机动车变更登记、备案申请表及填写模板 B
机动车变更登记备案申请表

机动车变更登记/备案申请表
理
填 表 说 明
2、“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时申请
多项变更登记/备案的可多选;
3、“邮寄地址”栏,填写可通过邮寄送达的地址;
4、“机动车所有人(代理人)签字”栏,机动车属于个人的,由变更后的现
机动车所有人签字,属于单位的,由单位的被委托人签字。
属于个人或者单位代
的,由代理人或者代理单位的经办人签字,并填写“代理人”栏;
5、“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通摩托车号牌、
轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号牌、领
汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽车号牌、
用摩托车号牌填写。
月 日
涂改;
□”中划“√”,同时申请
;
属于个人的,由变更后的现签字。
属于个人或者单位代理代理人”栏;
车号牌、普通摩托车号牌、号牌、使馆摩托车号牌、领馆托车号牌、警用汽车号牌、警。
机动车变更登记备案申请表

5.“机动车所有人签字”栏,机动车属于个人的,由变更后的现机动车所有人签字,属于单位的,由单位的被委托人签字。由代理人代为办理的,机动车所有人不签字;
6.“代理人签字”栏,属于个人代理的,填写代理人的姓名、邮寄地址、邮政编码、联系电话和电子信箱,在代理人栏内签名,不必填写经办人姓名等项目;属于单位代理的,应填写代理人栏的所有内容,代理单位的经办人签字;属于单位的机动车,由本单位被委托人办理的不需填写本栏;
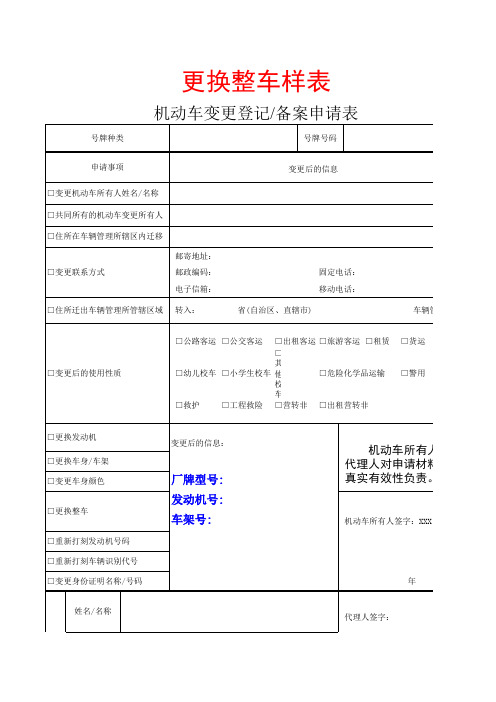
□幼儿校车 □小学生校车 □其他校车 □危险化学品运输 □警用 □消防
□救护 □工程救险 □营转非 □出租营转非
□更换发动机
变更后的信息:
机动车所有人及代理人对申请材料的真实有效性负责。
□更换车身/车架
□变更车身颜色
□更换整车
机动车所有人签字:
□重新打刻发动机号码
□重新打刻车辆识别代号
□变更身份证明名称/号码
年 月 日
代理人
姓名/名称
代理人签字:Biblioteka 邮寄地址邮政编码联系电话
电子信箱
经办人
姓名
联系电话
年 月 日
1.填写时请使用黑色或者蓝色墨水笔,字体工整,不得涂改;
2.“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时申请多项变更登记/备案的可多选;
3.“邮寄地址”栏,填写可通过邮寄送达的地址;
4.“电子信箱”栏,填写接收电子邮件的e-mail地址,尚未申请电子信箱的可以不填写;
机动车变更登记备案申请表填写样式
年
月
日
:
年
月
日
□更换整车
机动车所有人签字:XXX
□重新打刻发动机号码 □重新打刻车辆识别代号 □变更身份证明名称/号码 年 月 日
姓名/名称
代理人签字:
代 理 人
邮寄地址 邮政编码 联系电话
代 理 人 电子信箱 经办人姓名 联系电话 年 月真实 负责。
人签字:XXX
变更后的信息
□变更后的使用性质
□公路客运 □公交客运 □出租客运 □旅游客运 □租赁 □幼儿校车 □小学生校车 □其他校车 □危险化学品运输 □救护 □工程救险 □营转非 □出租营转非
□货运 □警用
□更换发动机 □更换车身/车架 □变更车身颜色
变更后的信息:
厂牌型号: 发动机号: 车架号:
机动车所有人及代 理人对申请材料的真实 有效性负责。
更换整车样表
机动车变更登记/备案申请表
号牌种类 申请事项 □变更机动车所有人姓名/名称 □共同所有的机动车变更所有人 □住所在车辆管理所辖区内迁移 邮寄地址: □变更联系方式 邮政编码: 电子信箱: □住所迁出车辆管理所管辖区域 转入: 省(自治区、直辖市) 固定电话: 移动电话: 车辆管理所 号牌号码
机动车变更登记备案申请表

联系电话
联系电话
.填写时请使用黑色或者蓝色墨水笔,字体工整,不得涂改;1.“申请事项”栏为选择项目,请在相应栏内“□”中划2备案的可多选;/,同时申请多项变更登记.“邮寄地址”栏,填写可通过邮寄送达的地址;3地址,尚未申e-mail4.“电子信箱”栏,填写接收电子邮件的.“机动车所有人签字”栏,机动车属于个人的,由变更后的5.“代理人签字”栏,属于个人代理的,填写代理人的姓名、6.“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通7
机动车变更登记备案申请表
号牌种类申请事项机动车所有人名称所有的机动车有人在车辆管理所迁移联系方式迁出车辆管理区域后的使用性质发动机车身/车架车身颜色整车打刻发动机号打刻车辆识别身份证明名称
号牌号码
变更后的信息
邮寄地址:邮政编码:电子信箱:
转入:省(自治区、直辖市)车辆管理所□出租客运□旅游客运□其他校车□危险化学品□营转非□出租营转非
代理人对申请ቤተ መጻሕፍቲ ባይዱ料真实有效性负责。机动车所有人签字
□公路客运□公交客运赁□货运□教练□幼儿校车□小学生校车□警用□消防
□救护□工程救险
变更后的信息:
固定移动机动车所有
名姓名/称邮寄地址代邮政编理码人电子信箱经办人姓名“√”请电子信箱的可以不填写;现机动车所有人签字,属于单位的,由单位的被委托人签字。由代理人代为办理的,机动车所有人不签字;邮寄地址、邮政编码、联系电话和电子信箱,在代理人栏内签名,不必填写经办人姓名等项目;属于单位代理的,应填写代理人栏的所有内容,代理单位的经办人签字;属于单位的机动车,由本单位被委托人办理的不需填写本栏;摩托车号牌、轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号牌、领馆汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽车号牌、警用摩托车号牌填写。
机动车变更登记备案申请表

线
订
装
附件2
机动车变更登记/备案申请表
填 表 说 明
1、填写时请使用黑色或者蓝色墨水笔,字体工整,不得涂改;
2、“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时申请多项变更登记/备案的可多选;
3、“邮寄地址”栏,填写可通过邮寄送达的地址;
4、“电子信箱”栏,填写接收电子邮件的e-mail地址,尚未申请电子信箱的可以不填写;
5、“机动车所有人签字”栏,机动车属于个人的,由变更后的现机动车所有人签字,属于单位的,由单位的被委托人签字。
由代理人代为办理的,机动车所有人不签字;
6、“代理人签字”栏,属于个人代理的,填写代理人的姓名、邮寄地址、邮政编码、联系电话和电子信箱,在代理人栏内签名,不必填写经办人姓名等项目;属于单位代理的,应填写代理人栏的所有内容,代理单位的经办人签字;属于单位的机动车,由本单位被委托人办理的不需填写本栏;
7、“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通摩托车号牌、轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号牌、领馆汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽车号牌、警用摩托车号牌填写。
车辆备案登记申请书模板

车辆备案登记申请书尊敬的车辆管理所:您好!我单位(个人)现需对以下车辆进行备案登记,请您予以审批。
一、车辆信息1. 车辆类型:(如小型轿车、货车、客车等)2. 车辆品牌:(如宝马、奥迪、东风等)3. 车辆型号:(如320、A6、货车A1等)4. 车辆颜色:(如黑色、白色、红色等)5. 车辆识别代码(VIN):6. 发动机号码:7. 车架号码:二、车辆所有人信息1. 单位名称(个人姓名):2. 单位地址(个人住址):3. 联系人:4. 联系电话:三、车辆使用情况1. 车辆用途:(如运输、商务、家用等)2. 车辆使用区域:(如市区、郊区、县区等)3. 车辆行驶证、驾驶证等相关证件是否齐全:四、备案原因及目的1. 原因:(如购新车、二手车过户、企业需求等)2. 目的:(如正常使用、租赁、销售等)五、承诺事项1. 车辆所有人承诺所提供的车辆信息真实、准确、完整。
2. 车辆所有人承诺遵守国家相关法律法规,合法使用车辆。
3. 车辆所有人承诺在车辆使用过程中,严格遵守交通规则,确保行车安全。
六、申请材料1. 车辆所有人的身份证明(单位证件、个人身份证等)。
2. 车辆所有人的营业执照(个人无需提供)。
3. 车辆购销合同、发票或其他证明车辆所有权的文件。
4. 车辆行驶证、驾驶证等相关证件。
5. 其他可能需要的材料。
请您予以审批,并给予办理车辆备案登记。
如有需要,我单位(个人)将积极配合贵所进行相关查验工作。
感谢您的关注与支持!此致敬礼!申请人:(单位名称或个人姓名)申请日期:年月日注意事项:1. 请根据实际情况填写车辆信息、车辆所有人信息、车辆使用情况等。
2. 请确保所提供的信息真实、准确、完整。
3. 如有疑问,请咨询车辆管理所工作人员。
4. 请按照当地车辆管理所的要求提交相关材料。
机动车变更登记备案申请表填写样式模板

号牌种类 申请事项
按大、小型汽车、挂车、摩 托车等种类填写
号牌号码
变更后的信息(请在相应□内打“√”)
□变更机动车所有人姓名/名称
□共同所有的机动车变更所有人
□住所在车辆管理所辖区内迁移
□变更联系方式
邮寄地址:浙江省嘉兴市XX(县、市、 区)XX路XX号
邮政编码:XXXXXX
手机号码:11位手机号码 固定电话:
□住所迁出车辆管理所管辖区域 转入:
省(自治区、直辖市) :XX
市(地、州)
□变更后的使用性质 (请在相应□内打“√”)
□更换发动机
□公路客运 □公交客运
□营转非
□出租营转 非
□救护
□接送幼儿
□出租客运 □
危 □接送小学生
□旅游客 □运
□租赁 □货运 □教练 □□
代 ቤተ መጻሕፍቲ ባይዱ 人
邮寄地址
按居住地详细地址填写(浙江省嘉兴市XX(县、市、区)XX路 XX号)
填写11位手机号码(相
邮政编码
XXXXXX
手机号码
当重要,请务必如实填
XX年XX月XX日
写)
填表说明
1、填写时请使用黑色或者蓝色墨水笔,字体工整,不得涂改; 2、“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时申 请多项变更登记/备案的可多选; 3、“邮寄地址”栏,填写可通过邮寄送达的地址; 4、“机动车所有人(代理人)签字”栏,机动车属于个人的,由变更后的 现机动车所有人签字,属于单位的,由单位的被委托人签字。属于个人或者单位 代理的,由代理人或者代理单位的经办人签字,并填写“代理人”栏; 5、“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通摩托车号牌 、轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号牌、 领馆汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽车号 牌、警用摩托车号牌填写。
机动车变更登记-变更备案申请表

机动车变更登记-变更备案申请表申请人信息:姓名:__________________性别:__________________车辆信息:车牌号码:__________________机动车所有人姓名:__________________发动机号码:__________________车辆识别代号:__________________变更前信息:变更前所有人姓名:__________________变更前车辆种类:__________________变更前车辆类型:__________________变更前使用性质:__________________变更前核定载人数:__________________变更前总质量:__________________变更前整备质量:__________________变更前外廓尺寸:__________________变更前核定载质量:__________________变更前准牵引总质量:__________________变更前轮胎数额:__________________变更前轴数:__________________变更前检验记录情况:__________________变更后信息:变更后所有人姓名:__________________变更后车辆种类:__________________变更后车辆类型:__________________变更后使用性质:__________________变更后核定载人数:__________________变更后总质量:__________________变更后整备质量:__________________变更后外廓尺寸:__________________变更后核定载质量:__________________变更后准牵引总质量:__________________变更后轮胎数额:__________________变更后轴数:__________________变更后检验记录情况:__________________申请事项:变更原因:__________________办理地点:__________________申请日期:__________________附件清单:2.机动车行驶证复印件:_______份3.机动车登记证书复印件:_______份6.其他附件(如涉及车辆过户,则需要提供过户协议等相关文件):_______份本人保证所提供的信息真实、准确,如有虚假,愿意承担由此产生的法律责任。
变更备案申请表
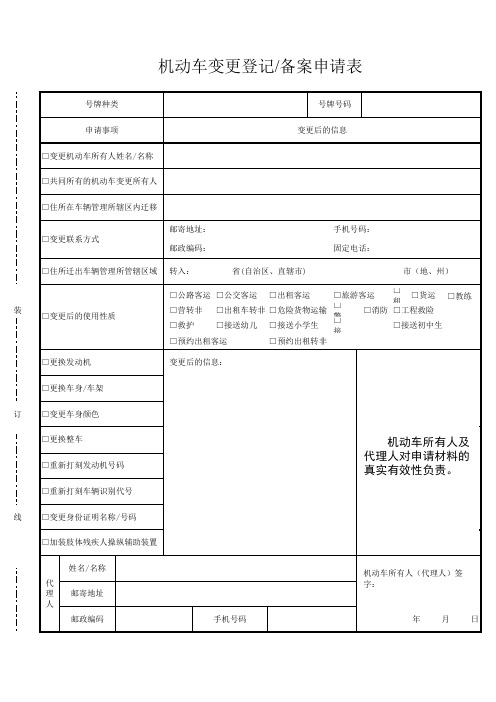
□公交客运 □出租客运 □出租车转非 □危险货物运输 □接送幼儿 □接送小学生
□旅游客运
□ 警□
□消防
接
□ 租
□货运
□工程救险
□接送初中生
□教练
□预约出租客运
□预约出租转非
□更换发动机
变更后的信息:
□更换车身/车架
订 □变更车身颜色
□更换整车 □重新打刻发动机号码 □重新打刻车辆识别代号
机动车所有人及 代理人对申请材料的 真实有效性负责。
线 □变更身份证明名称/号码
Hale Waihona Puke □加装肢体残疾人操纵辅助装置
姓名/名称
代 理 邮寄地址 人
邮政编码
手机号码
机动车所有人(代理人)签 字:
年
月
日
机动车变更登记/备案申请表
号牌种类
号牌号码
申请事项
变更后的信息
□变更机动车所有人姓名/名称
□共同所有的机动车变更所有人
□住所在车辆管理所辖区内迁移
□变更联系方式
邮寄地址: 邮政编码:
手机号码: 固定电话:
□住所迁出车辆管理所管辖区域 转入:
省(自治区、直辖市)
市(地、州)
装 □变更后的使用性质
□公路客运 □营转非 □救护
机动车变更登记备案申请表填写样式模板

机动车变更登记备案申请表填写样式模板申请表编号:
填表日期:
申请人姓名:
变更事项:
1.车辆基本信息变更:
(1)变更前车辆基本信息:
车辆牌照号码:
车辆类型:
车辆识别代码:
发动机号码:
(2)变更后车辆基本信息:
车辆牌照号码:
车辆类型:
车辆识别代码:
发动机号码:
2.车主信息变更:
(1)变更前车主信息:
车主姓名:
(2)变更后车主信息:
车主姓名:
3.车辆使用性质变更:
(1)变更前使用性质:
(2)变更后使用性质:
4.车辆注销与重新登记:
(1)变更前注销原因:
(2)变更后重新登记原因:
5.其他变更事项:
本人承诺所填写信息准确无误,如有虚假陈述,愿意承担相应的法律责任。
申请人签名:日期:
注意事项:
1.请如实填写申请表,并加盖单位公章或个人签名。
以上为机动车变更登记备案申请表填写样式,申请人应根据具体情况填写,并注意提供所需的相关证明材料。
请务必填写准确无误,以确保变更登记备案的顺利进行。
机动车变更登记备案申请表xls - 填写时请使用黑色或者蓝色墨水笔

机动车变更登记/备案申请表
填 表 说 明
1、填写时请使用黑色或者蓝色墨水笔,字体工整,不得涂改;
2、“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时申请多项变更登记/备案的可多选;
3、“邮寄地址”栏,填写可通过邮寄送达的地址;
4、“电子信箱”栏,填写接收电子邮件的e-mail地址,尚未申请电子信箱的可以不填写;
5、“机动车所有人签字”栏,机动车属于个人的,由变更后的现机动车所有人签字,属于单位的,由单位的被委托人签字。
由代理人代为办理的,机动车所有人不签字;
6、“代理人签字”栏,属于个人代理的,填写代理人的姓名、邮寄地址、邮政编码、联系电话和电子信箱,在代理人栏内签名,不必填写经办人姓名等项目;属于单位代理的,应填写代理人栏的所有内容,代理单位的经办人签字;属于单位的机动车,由本单位被委托人办理的不需填写本栏;
7、“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通摩托车号牌、轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号牌、领馆汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽车号牌、警用摩托车号牌填写。
机动车变更登记备案申请表

变更后的信息:
机动车所有人及代 理人对申请材料的真实 有效性负责。
江苏省无锡市
旧机动车交易服务公司 号
代理人签字:
江苏省无锡市苏锡路 214
@ .com
联系电话 联系电
请仔细阅读填表说明第 6 条,按照实际情况 填写;由机动车所有人本人办理的本栏目不用填 写本栏目;属于单位的机动车,由本单位被委托 人办理的不需填写本栏;属于个人代理的,不必 填写经办人姓名等项目
小型汽车号牌
号牌号码 变更后的信息
苏 B/
□小学生校车 □其他校车
□危险化学品运输
变更后的信息:
机动车所有人及代 理人对申请材料的真实 有效性负责。
李
四
号
代理人签字:
江苏省无锡市苏锡路 214
@ .com
联系电话 联系电话
8
李
四
年
月
日
请仔细阅读填表说明第 5 条,由代理人代为办理的,机动 车所有人不签字,属于单位的, 由单位的被委托人签字
固定电话: 移动电话: 省(自治区、直辖市) □公交客运 □工程救险 □出租客运 □营转非
8
车辆管理所
□旅游客运 □出租营转非
□租赁
□货运 □警用
□教练 □消防
□小学生校车 □其他校车
□危险化学品运输
请 先仔细阅读表格 反面的填表说明, 反面的填表说明,按第 7 填表说明 的要求填写 条的要求填写
机动车变更登记/备案申请表
小型汽车号牌
号牌号码 变更后的信息
请按实际号 牌填写
号牌种类 申请事项 □变更机动车所有人姓名/名称 □共同所有的机动车变更所有人 □住所在车辆管,同时申请多 项变更登记/备案的可多选
车辆的变更登记申请
变更后的使用性质
幼儿小车 小学生校车 危险化学品运输 警用 消防 救护
更换发动机
工程救险 营转非 出租营转非 变更后的信息:
更换车身/车架
变更车身颜色
更换整车 重新打刻发动机号码
机动车所有人及代理 人对申请材料的真实 有效性负责。
重新打刻车辆识别代号
变更身份证明名称/号码
加装肢体残疾人操纵辅助装置
姓名/名称
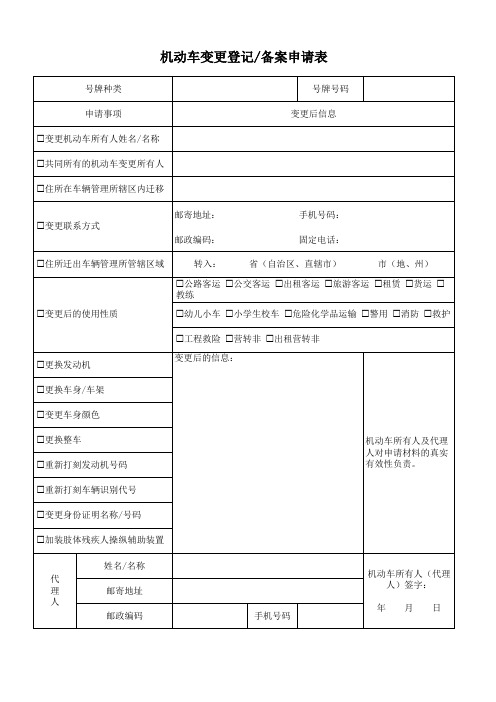
机动车变更登记/备案申请表
号牌种类
ቤተ መጻሕፍቲ ባይዱ
号牌号码
申请事项
变更后信息
变更机动车所有人姓名/名称
共同所有的机动车变更所有人
住所在车辆管理所辖区内迁移
变更联系方式
邮寄地址: 邮政编码:
手机号码: 固定电话:
住所迁出车辆管理所管辖区域
转入:
省(自治区、直辖市)
市(地、州)
公路客运 公交客运 出租客运 旅游客运 租赁 货运 教练
代
理
邮寄地址
人
邮政编码
手机号码
机动车所有人(代理 人)签字:
年月日
机动车驾驶证变更备案申请表

表一
机动车驾驶证变更备案申请表
姓名
性别
驾驶证证号
现住所地址
需变更 的内容
联系地址
住宅电话
驾驶证档案编号 出生日期
小灵通
手机邮编 邮编来自申请人签字:年月日
表二
机动车登记证书编号
机动车所有人
身份证号码
现住所地址
需变更 的内容
联系地址
住宅电话
机动车变更备案申请表
号牌号码
小灵通
手机
邮编 邮编
申请人签字:
年月日
注:根据《中华人民共和国道路交通安全法实施条例》的相关规定,机动车所有人、驾驶人的住 所地址、联系方式等变更的,应当向车辆管理所备案(可通过邮寄、传真、电子邮件等方式)。
机动车变更登记备案申请表

线
订
装
附件2
机动车变更登记/备案申请表
理
填 表 说 明
2、“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时
请多项变更登记/备案的可多选;
3、“邮寄地址”栏,填写可通过邮寄送达的地址;
4、“电子信箱”栏,填写接收电子邮件的e-mail地址,尚未申请电子信
的可以不填写;
5、“机动车所有人签字”栏,机动车属于个人的,由变更后的现机动车
有人签字,属于单位的,由单位的被委托人签字。
由代理人代为办理的,机
所有人不签字;
6、“代理人签字”栏,属于个人代理的,填写代理人的姓名、邮寄地址
邮政编码、联系电话和电子信箱,在代理人栏内签名,不必填写经办人姓名
目;属于单位代理的,应填写代理人栏的所有内容,代理单位的经办人签字
于单位的机动车,由本单位被委托人办理的不需填写本栏;
7、“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通摩托车号
、轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号
领馆汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽
牌、警用摩托车号牌填写。
月 日
得涂改;
□”中划“√”,同时申
;
地址,尚未申请电子信箱
,由变更后的现机动车所代理人代为办理的,机动车
理人的姓名、邮寄地址、,不必填写经办人姓名等项代理单位的经办人签字;属本栏;
车号牌、普通摩托车号牌车号牌、使馆摩托车号牌、练摩托车号牌、警用汽车号。
机动车变更登记备案申请表

附件2
机动车变更登记/备案申请表
线
订
装
填 表 说 明
1、填写时请使用黑色或者蓝色墨水笔,字体工整,不得涂改;
2、“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时申请多项变更登记/备案的可多选;
3、“邮寄地址”栏,填写可通过邮寄送达的地址;
4、“电子信箱”栏,填写接收电子邮件的e-mail地址,尚未申请电子信箱的可以不填写;
5、“机动车所有人签字”栏,机动车属于个人的,由变更后的现机动车所有人签字,属于单位的,由单位的被委托人签字。
由代理人代为办理的,机动车所有人不签字;
6、“代理人签字”栏,属于个人代理的,填写代理人的姓名、邮寄地址、邮政编码、联系电话和电子信箱,在代理人栏内签名,不必填写经办人姓名等项目;属于单位代理的,应填写代理人栏的所有内容,代理单位的经办人签字;属于单位的机动车,由本单位被委托人办理的不需填写本栏;
7、“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通摩托车号牌、轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号牌、领馆汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽车号牌、警用摩托车号牌填写。
机动车变更备案申请表

机动车登记证书编号
号牌号码
变更备案事项
□住所在车辆管理所管辖区域内迁移□变更机动车所有人姓名/名称□变更公章样式□变更联系方式□变更车辆识别代号拓印膜样式□变更机动车所有人身份证明名称/号码
机动车所有人信息
姓名/名称
联系电话
住所地址
邮政编码
装
身份证明名称
号码
□常住人口□暂住人口
居住/暂住证
明名称
号码
变更后信息
机动车所有人:
订
申请方式
□由机动车所有人申请
(个人签字/单位盖章)
□机动车所有人委托___________________________________代理申请
年月日
线
代理人
姓名/名称
联系电话
住所地址
身份证明名称
号码
代理人:
经办人
住所地址
(个人签字/单位盖章)
签字
年月日
年月日
机动车变更备案申请书模板

机动车变更备案申请书尊敬的车管所:我谨代表本人/本单位,向贵所提交机动车变更备案申请。
请贵所予以审核并办理相关手续。
具体情况如下:一、机动车信息1. 机动车登记证书编号:____________________2. 机动车号牌号码:____________________3. 机动车所有人姓名/名称:____________________4. 机动车所有人联系电话:____________________5. 机动车所有人住所地址:____________________6. 机动车所有人邮政编码:____________________7. 机动车身份证明名称:____________________8. 机动车身份证明号码:____________________二、变更事项1. 变更机动车车身颜色:____________________2. 更换发动机:____________________3. 更换车身或者车架:____________________4. 因质量问题更换整车:____________________5. 营运机动车改为非营运机动车或者非营运机动车改为营运机动车等使用性质改变的:____________________6. 发动机号码、车辆识别代号因磨损、锈蚀、事故、被盗抢等原因辨认不清或者损坏的:____________________7. 已注册登记机动车加装或拆除肢体残疾人操纵辅助装置的:____________________8. 已注册登记的机动车,机动车所有人的住所迁出或者迁入车辆管理所管辖区域的:____________________9. 机动车所有人为两人以上,需要将登记的所有人:____________________三、变更后信息1. 变更后的机动车车身颜色:____________________2. 变更后的发动机型号:____________________3. 变更后的车身或者车架号码:____________________4. 变更后的机动车所有人姓名/名称:____________________5. 变更后的机动车所有人联系电话:____________________6. 变更后的机动车所有人住所地址:____________________7. 变更后的机动车所有人邮政编码:____________________四、其他说明1. 本次变更申请系依法办理,符合国家相关政策规定。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
机动车变更登记/备案申请表
号牌种类 号牌号码
申请事项
变更后的信息
□变更机动车所有人姓名/名称 □共同所有的机动车变更所有人
□住所在车辆管理所辖区内迁移
□变更联系方式
邮寄地址:
邮政编码: 固定电话: 电子信箱:
移动电话:
□住所迁出车辆管理所管辖区域 转入
省(自治区、直辖市 )
车辆管理所
□变更后的使用性质
□公路客运
□公交客运
□出租客运 □旅游客运 □租赁 □货运 □教练
□幼儿校车 □小学生校车 □其他校车 □危险化学品运输 □警用 □消防 □救护
□工程救险
□营转非
□出租营转非
□更换发动机
变更后的信息:
机动车所有人及代理人对申请材料的真实有效性负责。
□更换车身/车架 □变更车身颜色 □更换整车
机动车所有人签字:
年 月 日
□重新打刻发动机号码
□重新打刻车辆识别代号 □变更身份证明名称/号码 姓名/名称
代理人签字:
代 理 人 邮寄地址
邮政编码 联系电话
电子信箱
装
订 线
填表说明
1、填写时请使用黑色或者蓝色墨水笔,字体工整,不得涂改;
2、“申请事项”栏为选择项目,请在相应栏内“□”中划“√”,同时申请多项变更登记/备案的可多选;
3、“邮寄地址”栏,填写可通过邮寄送达的地址;
4、“电子信箱”栏,填写接收电子邮件的e-mail地址,尚未申请电子信箱的可以不填写;
5、“机动车所有人签字”栏,机动车属于个人的,由变更后的现机动车所有人签字,属于单位的,由单位的被委托人签字。
由代理人代为办理的,机动车所有人不签字;
6、“代理人签字”栏,属于个人代理的,填写代理人的姓名、邮寄地址、邮政编码、联系电话和电子信箱,在代理人栏内签名,不必填写经办人姓名等项目;属于单位代理的,应填写代理人栏的所有内容,代理单位的经办人签字;属于单位的机动车,由本单位被委托人办理的不需填写本栏;
7、“号牌种类”栏,按照大型汽车号牌、小型汽车号牌、普通摩托车号牌、轻便摩托车号牌、低速车号牌、挂车号牌、使馆汽车号牌、使馆摩托车号牌、领馆汽车号牌、领馆摩托车号牌、教练汽车号牌、教练摩托车号牌、警用汽车号牌、警用摩托车号牌填写。
样表:机动车变更登记/
备案申请表。