中他克莫司含量谢以清1,2,吕悦广2,孟宪双2,雷海民1*,马强2*(1.北京中医药大学中药学院,北京102488;2.中国检验检疫科学研究院,北京100176)摘要:该文建立了血浆中免疫抑制剂他克莫司(TAC)的超分子溶剂(SUPRAS)萃取/超高效液相色谱-串联质谱分析方法。
通过单因素实验结合响应面设计对超分子溶剂组成、用量及涡旋萃取时间等关键因素进行优化后,血浆样本以正戊醇、四氢呋喃和水形成的超分子溶剂进行高效萃取。
萃取液经Waters ACQUITY UPLC BEH C18(50 mm × 2.1 mm,1.7 μm)色谱柱分离后,在电喷雾质谱正离子模式下,以多反应监测(MRM)模式对他克莫司进行测定,内标法定量。
结果表明,他克莫司在0.5 ~ 30 ng/mL质量浓度范围内的线性关系良好,相关系数(r)为0.998 6;方法检出限和定量下限分别为0.1、0.5 ng/mL;在低、中、高3个加标水平下,平均回收率(n = 3)为91.9% ~ 99.9%,相对标准偏差(RSD)为1.7% ~ 5.7%。
所建立的方法快速、灵敏、稳定,适用于血浆中他克莫司的准确测定。
关键词:他克莫司;免疫抑制剂;超分子溶剂;血浆;超高效液相色谱-串联质谱中图分类号:O657.7;R917文献标识码:A 文章编号:1004-4957(2023)05-0559-09Determination of Tacrolimus in Plasma by Supramolecular Solvent Extraction/Ultra-high Performance Liquid Chromatography-Tandem Mass SpectrometryXIE Yi-qing1,2,LÜ Yue-guang2,MENG Xian-shuang2,LEI Hai-min1*,MA Qiang2*(1.School of Chinese Materia Medica,Beijing University of Chinese Medicine,Beijing 102488,China;2.Chinese Academy of Inspection and Quarantine,Beijing 100176,China)Abstract:An analytical method for the determination of tacrolimus(TAC) in blood plasma was estab⁃lished by supramolecular solvent(SUPRAS)extraction combined with ultra-high performance liquid chromatography-tandem mass spectrometry.After optimizing the key factors such as the composition and amount of SUPRAS,and vortex extraction time through single factor experiment and response sur⁃face design,blood plasma samples were extracted efficiently with SUPRAS formed by pentanol,tetra⁃hydrofuran and water.The extract was separated on a Waters ACQUITY UPLC BEH C18column (50 mm × 2.1 mm,1.7 μm),analyzed by electrospray ionization mass spectrometry in positive ion mode under multiple reaction monitoring(MRM) mode,and quantified by internal standard method.Experimental results demonstrated that there was a good linear relationship for TAC in the concentration range of 0.5-30 ng/mL,with a correlation coefficient(r) of 0.998 6.The limit of detection(LOD)and quantitation(LOQ) were 0.1 ng/mL and 0.5 ng/mL,respectively.The average recoveries(n = 3)at low,medium and high spiked concentration levels ranged from 91.9% to 99.9%,with relative stan⁃dard deviations(RSDs) of 1.7%-5.7%.The proposed method is rapid,sensitive and stable,and it was suitable for the accurate determination of TAC in blood plasma.Key words:tacrolimus;immunosuppresive agent;supramolecular solvent;plasma;ultra-high performance liquid chromatography-tandem mass spectrometry免疫抑制剂是用于抑制机体免疫力的药物,多用于抑制肝肾移植术后的免疫反应,以及治疗变态反应性和自身免疫性疾病,如类风湿关节炎、红斑狼疮等[1-3]。
植物科学领域最全的“方法步骤软件”

植物科学领域最全的“⽅法步骤软件”该⽹盘所有内容也将根据⼤家的补充逐渐更新⽅法步骤1Western_blot_protocol.pdf2Yeast transformation tips.pdf3标签蛋⽩简单介绍.pdf4采⽤本⽒烟草瞬时表达系统进⾏蛋⽩质免疫共沉淀实验 Protein Immuno.. (2).pdf5超级感受态制备.pdf6超级感受态制作⽅法ULTRA.pdf7从纯化到星⾠.pdf8蛋⽩质电泳相关试剂、缓冲液的配制⽅法 - 副本.pdf9分⼦克隆实验指南第三版上册.pdf10分⼦克隆实验指南第三版下册.pdf11分⼦克隆实验指南上.pdf12分⼦⽣物学实验技术简册.pdf13分⼦⽣物学实验室⼯作⼿册.pdf14国内重点实验室分⼦⽣物学实验⽅法汇总实验室常⽤实验⽅法.pdf15核酸电泳相关试剂、缓冲液的配制⽅法 - 副本.pdf16核酸电泳相关试剂、缓冲液的配制⽅法.pdf17核算蛋⽩互算.pdf18基因northern blot⽅法.pdf19基因X 中⽂版.pdf20基因⼯程原理(吴乃虎编著).pdf21基因芯⽚⾼质量-CTAB提DNA.pdf22基于Gateway体系⼀步PCR法构建CRISPR终载体.pdf23酵母单杂说明书.pdf24酵母⾼效转化⽅法.pdf25酵母双杂交操作.pdf26酵母双杂交技术 - 副本.pdf27酵母相关实验⽅法.pdf28芥菜幼苗中超氧化物和H2O2累积的组织化学检测.pdf29抗⽣素的贮存溶液及其⼯作浓度 - 副本.pdf30类胡萝⼘素测定-Functional analysis of beta- and epsilon-ring carotenoid hydroxylases in Arabidopsis.pdf31免疫共沉淀Co-IP实验操作步骤.pdf32拟南芥的实验室⼿册.pdf33拟南芥提DNA.pdf34拟南芥叶⽚过氧化氢DAB染⾊.pdf35凝胶迁移实验(EMSA).pdf36农杆菌注射烟草叶⽚进⾏体内瞬时表达的⽅法.pdf37染⾊质免疫共沉淀(ChIP)实验.pdf38如何使⽤DNAMAN软件制作序列⽐对.pdf39实时荧光pcr中⽂.pdf40实时荧光定量PCR全⽅位解析.pdf41实验室常⽤培养基的配制⽅法 - 副本.pdf42实验室常⽤培养基的配制⽅法.pdf43实验室常⽤试剂、缓冲液的配制⽅法 - 副本.pdf44⼿把⼿教你使⽤EndNote_X8.pdf45数量遗传学、基因组学和植物分⼦育种.pdf46双酶切连接反应之全攻略.pdf47⽔稻多基因敲除系统.pdf48⽔稻原⽣质体的分离转化.pdf49体外降解.pdf50⽂献综述的写法详细.pdf51现代分⼦⽣物学第4版_朱⽟贤2013.pdf52原位杂交procotol.pdf53植物病原体感染期间的植物组织台盼蓝染⾊Bio-protocol2078.pdf 54植物组织电镜基本制样⽅法20171117.pdf55转基因⼤麦中gfp基因的染⾊体位置及其表达.pdf56转录组分析软件上机指南.pdf57综述的写法.pdf58总-常⽤逆境⽣理指标测定⽅法-LCL.pdf59[]毕⾚酵母表达操作⼿册(精译版).pdf609页的操作yeast Transformation.pdf612003 pMDC100 pDMC32 A Gateway Cloning Vector Set for High-Throughput Functional Analysis of Genes in Planta.pdf622007 pMDC100 pDMC32 Recombinational Cloning with Plant Gateway Vectors.pdf 63630491 中⽂版-单杂载体说明.pdf64 A Foxtail mosaic virus Vector for Virus-Induced Gene silencing in maize.pdf65 a loop-mediated isothermal amplification method.pdf66 A Toolkit for Illustrating Heatmap.PDF67Alexander_staining(⼀种花粉活性染⾊法).pdf68arabidopsis mesophyll bifc.pdf69Arabidopsis mesophyll protoplasts.pdf70BiFC.pdf71Bio-Rad荧光定量PCR应⽤指南.pdf72bp_clonaseii_man.pdf73CAPS_and_dCAPS.pdf74CapsaicinHPLC-UV-MS-PP.pdf75DUAL?Membrane?System?Protocol.pdf76EndNote.pdf77FISH Protocols.pdf78GAPDH酶活测定.pdf79Gateway Protocol.pdf80gateway system mannual.pdf81gateway_pdonr_vectors.pdf82GenStat统计⽅法与数据分析(1).pdf83GUS染⾊.pdf84GUS染⾊-杜长青.pdf85GV3101电击.pdf86HiTail PCR BTN_A_000112601_O_1421a.pdf87imageJ使⽤⽅法-叶⽚⾯积统计.pdf88ImpGWB manual E (pGWBnxx).pdf89infusion protocol.pdf90JoinMapML作图⽅法.pdf91Khalid Meksem, Guenter Kahl-The Handbook of Plant Genome Mapping_ Genetic and Physical Mapping (v. 1)-Wiley-VCH (2005).pdf92KOD –Plus- Neo (KOD-401).pdf93KOD-401.pdf94lr_clonase_man.pdf95LSM brief manual_final.pdf96Matchmaker Gold Yeast Two-Hybrid System User Manual (PT4084-1)_092413.pdf 97Maxent简明教程(翻译).pdf98MEGA3.1 中⽂使⽤说明.pdf99miRNA northern blot ⽅法.pdf100Molecular Plant Pathology.pdf101MultiExperiment_Viewer_Quickstart_Guide_MeV软件教程-1.pdf102Oligo的使⽤⽅法介绍.PDF103One-step enzymatic assembly of DNA-YubingHe.pdf104PCR Mutation Detection Protocols.pdf105Pcr Protocols-Methods In Molecular Biology.pdf106pDONR201.pdf107pDONR201 207 info..pdf108pgwb pdf.pdf109plant metabolite protocol and method.pdf110PrimeSTAR - 副本.pdf111R for Beginners 中⽂版.pdf112R050A PrimeSTAR? GXL DNA Polymerase.pdf113Rice Protocols_Methods in Molecular Biology, 2013, Vol. 956(Non-commercial use permitted).pdf114ROS染⾊⽅法.pdf115RPA III Kit.pdf116SDS PAGE.pdf117SPSS与多元统计分析⽅法讲义.pdf118Tai Te Wu Analytical Molecular Biology .pdf 119TAK-101_201.pdf120Vector_NTI_简体中⽂使⽤教程PDF版本.pdf 121Western blot详细操作步骤.pdf122western_blot - 副本.pdf123实验protocol⼤全124VIGS125endnote x8破解.rar.xltd.cfg126Endnote_X7实⽤教程.pptx127ij.jar128mmexport1510878447197.jpg129PastedGraphic-1.png130烟草遗传转化.jpg131可溶性糖和可溶性蛋⽩测定⽅案.wps132Adobe Acrobat XI Pro 11.0.1.rar.xltd.cfg 133SeqScannerInstall.exe.重命名134蛋⽩的相互作⽤ - 副本.ppt135蛋⽩质结构与功能预测.ppt136实验技术.zip.xltd.cfg137⽔稻花药花粉发育过程.jpg138注册账号.txt139载体构建.ppt140科研论⽂作图141⽟⽶营养液.xlsx142LI6400143protoplast_workshop.wmv144SPSS介绍与应⽤.pptx145⽂献管理教学-zd.pptx146拟南芥蜡质测定⽅法.jpg147软件148国家基础地理信息系统数据.zip149分⼦⽣物学150克隆构建载体知识积累151植物实验⽅法152转录组培训资料153补充的软件和试验⽅法154酵母双杂155实验技术156原⽣质体制备157备份word158⼦叶节培养程序2008整理.docx1594.5h Immunolocalization .doc160Arlequin操作说明.doc161change solution on August 2nc.doc162ChIP for Arabidopsis.doc163CHIP protocol.doc164CHIP实验⽅法.doc165CHIP实验⽅法2.doc166co-IP in vivo.doc167CTAB 提取DNA.doc168CTAB-96孔法⽔稻基因组DNA提取.doc169CTAB法提前DNA及其检测.doc170DNA粗提.doc171DNA提取过程中各种试剂的作⽤.doc172EMSA实验步骤.doc173FISH protocol.DOC174FISH药品配置.doc175H2O2的测定-KI法.doc176Hydrogen proxide_CN.doc177IAA treatment used for detecting IAA induced genes.doc 178In Situ hybridization in plants- protocol.doc179IPCR确定T-DNA突变体插⼊位点 - 副本.doc180Leica⽯蜡切⽚机操作规程.doc181LYM基因克隆构建PAbAi载体.doc182LYM酵母单杂流程.doc183Megazyme_总淀粉测定试剂盒翻译.doc184MS母液配制⽅法及⽤量.doc185MS培养基及配制注意事项 - 副本.doc186NEB mRNA 建库protocol.doc187N-P-K联合消煮测定.doc188PAGE胶流程.doc189PFP PFK ADH PDC enzyme activities.doc190POD 电泳⽅法.doc191Rice Leaf Protoplast Protocol-Modified by WM.doc192RT-PCR.doc193RuBP-PEP-Pr测定.doc194SEM.doc195SOD 电泳⽅法.doc196SOD、POD、CAT、MDA和可溶性蛋⽩测定.doc197VIGS实验⽅案.doc198WB实验步骤.doc199Y1H.doc200yongjiang氮代谢酶.doc201矮牵⽜转化.doc202半薄切⽚.doc203曹建康-果蔬采后⽣理⽣化实验指导.doc204测酶时需要配的溶液.doc205常⽤试剂配⽅.doc206超级感受态制备 (2).doc207超级感受态制备.doc208成熟胚⾼效转基因系统的建⽴(Ver_2011.03.21).doc209⼤肠杆菌电击感受态制备⽅法-曹⾼燚.doc210蛋⽩各组分含量测定⽅法.doc211蛋⽩酶活性的测定⽅法.doc212蛋⽩透析复性⽅法.doc213蛋⽩质胶内酶解及同位素标记实验步骤.doc214蛋⽩组学实验流程.doc215分⼦克隆技术实验操作⼿册2005.doc216分⼦⽣物学实验技术实验操作指南.doc217柑橘原⽣质体提取及转化.doc218根系活⼒测定-NRA测定.doc219过氧化氢测定⽅法.doc220花粉液体纯体外萌发⽅法.doc221花序浸染法转化拟南芥 - 副本.doc222基因枪法瞬时转化花粉.doc223甲基化⽔平重硫酸盐测序.doc224减数分裂染⾊体观察⽅法.doc225酵母感受态制备及转化.doc226磷酸盐缓冲液配制⽅法.doc227酶活测定⽅法.doc228免疫⾦⼿册.doc229茉莉酸JA提取.doc230拟南芥⾓质测定⽅法.doc231拟南芥解剖学图 - 副本.doc232拟南芥茎切⽚.doc233拟南芥原⽣质体转化系统.doc234凝胶迁移滞后实验 - 副本.doc235农杆菌感受态的制备.doc236农杆菌侵染拟南芥花序的转化⽅法 - 副本.doc237农杆菌转化番茄.doc238农杆菌转化烟草.doc239切⽚技术.doc240琼脂糖切⽚.doc241设计引物如何设计酶切位点.doc242⽯蜡包埋机(paraffin-embedding-station)Leica-EG1150H.doc 243⽯蜡切⽚制作⽇程(新).doc244实验室常⽤试剂、缓冲液的配制⽅法.doc245实验室⽣理⽣化指标实验简略步骤.doc246实验室原核表达步骤.doc247史上最全限制性内切酶酶切位点汇总.doc248树⽊DNA的提取.doc249双分⼦荧光互补实验原理 - 副本.doc250⽔稻根尖压⽚-2015-7-13.doc251⽔稻叶中CAT、POD和SOD活性的测定.doc252⽔稻营养液配⽅.doc253透明comfocs.doc254⼟壤速效钾测定.doc255⼟壤有机质测定.doc256⼟壤有效氮测定.doc257⼟壤有效磷测定.doc258⽡村单倍体诱导protocol.doc259我们实验室⽊本果树RNA提取⽅法.doc260烟草注射.doc261叶绿素含量测定.doc262液泡分离及V-PPase酶活和H 转运检测.doc263仪器操作规范-2017.11.17.doc264荧光定量引物⾃动化设计教程.doc265营养品质测定⽅法.doc266⽤于质谱鉴定的蛋⽩质凝胶样品酶解⽅法.doc267油菜转基因技术体系终结版.doc268原位.doc269中药鉴定学重点整理.doc270最新实验原理与⽅法2009.doc271[]进化树软件MEGA最新6.06说明书.docx2722个试验⽅法分享.docx2731003总结载体构建说明书.docx27420170106电击法农杆菌感受态的制备及其转化-曹⾼燚.docx275A simple and rapid method for the preparation of plant genomic DNA for PCR analysis..docx 276Benzoxazinoids extraction and quantification.docx277BSMV基因沉默相关.docx278ChIP for Plant.docx279circos画共线性图⽚.docx280CO-IP (2).docx281co-IP.docx282CoIP-Western blot.docx283CTAB法提DNA.docx284CTAB法提取⽔稻DNA.docx285Dellaporta法⼩量快速提取植物全基因组DNA.docx286EMSA protocol.docx287EST-SSR实验流程及软件使⽤.docx288FAA固定protocol.docx289FNPI-PCR⽅案.docx290GISH分析.docx291His-tag原核蛋⽩提取.docx292Histochemical detection of ROS.docx293Hoagland营养液配制⽅法.docx294IAA、 JA、 ABA、 SA 定量检测分析.docx295In vitro Pollen germination media.docx296IP MASS.docx297JASA LC-MS extraction.docx298Native chip.docx299Northern杂交.docx300PAGE 银染.docx301PCR平端加A流程.docx302Pi含量测定.docx303Protein expressed in yeast microsome.docx304Protein extracted from plant tissues__ZT.docx305Protein extraction protocol.docx306PS 、AI、CDR设计视频教程.docx307SEM实验⽅法.docx308Sls法提DNA.docx309TPS法提取植物DNA.docx310trizol法⼤量提取油菜RNA.docx311Trizol法提取RNA.docx312VIGS流程-汪汪-20161102.docx313Western Blot protocol .docx314Western Blot.docx315Western blot.docx316保护酶的测定⽅法.docx317便携式调制叶绿素荧光仪PAM-2500简明操作⼿.docx318表达蛋⽩的分离与纯化;EMSA.docx319打烟草coip或看荧光⽐较常⽤.docx320⼤肠杆菌感受态细胞制备实验(CaCl2法) _1510835367078.docx321⼤⾖蛋⽩提取.docx322⼤⾖⼦叶节遗传转化流程.docx323蛋⽩提取 CO-ip WESTERN BLOT.docx324多靶点pCRISPR载体(单⼦叶植物⽤)使⽤⽅法2014-05-19.docx325多靶点pCRISPR载体改良版(双⼦叶植物⽤)使⽤⽅法2014-10-26(P).docx 326改良的⽟⽶SLS法提取DNA.docx327柑橘DNA和RNA提取⽅法.docx328感受态细胞制备.docx329花青素原花⾊素相关protocol.docx330活性氧,胼胝质,叶⽚⽓孔的检测实验.docx331基础分⼦⽣物学实验操作指南.docx332基因枪轰击洋葱表⽪观察.docx333基因枪瞬时转化矮牵⽜花瓣.docx334简单粗暴提取拟南芥DNA-1.docx335酵母双杂筛库1.docx336酵母双杂实验⽅法总结.docx337酵母双杂最终版.docx338酵母转化⽅法及⽔稻组织培养液配⽅-1.docx339聚丙烯酰胺凝胶电泳检测PCR产物.docx340菌落PCR(⾃⼰总结).docx341可溶性糖测定.docx342临界点⼲燥.docx343拟南芥材料快速PCR鉴定.docx344拟南芥可溶性全蛋⽩的双向电泳.docx345拟南芥原⽣质体.docx346拟南芥原⽣质体的提取和转化.docx347拟南芥原⽣质体亚细胞定位Copy of Localization of procedure.docx348拟南芥原⽣质体制备.docx349拟南芥杂交.docx350拟南芥总蛋⽩简易提取.docx351凝胶迁移实验(EMSA)实验⽅法 - 副本.docx352农杆菌注射烟草顺时转化.docx353全基因组甲基化WGBS和转录组分析过程.docx354试验⽅法.docx355⽔稻缺素处理配⽅及步骤.docx356⽔稻原⽣质体分离及转化.docx357⽔稻原⽣质体提取及转化.docx358⽔稻原⽣质体制备和瞬时转化.docx359体外泛素连接酶活性测定.docx360⼟壤基本指标检测.docx361兔源抗体制备.docx362新Y1H载体构建及转化步骤.docx365亚细胞定位.docx366烟草瞬时表达.docx367叶⽚碳代谢相关物质和酶试验⽅法-棉花.docx368油菜下胚轴暗光培养转化.docx369原核表达.docx370原核表达protocol.docx371原核表达步骤.docx372原核蛋⽩的提取.docx373原⽣质体叶⾁细胞瞬时表达.docx374原位杂交.docx375真核细胞表达外源基因技术⽅法及操作步骤.docx376植物常见外援激素的作⽤.docx377植物⽣理⽣化指标.docx378植物⽯蜡切⽚.docx379植物有效磷测定.docx380植物总RNA提取与琼脂糖凝胶电泳分析.docx381注射烟草看BiFC.docx382SA测定⽅法3832007_Dolezel_NATURE_PROTOCOLS_2233.pdf384【代码M0101】常⽤溶液的配制.docx385AGROBACTERIUM COMPETENT CELLS.doc386Agrobacterium growth and transformation.doc387AutoDock&ADT教程(1).doc388cellulose content detection method.doc389CHIP protocal.docx390CRISPR-Cas9载体构建(⽔稻).docx391CTAB-96孔法⽔稻基因组DNA提取.doc392CTAB提取真菌DNA.docx393DAB染⾊,台盼蓝染⾊-钱礼超.docx394【代码M0201】PCR引物设计⽅法.docx395【代码M0202】PCR引物设计11条黄⾦原则.docx396ELISA的原理和技术要点.pdf397EMS mutagenesis.docx398Endnote简易教程-郑夏⽣.ppt399Floral dip transformation of Arabidopsis.pdf400【代码M0203】RACE技术扩增构建全长cDNA⽂库.docx 401【代码M0204】逆转录-聚合酶链式反应(RT-PCR).docx 402GPX测定⽅法.docx403【代码M0205】常规PCR.docx404GUS染⾊及GUS蛋⽩提取及GUS蛋⽩酶活性测定.doc405GUS组织化学染⾊.docx406【代码M0206】原位PCR.docx407Hoagland & Kimura营养液配⽅.docx408Hplc测定内源激素[1].doc409Image-Pro Plus官⽅简体中⽂快速⼊门指南.pdf410IN SITU AND DOUBLE IN SITU HYBRIDIZATION.doc411LIC 克隆中⽂版.docx412LIC-QLC.docx413lignin content detection method.docx414microRNA的stem-loop-Qrt-PCR引物设计.doc415mRNA的分离纯化.docx416【代码M0207】实时定量PCR.docx417【代码M0208】关于PCR的⼏个经验之谈.docx418【代码M0209】PCR扩增产物鉴定与纯化.docx419Phos-tag实验指导⼿册.pdf420Plant Genomic DNA Extraction using CTAB.docx421Poisoned Primer Extension.pdf422【代码M0301】酶切.docx42317. 扫描电镜.docx424Protocol for staining of Arabidopsis leaves with trypan blue and aniline blue.pdf425Protocol -EMSA.docx426Protocol-拟南芥转基因.docx427【代码M0302】质粒DNA的电泳检测.docx428【代码M0303】DNA⽚段回收.docx429【代码M0304】⽬的基因⽚段与载体连接.docx430SDS-PAGE胶观察.doc431AC双⾊原位杂交.doc432SOD、POD、CAT、辅氨酸测定⽅法.docx433Southern blot hybridization.doc434Southern-blotting Protocal正确.doc435AFLP实验流程.doc436staining for pollen viability.pdf437Taqman设计总结005.doc438【代码M0305】质粒DNA的转化.docx439【代码M0306】菌落PCR.docx440⼆代测序信息分析.pdf441亚细胞定位.doc442免疫荧光004.doc443全氮、有机质测定⽅法.doc444农杆菌侵染烟草叶⽚表⽪细胞实验步骤.doc445农杆菌感受态制作.docx446分⼦克隆指南(上)().pdf447分⼦克隆指南(下)().pdf448分⼦植物育种-徐云碧-中⽂版.pdf449半薄切⽚流程.doc450基因枪瞬时转化洋葱表⽪.docx451实验室常⽤分⼦⽣物学实验技术.docx452实验室常⽤培养基的配制⽅法.pdf453AsA测定⽅法.doc454实验报告RRF纯化.xls455【代码M0307】细菌的转化与平板筛选.docx456【代码M0402】蛋⽩电泳.docx457快速提取酵母基因组DNA.docx458⼿动进样HPLC-RID使⽤⽅法002.docx459拟南芥成花⽹络2015.pdf460拟南芥转基因试验流程.pdf461提取DNA和制种聚丙烯酰胺凝胶.docx462【代码M0308】E.coli感受态细胞的制备(CaCl2法).docx463⽊质素测定⽅法.docx464根系测定.doc465【代码M0405】双向电泳.docx466植物Western Blot.docx467植物分⼦遗传研究室实验技术.docx468植物病毒 dot-ELISA 检测⽅法.docx469植物组织⽯蜡切⽚.docx470⽔果果实(淀粉糖含量⾼)RNA提取⾃⼰配药品⽅法(经济性).doc 471⽔稻茎结培养操作流程.doc472泛素化蛋⽩质组.docx473液相⾊谱操作指南003.docx474烟草瞬时转化.docx475⽟⽶原⽣质体转化及原核表达⽅案.docx476⽟⽶叶⽚原⽣质体拟制备转化操作流程.doc477cDNA⽂库构建.doc478电镜切⽚制样流程.doc479直径测量.pdf480短柄草农杆菌转化流程.doc481⽯蜡切⽚制作流程.docx482⽯蜡切⽚染⾊.docx483⽯蜡切⽚的制作.doc484⽯蜡包埋切⽚操作程序.doc485碱煮法快速提取⽟⽶基因组DNA.docx486种⼦发芽流程.docx487简易CTAB法提取植物基因组DNA.docx488组培苗操作流程.doc489【代码M0406】多克隆抗体的免疫电泳技术.docx490草莓⾼效稳定转化.docx491蛋⽩实验相关操作及注意事项.doc492蛋⽩质电泳实验技术-第2版 (1).pdf493DAB和NBT染⾊.doc494超级感受态细胞的制备.docx495转基因技术.ppt496转基因拟南芥.doc497酵母单杂.docx498酵母单杂交系统及其应⽤.doc499酵母双杂交张娟娟.doc500酵母双杂交⽂库筛选流程_mating法.docx501酵母双杂实验操作.WMV502酵母⽂库筛选-Mating篇.doc503酶法催化⽣产母乳脂肪替代物001.doc504酶活测定基本⽅法.doc505醇溶蛋⽩的提取与电泳.doc506限制性内切酶酶切位点汇总.docx507⾼通量测序原理,⽂库构建及质量评价-1108.pdf508鲍鱼酶裂解海带配⼦体细胞壁步骤-卞曙光-中国科学院海洋所.doc509麦⾕蛋⽩亚基的粗提与电泳.doc510(原创)傻⽠软件进⾏简并引物设计.ppt511【代码M0401】Western Blotting.docx51215. 琼脂糖切⽚.docx51316. 叶原基离体培养和活体成像.docx514AFLP原理和操作.docx515AI_及SS活性的测定.doc516DAPI staining of mature pollen nuclei.pdf517Design degenerate primer ——Important.docx518【代码M0407】单细胞凝胶电泳标准操作.docx519H2O2的测定-KI法.doc520His标签蛋⽩纯化试剂盒(可溶性蛋⽩).pdf521【代码M0403】蛋⽩电泳后的凝胶染⾊(考马斯亮蓝、银染、铜染).docx 522mRNA差异显⽰之变性聚丙烯酰胺凝胶电泳及银染.docx523NSC测定⽅法⽐较.docx524P32放射性同位素测定注意事 (2016修改版)(1).docx525Rubisco含量测定.docx526SS,SPS,R酶,Q酶,ADPGase酶活性.docx527【代码M0408】明胶酶谱分析法.docx528第七期⽣物数据分析技能实训会.doc529多靶点pCRISPR载体改良版(单⼦叶植物⽤)使⽤⽅法2014-10-31(P).docx 530发给⼤家后⼩艾⼜修改的---⽔稻转基因实验操作.doc531⽢薯花青素提取与测定⽅法2.doc532DAB染⾊.doc533国内重点实验室分⼦⽣物学实验⽅法汇总实验室常⽤实验⽅法.pdf534GSH 测定⽅法.doc535全国Python科研应⽤专题实操班.doc536实时荧光定量PCR技术.ppt537⽔稻多基因敲除系统.pdf538⽔果抗性相关指标的测定⽅法.docx539⼀种快速鉴定转基因植物纯合体的新⽅法.pdf540软件541【代码M0404】RNA电泳(甲醛变性电泳).docx542【代码M0409】对流免疫电泳.docx543【代码M0410】Southern Blotting实验步骤.docx544【代码M0411】Northern Blotting实验步骤.docx545【代码M0501】细胞膜蛋⽩的分离.docx546【代码M0503】蛋⽩质在原核⽣物中的表达.docx547植物RNA提取⽅法资料548液相测维⽣素E549ddRT-PCR(差⽰反转录PCR).docx550ec值.pdf551GLDH含量测定.pdf552Griess试剂测NO含量.doc553H2O2测定⽅法.doc554vigs侵染⽅法.doc555Western blot(聂).doc556苯丙氨酸解氨酶(PAL)活性的测定.doc557丙酮酸的测定.pdf558酚类和⽊质素.doc559过氧化氢测定⽅法(廖).doc560抗氧化酶和MDA以及蛋⽩测定指南.doc561类胡萝⼘素提取⽅法⽂档.doc562酶联免疫测ABA .doc563台盼蓝染⾊.doc564糖含量的测定1.pdf565糖含量的测定2.pdf566叶黄素提取.doc567叶绿素⽅法.doc568载体构建⽅法.doc569MDA含量测定.doc570POD_测定⽅法.doc571POD活性测定.doc572SOD活性测定.doc573sss测定.doc574标准曲线.xls575不同光质的H2O2和超氧(1).xls576根系活⼒测定⽅法.docx577脯氨酸标准曲线.xls578脯氨酸含量.doc579⽣理实验数据(SOD,POD,DPPH,超氧).xls580植物体内氧⾃由基含量测定.docx581植物体内游离脯氨酸含量的测定.doc582本⽣烟草种植和管理.docx583病毒接毒及其后续管理.docx584植物病毒dot-ELISA检测⽅法.doc585AKTA FPLC 蛋⽩纯化操作.doc586CTAB法提取RNA.doc587CTAB提取植物DNA.docx588DNA的琼脂糖凝胶电泳.doc589Greiss试剂测定NO的⽅法.doc590变性梯度凝胶电泳.doc591实验⽅法整理 .docx592线粒体测定最终版.docx593叶绿体提取.doc594【代码M0502】细菌总蛋⽩和膜蛋⽩提取⽅法.docx 595【代码M0504】免疫复合物的纯化.docx596【代码M0602】动物组织块DNA的提取.docx597半薄切⽚技术.ppt598⽯蜡切⽚流程.doc599透射电镜切⽚的步骤.doc600荧光参数.doc601植物RNA的提取及其电泳鉴定.doc6021-1 ⽯蜡切⽚的制作.pdf603N-P-K联合消煮测定.doc604SOD、POD、CAT、MDA和可溶性蛋⽩测定.doc 605沉降值测定⽅法.doc606FISH procedure-2.doc607PCR-SSCP实验操作详细步骤.doc608Promega公司的TA载体克隆与转化.doc609shotgun_⽂库的构建.doc610southern 杂交实验流程.doc611毕业⽣离室前注意事项.doc612基因定点突变.doc613基于⽑细管电泳的荧光检测SSR实验程序.doc614拟南芥培养转化的流程[1].doc615庞斌双的蛋⽩电泳和连接转化程序.doc616实时荧光定量PCR技术.doc617⼩麦成熟胚标准技术规程.doc618⼩麦基因组DNA的提取.doc619⼩麦⽥间记载和室内考种项⽬与标准.doc620怎样记录实验结果.doc621重点实验室protocol-CWF全长cDNA⽂库构建流程.doc622重点实验室protocol-总RNA提取(Trizol法)和mRNA提取[Oligo(dT)cellulose]流程zgy.doc 623ABI3730XL测序实验流程-2007-3-15.doc624BAC⽂库构建与筛选实验程序-2007-3-10.doc625PCR产物的TA克隆(DNA的胶回收和连接).docx626基因X 中⽂版.pdf627植物中基因编辑.docx628【代码M0604】CTAB法提取植物总DNA.docx629【代码M0605】磁珠法提纯mRNA.docx630【代码M0606】连续梯度法纯化闭环DNA.docx631【代码M0607】不连续梯度法纯化闭环DNA.docx632【代码M0608】从外周⾎淋巴细胞中提取RNA.docx633【代码M0609】饱和氯化钠法提取外周⾎DNA.docx634【代码M0701】荧光原位杂交(FISH)探针的制备.docx635【代码M0801】Conditional gene knockout in C. elegans- CRISPR-Cas9.docx636【代码M0802】Target Sequence Cloning Protocol- CRISPR-Cas9.docx637【代码M0803】SAM target sgRNA cloning protocol- CRISPR-Cas9.docx638【代码M0901】酵母双杂交筛选简介和流程.docx639【代码M0902】构建于 AD 载体的 cDNA ⽂库的扩增.docx640【代码M0903】构建于 AD 载体的 cDNA ⽂库的纯化.docx641【代码M0904】pGBKT7bait ⼩规模转化酵母菌 AH109.docx642【代码M0905】钓饵蛋⽩⾃⾝激活报道基因的活性检测.docx643【代码M0906】⽂库质粒⼤规模转化已携带钓饵质粒的酵母.docx644【代码M0907】转化⼦的筛选与库质粒纯化.docx645【代码M0908】酵母质粒 DNA 的提取.docx646【代码M0909】蛋⽩相互作⽤的验证—pGBKT7bait 与 pACT2 ⼩规模共转化酵母.docx 647【代码M1001】分⼦⽣物学实验常⽤培养基的配制⽅法.docx648【代码M0601】⼩量制备质粒DNA(强碱法).docx649【代码M0603】动物组织RNA提取及纯化.docx650Southern Blot原理及实验⽅法.doc651SSR分⼦标记实验操作及其注意事项.doc652单核苷酸多态性(SNP)实验.docx653动物基因组DNA的提取.doc654甲基化检测原理及步骤.doc655降落PCR.docx656聚合酶链式反应(PCR)基本操作步骤.doc657菌落PCR.doc658⽬的基因PCR扩增产物的克隆.doc659逆转录-聚合酶链反应(RT-PCR).doc660凝胶迁移实验(EMSA).docx661普通PCR、原位PCR、反向PCR和反转录PCR的基本原理和操作步骤.doc 662染⾊质免疫共沉淀(ChIP)实验.docx663实时荧光定量PCR具体实验步骤.doc664限制性⽚段长度多态性实验(RFLP).doc665原位PCR技术基本原理.doc666质粒DNA的提取.doc667重叠延伸PCR实验.docx668重组质粒的连接、转化及筛选.doc669(原创)实时定量神器Allele ID 5破解教程.doc670分⼦⽣物学⼤实验2-L.pptx671分⼦⽣物学实验II新教案.pdf672分⼦⽣物学实验673实验技术视频674现科课件675研究⽣课件676研究⽣分⼦⽣物学实验I讲义-2017.doc677⼤肠杆菌感受态细胞的制备和转化.doc678pGEMT.pdf679TaKaRa_LA_Taq说明书.pdf680TAKARA双酶切表.doc681分⼦克隆技术实验操作⼿册.pdf682宝⽣物去磷酸化说明书.pdf683构建载体知识查询.docx684载体图谱685载体图谱序列686上海硕盟_TOYOBO说明书.pdf687分⼦克隆实验指南第三版上册.pdf688分⼦克隆实验指南第三版下册.pdf6891 PCR实验技术6902 DNA实验技术6913 RNA实验技术6924 蛋⽩质实验技术6935 补充694DH5α感受态制备⽅法.pdf695EMSA696Easy Yeast plasmid isolation kit user mannual.pdf697GM配⽅.pdf698GST pull down.pdf699word700Immobilized Glutathione.pdf701MagneGST Pull-Down System.pdf702Ni-NTA handbook.pdf703PCR704PHARMACIA_GST_Gene_Fusion_System_Handbook.pdf705Pull-Down GST kit.pdf706RNAi707RNA提取⽅法——TRIZOL.pdf708SDS_PAGE胶配⽅表.pdf709Y2H Membrane Protein Kit.pdf710activation tagging.pdf711chip EZ-ChIP_manual.pdf712chromas.rar713his tag pull down assay .pdf714iTILLING.pdf715pfu.pdf716promega_GloMax2020完整版.pdf717western breeze chromo_man.pdf718western_blot技术集锦.rar719免疫共沉淀与Western Blot.pdf720原位杂交⽅法721国内重点实验室分⼦⽣物学实验⽅法汇总实验室常⽤实验⽅法.pdf 722常⽤缓冲液的配制.pdf723普通表达载体724植物细胞组织学⾼⽼师课件725荧光定量PCR.pdf726荧光激发及吸收波长.PDF727酵母双杂交728酵母双杂交技术研究与应⽤进展_李先昆.pdf729AxyPrep_DNA_Gel_DYE_Ext_Kit_ver20090727.pdf730His Spin Trap.pdf731In-Fusion Kit User manual.pdf732P020091009585923799380.ppt733PPICZalpha.pdf734[]基因克隆研究⽅法综述.pdf735matchmaker gold yeast one-hybrid library screening system.pdf736pET28_map.pdf737ppic9k_man.pdf738分⼦克隆实验指南上册v3.pdf739分⼦克隆实验指南下册v3.pdf740拟南芥T-DNA插⼊突变纯合体的鉴定.ppt741EMSA.pdf742淀粉检测说明书.pdf743RT-PCR744DNA kit.pdf745DP441 RNAprep pure多糖多酚植物总RNA提取试剂盒.pdf746Gateway_载体构建.ppt747RNAprep Pure 多糖多酚植物总RNA提取试剂盒--天根.pdf748系统发育分析课件2017完整版.pdf749Real-time PCR750office2013完美激活.rar751[3]实时荧光定量PCR技术_天根.ppt752[4]国内重点实验室分⼦⽣物学实验⽅法汇总实验室常⽤实验⽅法.PDF 753[7]PCR技术讲座.pdf754[8]国内重点实验室分⼦⽣物学实验⽅法汇总实验室常⽤实验⽅法(1).pdf755[11]荧光定量PCR技术讲座:理论基础、引物及探针设计、体系优化、实验⽅案、数据分析、污染防控 2.ppt756R语⾔绘图-实操训练.pptx757R语⾔绘图⼊门.pptx758基因功能分析.pptx759基因聚类分析和样品相关性分析.pptx 760基因表达定量.pptx761差异表达分析.pptx762数据质控.pptx763⽆参转录组序列组装及实际操作.pptx764测序数据⽐对.pptx765测序数据统计.pptx766转录组数据库注释.pptx767转录组⽣物信息分析整体介绍.pptx下⾯是软件软件1最新版SCI-Endnote⽂献样式.zip2circos-0.69-5.tgz3hmmer-3.1b2-cygwin32.tar.gz4Adobe_Photoshop_CC.7z5BeoPlayer ⾳质最好的播放器.7z6EndNote X8.7z7genespring.7z8PRIMER 6破解版.7z9[]DPS数据处理系统.rar10[]primer5x64bit.rar11[]DNAMAN8_Registered.rar12[专业图像分析软件].ipp6.0.rar1364位系统所需要的primer5程序.rar14Adobe Acrobat XI Pro 11.0.1.rar15Adobe Photoshop CS6.rar16BeaconDesignerpjb.rar17blast.rar18CE Design V1.04.rar19chromas (2).rar20Chromas.rar21clustal.rar22clustalx.rar23Design-Expert.8.05b完全破解版.rar24DNAMAN.rar25DNAMAN7setup.rar26DNAMAN8_20150421.rar27DNAMAN8_Registered.rar28DNASTAR Lasergene v7.1.0.rarsergene.v7.1.rar30DNASTAR6.0.rar31dps7.05.rar32EditPlus_.rar33EndNote X7 批量授权中科⼤版.rar34EndNote X8.rar35EndnoteX8(含汉化⽂件).rar36EndNote教程.rar37FigTree v1.4.2.rar38GWAS常⽤软件及⼿册.rar39jionmap4.0注册版.rar40LI-6400XT OPEN6.2 中⽂操作⼿册(2014年2⽉校).rar41MapChart 2.2.rar42MapDraw.rar43MapMaker 3.0.rar44MapQTL6.03.187破解版 best.rar45MapQTL6补丁.rar46MEGA6 (2).rar47MEGA6.rar48Mindmanager15.rar49NoteExpress完美破解版.rar50Oligo.v7.53.rar51origin8.0破解版附安装图⽂教程.rar52OriginLab 8.0 绿⾊破解版.rar53PHOTOSHOP_CS_8.01_简体中⽂版.rar54POPGENE32.rar55PP600Installer.rar56Primer5.0 64wei.rar57Primer5.064.rar58Protocol.rar59Protocols.rar60python批量下载指定序列__.rar61RIP.rar62SAS Software Depot.rar63SAS.rar64ScienceSlides.rar65sigmaplot 10.0 破解版.rar66Simvector 4.5.rar67SnapGene2.3.2.rar68SnapGene各版本破解版-淼灵⽣物.rar69SPSS.rar70TBtools-crossplatform.rar71TeamViewer.rar72Trados翻译软件2014特别版(密码).rar 73treeview.rar74Uedit32.rar75Vector NTI 11.5.1 破解版.rar76Vector NTI 11.5.3破解版.rar77迅龙数据恢复.rar78[GraphPad.Prism.v5.0.注册版].prism.zip79Adobe Illustrator CS5.zip80Adobe Illustrator CS6.zip81AdobeIllustrator21_HD_win64安装包.zip82AMOS21.0.zip83BEAST.v2.3.0.Windows.zip84BioEdit (2).zip85bioedit.zip86BioMercator2-1.zip87BioXM2.6.zip88BLAST2GO - 复件.zip89BLAST2GO.zip90ChemOffice已破解解压即⽤.zip91cluster.zip92CRISPR PROTOCOL For Plant.zip93Diverge3.0B1(家族分歧软件).zip94DNAMAN.zip95DNAMAN_setup.zip96DNAMAN9_registered.zip97Dual_genomic_comparison.zip98EditPlus_3.3.0.715_XiaZaiBa.exe.zip99EMS-Mutagenesis-of-Arabidopsis.zip100Endnote X7 Windows.zip101EndNoteX7.zip102FV10-ASW_Viewer3_1.zip103Gateway Cloning Vectors.zip104GPS-SNO.zip105GraphPad Prism (2).zip106GraphPad Prism 5.zip107GraphPad Prism.zip108GraphPad.Prism.v5.01.zip109GraphPad Prism7.zip110GraphPadPrism.zip111GST_pull_down.zip112hmmer3.0_windows.zip113ICIMSetup V4.1.zip114IGV详细使⽤.zip115jionmap4.0.zip116MEGA6.zip117MrBayes-3.2.6_WIN32_x64.zip118Origin 9 32位专业破解版.zip119phylip-3.695.zip120Plant Vectors.zip121PRIMER 5.0含序列号.zip122Primer premier 5 - 32bit.zip123Primer Premier 5.zip124Primer Premier 5q.zip125Protocols.zip126python⼊门及数据分析.zip127SCI论⽂写作语⾔润⾊修改WhiteSmoke(破解).zip 128SigmaPlot_V12.5.zip129SIMCA 13.0 (破解版).zip130SIMCA 14.1 32-bit.zip131simca.14.1.crack.zip132SIMCA 13.0.zip133SnapGene 2(1).zip134snapgene 3.2.1 mac 版本.zip135SnapGene.zip136SPDBV_4.10_PC.zip137spss23 64.zip138TreeView_1.1.6r2_win.zip139UltraEdit.zip140VectorNTIAdvance11.5.1破解.zip141VIGS.zip142显微镜图像处理软件 - 复件.zip143MAGA6.0144SnapGene145EndNoteX6_16.0.0.6348_PortableSoft.zip146MEGA7.0.21_win64_setup.exe147SnapGene-.zip148SnapGene_pc().rar149Vector NTI 11.5.1 破解版.zip150Vector_NTI_简体中⽂使⽤教程PDF版本.pdf151DNAMAN汉化版.rar152software-win153⽣物信息分析软件与TCGA相关⽂献(2)154⽣物_PilotEdit_5.3.0.rar155⼀些软件 - 复件(1).rar156引物设计.rar157必应词典.zip158补充的软件和试验⽅法.zip159蛋⽩保守结构域分析与分⼦进化树构建.zip160蛋⽩三维结构处理raswin.zip161基因蛋⽩结构图绘制IBS_1.0_windows_20141228.zip 162实验protocol.zip163⽣物软件.zip164实验⼯具软件.zip165新建⽂件夹 (2).zip166英⽂⽂章语法⾃动纠错.exe167endnote x8破解.rar.xltd168ICIMSetup V4.1.exe169ij146-jdk6-64bit-setup.exe170Illustrator CS5_Lite.exe171MEGA5.05_Setup-系统树构建.exe172npp_6.9.2_Installer.exe173Pathway Builder-通路绘图.exe174Photoshop setup.exe175Photoshop_CS3.exe176R-3.4.1-win.exe177RStudio-1.0.143.exe178SETUNA.exe179iSlideTools.Setup.Online.2.4.1.exe180HemI_windows_1_0_win32_x64.exe181HemI_windows_1_0_win32_x64 (2).exe182ApE.exe183MeV_4_9_0_r2731_win(1).rar184GraphPad Prism 7 教程及软件185信号通路作图软件186winmd5free---检测数据完整性.zip187全基因组扫描-转录因⼦结合位点-⼯具.pdf188⽤origin软件绘制折线图和误差线(By 蜗⽜菌).pdf 189多序列⽐对软件clustalx-2.1-win.msi190dnasp51001.msi191Sigmaplot.rar192SPSS22.0.rar。
《纳米抗体研究进展综述》3300字

纳米抗体研究进展综述摘要:单域抗体因其独特的优势,如水溶性好、分子量小、稳定性好、免疫原性小等一系列特点,在生物研究和医学领域中的作用愈发广泛。
在疾病诊断、病原检测、癌症疾病治疗、药物残留检测分析,坏境检测,用作sdAbs分子探针、分子诊断和显影等等领域具有广阔的应用前景。
纳米抗体因其优势,可实现重组表达,从而使得生产周期和生产成本均可大幅下降,是目前国内外研发的热点。
作者重点介绍了纳米抗体的特点,然后简述了纳米抗体的制备流程,简述了纳米抗体在疾病诊断、疾病治疗、食品安全和环境监测等领域的应用,最后对纳米抗体的应用前景进行了分析和展望。
1 介绍自1890年,第一种抗体——抗毒素,这是在血清中发现的第一种抗体[1]。
这是一种可中和外毒素的物质,1975年,杂交瘤技术的诞生开始了抗体研究和应用快速发展的时代。
由于抗体可特异性识别和结合抗原的特性,使其在疾病诊断、疾病治疗、药物运载、病原、毒素和小分子化合物检测等领域具有广泛的应用[2]。
但通过单克隆抗体技术制备的传统单克隆抗体有其不可忽视的缺点:生产耗时长、成本高、在组织和肿瘤中穿透力差、长期使用会引起机体免疫排斥反应以及动物道德问题等。
相比于传统抗体,纳米抗体具备传统抗体不具备的分子质量小和穿透性强的优势而成为现在抗体研究的主要方向之一。
单链抗体(single chain antibody fragment,scFv)就是新型小分子抗体的一种,其穿透力更强、生产成本更低,但scFv抗体存在溶解度低、稳定性较差、表达量低、易聚合和亲和力低的缺点[3]。
1989年,比利时免疫学家Hamers-Casterman 在骆驼血清中的偶然发现一种天然缺失轻链的重链抗体(HcAbs)可以解决scFv所存在的问题,重链抗体只包含2个常规的CH2与CH3区和1个重链可变区(VHH),重链可变区具有与原重链抗体相当的结构稳定性以及与抗原的结合活性,是已知的可结合目标抗原的最小单位,其分子质量只有单克隆抗体的1/10,是迄今为止获得的结构稳定且具有抗原结合活性的最小抗体单位,因此也被称作纳米抗体(nanobody,Nb)[4]。
检验专业英语试题及答案

检验专业英语试题及答案一、选择题(每题2分,共20分)1. Which of the following is not a routine test in clinical laboratory?A. Blood countB. Urine analysisC. Liver function testD. DNA sequencing2. The term "hemoglobin" refers to:A. A type of proteinB. A type of enzymeC. A type of hormoneD. A type of lipid3. What is the primary function of the enzyme amylase?A. To break down proteinsB. To break down carbohydratesC. To break down fatsD. To break down nucleic acids4. The process of identifying the presence of a specific microorganism in a sample is known as:A. CulturingB. IsolationC. IdentificationD. Quantification5. Which of the following is a common method for measuring the concentration of glucose in blood?A. SpectrophotometryB. ChromatographyC. ElectrophoresisD. Enzymatic assay6. The term "ELISA" stands for:A. Enzyme-Linked Immunosorbent AssayB. Electrophoresis-Linked Immunosorbent AssayC. Enzyme-Linked Immunofluorescence AssayD. Electrophoresis-Linked Immunofluorescence Assay7. In medical diagnostics, what does "PCR" refer to?A. Polymerase Chain ReactionB. Protein Chain ReactionC. Particle Count ReactionD. Pathogen Characterization Reaction8. The process of measuring the amount of a specific substance in a sample is known as:A. TitrationB. CalibrationC. QuantificationD. Qualification9. Which of the following is a common type of clinical specimen?A. BloodB. SoilC. HairD. Water10. The term "antibodies" refers to:A. Proteins that recognize and bind to specific antigensB. Substances that neutralize toxinsC. Hormones that regulate immune responseD. Cells that produce immune responses二、填空题(每空1分,共10分)1. The process of separating molecules based on their size is known as __________.2. In clinical chemistry, the term "assay" refers to a__________ method.3. The unit of measurement for pH is __________.4. A common method for detecting the presence of antibodies in a sample is the __________ test.5. The process of identifying the type of bacteria in a sample is known as __________.6. The process of separating DNA fragments based on their size is known as __________.7. The term "ELISA" is used in __________ to detect the presence of specific antibodies or antigens.8. The process of identifying the genetic makeup of an organism is known as __________.9. The process of measuring the amount of a substance in a sample using a specific wavelength of light is called__________.10. The process of identifying the presence of specific microorganisms in a sample is known as __________.三、简答题(每题5分,共20分)1. Describe the principle of the Enzyme-Linked Immunosorbent Assay (ELISA).2. Explain the importance of maintaining aseptic technique ina clinical laboratory.3. What are the steps involved in performing a blood count?4. Discuss the role of antibodies in the immune response.四、论述题(每题15分,共30分)1. Compare and contrast the methods of Chromatography and Electrophoresis in terms of their applications in clinical diagnostics.2. Discuss the ethical considerations in the use of genetic testing for medical purposes.五、翻译题(每题5分,共10分)1. 将以下句子从中文翻译成英文:在临床实验室中,酶联免疫吸附测定法是一种常用的检测特定抗体或抗原的方法。
DAPI染DNA的原理及使用方法

DAPI染DNA的原理及使用方法DAPI 即4',6-二脒基-2-苯基吲哚(4',6-diamidino-2-phenylindole),分子式为C16H15N5·2C3H6O3,分子量457.48。
DAPI是一种能够与DNA强力结合的荧光染料。
它结合到双链DNA小沟的AT碱基对处,一个DAPI分子可以占据三个碱基对的位置。
结合到双链DNA上DAPI分子的荧光强度提高大约20倍,常用与荧光显微镜观测,根据荧光的强度可以确定DNA的量。
另外,因为DAPI可以透过完整的细胞膜,它可以用于活细胞和固定细胞的染色。
在荧光显微镜观察下,DAPI染剂是利用紫外光波长的光线激发。
单独DAPI的最大吸收波长为340nm,最大发射波长为488nm;当DAPI与双链DNA结合时,最大吸收波长为364nm,最大发射波长为454nm(10 mM Tris pH7.0,100 mM NaCl,10 mM EDTA),其发散光的波长范围含盖了蓝色至青绿色。
DAPI也可以和RNA 结合,但产生的荧光强度只有与DNA结合的荧光强度的1/5,其发散光的波长范围约在500nm左右。
DAPI的配制及贮存固体粉末:避光,2-8 ℃保存,3年没有问题。
贮存液:用无菌水配制成浓度1 mg/ml的贮存液,配好后用锡纸包起来,避光,可在-20 ℃下长期保存。
(DAPI易溶于水,在PBS中溶解度不高)使用浓度:贮存液用1xPBS稀释到终浓度100 ng/ml。
荧光封片液:0.5 mol/L碳酸盐缓冲液与甘油等体积混合,pH9.5染色与观察:制好的玻片上滴加几滴DAPI染液,染色10分钟,流水冲去染液,滤纸吸除多余水分,加一滴荧光封片液,置于荧光显微镜下观察,激发波长360 -400nm。
注意事项:DAPI可能具有致癌性,全部操作过程中必须带塑料或乳胶手套。
推荐厂商:Invitrogen,Sigma资料下载:点击下载结构式:DAPI结构式DAPI与DNA的复合物晶体结构以下英文介绍摘自Wikipedia。
大鼠皮质脊髓束和红核脊髓束定位损伤后运动皮质和红核神经细胞

山西医科大学硕士学位论文大鼠皮质脊髓束和红核脊髓束定位损伤后运动皮质和红核神经细胞凋亡及NT-3表达的变化姓名:王茂申请学位级别:硕士专业:骨外科学指导教师:孙天胜20100315 山Ifl『医科人学硕上学位论文大鼠皮质脊髓束和红核脊髓束定位损伤后运动皮质和红核神经细胞凋亡及NT-3表达的变化。
研究生:王茂专业:骨外科导师:孙天胜摘要目的:1.观察分别损伤大鼠双侧皮质脊髓背侧束和红核脊髓束后相应的运动皮质内的贝茨细胞和红核细胞凋亡的形态及数量的变化。
2.观察分别损伤大鼠双侧皮质脊髓背侧束和红核脊髓束后相应的运动皮质内的贝茨细胞和红核细胞神经营养因子NT一3的表达变化。
3.分析两者的变化是否能反映进化论的观点,即进化低的结构的修复能力大于进化高的结构。
方法:分别建立SD大鼠双侧皮质脊髓背侧束损伤模型和双侧红核脊髓束损伤模型,采用原位细胞凋亡TUNEL法检测两组术后1d、3d、7d、14d大脑运动皮质及红核中神经细胞凋亡的形态及凋亡指数的变化;采用免疫组织化学方法检测神经营养因子NT一3的阳性细胞数及表达变化。
结果:1.皮质脊髓背侧束损伤和红核脊髓束损伤后原位细胞凋亡TUNEL法检测神经细胞凋亡结果:不同时问点大脑皮质及红核均有凋亡细胞分布,细胞核呈紫色及黄褐色双重染色,形态不一;损伤后1d,皮质脊髓背侧束损伤组的运动皮质及红核脊髓束损伤组红核的神经细胞凋亡数量增加不明显;损伤后3d,两组相应的大脑皮质及红核有大量凋亡细胞出现,并持续至14d,RST损伤组凋亡指数较dCST损伤组低(P<0.05);各时间点凋亡细胞数量与假手术组相比均增加(P<0.05);凋亡神经细胞在伤后14d较7d少,RST损伤组凋亡指数在7d及14d均较dCST损伤组低(P<0.05)。
2.免疫组织化学方法检测神经营养因子NT一3结果:NT一3在两组损伤后不同时间点在相应的运动皮质和红核均有表达;与假手术组相比,皮质脊髓背侧束损伤组于术后7d,NT一3在运动皮质表达达高峰;红核脊髓束组于术后3d,NT一3在红核表达达高峰,后缓慢下降。
SiriusRedstaining天狼星红染色方法

SiriusRedstaining天狼星红染色方法1、来自丁香园Sirius red苦陈酸染色法[试剂配制](1)天狼星红饱和苦昧酸液 O(5,天狼星红10ml,苦味酸饱和液90ml。
(2)天青石蓝液天青石蓝B1(25g(铁明矾1(25g,蒸馏水250ml。
溶解煮沸、待冷却过滤加入甘油30ml,然后再加入浓盐酸0(5ml。
[染色步骤](1)中性甲醛液固定组织,石蜡切片,常规脱蜡至水。
(2)人大青石蓝液染5一lOmin。
(3)蒸馏水洗3次。
(4)天狼星红饱和苦昧酸浓染15-30min(5)无水乙醇直接分化与脱水。
二甲苯透明,中性树胶封固。
[注意事项](1)细胞核里染色可以用Harris苏木素染色液谈染。
(2)染色封固后的切片,须及时用偏光显微镜进行观察和照相已保持鲜艳的色彩注:在偏光显微镜下可以观察到四种类型的胶原纤维。
l型胶原纤维:紧密排列,显示很强的双折光性,呈黄色或红色的纤维oll型胶原纤维:显示弱的双折光,呈多种色彩的疏松网状分布。
皿型胶原纤维:显示弱的双折光,呈绿色的细纤维。
lv型胶原纤维:显示弱的双折光的基膜,呈淡黄色。
2、来自网页Sirius Red Staining Protocol for CollagenJohn A. KiernanDepartment of Anatomy & Cell Biology,The University of Western Ontario,LONDON, Canada N6A 5C1NovaUltra Special Stain KitsDescription: It's one of the best understood techniques of collagen histochemistry. Technical details follow, and are followed by some comments and a few references. You should come to grips with the theory, advantages and limitations of this method before using it on alarge scale. Picro-sirius red method (after Puchtler et al., 1973; Junqueira et al., 1979). Step 4 is an addition that prevents the loss of dye that happens if the stained sections are washed in water.Fixation: Fixation is not critical, The method is most frequently used on paraffin sections of objects fixed adequately (at least 24 hours but ideally 1 or 2 weeks) in a neutral buffered formaldehyde solution. This protocol has not been tested on frozen sections.Solutions and Reagents:Picro-sirius Red SolutionSirius red F3B (C.I. 35782) ------------------------- 0.5 gSaturated aqueous solution of picric acid ---------500 ml(Keeps for at least 3 years and can be used many times)Sirius Red is available from Sigma-Aldrich under the name of "Direct Red 80" Cat#365548 or Cat#43665. Saturated aqueous solution of picric acid (1.3% in water) is also available from Sigma, Cat# P6744-1GA.Acidified WaterAdd 5 ml acetic acid (glacial) to 1 liter of water (tap or distilled).Weigert's haematoxylinProcedure:1. De-wax and hydrate paraffin sections.2. Stain nuclei with Weigert's haematoxylin for 8 minutes, and then wash the slides for 10 minutes in running tap water).3. Stain in picro-sirius red for one hour (This gives near-equilibrium staining, which does not increase with longer times. Shorter times should not be used, even if the colors look OK.)4. Wash in two changes of acidified water.5. Physically remove most of the water from the slides by vigorous shaking.5. Dehydrate in three changes of 100% ethanol.6. Clear in xylene and mount in a resinous medium.Results:In bright-field microscopy collagen is red on a pale yellow background. (Nuclei, if stained, are ideally black but may often be greyor brown. The long time in picro-sirius red causes appreciable de-staining of the nuclei. This is not a problem with traditional van Gieson or with picro-aniline blue, with their 1-minute staining times.) When examined through crossed polars the larger collagen fibers are bright yellow or orange, and the thinner ones, including reticular fibers, are green. According to Junqueira et al. (1979) thebirefringence is highly specific for collagen. A few materials,including Type 4 collagen in basement membranes, keratohyaline granules and some types of mucus, are stained red but are not birefringent. It is necessary to rotate the slide in order to see all the fibres, because in any single orientation the birefringence of some fibres will be extinguished. This minor inconvenience can be circumvented by equipping the microscope for use with circularly rather than plane polarized light (Whittaker et al., 1994; Whittaker, 1995), but then you don't get a completely black background. Comments and References:Although this method is technically very easy, it is important for the person doing it and (if it's someone else) the person using the stained slides, to know what it does and how it works. Even without a polarizingmicroscope, picro-sirius red shows things like reticular fibres and the basal laminae of cerebral capillaries, which are missed by van Gieson and may be obscured by masses of other stained details in trichrome methods (Mallory, Masson, Heidenhain etc).To the best of my knowledge, most users of picro-sirius red aredoing research that exploits the enhancement by sirius red of the birefringence of collagen fibres, which is largely due to co-aligned molecules of Type I collagen. It is also used to stain amyloid.If you are using only polarized light it does not matter if you lose the "yellow background" of picric acid staining. If you use picro-sirius red as a "better" van Gieson and want to keep the yellow cytoplasm, be hasty with the dehydrating - even more so than with the original van Gieson method.About 4 years ago, someone (sorry, I've forgotten who, so I can't shout your name) posted to HistoNet an excellent bibliography ofstaining methods using sirius red F3B. This should be findable in the Archives()Nobody should do (or order to be done) a picro-sirius red stain without reading at least one of the first two items listed below.1. Junqueira LCU, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J 1979; 11, 447-4552. Puchtler H, Waldrop FS, Valentine LS. Polarization microscopic studies of connective tissue stained with picro-sirius red FBA. Beitr Path 1973; 150, 174-1873. Whittaker P. Polarized light microscopy in biomedical research. Microscopy and Analysis 1995; 44, 15-174. Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Research in Cardiology 1994; 89, 397-4105. Kiernan. J.A., (1999) Histological and Histochemical Methods: Theory and Practice, Ed. 3 Butterworth Heinemann, Oxford, UK.Finally, it's important to get the right dye. Sirius red F3B is C.I. 35782 (Direct red 80). There are other "sirius red"s that are quite different. At least one that I've used a lot is OK but does not carryany C.I. designation on the label. With this kind of dye (a tetra-azo direct cotton dye) the manufacturing process necessarily generates more than one coloured product, and other compounds are added to precipitate the dye and adjust its colour intensity. Test your sirius red onsections of muscle, brain andkidney before using it for research or diagnosis. In normal kidneythe glomerular basement membranes should be red but not birefringent. Every muscle fibre should be surrounded by red and birefringent collagen.I could continue, but this is already too long.。
Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease trans

Reproduction numbers and sub-threshold endemicequilibria for compartmental models of disease transmissionP.van den Driesschea,1,James Watmough b,*,2aDepartment of Mathematics and Statistics,University of Victoria,Victoria,BC,Canada V8W 3P4b Department of Mathematics and Statistics,University of New Brunswick,Fredericton,NB,Canada E3B 5A3Received 26April 2001;received in revised form 27June 2001;accepted 27June 2001Dedicated to the memory of John JacquezAbstractA precise definition of the basic reproduction number,R 0,is presented for a general compartmental disease transmission model based on a system of ordinary differential equations.It is shown that,if R 0<1,then the disease free equilibrium is locally asymptotically stable;whereas if R 0>1,then it is unstable.Thus,R 0is a threshold parameter for the model.An analysis of the local centre manifold yields a simple criterion for the existence and stability of super-and sub-threshold endemic equilibria for R 0near one.This criterion,together with the definition of R 0,is illustrated by treatment,multigroup,staged progression,multistrain and vector–host models and can be applied to more complex models.The results are significant for disease control.Ó2002Elsevier Science Inc.All rights reserved.Keywords:Basic reproduction number;Sub-threshold equilibrium;Disease transmission model;Disease control1.IntroductionOne of the most important concerns about any infectious disease is its ability to invade a population.Many epidemiological models have a disease free equilibrium (DFE)at whichtheMathematical Biosciences 180(2002)29–48/locate/mbs*Corresponding author.Tel.:+1-5064587323;fax:+1-5064534705.E-mail addresses:pvdd@math.uvic.ca (P.van den Driessche),watmough@unb.ca (J.Watmough).URL:http://www.math.unb.ca/$watmough.1Research supported in part by an NSERC Research Grant,the University of Victoria Committee on faculty research and travel and MITACS.2Research supported by an NSERC Postdoctoral Fellowship tenured at the University of Victoria.0025-5564/02/$-see front matter Ó2002Elsevier Science Inc.All rights reserved.PII:S0025-5564(02)00108-630P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–48population remains in the absence of disease.These models usually have a threshold parameter, known as the basic reproduction number,R0,such that if R0<1,then the DFE is locally as-ymptotically stable,and the disease cannot invade the population,but if R0>1,then the DFE is unstable and invasion is always possible(see the survey paper by Hethcote[1]).Diekmann et al.[2]define R0as the spectral radius of the next generation matrix.We write down in detail a general compartmental disease transmission model suited to heterogeneous populations that can be modelled by a system of ordinary differential equations.We derive an expression for the next generation matrix for this model and examine the threshold R0¼1in detail.The model is suited to a heterogeneous population in which the vital and epidemiological parameters for an individual may depend on such factors as the stage of the disease,spatial position,age or behaviour.However,we assume that the population can be broken into homo-geneous subpopulations,or compartments,such that individuals in a given compartment are indistinguishable from one another.That is,the parameters may vary from compartment to compartment,but are identical for all individuals within a given compartment.We also assume that the parameters do not depend on the length of time an individual has spent in a compart-ment.The model is based on a system of ordinary equations describing the evolution of the number of individuals in each compartment.In addition to showing that R0is a threshold parameter for the local stability of the DFE, we apply centre manifold theory to determine the existence and stability of endemic equilib-ria near the threshold.We show that some models may have unstable endemic equilibria near the DFE for R0<1.This suggests that even though the DFE is locally stable,the disease may persist.The model is developed in Section2.The basic reproduction number is defined and shown to bea threshold parameter in Section3,and the definition is illustrated by several examples in Section4.The analysis of the centre manifold is presented in Section5.The epidemiological ramifications of the results are presented in Section6.2.A general compartmental epidemic model for a heterogeneous populationConsider a heterogeneous population whose individuals are distinguishable by age,behaviour, spatial position and/or stage of disease,but can be grouped into n homogeneous compartments.A general epidemic model for such a population is developed in this section.Let x¼ðx1;...;x nÞt, with each x i P0,be the number of individuals in each compartment.For clarity we sort the compartments so that thefirst m compartments correspond to infected individuals.The distinc-tion between infected and uninfected compartments must be determined from the epidemiological interpretation of the model and cannot be deduced from the structure of the equations alone,as we shall discuss below.It is plausible that more than one interpretation is possible for some models.A simple epidemic model illustrating this is given in Section4.1.The basic reproduction number can not be determined from the structure of the mathematical model alone,but depends on the definition of infected and uninfected compartments.We define X s to be the set of all disease free states.That isX s¼f x P0j x i¼0;i¼1;...;m g:In order to compute R0,it is important to distinguish new infections from all other changes inpopulation.Let F iðxÞbe the rate of appearance of new infections in compartment i,Vþi ðxÞbe therate of transfer of individuals into compartment i by all other means,and VÀi ðxÞbe the rate oftransfer of individuals out of compartment i.It is assumed that each function is continuously differentiable at least twice in each variable.The disease transmission model consists of non-negative initial conditions together with the following system of equations:_x i¼f iðxÞ¼F iðxÞÀV iðxÞ;i¼1;...;n;ð1Þwhere V i¼VÀi ÀVþiand the functions satisfy assumptions(A1)–(A5)described below.Sinceeach function represents a directed transfer of individuals,they are all non-negative.Thus,(A1)if x P0,then F i;Vþi ;VÀiP0for i¼1;...;n.If a compartment is empty,then there can be no transfer of individuals out of the compartment by death,infection,nor any other means.Thus,(A2)if x i¼0then VÀi ¼0.In particular,if x2X s then VÀi¼0for i¼1;...;m.Consider the disease transmission model given by(1)with f iðxÞ,i¼1;...;n,satisfying con-ditions(A1)and(A2).If x i¼0,then f iðxÞP0and hence,the non-negative cone(x i P0, i¼1;...;n)is forward invariant.By Theorems1.1.8and1.1.9of Wiggins[3,p.37]for each non-negative initial condition there is a unique,non-negative solution.The next condition arises from the simple fact that the incidence of infection for uninfected compartments is zero.(A3)F i¼0if i>m.To ensure that the disease free subspace is invariant,we assume that if the population is free of disease then the population will remain free of disease.That is,there is no(density independent) immigration of infectives.This condition is stated as follows:(A4)if x2X s then F iðxÞ¼0and VþiðxÞ¼0for i¼1;...;m.The remaining condition is based on the derivatives of f near a DFE.For our purposes,we define a DFE of(1)to be a(locally asymptotically)stable equilibrium solution of the disease free model,i.e.,(1)restricted to X s.Note that we need not assume that the model has a unique DFE. Consider a population near the DFE x0.If the population remains near the DFE(i.e.,if the introduction of a few infective individuals does not result in an epidemic)then the population will return to the DFE according to the linearized system_x¼Dfðx0ÞðxÀx0Þ;ð2Þwhere Dfðx0Þis the derivative½o f i=o x j evaluated at the DFE,x0(i.e.,the Jacobian matrix).Here, and in what follows,some derivatives are one sided,since x0is on the domain boundary.We restrict our attention to systems in which the DFE is stable in the absence of new infection.That is, (A5)If FðxÞis set to zero,then all eigenvalues of Dfðx0Þhave negative real parts.P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–4831The conditions listed above allow us to partition the matrix Df ðx 0Þas shown by the following lemma.Lemma 1.If x 0is a DFE of (1)and f i ðx Þsatisfies (A1)–(A5),then the derivatives D F ðx 0Þand D V ðx 0Þare partitioned asD F ðx 0Þ¼F 000 ;D V ðx 0Þ¼V 0J 3J 4;where F and V are the m Âm matrices defined byF ¼o F i o x j ðx 0Þ !and V ¼o V i o x jðx 0Þ !with 16i ;j 6m :Further ,F is non-negative ,V is a non-singular M-matrix and all eigenvalues of J 4have positive real part .Proof.Let x 02X s be a DFE.By (A3)and (A4),ðo F i =o x j Þðx 0Þ¼0if either i >m or j >m .Similarly,by (A2)and (A4),if x 2X s then V i ðx Þ¼0for i 6m .Hence,ðo V i =o x j Þðx 0Þ¼0for i 6m and j >m .This shows the stated partition and zero blocks.The non-negativity of F follows from (A1)and (A4).Let f e j g be the Euclidean basis vectors.That is,e j is the j th column of the n Ân identity matrix.Then,for j ¼1;...;m ,o V i o x jðx 0Þ¼lim h !0þV i ðx 0þhe j ÞÀV i ðx 0Þh :To show that V is a non-singular M-matrix,note that if x 0is a DFE,then by (A2)and (A4),V i ðx 0Þ¼0for i ¼1;...;m ,and if i ¼j ,then the i th component of x 0þhe j ¼0and V i ðx 0þhe j Þ60,by (A1)and (A2).Hence,o V i =o x j 0for i m and j ¼i and V has the Z sign pattern (see Appendix A).Additionally,by (A5),all eigenvalues of V have positive real parts.These two conditions imply that V is a non-singular M-matrix [4,p.135(G 20)].Condition (A5)also implies that the eigenvalues of J 4have positive real part.Ã3.The basic reproduction numberThe basic reproduction number,denoted R 0,is ‘the expected number of secondary cases produced,in a completely susceptible population,by a typical infective individual’[2];see also [5,p.17].If R 0<1,then on average an infected individual produces less than one new infected individual over the course of its infectious period,and the infection cannot grow.Conversely,if R 0>1,then each infected individual produces,on average,more than one new infection,and the disease can invade the population.For the case of a single infected compartment,R 0is simply the product of the infection rate and the mean duration of the infection.However,for more complicated models with several infected compartments this simple heuristic definition of R 0is32P.van den Driessche,J.Watmough /Mathematical Biosciences 180(2002)29–48insufficient.A more general basic reproduction number can be defined as the number of new infections produced by a typical infective individual in a population at a DFE.To determine the fate of a‘typical’infective individual introduced into the population,we consider the dynamics of the linearized system(2)with reinfection turned off.That is,the system _x¼ÀD Vðx0ÞðxÀx0Þ:ð3ÞBy(A5),the DFE is locally asymptotically stable in this system.Thus,(3)can be used to de-termine the fate of a small number of infected individuals introduced to a disease free population.Let wi ð0Þbe the number of infected individuals initially in compartment i and letwðtÞ¼w1ðtÞ;...;w mðtÞðÞt be the number of these initially infected individuals remaining in the infected compartments after t time units.That is the vector w is thefirst m components of x.The partitioning of D Vðx0Þimplies that wðtÞsatisfies w0ðtÞ¼ÀV wðtÞ,which has the unique solution wðtÞ¼eÀVt wð0Þ.By Lemma1,V is a non-singular M-matrix and is,therefore,invertible and all of its eigenvalues have positive real parts.Thus,integrating F wðtÞfrom zero to infinity gives the expected number of new infections produced by the initially infected individuals as the vector FVÀ1wð0Þ.Since F is non-negative and V is a non-singular M-matrix,VÀ1is non-negative[4,p.137 (N38)],as is FVÀ1.To interpret the entries of FVÀ1and develop a meaningful definition of R0,consider the fate of an infected individual introduced into compartment k of a disease free population.The(j;k)entry of VÀ1is the average length of time this individual spends in compartment j during its lifetime, assuming that the population remains near the DFE and barring reinfection.The(i;j)entry of F is the rate at which infected individuals in compartment j produce new infections in compartment i. Hence,the(i;k)entry of the product FVÀ1is the expected number of new infections in com-partment i produced by the infected individual originally introduced into compartment k.Fol-lowing Diekmann et al.[2],we call FVÀ1the next generation matrix for the model and set R0¼qðFVÀ1Þ;ð4Þwhere qðAÞdenotes the spectral radius of a matrix A.The DFE,x0,is locally asymptotically stable if all the eigenvalues of the matrix Dfðx0Þhave negative real parts and unstable if any eigenvalue of Dfðx0Þhas a positive real part.By Lemma1, the eigenvalues of Dfðx0Þcan be partitioned into two sets corresponding to the infected and uninfected compartments.These two sets are the eigenvalues of FÀV and those ofÀJ4.Again by Lemma1,the eigenvalues ofÀJ4all have negative real part,thus the stability of the DFE is determined by the eigenvalues of FÀV.The following theorem states that R0is a threshold parameter for the stability of the DFE.Theorem2.Consider the disease transmission model given by(1)with fðxÞsatisfying conditions (A1)–(A5).If x0is a DFE of the model,then x0is locally asymptotically stable if R0<1,but un-stable if R0>1,where R0is defined by(4).Proof.Let J1¼FÀV.Since V is a non-singular M-matrix and F is non-negative,ÀJ1¼VÀF has the Z sign pattern(see Appendix A).Thus,sðJ1Þ<0()ÀJ1is a non-singular M-matrix;P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–483334P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–48where sðJ1Þdenotes the maximum real part of all the eigenvalues of the matrix J1(the spectral abscissa of J1).Since FVÀ1is non-negative,ÀJ1VÀ1¼IÀFVÀ1also has the Z sign pattern.Ap-plying Lemma5of Appendix A,with H¼V and B¼ÀJ1¼VÀF,we have ÀJ1is a non-singular M-matrix()IÀFVÀ1is a non-singular M-matrix:Finally,since FVÀ1is non-negative,all eigenvalues of FVÀ1have magnitude less than or equal to qðFVÀ1Þ.Thus,IÀFVÀ1is a non-singular M-matrix;()qðFVÀ1Þ<1:Hence,sðJ1Þ<0if and only if R0<1.Similarly,it follows thatsðJ1Þ¼0()ÀJ1is a singular M-matrix;()IÀFVÀ1is a singular M-matrix;()qðFVÀ1Þ¼1:The second equivalence follows from Lemma6of Appendix A,with H¼V and K¼F.The remainder of the equivalences follow as with the non-singular case.Hence,sðJ1Þ¼0if and only if R0¼1.It follows that sðJ1Þ>0if and only if R0>1.ÃA similar result can be found in the recent book by Diekmann and Heesterbeek[6,Theorem6.13].This result is known for the special case in which J1is irreducible and V is a positive di-agonal matrix[7–10].The special case in which V has positive diagonal and negative subdiagonal elements is proven in Hyman et al.[11,Appendix B];however,our approach is much simpler(see Section4.3).4.Examples4.1.Treatment modelThe decomposition of fðxÞinto the components F and V is illustrated using a simple treat-ment model.The model is based on the tuberculosis model of Castillo-Chavez and Feng[12,Eq.(1.1)],but also includes treatment failure used in their more elaborate two-strain model[12,Eq.(2.1)].A similar tuberculosis model with two treated compartments is proposed by Blower et al.[13].The population is divided into four compartments,namely,individuals susceptible to tu-berculosis(S),exposed individuals(E),infectious individuals(I)and treated individuals(T).The dynamics are illustrated in Fig.1.Susceptible and treated individuals enter the exposed com-partment at rates b1I=N and b2I=N,respectively,where N¼EþIþSþT.Exposed individuals progress to the infectious compartment at the rate m.All newborns are susceptible,and all indi-viduals die at the rate d>0.Thus,the core of the model is an SEI model using standard inci-dence.The treatment rates are r1for exposed individuals and r2for infectious individuals. However,only a fraction q of the treatments of infectious individuals are successful.Unsuc-cessfully treated infectious individuals re-enter the exposed compartment(p¼1Àq).The diseasetransmission model consists of the following differential equations together with non-negative initial conditions:_E¼b1SI=Nþb2TI=NÀðdþmþr1ÞEþpr2I;ð5aÞ_I¼m EÀðdþr2ÞI;ð5bÞ_S¼bðNÞÀdSÀb1SI=N;ð5cÞ_T¼ÀdTþr1Eþqr2IÀb2TI=N:ð5dÞProgression from E to I and failure of treatment are not considered to be new infections,but rather the progression of an infected individual through the various compartments.Hence,F¼b1SI=Nþb2TI=NB B@1C CA and V¼ðdþmþr1ÞEÀpr2IÀm Eþðdþr2ÞIÀbðNÞþdSþb1SI=NdTÀr1EÀqr2Iþb2TI=NB B@1C CA:ð6ÞThe infected compartments are E and I,giving m¼2.An equilibrium solution with E¼I¼0has the form x0¼ð0;0;S0;0Þt,where S0is any positive solution of bðS0Þ¼dS0.This will be a DFE if and only if b0ðS0Þ<d.Without loss of generality,assume S0¼1is a DFE.Then,F¼0b100;V¼dþmþr1Àpr2Àm dþr2;givingVÀ1¼1ðdþmþr1Þðdþr2ÞÀm pr2dþr2pr2m dþmþr1and R0¼b1m=ððdþmþr1Þðdþr2ÞÀm pr2Þ.A heuristic derivation of the(2;1)entry of VÀ1and R0are as follows:a fraction h1¼m=ðdþmþr1Þof exposed individuals progress to compartment I,a fraction h2¼pr2=ðdþr2Þof infectious individuals re-enter compartment E.Hence,a fractionh1of exposed individuals pass through compartment I at least once,a fraction h21h2passthroughat least twice,and a fraction h k 1h k À12pass through at least k times,spending an average of s ¼1=ðd þr 2Þtime units in compartment I on each pass.Thus,an individual introduced into com-partment E spends,on average,s ðh 1þh 21h 2þÁÁÁÞ¼s h 1=ð1Àh 1h 2Þ¼m =ððd þm þr 1Þðd þr 2ÞÀm pr 2Þtime units in compartment I over its expected lifetime.Multiplying this by b 1gives R 0.The model without treatment (r 1¼r 2¼0)is an SEI model with R 0¼b 1m =ðd ðd þm ÞÞ.The interpretation of R 0for this case is simpler.Only a fraction m =ðd þm Þof exposed individuals progress from compartment E to compartment I ,and individuals entering compartment I spend,on average,1=d time units there.Although conditions (A1)–(A5)do not restrict the decomposition of f i ðx Þto a single choice for F i ,only one such choice is epidemiologically correct.Different choices for the function F lead to different values for the spectral radius of FV À1,as shown in Table 1.In column (a),treatment failure is considered to be a new infection and in column (b),both treatment failure and pro-gression to infectiousness are considered new infections.In each case the condition q ðFV À1Þ<1yields the same portion of parameter space.Thus,q ðFV À1Þis a threshold parameter in both cases.The difference between the numbers lies in the epidemiological interpretation rather than the mathematical analysis.For example,in column (a),the infection rate is b 1þpr 2and an exposed individual is expected to spend m =ððd þm þr 1Þðd þr 2ÞÞtime units in compartment I .However,this reasoning is biologically flawed since treatment failure does not give rise to a newly infected individual.Table 1Decomposition of f leading to alternative thresholds(a)(b)Fb 1SI =N þb 2TI =N þpr 2I 0000B B @1C C A b 1SI =N þb 2TI =N þpr 2I m E 000B B @1C C A Vðd þm þr 1ÞE Àm E þðd þr 2ÞI Àb ðN ÞþdS þb 1SI =N dT Àr 1E Àqr 2I þb 2TI =N 0B B @1C C A ðd þm þr 1ÞE ðd þr 2ÞI Àb ðN ÞþdS þb 1SI =N dT Àr 1E Àqr 2I þb 2TI =N 0B B @1C C A F0b 1þpr 200 0b 1þpr 2m 0 V d þm þr 10Àm d þr 2d þm þr 100d þr 2 q (FV À1)b 1m þpr 2mðd þm þr 1Þðd þr 2Þffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffib 1m þpr 2mðd þm þr 1Þðd þr 2Þs 36P.van den Driessche,J.Watmough /Mathematical Biosciences 180(2002)29–484.2.Multigroup modelIn the epidemiological literature,the term‘multigroup’usually refers to the division of a het-erogeneous population into several homogeneous groups based on individual behaviour(e.g., [14]).Each group is then subdivided into epidemiological compartments.The majority of mul-tigroup models in the literature are used for sexually transmitted diseases,such as HIV/AIDS or gonorrhea,where behaviour is an important factor in the probability of contracting the disease [7,8,14,15].As an example,we use an m-group SIRS-vaccination model of Hethcote[7,14]with a generalized incidence term.The sample model includes several SI multigroup models of HIV/ AIDS as special cases[8,15].The model equations are as follows:_I i ¼X mj¼1b ijðxÞS i I jÀðd iþc iþ iÞI i;ð7aÞ_S i ¼ð1Àp iÞb iÀðd iþh iÞS iþr i R iÀX mj¼1b ijðxÞS i I j;ð7bÞ_Ri¼p i b iþc i I iþh i S iÀðd iþr iÞR i;ð7cÞfor i¼1;...;m,where x¼ðI1;...;I m;S1;...;S m;R1;...;R mÞt.Susceptible and removed individu-als die at the rate d i>0,whereas infected individuals die at the faster rate d iþ i.Infected in-dividuals recover with temporary immunity from re-infection at the rate c i,and immunity lasts an expected1=r i time units.All newborns are susceptible,and a constant fraction b i are born into each group.A fraction p i of newborns are vaccinated at birth.Thereafter,susceptible individuals are vaccinated at the rate h i.The incidence,b ijðxÞdepends on individual behaviour,which determines the amount of mixing between the different groups(see,e.g.,Jacquez et al.[16]). The DFE for this model isx0¼ð0;...;0;S01;...;S0m;R01;...;R0mÞt;whereS0 i ¼b i d ið1Àp iÞþr iðÞd iðd iþh iþr iÞ;R0 i ¼b iðh iþd i p iÞd iðd iþh iþr iÞ:Linearizing(7a)about x¼x0givesF¼S0i b ijðx0ÞÂÃandV¼½ðd iþc iþ iÞd ij ;where d ij is one if i¼j,but zero otherwise.Thus,FVÀ1¼S0i b ijðx0Þ=ðd iÂþc iþ iÞÃ:P.van den Driessche,J.Watmough/Mathematical Biosciences180(2002)29–4837For the special case with b ij separable,that is,b ijðxÞ¼a iðxÞk jðxÞ,F has rank one,and the basic reproduction number isR0¼X mi¼1S0ia iðx0Þk iðx0Þd iþc iþ i:ð8ÞThat is,the basic reproduction number of the disease is the sum of the‘reproduction numbers’for each group.4.3.Staged progression modelThe staged progression model[11,Section3and Appendix B]has a single uninfected com-partment,and infected individuals progress through several stages of the disease with changing infectivity.The model is applicable to many diseases,particularly HIV/AIDS,where transmission probabilities vary as the viral load in an infected individual changes.The model equations are as follows(see Fig.2):_I 1¼X mÀ1k¼1b k SI k=NÀðm1þd1ÞI1;ð9aÞ_Ii¼m iÀ1I iÀ1Àðm iþd iÞI i;i¼2;...;mÀ1;ð9bÞ_Im¼m mÀ1I mÀ1Àd m I m;ð9cÞ_S¼bÀbSÀX mÀ1k¼1b k SI k=N:ð9dÞThe model assumes standard incidence,death rates d i>0in each infectious stage,and thefinal stage has a zero infectivity due to morbidity.Infected individuals spend,on average,1=m i time units in stage i.The unique DFE has I i¼0,i¼1;...;m and S¼1.For simplicity,define m m¼0. Then F¼½F ij and V¼½V ij ,whereF ij¼b j i¼1;j6mÀ1;0otherwise;&ð10ÞV ij¼m iþd i j¼i;Àm j i¼1þj;0otherwise:8<:ð11ÞLet a ij be the(i;j)entry of VÀ1.Thena ij¼0i<j;1=ðm iþd iÞi¼j;Q iÀ1k¼jm kQ ik¼jðm kþd kÞj<i:8>>><>>>:ð12ÞThus,R0¼b1m1þd1þb2m1ðm1þd1Þðm2þd2Þþb3m1m2ðm1þd1Þðm2þd2Þðm3þd3ÞþÁÁÁþb mÀ1m1...m mÀ2ðm1þd1Þ...ðm mÀ1þd mÀ1Þ:ð13ÞThe i th term in R0represents the number of new infections produced by a typical individual during the time it spends in the i th infectious stage.More specifically,m iÀ1=ðm iÀ1þd iÀ1Þis the fraction of individuals reaching stage iÀ1that progress to stage i,and1=ðm iþd iÞis the average time an individual entering stage i spends in stage i.Hence,the i th term in R0is the product of the infectivity of individuals in stage i,the fraction of initially infected individuals surviving at least to stage i,and the average infectious period of an individual in stage i.4.4.Multistrain modelThe recent emergence of resistant viral and bacterial strains,and the effect of treatment on their proliferation is becoming increasingly important[12,13].One framework for studying such sys-tems is the multistrain model shown in Fig.3,which is a caricature of the more detailed treatment model of Castillo-Chavez and Feng[12,Section2]for tuberculosis and the coupled two-strain vector–host model of Feng and Velasco-Hern a ndez[17]for Dengue fever.The model has only a single susceptible compartment,but has two infectious compartments corresponding to the two infectious agents.Each strain is modelled as a simple SIS system.However,strain one may ‘super-infect’an individual infected with strain two,giving rise to a new infection incompartment。
基因毒性杂质之结构警示(欧洲)

EUR 23844 EN -2009Development of structural alerts for the in vivo micronucleus assay inrodentsRomualdo Benigni a , Cecilia Bossa a , Olga Tcheremenskaia aand Andrew Worthb aIstituto Superiore di Sanita’, Environment and Health Department,Rome, Italy b Institute for Health & Consumer Protection, European Commission -Joint Research Centre, Ispra, ItalyThe mission of the IHCP is to provide scientific support to the development and implementation of EU policies related to health and consumer protection.The IHCP carries out research to improve the understanding of potential health risks posed by chemical, physical and biological agents from various sources to which consumers are exposed.European CommissionJoint Research CentreInstitute for Health and Consumer ProtectionContact informationAddress: TP 582E-mail: andrew.worth@ec.europa.euTel.: +39 0332 789566Fax: +39 0332 786717http://http://ecb.jrc.ec.europa.eu/qsar/http://ec.europa.eu/dgs/jrc/Legal NoticeNeither the European Commission nor any person acting on behalf of the Commission is responsible for the use which might be made of this publication.A great deal of additional information on the European Union is available on the Internet.It can be accessed through the Europa serverhttp://europa.eu/JRC 52274EUR23844 ENISSN 1018-5593Luxembourg: Office for Official Publications of the European Communities© European Communities, 2009Reproduction is authorised provided the source is acknowledgedPrinted in ItalyABSTRACTIn vivo mutagenicity and carcinogenicity studies are posing a high demand for test-related resources. Among these studies, the micronucleus test in rodents is the most widely used, as follow up to positive in vitro mutagenicity results. A recent survey of the (Q)SAR models for mutagenicity and carcinogenicity has indicated that no (Q)SAR models for in vivo micronucleus are available in the public domain. Therefore, the development and extensive use of estimation techniques such as (Q)SARs, read-across and grouping of chemicals, promises to have a huge animal saving potential for this endpoint. In this report, we describe the identification of structural alerts for the in vivo micronucleus assay, and provide the list of underlying chemical structures. These structural alerts provide a coarse-grain filter for the preliminary screening of potential in vivo mutagens.LIST OF ABBREVIATIONSEPA Environmental Protection AgencyEU European UnionFDA Food and Drug AdministrationHOMO Highest Occupied Molecular OrbitalISS Istituto Superiore di Sanita’JRC Joint Research CentreLUMO Lowest Unccupied Molecular OrbitalOECD Organisation for Economic Cooperation and Development(Q)SAR(Quantitative)Structure-Activity RelationshipREACH Registration Evaluation and Authorisation of CHemicalsROC Receiver Operating CurveSA Structural AlertSA_BB Benigni-Bossa structural alerts for mutagnicity /carcinogenicity in ToxtreeSA_Mic Structural alerts refers for the in vivo micronucleus assay inToxtreeSA_Prot Structural alerts for protein binding in the OECD QSAR ToolboxCONTENTS1.Introduction (6)2.Structural alerts (8)3.Development of structural alerts for the in vivo micronucleus assay (10)4. Final considerations (20)5.References (21)Appendix 1 (23)1.IntroductionMutagenicity testing is an important part of the regulatory hazard assessment of chemicals. It is undertaken for two main reasons: a) to detect chemicals that might cause genetic damage in germ cells, and thus increase the burden of heritable (genetic) disease in the human population; and b) to detect chemicals that might be carcinogenic (based on the assumption that mutagenesis, for example in somatic cells, is a key event in the process of carcinogenesis). Since no method is able alone to detect all possible genotoxic events, a wide array of test systems has been developed and accepted internationally in regulatory schemes.Most often, these methods are used within a 2-tiered integrated testing approach: Tier 1 includes in vivo assays, and Tier 2 includes in vivo assays. As a matter of fact, mutagenicity testing was the first toxicity endpoint for which in vivo assays were accepted for regulatory testing, some 25 years ago. The latter usually comprise bacterial mutagenicity and cytogenetics tests, although gene mutation testing in cultured mammalian cells is sometimes also undertaken.Tier 2 of the testing strategy involves the use of short-term in vivo studies (usually a bone-marrow cytogenetics assay) to assess whether any potential for genotoxicity detected at the Tier 1 in vivo stage is actually expressed in the whole animal. Thus, negative results in vivo are usually considered sufficient to indicate lack of mutagenicity, whereas a positive result is not considered sufficient to indicate that the chemical represents a mutagenic hazard (i.e. it could be a false positive). The above approach to genotoxicity testing has been adopted throughout the EU1,and has been recommended internationally as part of the strategy for predicting and quantifying mutagenic and carcinogenic hazard (Ashby et al.,1996; Combes et al.,2007; Kirkland and Speit,2008; Lilienblum et al.,2008).1http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_r7a_en.p df?vers=20_08_08According to an assessment carried out by the former European Chemicals Bureau (ECB), the in vivo mutagenicity studies, shortly followed by carcinogenicity, are posing high demand for test-related recourses (Pedersen et al.,2003; Van der Jagt et al.,2004). Among those, the micronucleus test in rodents is the most widely used, as follow up to positive in vivo mutagenicity results. A recent survey of the (Q)SAR models for mutagenicity and carcinogenicity (performed jointly by ISS and the JRC) has indicated that no (Q)SAR models for in vivo micronucleus are available in the public domain (Benigni et al.,2007): therefore, the development and extensive use of estimation techniques such as (Q)SARs, read-across and grouping of chemicals, might have a huge saving potential for this endpoint.In this report, we describe: a) the collection of data on chemicals tested with the in vivo micronucleus assay; b) preliminary analyses of the data; c) the identification of Structural Alerts (SA) proper to this toxicological endpoint. First, some background information on the concept of SA is provided.2.Structural alertsThe SAs for a toxicological endpoint are molecular functional groups or substructures known to be linked to that type of toxicity.The SAs are a coarse-grained approach to the use of Structure-Activity Relationships (SAR) to understand the toxicity mechanisms and to predict the toxic activity of chemicals. Because of their nature, the SAs have the role of pointing to chemicals potentially toxic, whereas no conclusions or indications about nontoxic chemicals are possible (except by exclusion) (Benigni and Bossa,2006; Benigni and Bossa,2008).A set of chemicals characterized by the same SA constitute a family (class) of compounds that share the same mechanism of action. The reactivity of a SA can be modulated or abolished by the remaining part of the molecule in which the SA is embedded. At a coarse-grain level, such modulating effects can be represented by other molecular substructures (e.g., bulky groups ortho to an aromatic amine group) that are known to have an influence on the reactivity of the SA. Usually, the knowledge on the modulating substructures is quite limited for most of the SAs, thus it provides limited help in deciding which chemicals in a class will actually be toxic and viceversa. A powerful generalization of the Structure-Activity Relationships is provided by the Quantitative Structure-Activity Relationship (QSAR) analysis, which produces a mathematical model that links the biological activity to a limited number of physical chemical or other molecular properties (descriptors) with general relevance. Since most of the descriptors have continuous values, the QSARs provide fine-tuned models of the biological activity,and can give account of subtle differences. General introductions on QSAR are given elsewhere (Hansch and Leo, 1995, Hansch et al.,2002). Thus the SAs are not a discriminant model on the same ground of the QSAR models: the latter produce estimates for both positive and negative chemicals, as well as for the gradation of toxic potency.The main role of the SAs is that of preliminary, or large-scale screenings. They are excellent tools for coarse-grain characterization of chemicals, including: description of sets of chemicals, preliminary hazard characterization, category formation and priority setting (enrichment). Since fine-tuned QSARs do not exist for many types of chemicals, the models based on SAs hold a special place in predictive toxicology. Theknowledge on the action mechanisms as exemplified by the SAs is routinely used in SAR assessment in the regulatory context (see, for example, the mechanistically-based reasoning as presented in Woo et al. (2002). In addition, the SAs are at the basis of popular commercial (e.g., DEREK, by Lhasa Ltd.2) and non-commercial software systems (e.g., Oncologic, by US Environmental Protection Agency[EPA]3).Recently, as follow-up of the collaboration between ISS and JRC,a rulebase for mutagens and carcinogens has been designed and implemented in the software Toxtree 1.51. It uses a structure-based approach consisting of a new compilation of SAs for carcinogenicity and mutagenicity. It also offers three mechanistically based QSARs for congeneric classes (aromatic amines and aldehydes) (Benigni et al., 2008a). Toxtree 1.51 is freely available from the JRC website.42/3/oppt/newchems/tools/oncologic.htm4http://ecb.jrc.ec.europa.eu/qsar/qsar-tools/index.php?c=TOXTREE3.Development of structural alerts for the in vivomicronucleus assay3.1 DataThe compilation of SAs for the in vivo micronucleus assay in rodents provided here, is based on both the existing knowledge on the mechanisms of toxic action and a structural analysis of the chemicals tested in the assay.The in vivo micronucleus data in the public domain is quite limited. A search of the Chemical Carcinogenesis Research Information System(CCRIS) at the Toxnet website with the query: “in vivo micronucleus” points only to 240 chemicals.5For this work, the remarkably larger commercial database by Leadscope Inc., called “FDA SAR Genetox Database” was used.6This database contains more than 700 chemicals tested in in vivo micronucleus with rodents, and includes data from both the public domain and the US Food and Drug Administration (FDA) files. A large majority of data were based on the analysis of micronuclei in bone marrow cells; for details on the technique, see for example, Krishna and Hayashi (2000).3.1 Preliminary analysesSince the main role of the in vivo micronucleus assay in regulatory schemes is that of confirming (or disproving) the positive in vitro results, it is of interest to check how the in vivo micronucleus results relate to the rodent carcinogenicity data and to the primary in vitro prediction test, i.e., the Salmonella typhimurium(Ames) test.Tables I and II display the relationships between the in vivo micronucleus ad the two reference tests. The results for rodent carcinogenicity and the Ames test were retrieved from the freely available ISSCAN v3a database,7which is characterized by:5/cgi-bin/sis/search6/product_info.php?products_id=777http://www.iss.it/ampp/dati/cont.php?id=233&lang=1&tipo=7a) the high quality of both chemical and biological information; b) the QSAR-ready format (Benigni et al.,2008b). Obviously, the total numbers of chemicals in the two tables are relative only to those chemicals tested in both systems.Table I. Contingency table comparing the results of the rodent carcinogenicity testwith the micronucleus testTable II:Contingency table comparing the results of the Salmonella typhimuriumassay with the micronucleus testTable I shows that is the in vivo micronucleus assay is poorly sensitive to the rodent carcinogens: about 60% of the rodent carcinogens are not detected by the micronucleus. The poor sensitivity of the micronucleus assay to potential genotoxins is also apparent from Table II.It should be emphasized that the present results obtained with the large Leadscope micronucleus database are in agreement with previous analyses based on smaller datasets in the public domain (Benigni,1995).In a second round of analyses, the extent to which the micronucleus data are related to well established indicators of DNA and protein binding was checked. This in view of the plethora of the reported mechanisms of micronucleus induction. As a matter of fact, micronuclei are markers of both aneugenic (change in the chromosomes number, usually by loss) and clastogenic (chromosome breakage) effects. It is generally assumed that such effects are generated through a range of different pathways. Evidence (mainly gathered from in vitro studies) indicates that micronuclei can be induced e.g., by typical DNA-attacking agents (e.g., alkylating agents like methylmethane sulfonate), by mitotic spindle poisons (e.g., colcemide, vincristine), or by inhibitors of cytokinesis (e.g., cytochalasin B). The latter effects are probably due to interference with proteins. Other chemicals are thought to be clastogenic through aspecific disturbance of cytokinesis due to lipophilicity (Dorn et al.,2007).The relative influence of DNA and protein binding on micronucleus generation was checked by recording the distribution of structural alerts for the two effects in the Leadscope in vivo micronucleus database. As probes for DNA binding, we used the structural alerts for carcinogenicity / mutagenicity implemented in Toxtree 1.51. As a matter of fact, the large majority of these alerts refer to genotoxic carcinogenicity, which is assumed to be caused through direct interaction with DNA (Benigni and Bossa, 2008). As probes for protein binding, we used the alerts implemented in the Organisation for Economic Cooperation and Development (OECD) QSAR Toolbox.8 These alerts were mainly developed from the mechanistic knowledge on skin sensitization, and model the covalent binding to proteins.The results of the above analysis is displayed in Figure 1 as a ROC graph. It appears that the structural alerts for carcinogenicity /mutagenicity correlate to some extent with the induction of micronuclei, whereas those for protein covalent binding show no correlation (in the graph, they are on the diagonal line which represents random results).8/document/23/0,3343,en_2649_34379_33957015_1_1_1_1,00.htmlFigure 1. Receiver Operating Curve showing the concordance of two sets of structural alerts with the results of the in vivo micronucleus assay(SA_BB refers to the Benigni-Bossa alerts in Toxtree; SA_Prot refers to the alerts for proteinbinding in the OECD QSAR Toolbox)3.3 Structural Alerts for in vivo micronucleus assaySince the above analyses pointed to genotoxic effects as an important determinant of micronuclei induction, we developed the list of Structural alerts for in vivo micronucleus using the carcinogenicity / mutagenicity alerts in Toxtree as a core , and then searching for additional substructures specific to the micronucleus-positive chemicals. From the Toxtree alerts for carcinogenicity / mutagenicity,we excluded four alerts specific for non-genotoxic mechanisms of carcinogenicity.Using linear discriminant analysis as an analytical tool and ROC plots as a graphical tool, a series of additional substructures were added / removed to / from the Toxtreealerts in order to increase sensitivity and specificity.In these exploratory analyses, wescreened the very large collection of substructural patterns and functional groups (more than 27,000) contained in the software Leadscope Enteprise 2.4.15-6. We also re-checked the Toolbox protein binding alerts for individual substructures related with micronucleus induction.The result is the optimized list of alerts in Appendix 1. Together with the Toxtree alerts, it contains five additional substructures identified in the course of this research. For the sake of clarity,the codes of the alerts in Toxtree are maintained, whereas the five additional alerts have new codes.Figure 2 displays the agreement between the alerts for in vivo micronucleus, and the experimental results for this endpoint. Out of 547 negatives, the specificity of the SAs is 0.57. The sensitivity is 0.65 out of 182 positives. The overall accuracy is 0.59. For a comparison, the ROC graph shows the newly developed alerts for micronucleus together with those for DNA and protein binding. It appears that the performance of the final list of alerts is considerably higher than that of the DNA binding and Protein binding alerts.Table III gives the true positive rate for the individual alerts.Figure 2 Receiver Operating Curve showing the concordance of structural alerts for the in vivo micronucleus assay with the experiemtnal results for this assay(SA_Mic refers to the in vivo micronucleus alerts in Toxtree)Table III: Characterisation of Structural Alerts.3.4 Further analyses on the alerts for micronucleusA striking evidence in Table III is the relatively low percentage of true positives identified by many SAs. In other words, often the toxic potential of the alerts is not translated into actual toxicity in the experimental system. For a comparison, the True Positive Rate of the various alerts for mutagenicity /carcinogenicity in Toxtree is remarkably higher, ranging from 70 to 100% (Benigni and Bossa,2008).The above result contributes to better understand the evidence in Tables I and II, where it appears that the micronucleus assay has many more negatives than the carcinogenicity bioassay and the Salmonella mutagenicity test. Table III indicates that the low sensitivity of the micronucleus assay is largely due to the fact that often,chemical functionalities and substructures which are supposed to be reactive do not exert their potential reactivity in this experimental system.The issue of the low sensitivity of the micronucleus assay has been recognized by scientists involved in research aimed at improving the available short-term mutagenicity assays; as a matter of fact, validation of further, more sensitive in vivo assays (e.g., in vivo Comet assay) is presently in progress (Kirkland and Speit,2008).In the context of this research, we investigated if a general effect of bioavailability on the limited sensitivity of micronucleus was apparent. To this aim, we considered two chemical descriptors well known as to be linked to bioavailability: logP (hydrophobicity) and Molar Refractivity (MR) (Hansch and Leo,1995). The two descriptors were calculated with the C-QSAR software (Daylight, Inc.)9for all the chemicals in the micronucleus database. For the two parameters, Table IV reports the ranges of values for positive and negative micronucleus results.Table IV: Ranges of C-logP and C-MR in chemicals assayed withthe micronucleus testIt appears that the micronucleus positives cover a more limited range of logP values than the micronucleus negatives; however, the consideration of exclusion values for logP in combination with the SAs did not improve the overall performance (results not shown).Whereas no general effect of logP (or MR) was found, analyses on the individual chemical classes showed that logP cut-offs can be identified for the classes of Nitroaromatics (Negatives at logP > 0.0), Aromatic Diazo (Negatives at logP < 3.7),9/about/index.htmland Oxolanes (Negatives at logP > 1.5). The consideration of these cut-offs increases the specificity of the SAs from0.57 to 0.60.The above result suggests a possible strategy to understand and modeling the many negative results observed with the micronucleus. Since the bone marrow (main target of the test) is an organ easily accessible by the blood stream, it can be hypothesized that the lack of effect shown by several chemicals with SAs (hence potentially reactive) is due to the many possible targets for reaction encountered in the in vivo situation; this diminishes the probability for the chemicals of reaching, and interacting with the molecular target(s) of the micronucleus test. For example, highly reactive chemicals will probably react with any target encountered in their way (e.g., proteins, water)before reaching the bone marrow. Thus it can be envisaged that QSARs for individual chemical classes should be developed, and that they should consider parameters linked to chemical reactivity(such as HOMO and LUMO energies). It can be hypothesized that the models derived from these QSARs will contribute to modulate the individual SAs.4. Final considerationsStructural alerts point to classes of chemicals with the potential to cause toxic effects (here, in vivo micronucleus). Since this potential is modulated in each molecule by the rest of the structure (e.g., other functional groups, electronic structure, bulky groups), not all chemicals in a class are equally toxic. In the case of the SAs identified in the present study for the in vivo micronucleus test, the percentage of chemicals that have SAs but are not active in the test system is particularly high. This evidence agrees with, and rationalizes the notion that this test system is sensitive to genotoxins to a limited extent, and does not respond to a large number of recognized carcinogens and mutagens. For this reason, a positive in vivo micronucleus result adds a strong weight to an in vivo positive mutagenicity result, whereas a negative in vivo micronucleus result has a much lower relevance. The availability of a wider range of in vivo mutagenicity assays is a priority for the present regulatory strategies.Within the above perspective, the SAs identified in this study provide a coarse-grain filter for a preliminary screening of potentially in vivo mutagens. In a risk assessment process, further information(e.g., QSARs for individual classes, experiments) is necessary to complete this initial screening step.5.ReferencesAshby,J., M.D.Waters, J.Preston, I.D.Adler, G.R.Douglas, R.Fielder, M.D.Shelby,D.Anderson, T.Sofuni, H.N.B.Gopalan, G.Becking and C.Sonich-Mullin(1996). IPCS harmonization of methods for the prediction and quantification of human carcinogenic/mutagenic hazard, and for indicating the probable mechanism of action of carcinogens. Mutat.Res./Fundamental and Molecular Mechanisms of Mutagenesis. 352:153-157.Benigni,R. (1995). Mouse bone marrow micronucleus assay: relationships with in Vitro mutagenicity and rodent carcinogenicity. J.Toxicol.Environ.Health.45:337-347.Benigni,R. and C.Bossa(2006). Structural alerts of mutagens and carcinogens.put.-Aid.Drug Des.2:169-176.Benigni,R. and C.Bossa(2008). Structure Alerts for carcinogenicity,and the Salmonella assay system: a novel insight through the chemical relational databases technology. Mutat.Res.Revs.659:248-261.Benigni,R., C.Bossa, N.G.Jeliazkova, zeva and A.P.Worth(2008a). The Benigni / Bossa rulebase for mutagenicity and carcinogenicity -a module of Toxtree. JRC Report EUR 23241 EN. European Commission Joint Research Centre, Ispra, Italy.http://ecb.jrc.ec.europa.eu/DOCUMENTS/QSAR/EUR_23241_EN.pdfBenigni,R., C.Bossa, A.M.Richard, and C.Yang (2008b). A novel approach: chemical relational databases, and the role of the ISSCAN database on assessing chemical carcinogenicity.Ann.Ist.Super.Sanità. 44:48-56.Benigni,R., C.Bossa, zeva and A.P.Worth(2007).Collection and evaluation of (Q)SAR models for mutagenicity and carcinogenicity. JRC Report EUR 22772 EN. European Commission Joint Research Centre, Ispra, Italy.http://ecb.jrc.ec.europa.eu /documents/QSAR/EUR_22772_EN.pdfCombes,R., C.Grindon, M.T.D.Cronin, D.W.Roberts and J.Garrod(2007). Proposed integrated decision-tree testing strategies for mutagenicity and carcinogenicity in relation to the EU REACH legislation.ATLA. 35:267-287.Dorn,S.B., G.H.Degen, T.Müller, D.Bonacker, H.F.P.Joosten, J.van der Louw,F.A.A.van Acker and H.M.Bolt(2007). Proposed criteria for specific and non-specific chromosomal genotoxicity based on hydrophobic interactions.Mutat.Res./Genetic Toxicology and Environmental Mutagenesis. 628:67-75. Hansch,C., D.Hoekman, A.Leo, D.Weininger and C.D.Selassie(2002).Chem-bioinformatics: comparative QSAR at the interface between chemistry and biology.Chem.Revs.102:783-812.Hansch,C. and A.Leo (1995). Exploring QSAR. 1. Fundamentals and applications in chemistry and biology.American Chemical Society. Washington, D.C.Kirkland,D. and G.Speit(2008). Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens III.Appropriate follow-up testing in vivo.Mutat.Res.654:114-132.Krishna,G. and M.Hayashi(2000). In vivo rodent micronucleus assay: protocol, conduct and data interpretation.Mutat.Res.455:155-166.Lilienblum,W., W.Dekant, H.Foth, T.Gebel, J.G.Hengstler, R.Kahl, P.J.Kramer,H.Schweinfurth and K.M.Wollin(2008). Alternative methods to safety studiesin experimental animals: role in the risk assessment of chemicals under the new European Chemicals Legislation (REACH).Regulat.Toxicol.82:211-236.Pedersen,F., J.de Brujin, S.J.Munn and K.Van Leeuwen(2003).Assessment of additional testing needs under REACH. Effects of (Q)SARs, risk based testing and voluntary industry initiatives. JRC report EUR 20863 EN. European Commission Joint Research Centre, Ispra, Italy.http://ecb.jrc.ec.europa.eu/home.php?CONTENU=/DOCUMENTS/REACH/PUBLICATIONS/ Van der Jagt,K., S.J.Munn, J.Torslov and J.de Brujin (2004).Alternative approaches can reduce the use of test animals under REACH. Addendum to the Report "Assessment of addtional testing needs under REACH. Effects of (Q)SARs, risk based testing and voluntary industry initiatives. JRC Report EUR 21405 EN.European Commission Joint Research Centre, Ispra, Italy.http://ecb.jrc.ec.europa.eu/home.php?CONTENU=/DOCUMENTS/REACH/PUBLICATIONS/ Woo,Y.T., i, J.L.McLain, M.Ko Manibusan and V.Dellarco(2002). Use of mechanism-based structure-activity relationships analysis in carcinogenic potential ranking for drinking water disinfection by-products. Environ.Health Perspect.110:75-87.Appendix 1European CommissionEUR 23844 EN–Joint Research Centre –Institute for Health and Consumer ProtectionTitle: Development of Structural alerts for the in vivo micronucleus assay in rodentsAuthor(s): Benigni R, Bossa C, Tcheremenskaia O and Worth A Luxembourg: Office for Official Publications of the European Communities 2009–42pp. –21x 29.7cmEUR –Scientific and Technical Research series –ISSN 1018-5593AbstractIn vivo mutagenicity and carcinogenicity studies are posing a high demand for test-related resources. Among these studies, the micronucleus test in rodents is the most widely used, as follow up to positive in vitro mutagenicity results. A recent survey of the (Q)SAR models for mutagenicity and carcinogenicity has indicated that no (Q)SAR models for in vivo micronucleus are available in the public domain. Therefore, the development and extensive use of estimation techniques such as (Q)SARs, read-across and grouping of chemicals, promises to have a huge animal saving potential for this endpoint. In this report, we describe the identification of structural alerts for the in vivo micronucleus assay, and provide the list of underlying chemical structures. These structural alerts provide a coarse-grain filter for the preliminary screening of potential in vivo mutagens.。
博士复试英文PPT

3. PTBP1 enhances exon11a skipping of Mena premRNA in lung cancer cells
Results
1. PTBP1 is highly expressed in lung adenocarcinoma (LUAD) tissues and 95-D cells and upregulation of PTBP1 is associated with EMT progress
2. PTBP1 promotes migration and invasion of lung cancer cells
Master Research
PTBP1 enhances exon11a skipping in Mena premRNA to promote migration and invasion in lung
carcinoma cells
Background Objectives Technology Methods Results Conclusions
5. PTBP1-mediated migration and invasion of 95-D cells are partially dependent on MenaINV
Results
2.1. Overexpressed PTBP1 promotes levels of EMT-related proteins in lung cancer cells
Technology Methods
Results
1. PTBP1 is highly expressed in lung adenocarcinoma (LUAD) tissues and 95-D cells and upregulation of PTBP1 is associated with EMT progress
学术英语(第二版)医学教师用书Unit 8
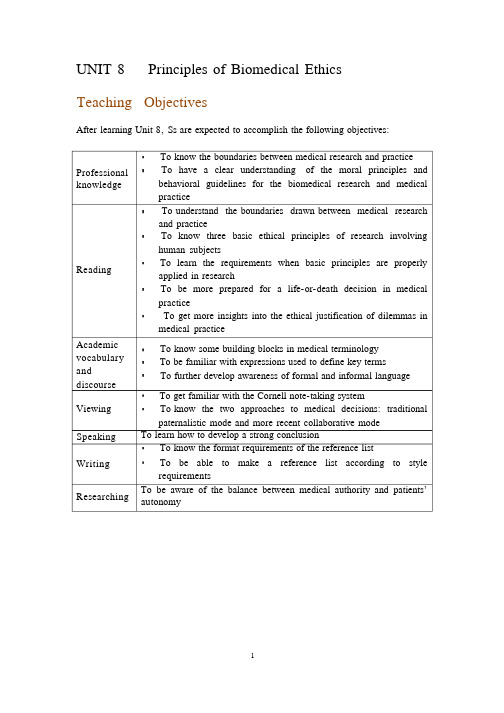
UNIT 8 Principles of Biomedical EthicsTeaching ObjectivesAfter learning Unit 8, Ss are expected to accomplish the following objectives:To know the boundaries between medical research and practice To have a clear understanding of the moral principles and behavioral guidelines for the biomedical research and medical practice To understand the boundaries drawn between medical research and practice To know three basic ethical principles of research involving human subjects To learn the requirements when basic principles are properly applied in research To be more prepared for a life-or-death decision in medical practice To get more insights into the ethical justification of dilemmas in medical practice To know some building blocks in medical terminologyTo be familiar with expressions used to define key termsTo further develop awareness of formal and informal language To get familiar with the Cornell note-taking system To know the two approaches to medical decisions: traditional paternalistic mode and more recent collaborative modeTo learn how to develop a strong conclusion To know the format requirements of the reference listTo be able to make a reference list according to style requirements To be aware of the balance between medical authority and patients ’ autonomyProfessionalknowledgeReadingAcademic vocabulary anddiscourseViewingSpeakingWritingResearchingTeaching Activities and ResourcesPart 1 ReadingText ALead-inSuggested teaching plan1. To draw Ss’ attention and to raise their awareness of the importance ofbiomedical ethics, T is advised to relate the discussion of this unit to the real-world happenings.Before starting the class,search the media for the latest news reports,either at home or abroad,about controversial events in medicine community or healthcare settings.2. Start the class by doing Task / Lead-in and relate the content of the video clip toyour findings in the pre-class searching.Key to the task2) Death4) Patient rightsScriptWell,advancements in medical science have afforded us the opportunity to live decades longer than in previous generations.For every new possibility offered, we now face an equal number of challenges and we find ourselves confronting decisions that are unprecedented in human history.When does life begin?When should life end? How do we define death when we have the ability to keep people technically alive,or we should say,technologically alive long after their discrete body parts no longer function? Welcome to “Matter and Beyond . ” I’m your host MaryLynn Schiavi.In this program we’re going to explore issues around medical science that are forcing us to define life, death, quality of life, patient rights, and confront the moral and ethical questions that arise when facing critical healthcare decisions.3. Introduce the topic of Text A as a natural continuum of Lead-in .Text Comprehension1. Make good use of Lead-in video clip as it serves as a perfect introduction to thetopic of this unit. Elaborate on the connection of its content with the latest events in the real world. Naturally, ask Ss how medicine differs from other branches of natural science, especially when human subjects are involved in the research. Here are some hints:2. Analyze the text and lead Ss to discuss, integrating Task 2 / Critical reading andthinking / Text A into analysis and discussion. The presentation topics should be assigned to individual Ss for preparation at least one week in advance. Ask other Ss to preview the text with the guidance of presentation topics.3. Integrate Task 2 / Language building-up / Text A when a careful definition ofkey terms is covered.4. When analyzing the text, ask Ss to pay special attention to the sentences listed inLanguage focus below.5. If time allows, ask Ss to do Task 1 / Critical reading and thinking / Text A inabout five minutes. Check out the task by asking one or two Ss to read their answers. This is done to get an overview about the text.Language focus 1. … described in a formal protocol that sets forth an objective … (P185, Para.2)set forth 是动词词组,表示用清晰、具体的方式解释或描述,多用于正式的 书面语中。
Apparatus and methods for reducing tool-induced sh

专利名称:Apparatus and methods for reducing tool-induced shift during overlay metrology发明人:Vladimir Levinski,Ilan Sela申请号:US10913188申请日:20040806公开号:US07433039B1公开日:20081007专利内容由知识产权出版社提供专利附图:摘要:Disclosed are apparatus and methods for determining a minimum tool-induced shift (TIS) during an overlay metrology procedure. In a specific embodiment, a method of determining overlay or misalignment error on a target is disclosed. For a predefinednumber of positions of a target within a field of view (FOV) of a metrology tool, the following operations are performed: (i) determining a tool-induced shift (TIS) parameter value for each predefined position of the target within the FOV based on at least one overlay measurement obtained from the target at the each position (for example, based on overlay measurements at 0 and 180 degrees of wafer orientation) and (ii) determining a minimum TIS parameter value and its corresponding FOV position from the plurality of determined TIS parameters values at the predefined positions of the target within the FOV. The FOV position that corresponds to the minimum TIS is then defined as an appropriate position for the actual overlay measurement and the value of minimum TIS is used for overlay correction.申请人:Vladimir Levinski,Ilan Sela地址:Nazareth Ilit IL,Haifa IL国籍:IL,IL代理机构:Weaver Austin Villeneuve & Sampson LLP更多信息请下载全文后查看。
mendelian randomization-based methods

Mendelian randomization (MR) is a statistical method used in genetic epidemiology to estimate the causal effect of an exposure on an outcome using genetic variants as instrumental variables. The method is based on the principles of Mendelian inheritance, which state that genetic variants are randomly allocated at conception and remain constant throughout life. This random allocation creates a natural experiment that can be used to estimate the causal effect of an exposure on an outcome.MR-based methods involve three main steps:1. Selection of genetic variants: Genetic variants that are strongly associated with the exposure of interest are selected as instrumental variables. These variants should also be independent of any confounding factors that may affect the outcome.2. Estimation of the effect of the genetic variant on the outcome: The effect of each genetic variant on the outcome is estimated using regression analysis.3. Estimation of the causal effect of the exposure on the outcome: The causal effect of the exposure on the outcome is estimated by combining the effects of all genetic variants on the outcome using meta-analysis techniques.MR-based methods have several advantages over traditional observational studies, including the ability to control for confounding factors and reverse causation bias. However, they also have some limitations, such as the assumption that the genetic variants are valid instrumental variables and that they have nopleiotropic effects (i.e., they only affect the exposure and not the outcome through other pathways).中文翻译如下:孟德尔随机化(Mendelian randomization,MR)是一种用于遗传流行病学的统计方法,它利用遗传变异作为工具变量来估计某个暴露因素对结果的因果效应。
不同级别胶质瘤患者MEK、ERK、CHKα蛋白的阳性表达情况及临床意义

㊃论著㊃D O I:10.3969/j.i s s n.1672-9455.2024.08.019不同级别胶质瘤患者M E K㊁E R K㊁C H Kα蛋白的阳性表达情况及临床意义甘杰1,文擘彬1,杨晗2,王争1ә1.湖南省长沙市第四医院神经外科,湖南长沙410000;2.中南大学湘雅医院神经外科,湖南长沙410000摘要:目的探讨不同级别胶质瘤患者胆碱激酶α(C H Kα)㊁细胞外信号调节激酶(E R K)及丝裂原激活蛋白激酶(M E K)蛋白的阳性表达情况及临床意义㊂方法回顾性分析2019年1月至2022年12月长沙市第四医院神经外科收治的124例胶质瘤患者的临床资料㊂检测㊁统计不同级别胶质瘤患者M E K㊁E R K㊁C H Kα蛋白的阳性表达情况㊂结果不同肿瘤最大径㊁年龄㊁WHO分级及预后胶质瘤患者M E K㊁E R E㊁C HKα蛋白的高㊁低表达情况比较,差异均有统计学意义(P<0.05)㊂Ⅰ㊁Ⅱ㊁Ⅲ㊁Ⅳ级胶质瘤患者M E K蛋白阳性表达率分别为9.62%㊁7.69%㊁94.08%和92.04%,E R K蛋白阳性表达率分别为9.57%㊁7.49%㊁94.52%和92.18%, C HKα蛋白阳性表达率分别为9.60%㊁7.53%㊁94.71%和92.23%,高㊁低级别胶质瘤患者M E K㊁E R K㊁C H Kα蛋白阳性表达率比较,差异均有统计学意义(P<0.05)㊂随访结束后结果显示,124例患者中死亡52例,失访12例,获得随访112例,随访率为90.32%㊂结论不同级别胶质瘤患者M E K㊁E R K㊁C H Kα蛋白的阳性表达情况有明显区别,并且与患者的疾病预后明显相关,对疾病的诊断和预后判断具有重要价值㊂关键词:胶质瘤;丝裂原激活蛋白激酶;细胞外信号调节激酶;胆碱激酶α;免疫组织化学法中图法分类号:R739.4文献标志码:A文章编号:1672-9455(2024)08-1118-05 P o s i t i v e e x p r e s s i o n a n d c l i n i c a l s i g n i f i c a n c e o f M E K,E R K a n d C H Kαp r o t e i n si n p a t i e n t s w i t h g l i o m a o f d i f f e r e n t g r a d e sG A N J i e1,WE N B o b i n1,Y A N G H a n2,WA N G Z h e n g1ә1.D e p a r t m e n t o f N e u r o s u r g e r y,T h e F o u r t h H o s p i t a l o f C h a n g s h a i n H u n a n P r o v i n c e,C h a n g s h a,H u n a n410000,C h i n a;2.D e p a r t m e n t o f N e u r o s u r g e r y,X i a n g y a H o s p i t a l,C e n t r a l S o u t hU n i v e r s i t y,C h a n g s h a,H u n a n410000,C h i n aA b s t r a c t:O b j e c t i v e T o i n v e s t i g a t e t h e p o s i t i v e e x p r e s s i o n a n d c l i n i c a l s i g n i f i c a n c e o f c h o l i n e k i n a s eα(C H Kα),e x t r a c e l l u l a r s i g n a l-r e g u l a t e d k i n a s e(E R K)a n d m i t o g e n-a c t i v a t e d p r o t e i n k i n a s e(M E K)p r o t e i n s i n p a t i e n t s w i t h d i f f e r e n t g r a d e s o f g l i o m a.M e t h o d s T h e c l i n i c a l d a t a o f124g l i o m a p a t i e n t s a d m i t t e d t o t h e D e p a r t m e n t o f N e u r o s u r g e r y,t h e F o u r t h H o s p i t a l o f C h a n g s h a f r o m J a n u a r y2019t o D e c e m b e r2022w e r e r e t-r o s p e c t i v e l y a n a l y z e d.T h e p o s i t i v e e x p r e s s i o n s o f M E K,E R K a n d C H Kαp r o t e i n s i n p a t i e n t s w i t h g l i o m a o f d i f f e r e n t g r a d e s w e r e d e t e c t e d a n d a n a l y z e d.R e s u l t s T h e m o r t a l i t y r i s k o f p a t i e n t s w i t h g l i o m a w a s s t a t i s t i-c a l l y s i g n i f i c a n t c o m p a r e d w i t h t h e m a x i m u m d i a m e t e r o f d i f f e r e n t t u m o r s,t h e a g e,WHO g r a d e a n d p r o g n o s i s o f g l i o m a p a t i e n t s w i t h h i g h a n d l o w e x p r e s s i o n o f M E K,E R K a n d C HKαp r o t e i n s(P<0.05).T h e p o s i t i v e e x p r e s s i o n r a t e s o f M E K p r o t e i n i n g r a d eⅠ,Ⅱ,Ⅲa n dⅣg l i o m a s w e r e9.62%,7.69%,94.08%a n d92.04% r e s p e c t i v e l y.T h e p o s i t i v e e x p r e s s i o n r a t e s o f E R K p r o t e i n w e r e9.57%,7.49%,94.52%a n d92.18%r e s p e c-t i v e l y.T h e p o s i t i v e e x p r e s s i o n r a t e s o f C H Kαp r o t e i n w e r e9.60%,7.53%,94.71%a n d92.23%r e s p e c t i v e l y. T h e r e w e r e s i g n i f i c a n t d i f f e r e n c e s i n t h e p o s i t i v e e x p r e s s i o n r a t e s o f M E K,E R K a n d C H Kαp r o t e i n s b e t w e e n h i g h a n d l o w g r a d e p a t i e n t s w i t h g l i o m a(P<0.05).T h e f o l l o w-u p r e s u l t s s h o w e d t h a t52p a t i e n t s d i e d,12 p a t i e n t s w e r e l o s t t o f o l l o w-u p,a n d112p a t i e n t s w e r e r e c o v e r e d,w i t h a f o l l o w-u p r a t e o f90.32%.C o n c l u s i o n T h e p o s i t i v e e x p r e s s i o n o f M E K,E R K a n d C H Kαp r o t e i n s i n p a t i e n t s w i t h g l i o m a o f d i f f e r e n t g r a d e s a r e s i g n i f i c a n t l y d i f f e r e n t,a n d a r e s i g n i f i c a n t l y c o r r e l a t e d w i t h t h e p r o g n o s i s o f t h e p a t i e n t s,w h i c h i s o f g r e a t v a l u e f o r t h e d i a g n o s i s a n d p r o g n o s i s o f t h e d i s e a s e.K e y w o r d s:g l i o m a; m i t o g e n-a c t i v a t e d p r o t e i n k i n a s e;e x t r a c e l l u l a r s i g n a l-r e g u l a t e d k i n a s e;c h o l i n e k i n a s eα;i mm u n e h i s t o c h e m i c a l m e t h o d脑胶质瘤是一种生长迅速㊁侵袭性较强的原发性颅内肿瘤,其发病机制涉及多个信号通路的异常激活和调控失衡[1]㊂胆碱激酶α(C H Kα)㊁细胞外信号调节激酶(E R K)及丝裂原激活蛋白激酶(M E K)是细胞信号通路中的关键蛋白,它们均参与了细胞的增殖㊁生存㊁分化等多个生物学过程,恶性肿瘤中异常活化㊃8111㊃检验医学与临床2024年4月第21卷第8期 L a b M e d C l i n,A p r i l2024,V o l.21,N o.8作者简介:甘杰,男,主治医师,主要从事脑血管疾病方面的研究㊂ә通信作者,E-m a i l:30694283@q q.c o m㊂的信号通路通常与肿瘤的发生和发展密切相关[2]㊂M E K /E R K 信号通路是一个主要的细胞增殖和生存信号传导通路,而C H K α则在细胞周期调控和D N A 损伤修复中发挥关键的调节作用,这些蛋白的表达水平是否可以作为胶质瘤的生物标志物,且是否有助于对不同级别的胶质瘤进行分类和预测患者的生存状况还有待考察㊂临床通常使用基因表达水平的定量数据来定义M E K ㊁E R K 和C H K α等基因的高表达和低表达状态㊂基于此,本研究采用免疫组织化学法检测不同级别胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白的阳性表达情况,判断其预后价值,现报道如下㊂1 资料与方法1.1 一般资料 回顾性分析2019年1月至2022年12月长沙市第四医院神经外科收治的124例胶质瘤患者的临床资料,其中男64例(51.61%),女60例(48.39%);年龄10~75岁,平均(43.58ʃ11.03)岁㊂根据世界卫生组织(WHO )胶质瘤的分级标准将其划分为Ⅰ~Ⅳ级[3]㊂纳入标准:(1)所有患者均经病理学检查诊断符合胶质瘤诊断标准[4];(2)血液㊁尿液㊁大便㊁肝功能㊁肾功能㊁心功能检查均无异常㊂排除标准:(1)疾病类型为复发性胶质瘤或接受过分子靶向药物及放化疗治疗的患者;(2)合并心㊁肝㊁肺㊁肾等实质性脏器病变或其他恶性肿瘤患者㊂本研究经长沙市第四医院医学伦理委员会审核批准(201810-013)㊂1.2 M E K ㊁E R K ㊁C H K α蛋白表达情况检测方法 根据患者胶质瘤发生的不同功能区进行切除,并采用3.7%甲醛溶液固定和脱水包埋切除组织,随后进行染色㊂采用免疫组织化学法对上述组织实施抗原修复及脱蜡,再用封闭液封闭2h ,加入M E K ㊁E R K ㊁C HK α抗体温育2h ,使用磷酸盐缓冲液洗净后加入相应种属二抗㊂并将苏木精和二氨基联苯胺显色液加入实施常规复染,随后给予封片和干燥,同时观察M E K ㊁E R K ㊁C H K α蛋白的表达情况㊂在400倍光学显微镜下任意选取6个视野,每个视野观察100个细胞㊂M E K ㊁E R K ㊁C H K α蛋白表达情况以细胞膜/细胞核/细胞质中出现着色视为阳性细胞㊂阳性细胞百分数评分标准:阳性细胞数ɤ5%为0分;>5%~20%为1分;>20%~50%为2分;>50%为3分㊂着色程度强弱评分标准:无着色为0分;轻度着色为1分;中度着色为2分,重度着色为3分㊂M E K ㊁E R K ㊁C H K α蛋白表达情况评估:阳性细胞百分数评分和着色程度强弱评分总分为0分为 -,总分为1~2分为 + ,定义为M E K ㊁E R K ㊁C HK α蛋白表达阴性(低表达);总分为3~4分为 ++ ,总分为5~6分为 +++ ,定义为M E K ㊁E R K ㊁C H K α蛋白表达阳性(高表达)㊂1.3 免疫组织化学法判读标准 采用C a pt u r e 2.1显微镜图片采集软件选取3个染色均匀且不相邻的高倍视野(ˑ400)进行拍照,以阳性细胞百分数和着色强度作为标准,对所有观察到的阳性细胞实施计数,并对阳性细胞的百分数进行计算,取其平均值㊂1.4 随访情况 术后医务人员需指导患者准时来院进行放化疗,注意饮食营养补充,避免进食辛辣生冷食物,并以电话的方式对参加本研究的患者进行追踪随访㊂主要观察患者有无新并发症出现等㊂患者生存时间以住院手术后经病理检查证实时间为起始,停止时间为患者死亡或随访终止时㊂1.5 统计学处理 采用S P S S 22.0统计软件进行数据分析处理㊂计数资料以例数或百分率表示,组间比较采用χ2检验㊂以P <0.05为差异有统计学意义㊂2 结 果2.1 不同临床病理特征胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白表达情况比较 不同年龄㊁WHO 分级㊁肿瘤最大径及预后胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白高㊁低表达情况比较,差异均有统计学意义(P <0.05);不同性别㊁肿瘤位置㊁吸烟情况胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白高㊁低表达情况比较,差异均无统计学意义(P >0.05)㊂见表1㊂表1 不同临床病理特征胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白表达情况比较[n (%)]病理特征nM E K 蛋白低表达高表达χ2PE R K 蛋白低表达高表达χ2PC H K α蛋白低表达高表达χ2P性别0.5480.1440.6100.2030.9430.621男6438(59.38)26(40.62)37(57.81)27(42.19)40(62.50)24(37.50) 女6025(41.67)35(58.33)26(43.33)34(56.67)24(40.00)36(60.00)年龄(岁)6.8210.0385.4930.0246.2580.010ȡ659843(43.88)55(56.12)44(44.90)54(55.10)45(45.92)53(54.08) <652620(76.92)6(23.08)18(69.23)8(30.77)19(73.08)7(26.92)W H O 分级10.3640.0029.2410.0038.4920.004Ⅰ~Ⅱ级6048(80.00)12(20.00)46(76.67)14(23.33)49(81.67)11(18.33) Ⅲ~Ⅳ级6414(21.88)50(78.13)15(23.44)49(76.56)12(18.75)52(81.25)肿瘤最大径(m m)6.513<0.0016.1020.0087.2580.006ɤ610049(49.00)51(51.00)49(49.00)51(51.00)50(50.00)50(50.00) >62414(58.33)10(41.67)14(58.33)10(41.67)13(54.17)11(45.83)㊃9111㊃检验医学与临床2024年4月第21卷第8期 L a b M e d C l i n ,A pr i l 2024,V o l .21,N o .8续表1 不同临床病理特征胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白表达情况比较[n (%)]病理特征nM E K 蛋白低表达高表达χ2PE R K 蛋白低表达高表达χ2PC H K α蛋白低表达高表达χ2P肿瘤位置0.6810.4190.4890.0800.5410.108额叶4823(47.92)25(52.08)23(47.92)25(52.08)20(41.67)28(58.33) 颞叶3616(44.44)20(55.56)17(47.22)19(52.78)16(44.44)20(55.56) 顶叶1410(71.43)4(28.57)10(71.43)4(28.57)9(64.29)5(35.71) 枕叶128(66.67)4(33.33)8(66.67)4(33.33)10(83.33)2(16.67) 其他146(42.86)8(57.14)4(28.57)10(71.43)9(64.29)5(35.71)吸烟0.2360.0970.5820.0960.7150.072是1912(63.16)7(36.84)11(57.89)8(42.11)13(68.42)6(31.58) 否10551(48.57)54(51.43)52(49.52)53(50.48)51(48.57)54(51.43)预后7.0150.0057.8460.0046.5080.009死亡524(7.69)48(92.31)6(11.54)46(88.46)7(13.46)45(86.54) 存活6054(90.00)6(10.00)53(83.33)7(11.67)51(85.00)9(15.00) 失访125(41.67)7(58.33)4(33.33)8(66.67)6(50.00)6(50.00)2.2 Ⅰ~Ⅳ级胶质瘤患者M E K ㊁E R K ㊁C HK α蛋白阳性表达率比较 124例胶质瘤患者中Ⅰ级33例,Ⅱ级27例,Ⅲ级31例,Ⅳ级33例㊂M E K ㊁E R K ㊁C H K α蛋白主要分布在细胞质内,Ⅰ~Ⅳ级胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白阳性表达率见表2㊂高级别胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白阳性表达率较低级别胶质瘤患者更高,差异均有统计学意义(P <0.05)㊂见图1㊁2㊁3㊂2.3 随访情况 随访时间截止于2022年12月30日,124例患者中失访12例(9.68%),获得随访112例(90.32%),死亡52例(41.94%)㊂2.4 K a p l a n -M e i e r 生存曲线分析 K a pl a n -M e i e r 生存曲线分析结果显示,M E K ㊁E R K ㊁C H K α蛋白低表达胶质瘤患者均较M E K ㊁E R K ㊁C H K α蛋白高表达患者累计生存率更高,差异有统计学意义(P <0.05),见图4㊁5㊁6㊂表2 Ⅰ~Ⅳ级胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白阳性表达率比较(%)W H O 分级nM E K 蛋白高表达低表达E R K 蛋白高表达低表达C H K α蛋白高表达低表达Ⅰ级33-9.62-9.57-9.60Ⅱ级27-7.69-7.49-7.53Ⅲ级3194.08-94.52-94.71-Ⅳ级3392.04-92.18-92.23-注:WH O 分级为Ⅰ级其特征为毛细胞性星形细胞瘤;Ⅱ级为弥漫性星形细胞瘤;Ⅲ级为间变性星形细胞瘤;Ⅳ级为多形变胶质母细胞瘤;-表示无数据㊂图1 胶质瘤患者C H K α蛋白的表达(从左至右依次为Ⅰ㊁Ⅱ㊁Ⅲ㊁Ⅳ级,ˑ400)图2 胶质瘤患者E R K 蛋白的表达(从左至右依次为Ⅰ㊁Ⅱ㊁Ⅲ㊁Ⅳ级,ˑ400)㊃0211㊃检验医学与临床2024年4月第21卷第8期 L a b M e d C l i n ,A pr i l 2024,V o l .21,N o .8图3 胶质瘤患者M E K 蛋白的表达(从左至右依次为Ⅰ㊁Ⅱ㊁Ⅲ㊁Ⅳ级)图4 M E K蛋白低表达和高表达胶质瘤患者生存曲线图5 E R K蛋白低表达和高表达胶质瘤患者生存曲线图6 C H K α蛋白低表达和高表达胶质瘤患者生存曲线3 讨 论胶质瘤是中枢神经系统最常见的原发性恶性肿瘤之一,具有高度异质性和侵袭性,严重影响患者的生活质量和预后㊂目前根据病理学特征及分子生物学标志,胶质瘤主要分为多种亚型,其中Ⅰ级为良性肿瘤,Ⅳ级为高度恶性肿瘤㊂不同级别的胶质瘤具有明显的生物学差异,包括细胞增殖速度㊁血管生成㊁细胞凋亡等,这种差异可能与细胞内信号通路的激活状态有关[5-6]㊂M E K /E R K 信号通路和C H K α是研究的焦点,它们在细胞生存㊁增殖和凋亡等方面发挥关键作用㊂M E K /E R K 信号通路作为一个重要的细胞信号传导通路,参与调控细胞的增殖㊁分化㊁生存和凋亡等生命活动,在胶质瘤中,M E K /E R K 信号通路的异常激活与肿瘤的发生和发展密切相关㊂有研究发现,不同级别的胶质瘤患者M E K 和E R K 蛋白表达存在差异,提示这一通路在胶质瘤分级过程中可能起关键的调控作用[7]㊂此外,C H K α是细胞周期检查点激酶之一,参与细胞周期的调控和D N A 损伤修复,胶质瘤中C H K α的异常表达与细胞的D N A 损伤应答和凋亡抵抗性有关,其在维持细胞染色体稳定性和基因组完整性中扮演重要角色[8]㊂然而,目前针对不同级别的胶质瘤,M E K ㊁E R K 和C H K α的表达及其在肿瘤生物学过程患者的作用尚未得到充分研究㊂了解不同级别胶质瘤中这些分子的表达情况及它们在调控细胞生物学功能中的作用,对于深入理解胶质瘤的分子机制㊁寻找潜在的治疗靶点及制订个性化治疗策略具有重要临床意义㊂基于此,本研究以不同级别胶质瘤患者作为研究对象,对其肿瘤组织中M E K ㊁E R K ㊁C H K α蛋白表达进行观察分析,通过对不同级别胶质瘤组织样本的免疫组织化学染色和分子生物学试验,系统探讨它们对胶质瘤发生㊁发展及预后的潜在影响,结果显示,其各亚型胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白的表达在弥漫性星形细胞瘤与少突胶质瘤或同级别之间无明显差别㊂基于此,本研究只选择了其中一种类型的胶质瘤作为研究方向,结果显示,高级别胶质瘤患者M E K ㊁E R K ㊁C H K α蛋白阳性表达率明显高于低级别患者㊂可能与高级别胶质瘤通常具有更高的细胞增殖能力㊁侵袭性和治疗抵抗性有关,高级别胶质瘤的细胞增殖速度更快,具有更强的侵袭性,而M E K ㊁E R K 和C H K α信号通路在细胞增殖和侵袭过程中发挥重要作用,因此其表达增加可能支持这些恶性特征㊂本研究结果还显示,M E K ㊁E R K ㊁C H K α蛋白的表达与肿瘤最大径㊁年龄㊁WHO 分级存在明显相关性,肿瘤最大径ɤ6mm ㊁WHO 分级为Ⅲ~Ⅳ级㊁年龄ȡ65岁胶质瘤患者肿瘤组织中M E K ㊁E R K ㊁C H K α㊃1211㊃检验医学与临床2024年4月第21卷第8期 L a b M e d C l i n ,A pr i l 2024,V o l .21,N o .8蛋白表达明显高于肿瘤最大径>6c m㊁WHO分级为Ⅰ~Ⅱ级㊁年龄<65岁的患者㊂原因可能包括以下几点:(1)肿瘤最大径较大常表示肿瘤已经发展到了更晚期,其细胞增殖和侵袭性更强;(2)WHO分级反映了肿瘤的分化程度和组织学特征,Ⅲ~Ⅳ级肿瘤通常是高度恶性的,细胞分化程度低,而Ⅰ~Ⅱ级肿瘤的恶性程度则相对较低,不同级别的肿瘤细胞可能在蛋白表达方面存在差异,高级别肿瘤可能表现出更高的M E K㊁E R K㊁C H Kα蛋白表达;(3)年龄是一个重要的生理特征,不同年龄段的个体在肿瘤发展和生物学特征方面可能存在差异,年龄ȡ65岁的患者可能受到年龄相关的生理变化和免疫功能下降的影响,导致肿瘤细胞中M E K㊁E R K㊁C H Kα蛋白表达升高㊂有研究表明,M E K㊁E R K㊁C H Kα蛋白的高表达在一定程度上会使患者发生不良预后的可能性增加[9-10]㊂也有研究发现,M E K㊁E R K㊁C H Kα蛋白的高表达与肿瘤患者的复发和死亡风险有关[11-12]㊂从上述不同研究结论来看,目前临床尚不完全清楚M E K㊁E R K㊁C H Kα蛋白在胶质瘤患者中的高表达对其预后及作用机制的影响㊂本研究发现,高级别胶质母细胞瘤中M E K㊁E R K㊁C H Kα蛋白高表达较多,但在Ⅰ级星形细胞瘤中较少(P<0.05)㊂这可能是因为M E K/E R K信号通路的过度活化可能会导致细胞增殖和生长异常加速,高级别胶质母细胞瘤中C H Kα蛋白低表达可能会导致D N A损伤修复机制失调,使细胞更容易发生不稳定和突变,从而加速肿瘤发展,并且其表面形态与患者的疾病预后可能有一定关系[13-14]㊂本研究结果显示,C H Kα蛋白低表达患者累计生存率更高,差异有统计学意义(P<0.05),可能与C H Kα蛋白的低表达降低了肿瘤细胞的生存优势及减缓了肿瘤的进展有关,C H Kα作为一个细胞周期调控的关键分子,它的低表达使肿瘤细胞更容易受到治疗的影响[15]㊂WHO分级为Ⅲ级和Ⅳ级是高级别胶质瘤,并且通常会采取术中全切与术后放化疗联合进行治疗,尽管现如今已有多种对高级别胶质瘤进行治疗的方案,但效果均不是特别理想,且患者术后复发率仍居高不下[16]㊂此外,大多数患者在被确诊或发觉异常时已处于疾病中晚期或肿瘤已转移或已扩散,极难再通过外科手术完全切除[17]㊂所以,早期诊断和更精确地观察对于改善胶质瘤患者的预后十分关键㊂但目前由于临床样本量太少,还需扩大样本量实施进一步研究㊂综上所述,M E K㊁E R K㊁C H Kα蛋白在不同级别胶质瘤患者中的阳性表达情况有明显区别,且与患者的疾病预后明显相关,对其疾病的诊断和预后判断具有重要价值㊂参考文献[1]P L A T T E N M,B U N S E L,W I C K A,e t a l.A v a c c i n e t a r-g e t i n g m u t a n t I D H1i n n e w l y d i a g n o s e d g l i o m a[J].N a-t u r e,2021,592(7854):463-468.[2]黄灵,邹有瑞,李琢琦,等.C H K A基因沉默对胶质瘤细胞生物学行为的影响及其机制[J].中国癌症杂志,2021,31(11):1041-1049.[3]薛彩强,杜晓灏,金龙,等.基于M R I深度学习模型预测WHOⅡ㊁Ⅲ级胶质瘤MGM T启动子甲基化状态[J].中华放射学杂志,2021,55(7):734-738.[4]蔡志超,池琦.磁共振扩散张量成像定量参数对脑胶质瘤分级的诊断价值[J].现代肿瘤医学,2019,27(11):2005-2009.[5]M E L L I N G HO F F I K,E L L I N G S O N B M,T O U A T M,e ta l.I v o s i d e n ib i n i s oc i t r a t ede h y d r o g e n a s e1-m u t a t e d a d-v a n c e d g l i o m a[J].J C l i n O n c o l,2020,38(29):3398-3406.[6]V A N D E N B E N T M J,T E S I L E A N U C M,W I C K W,e ta l.A d j u v a n t a n d c o n c u r r e n t t e m o z o l o m i d e f o r1p/19q n o n-c o-d e l e t e d a n a p l a s t i c g l i o m a(C A T N O N;E O R T C s t u d y26053-22054):s e c o n d i n t e r i m a n a l y s i s o f a r a n d o m-i s e d,o p e n-l a b e l,p h a s e3s t u d y[J].L a n c e t O n c o l,2021,22(6):813-823.[7]陈燕伟,马善波,张蕊.血府逐瘀汤联合替莫唑胺对脑胶质瘤大鼠E R K/MA P K信号通路的调节作用[J].西部医学,2021,33(9):1258-1263.[8]杨彬,于晶,刘晓智,等.m i R-209下调M E K/E R K信号通路抑制胶质瘤U87细胞增殖㊁增强放疗敏感性的研究[J].实用医学杂志,2021,37(13):1670-1673. [9]李斌,李娜.3项指标在不同病理分级脑胶质瘤中的表达及临床意义[J].检验医学与临床,2020,17(1):113-115.[10]张莞萍,李长生.P A X8㊁A S A H1㊁C u l l i n7在脑胶质瘤患者癌组织与癌旁组织中的蛋白表达及与生存状况的相关性分析[J].医学临床研究,2021,38(3):413-415. [11]张慧,范月超,陈洪福,等.胶质瘤组织中G R K5和K i-67的表达情况及临床意义[J].现代肿瘤医学,2021,29(2): 215-219.[12]丁大领,张风江,郭宗泽,等.H C MV p p65蛋白在脑胶质瘤组织中的表达及临床意义[J].现代肿瘤医学,2019,27(7):1145-1148.[13]周杰,徐聃,李波,等.基于生物信息学分析M E T T L7B在胶质瘤组织中的表达及其临床意义[J].中国肿瘤生物治疗杂志,2022,29(1):55-62.[14]王伟,王茂德,姜海涛,等.基于T C G A与G T E x数据库分析A E B P1基因在不同分型胶质瘤中的表达及对预后的影响[J].西安交通大学学报(医学版),2022,43(4): 503-508.[15]张东旭,曲彦明,张明山,等.WHOⅡ级脑胶质瘤患者无进展生存期的影响因素分析[J].中华神经外科杂志, 2019,35(5):469-473.[16]孙莹,李相军,于鹏跃,等.组织因子在人胶质瘤组织中的表达水平及其临床意义[J].中国实验诊断学,2021,25(3):317-321.[17]王小刚,刘强,王伯栋,等.人脑胶质瘤组织的I M P3和I G F2的表达水平及其临床意义[J].临床和实验医学杂志,2019,18(19):2063-2066.(收稿日期:2023-09-26修回日期:2023-12-28)㊃2211㊃检验医学与临床2024年4月第21卷第8期 L a b M e d C l i n,A p r i l2024,V o l.21,N o.8。
雪旺细胞源运动神经营养因子的亚细胞定位和初步分布对策

雪旺细胞源运动神经营养冈子的弧细胞定位雨l初步分布研究
幽2._【_I』ABC法对SC进行S-100染色的阴性对照.(10X40)
雪旺细胞源运动神经营养闪子的Ⅱ细胞定位和初步分布1iJ|_究
削4.阴性对照组l:川TBS代替SDNF.McAb(10X40)
雪址E细胞源运动神经营养闪,的,lEVI胞定佗年¨们步分m,圳究
|璺|5.阴性对照组2:/4jTBS代替胶体金结合羊抗鼠IgG(10>t10)
雪旺细胞源运动神经营养闪子的i『F细胞定位和考u步分,f'liliJf究
图7.内质网上的金颗粒(I4x,11.Jjf商,)
33
雪旺细胞源运动神经营养Ⅲf的qEfltl胞定,ftz,Fin初步分柑Ⅲ究
幽8.高尔基体上人耸金颗粒密集(1.4×3万倍)
雪旺细胞源运动神经营养闪子的弧细胞定位平||初步分布{{J|_究
幽10·金银染色法对照组2:变性神经,用TBS代替胶体金结合羊抗鼠IgG(10X4)
39
幽12.IGSS法对变性坐骨神经染色(10×10)
40
图14.IGSS法对失神经支配皮肤染色(10×10)
41
雪旺细胞源运动神经营养冈子的Ⅱ细胞定位和初步分布研究
图16.1GSS法对火神经支配肌肉染色(10×10)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Enchondral calcification and ossification is of particular interest in developmental biology, growth, and fracture healing. The histochemical demonstration of calcifying cartilage in enchondral bone formation is not commonly done due to methodological difficulties. These technical problems include providing sufficient embedding medium support for sectioning of relatively hard tissue and appropriate staining to demonstrate histochemical characteristics of both calcified and uncalcified cartilage while preserving cell detail. The availability of polymethylmethacrylate embedding for sectioning of calcified tissue has largely obviated the problem
Key words: Enchondral bone formation - - Histochemical - - Staining - - Mineralized, unmineralized - - Embedding.
of providing sufficient support. In this communication we describe our method of processing calcifying cartilage and our technique of histochemical staining which permits the examination of calcification in histochemically distinct cartilage while at the same time preserving chondrocyte histologic detail. The induction of enchondral ossification by the implantation of decalcified bone matrix was initially described by Reddi and Huggins [1]. We, and others, have applied this model as a test system for physical and pharmacological agents that affect the enchondral ossification process [2, 3]. The developmental sequence of the model is biochemically well characterized. The application of our histological techniques has permitted a detailed morphological description of the system (R. K. Aaron et al., unpublished data).
Calcif Tissue Int (1988) 43:244-249
Calcified Tissue International
9 1988Springer-VerlagNew YorkInc.
A Histochemical Method for the Demonstration of Calcifying Cartilage
Embedding and Fixation
The specimens were fixed in 10% formaldehyde in 0.1 M sodium cacodylate at pH 7.2 for 24 hours at 4~ They were then dehydrated in an upgraded series of alcohols of 30, 70, and 85% for 1 hour each with two more changes in 100% alcohol for 1 hour each. The specimens were placed into equal volumes of absolute alcohol and cellosolve for 1 hour and then in cellosolve overnight. They were then transferred to freshly prepared methylmethacrylate containing 15% dibutyl phthalate and 2.5% benzol peroxide and were placed in a vacuum of 20" Hg at room temperature for 30 min, this process was repeated three times. After the last change, the specimens were stored at 4~ overnight. Freshly prepared embedding media was partially polymerized by heating to 85-90~ with constant stirring. When small bubbles appeared that were not caused by stirring, it was immersed in ice water to lower the temperature to 40-45 ~. The process was repeated until there was a slight increase in the viscosity of the methylmethacrylate. The specimens were then placed in this partially polymerized medium at 4~ for 24 hours after which time they were allowed to polymerize at room temperature for 2 days. The specimens were cut on a Polycut microtome (Reichert-Jung) with a tungsten carbide knife moistened with 40% alcohol, and 5 ixm sections were prepared. The sections were placed with 95% alcohol on slides that had been pretreated with 0.4% gelatin, and pressed fiat. A total of 6 days was required for processing and full polymerization
Send reprint requests to Roy K. Aaron, M.D., 154 Waterman Street, Providence, RI 02906, USA.
Decalcified Bone Matrbc-Induced Enchondral Ossification
Decalcified bone matrix was prepared from the diaphyses of femurs and tibias of adult CD rats [1]. The bones were defatted with ethanol and ether, milled frozen in liquid nitrogen to a particle size of 75-400~m, decalcified with 0.5 N hydrochloric acid, and extracted with ethanol and ether and air dried. Twenty-five milligrams of the decalcified bone matrix powder was implanted subcutaneously along the thoracic musculature in immature CD rats. Animals were sacrificed periodically and ossicles were taken for biochemical and histological study. Prior to sacrifice, animals were injected intraperitoneally with 1 i~Ci/g body weight of 35sulfate for proteoglycan analysis. On day 2 following implantation, mesenchymal cells migrated into the implant. On days 4 through 8, differentiation to chondrocytes occurred primarily on the surfaces of the bone chips. Chondrogenesis was maximal on days 8-10. Calcification of the cartilage began on about day l0 with the formation of bony trabeculae from days 16 through 20.