17 气相色谱法
职业卫生检测新题库判断题300题

职业卫生检测新题库判断题300题1.职业病危害因素,是指在职业活动中产生和(或)存在的、可能对职业人群健康、安全和作业能力造成不良影响的因素或条件,包括化学、物理、生物等因素。
(√)2。
目前,我国职业病危害因素检测主要以有毒物质的空气检测和作业环境中物理因素的检测为主。
(√)3.按照检测目的分类,职业病危害因素检测可分为评价检测、日常检测、监督检测和事故性检测四类。
(√)4.评价检测适用于建设项目职业病危害因素预评价、建设项目职业病危害因素制效果评价和职业病危害因素现状评价。
(√)5.开展评价检测过程中,如果评价职业接触限值为时间加权平均容浓度时,应选择有代表性的采样点,连续采样2个工作日,其中应包括空气中有害物质浓度最高的工作日(×)6.在监督检测过程中,如果评价职业接触限值为短时间接触容许浓度或最高容许浓度时,应选择有代表性的采样点,在两工作班内空气中有害物质浓度最高的时段进行采样。
(×)7.事故检测应根据现场情况确定采样点进行检测。
检测时应对空气中有害物质进行逐时监测至其浓度低于短时间接触容许浓度或最高容许浓度为止。
(√)8、按照检测方法及仪器类型分类,职业病危害因素检测可分为现场检测和实验室检测两类。
(√)9.现场检测是指利用便携直读式仪器设备在工作场所进行实时检测、快速给出检测结果,适用于对工作场所的职业卫生状况作出迅速判断。
(√)10。
事故检测一般采取实验室检测。
(×)11.现场检测常用方法有检测气管法、便携式气体分析仪测定法、物理因素的现场检测等。
(√)12.检测气管法是将浸渍过化学试剂的固体吸附剂制成指示剂,装在玻璃管内,当空气通过时,有害物质与化学试剂反应引固体吸附剂变色,根据颜色深浅、或变色柱的长度,并与事先制备好的标准色板或浓度标尺比较后,即时作出定性或定量的检测。
(√)13.检测气管的保存时间较短,一般为半年左右。
(×)14便携式气体分析仪测定法是指采用以红外线、半导体、电化学、色谱分析、激光等检测原理制成的便携式直读仪器在工作现场进行的快速检測。
第17章气相色谱法

二、气相色谱法的一般流程 载气(高压瓶) ,经降压、脱水及净 化,调至适宜的流量而进入色谱柱,经 检测器流出色谱仪。待流量、温度及基 线稳定后,即可进样。液态样品用微量 注射器吸取,由进样器注入,气态样品 可用六通阀或注射器进样,样品被载气 带入色谱柱。
图19-1气相色谱仪示意图
1.载气瓶 2.减压阀 3.净化器 4.稳压阀 5.柱前压力表 6.转子流量计 7.进样器 8.色谱柱 9.色谱柱恒温箱 10.馏分收集口(柱后分流阀) 11.检测器 12.检测器恒 温箱 13.记录器 14.尾气出口
第十七章 气相色谱法(GC) 第一节 气相色谱法分类和一般程序 一、气相色谱法分类和特点 1.分类 气相色谱法属于柱色谱法 按固定相的物态可分为 气一固(GSC)和气一液 (GLC)
按柱的粗细和填充情况可分为: 填充柱和毛细管柱。 (1)填充柱由不锈钢或玻璃材料制成, 内装固定相。填充柱的形状有U型和螺 旋型二种。内径一般为4 ~ 6mm,长1 ~3m (2)毛细管柱又叫空心柱,分为涂壁、 多孔层和涂载体空心柱。空心毛细管柱 材质为玻璃或石英,呈螺旋型。内径一 般为0.1 ~ 0.5mm,长度30 ~ 300m。
2.噪声和漂移 无样品通过检测器时,由仪器本身和 工作条件等的偶然因素引起的基线起 伏称为噪声(noise;N)。噪声的大小用 噪声带(峰一峰值)的宽度来衡量(下图)。 例如,不进样时,记录某检测器的基 线带宽为0.02mV,即噪声为0.02mV。
图19-3检测器的噪声和检测限 基线随时间朝某一方向的缓慢变化称为 漂移(drift;d)。通常用一小时内基线水 平的变化来衡量。
高分子多孔微球有如下优点: ①可以合成各种比表面及孔径的聚合物。 ②无有害的吸附活性中心 ③无流失现象,有利于大幅度程序升温。 ④具有强疏水性能,适于分析混合物中的微 量水分。适于多元醇、脂肪酸、腈类和胺类 的分析 ⑤粒度均匀,机械强度高,具有耐腐蚀性能。 ⑥热稳定性好,最高使用温度为200~300C。
第十七章-气相色谱法..

相对极性Px分类:
β,β ' -氧二丙腈:P =100
角鲨烷:
P =0
其余:Px=0---100 分为5级 0—20 0或1 非和弱极性
角鲨烷、甲基硅 橡胶
21—40 2 中等极性 DNP、OV-17
41—60 3 中等极性 氰基硅橡胶
61—80 4 极性
二、实验条件的选择性
R(
n 4
)
r2r,12,1 1
k2k2 1
(a) ( b) (c)
a:柱效项 b:柱选择性项 c:柱容量项
1.提高r21和k
(1) r21 反映了固定液的选择性, r21值越大(即固定液选 择性 好),分离度就越大,分离越好。若r21 =1 ,则 R = 0 ,两组分不能分离。
1. 填充柱
2. 毛细管柱(开管柱)
四、检测系统 1. 热导池检测器 2. 氢火焰离子化检测器 3. 电子捕获检测器 4. 火焰光度检测器
五、温度控制系统 控制和测量汽化室、色谱柱、检测器温度。
色谱柱温控方式:恒温分析、程序升温。
三、气相色谱法的特点
1. 高选择性 2. 高效能 3. 高灵敏度 4. 分析速度快 5. 应用范围广 沸点在500℃以下,热稳定好,分 子量在400以下的组分的分离和测定
对于气体、气态烃等低沸点混合物,柱温选在比平均沸 点低50℃至平均沸点范围内分析。
对于沸点范围较宽的试样,宜采用程序升温。
程序升温:指在一个分析周期里,色谱柱的温度随时 间由低温到高温呈线性或非线性地变化,使沸点不同的组分, 在其最佳柱温下流出,从而改善分离效果,缩短分析时间。
总结:范第姆特方程 H=A+B/U+CU
气相色谱法分析天然气的组成

气相色谱法分析天然气的组成张秋萍【摘要】使用一种新型气相色谱仪准确分析天然气的组成.以天然气标准物质为样品,对色谱柱、阀切换时间、柱箱温度控制等方面进行优化,建立了良好的色谱分析条件,利用外标法确定了天然气中各组分的保留时间.在同一最佳色谱分析条件下,标准物质中各组分连续检测两次测定结果的差值不大于0.11%,满足国家标准GB/T 13610–2014的要求,且与标准值相对误差的绝对值小于5%.测定结果组分含量应与所用标准物质浓度的单位保持一致.所建立分析方法准确可靠,适用于天然气的常规分析.%A new type gas chromatograph was employed to accurately analyze the compositions of natural gas. The standard material of natural gas was applied as the sample. The best chromatographic conditions were established by optimizing chromatographic column, valves changing-over time and the temperature of column oven. The external standard method was utilized to verify the remain time of the compositions of natural gas. The difference between the two testing results met the requirements of GB/T 13610–2014 under the same optimum gas chromatographic analysis conditions.The relative error between the testing results and the standard value of the standard material was less than 5%, which confirmed the accuracy and repeatability of this analyzing process. The testing result was consistent with that of the applied standard material. The established method is accurate, reliable and suitable for the conventional analysis of natural gas.【期刊名称】《化学分析计量》【年(卷),期】2018(027)001【总页数】6页(P77-82)【关键词】天然气组成;气相色谱法;气路流程;保留时间【作者】张秋萍【作者单位】武汉市度量衡管理所,武汉 430000【正文语种】中文【中图分类】O657.7天然气是一种以甲烷为主要组分的多组分烃类混合物。
气相色谱法工作原理

气相色谱法工作原理
气相色谱法(Gas chromatography, GC)是一种常用的分离和
分析技术,其工作原理基于样品分子在固定相和流动相之间的分配平衡。
在气相色谱法中,样品首先被注入进色谱柱,色谱柱通常是由具有高表面活性的固定相填充的长管状物质构成。
接下来,通过使用一个称为载气的流动相,样品组分被推送通过色谱柱。
在色谱柱内,样品组分与固定相发生相互作用。
具有极性的组分会与固定相之间的化学吸附力发生作用,而非极性的组分则会通过色谱柱的惰性表面发生物理吸附作用。
这些作用力会导致样品组分在色谱柱内以不同的速度进行分离。
最终,在色谱柱的出口处,各个组分将会陆续出现。
为了检测和分析这些组分,常常会使用一种称为检测器的设备。
检测器可以根据被分离组分的特性,如折射率、导电性或化学反应性,对它们进行识别和测量。
由于气相色谱法的灵敏度高、分离效果好、分析速度快等优点,因此在许多领域得到了广泛应用。
无论是在环境监测、食品质量控制还是药物分析等方面,气相色谱法都扮演着重要的角色。
第十七章气相色谱法

第十七章气相色谱法思考题和习题1.名词解释:噪音检测限死体积分离度程序升温保留温度分流进样分流比线性分流相对重量校正因子麦氏常数2.说出下列缩写的中文名称:TCD FID ECD TID FPD WCOT柱PLOT柱SCOT柱FSOT柱3.简述范氏方程在气相色谱中的表达式以及在分离条件选择中的应用。
4.某色谱柱理论塔板数很大,是否任何两种难分离的组分一定能在该柱上分离?为什么?5.气相色谱仪主要包括哪几部分?简述各部分的作用。
6.在气相色谱中,如何选择固定液?7.说明氢焰、热导以及电子捕获检测器各属于哪种类型的检测器,它们的优缺点以及应用范围。
8.在气相色谱分析中,应如何选择载气流速与柱温?9.气相色谱定量分析的依据是什么?为什么要引入定量校正因子?常用的定量方法有哪几种?各在何种情况下应用?10.毛细管柱气相色谱有什么特点?毛细管柱为什么比填充柱有更高的柱效?11.当出现下列三种情况时,Van Deemter曲线是什么形状?(1)B/u=Cu=0;(2)A=Cu=0;(3)A=B/u=012.用气相色谱法分离某二元混合物时,当分别改变下列操作条件之一时,推测一下对t R、H、R的影响(忽略检测器、气化室、连接管道等柱外死体积)。
(a)流速加倍,(b)柱长加倍,(c)固定液液膜厚度加倍,(d)色谱柱柱温增加。
13.当色谱峰的半峰宽为2mm,保留时间为4.5min,死时间为1min,色谱柱长为2m,记录仪纸速为2cm/min,计算色谱柱的理论塔板数,塔板高度以及有效理论塔板数,有效塔板高度。
(11200 ,0.18mm;6790,0.29mm)14.在某色谱分析中得到如下数据:保留时间t R =5.0min ,死时间t 0=1.0min ,固定液体积V s =2.0ml ,载气流速F =50ml/min 。
计算:(1)容量因子;(2)分配系数;(3)死体积;(4)保留体积。
(4.0,100,50ml ,250ml )15.用一根2米长色谱柱将两种药物A 和B 分离,实验结果如下:空气保留时间30秒,A 与B 的保留时间分别为230秒和250秒,B 峰峰宽为25秒。
气相色谱柱“-17”和“-1701”的区别

2020版《中国药典》于今年6月份正式发布,计划2020年12月30日开始实施。
其中药典四部中最大的一个变化就是通则0212中关于药材及饮片(植物类)中33种禁用农残不得检出的规定。
针对此项规定,通则2341新增了第五法药材及饮片(植物类)中禁用农药多残留测定法。
相信很多中药行业的老师已经开始着手准备这个方法的扩项了,但是在开展这个实验过程中,在选购符合药典要求的气相毛细管色谱柱时,遇到了一个问题:实验室没有“-17”的色谱柱,之前按照2341第一法做甘草黄芪中有机氯检测有用到“-1701”的色谱柱,它们两个一样吗?可以使用吗?针对这个问题,小编今天给大家做一些讲解。
“-17”和“-1701”是不一样的,它们两个的固定液是完全不同的,是不能够混用的。
气相毛细管色谱柱的常见参数01色谱柱型号毛细管色谱柱通常由3层组成,分别是中间层的熔融石英骨架,外层的聚酰亚胺图层,以及内层的固定液涂层。
固定相起到分离不同目标化合物的作用,其种类多种多样,不同的固定液组成也就导致了不同的色谱柱型号。
商品化的色谱柱型号主要包括两部分,即品牌名-固定液种类,在其包装盒和色谱柱铭牌上均可看到,如:HP-5,C D-1,T G-WAX等等,HP、C D和TG是商品的品牌名,每家公司的名称都不一样;品牌名之后的数字或英文标记“-1”“-5”“WAX”等是表示色谱柱的固定液类型,各家公司的表示总体上相同,但也有不同。
比如“-1”通常都表示100%聚二甲基硅氧烷的固定液,“-5”通常都表示5% 苯基/ 95% 甲基聚硅氧烷的固定液。
在药典通则2341 新增第五法气相色谱-串联质谱法中要求的50%苯基-甲基聚硅氧烷是我们的固定液种类,针对这种固定液,有的厂商商品型号中将其标记为“-17”,如DB-17,有的厂商商品型号将其标记为“-50”,如C D-50;小明同学说到的“-1701”的标记通常代表的固定液种类是14% 氰丙基苯基/ 86% 甲基聚硅氧烷,而不是标准中要求的50%苯基-甲基聚硅氧烷。
一种聚乳酸产品中丙交酯含量的测定方法

摘 要 : -丙 FID 检 DB - 17 ( 30m 0. 25mm) 采 用 气 相 色 谱 法 测 定 聚 乳 酸 材 料 中 残 留 单 体 交 酯 的 含 量 。采 用 测 器 ; 色 谱 柱 为 ; 进 样 口 温 度 220 ħ , 100 ħ 保 2 min, 10 ħ / min 增 150 ħ 并 2 min; 10 : 1; , 检 测 器 温 度 柱 温 采 用 梯 度 升 温 程 序 : 先 持 然 后 以 加 到 保 持 分 流 比 载 10mL / min; 1 μL。在 0. 50 250 mg / L 浓 气 为 高 纯 氮 , 流 速 进 样 量 为 此 条 件 下 , 丙 交 酯 峰 与 杂 质 峰 分 离 度 良 好 , 在 度 范 围 内 , 线 性 关 系 良 101. 6 % , 1. 8 % 。该 好 ; 样 品 的 加 标 回 收 率 为 相 对 标 准 偏 差 为 方 法 简 单 、 稳 定 、 准 确 , 可 以 作 为 丙 交 酯 含 量 的 分 析 方 法 。 关 键 词 : 气 相 色 谱 法 ; 测 定 ; 丙 交 酯 O657. 7 1㊀ ㊀ ㊀ ㊀ ㊀ 文 A㊀ ㊀ ㊀ ㊀ 文 1008 - 021X( 2017 ) 20 - 0085 - 01 中 图 分 类 号 : 献 标 识 码 : 章 编 号 :
+
Method for Measuring Lactide Content in Polylactic Acid Product
Zhuang Zhiwen Qin Xiaona Wang Caixiao
, ) Abstract: The content of residual monomer lactide in polylactic acid was determined by GC. Using the FID detector; the column 30m0. 25mm) was DB - 17 ( ;the inlet temperature is 260 ħ ,the detector temperature is 220 ħ ,the column temperature is 100 ħ temperature gradient procedure:first 2 min,then increased to 150 to 10 ħ / min ħ and 2 min;split ratio 10 : 1 ;high the injection volume was 1 L. Under these conditions, the separation degree of purity nitrogen carrier gas flow rate. 10 mL / min; lactide peak and impurity peak is good. In the range of 0. 50 250mg / L concentration,the linear relationship is good. The stable and accurate. It recovery rate of sample is 101. 6% ,and the relative standard deviation is 1. 8% . The method is simple, can be used as an analytical method for the content of lactide. Key words:GC; measuring; lactide ㊀ ㊀ 聚 PLA )是 乳 酸 ( 一 种 具 有 良 好 的 生 物 相 容 性 和 生 物 降 解 1. 3㊀ 分 析 步 骤 性 , 可 广 泛 用 于 生 物 医 学 领 域 、 服 装 行 业 及 农 业 环 保 材 料 。现 精 PLA 样 0. 0100 g, 100 mL 离 密 称 取 品 置 于 心 管 中 , 加 入 - 阶 段 生 产 聚 乳 酸 的 方 法 通 常 是 先 将 乳 酸 脱 水 缩 合 成 低 聚 物 10. 0 mL 二 1 min。再 30. 0 mL 乙 氯 甲 烷 , 涡 旋 震 荡 加 入 醇 , 再 丙 交 酯 , 再 在 高 温 、 真 空 的 条 件 下 开 环 聚 合 。在 合 成 过 程 中 , 难 次 1 min。待 5000 r / 沉 淀 析 出 之 后 放 置 离 心 机 中 , 以 涡 旋 震 荡 免 会 混 入 中 间 体 丙 交 酯 , 残 留 的 丙 交 酯 会 水 解 成 乳 酸 , 在 酸 性 min 离 2min。取 0. 45 μm 微 心 上 清 液 , 以 孔 滤 膜 过 滤 , 等 待 上 条 件 下 会 加 速 聚 乳 酸 的 分 解 。因 此 , 残 留 丙 交 酯 的 含 量 是 衡 量 机 。 聚 乳 酸 材 料 性 能 的 重 要 指 标 。 果 与 讨 论 2㊀ 结 目 前 , 测 定 丙 交 酯 的 方 法 有 氢 核 磁 法 、 光 度 法 、 红 外 法 、 高 2. 1㊀ 样 品 前 处 理 效 液 相 色 谱 法 及 气 相 色 谱 法 。其 中 氢 核 磁 法 、 光 度 法 、 红 外 法 本 PLA、 方 法 先 用 二 氯 甲 烷 将 样 品 充 分 溶 解 , 使 丙 交 酯 、 杂 定 量 不 准 而 很 少 采 用 。在 用 高 效 液 相 色 谱 法 时 发 现 , 纯 有 机 流 质 、 乳 酸 及 乳 酸 的 低 聚 物 全 部 溶 解 , 接 着 加 入 适 当 比 例 的 乙 醇 , 动 相 无 法 使 丙 交 酯 与 杂 质 分 离 , 只 有 在 流 动 相 中 加 入 水 才 能 达 使 溶 液 中 的 大 分 子 聚 合 物 沉 淀 析 出 , 起 到 除 杂 作 用 。经 预 实 验 到 分 离 要 求 , 但 丙 交 酯 与 水 接 触 会 发 生 水 解 , 是 结 果 测 定 不 准 发 1: 3时 , 沉 淀 效 果 最 好 。 现 , 当 二 氯 甲 烷 与 乙 醇 的 比 例 为 确 。就 此 , 本 文 提 出 了 一 种 使 用 气 相 色 谱 法 测 定 丙 交 酯 含 量 , 2. 2㊀ 色 谱 条 件 的 选 择 有 效 避 免 了 以 上 方 法 出 现 的 弊 端 , 操 作 简 单 , 结 果 准 确 。 2. 2. 1㊀ 进 样 口 温 度 的 选 择 1㊀ 试 验 部 分 样 品 经 法 处 理 之 后 , 溶 液 中 含 有 乳 酸 及 乳 酸 低 聚 2. 1 方 1. 1㊀ 仪 器 与 试 剂 物 , 影 响 丙 交 酯 峰 的 分 离 度 , 故 利 用 乳 酸 的 热 不 稳 定 性 , 采 用 较 相 色 谱 仪 , 配 测 器 ; 色 谱 柱 : Agilent 7890B 气 FID 检 DB - 17 高 的 进 样 口 温 度 , 使 乳 酸 分 解 , 从 而 达 到 除 杂 的 目 的 。经 过 预 30m ˑ 0. 25mm ˑ 0. 25 μm ) 98% )( Sigma - ( ; 丙 交 酯 (标 准 品 , 260ħ 时 , 乳 酸 分 解 彻 底 且 丙 交 酯 不 被 实 验 发 现 , 进 样 口 温 度 为 Aldrich 公 Fisher 公 司 ) ; 二 氯 甲 烷 (色 谱 纯 , 司 ) ; 乙 醇 (色 谱 纯 , 破 坏 。 司 ) ;水 (一 级 水 ,实 验 室 自 制 ) ;载 气 :高 纯 氮 (99. 2. 2. 2㊀ 升 Fisher 公 温 曲 线 的 选 择 。 9% ) 1. 2 升 采 用 温 程 序 , 由 于 前 期 温 度 低 , 分 离 度 低 , 杂 质 峰 与 谱 条 件 1. 2㊀ 色 3min 之 溶 剂 峰 重 合 明 显 , 均 在 前 出 峰 。随 着 温 度 升 高 , 丙 交 酯 FID 检 DB - 17 ( 30m ˑ 0. 25mm) 测 器 ; 色 谱 柱 为 ; 进 样 口 温 在 7min 出 1所 峰 , 如 图 示 , 峰 型 较 好 , 与 杂 质 峰 分 离 效 果 好 , 样 260 ħ , 220 ħ ,柱 度 检 测 器 温 度 温 采 用 梯 度 升 温 程 序 :先 品 10 min。 测 定 平 均 周 期 小 于 100ħ 保 2 min,然 10 ħ / min 增 150 ħ 并 2 持 后 以 加 到 保 持 (下 转 第 ) 88 页 分 流 比 载 气 为 高 纯 氮 , 流 速 10 : 1; 10 mL / min。 min;
第十七章 色谱分析法概论-分析化学

I X 100 [Z n
' ' lg t R lg t ( x) R( z )
lg t
' R( z n)
lg t
' R( z )
]
Ix为待测组分的保留指数,z 与 z+n 为
正构烷烃对的碳原子数。
P
16
乙酸正丁酯的保留指数测定
xie 仪 器 分 析
第 十 七 章 色 谱 分 析 法 概 论
xie 仪 器 分 析
第 十 七 章 色 谱 分 析 法 概 论
第十七章 色谱分析法概论
P
1
第一节 色谱法的分类和发展
xie 仪 器 分 析
第 十 七 章 色 谱 分 析 法 概 论
色谱分析法是一种物理或物理化学分离分 析方法。 始于20世纪初; 30与40年代相继出现了薄层色谱与纸色谱; 50年代气相色谱兴起、色谱理论、毛细管色 谱; 60年代气相色谱-质谱联用; 70年代高效液相色谱; 80年代末超临界流体色谱、高效毛细管电泳 色谱。
• R=1 4σ分离 • R=1.5 6σ分离 95.4% 99.7%
w1
w1
tR2-tR1
P
21
三、分配系数与色谱分离
xie 仪 器 分 析
第 十 七 章 色 谱 分 析 法 概 论
1、分配系数 在一定温度和压力下,达到分配平衡 时,组分在固定相和流动相中的浓度之比 CS K Cm 2、容量因子
m
X+
H+
SO3-R
S
X+ SO -R 3 H+
P
30
阳离子交换树脂
xie 仪 器 分 析
仪器分析习题及答案

第8章电位法和永停滴定法1.是否能用普通电位计或伏特计测量参比电极和pH玻璃电极所组成电池的电动势?简述原因。
答:不能。
因为玻璃电极的内阻(50MΩ~500MΩ)很高,假设采用普通电位计或伏特计测量其电位,会引起较大的测量误差。
用普通电位计或伏特计测量玻璃电极所组成电池的电动势时,假设检流计的灵敏度为10-9A〔测量中有10-9A电流通过〕,玻璃电极的内阻108Ω,当这微小电流流经电极时,由于电压降所引起的电动势测量误差可达:△E=IV=10-9×108=0.1V,它相当于1.7个pH单位的误差。
因此不能用普通电位计或伏特计测量参比电极和pH玻璃电极所组成电池的电动势。
2.=0.10,假设试样溶液中F-浓度为1.0×10-2mol/L时,允许测定误差为5%,问溶液允许的最大pH〔以浓度代替活度计算〕为多少?解:离子电极选择性误差用下式表示和计算:即:-离子选择电极插入50.00ml*高氯酸盐待测溶液,与饱和甘汞电极〔为3.将一支ClO4标准溶液负极〕组成电池。
25℃时测得电动势为358.7mV,参加1.00ml NaClO4-浓度。
〔0.0500mol/L〕后,电动势变成346.1mV。
求待测溶液中ClO4解:-为阴离子,但该离子选择电极为电池的正极,因此S为负值。
注意:此题中虽然ClO44. 用离子选择电极校正曲线法进展定量分析有什么优点?需注意什么问题?使用TISAB有何作用?最正确分辨率:1024*768第10章紫外1.钯〔Pd〕与硫代米蚩酮反响生成1:4的有色配位化合物,用1.00cm 吸收池在520nm处测得浓度为0.200×10-6g/ml的Pd溶液的吸光度值为0.390,试求钯-硫代米蚩酮配合物的及ε值。
〔钯-硫代米蚩酮配合物的分子量为106.4〕解:2.取咖啡酸,在105°C枯燥至恒重,精细称取10.00mg,加少量乙醇溶解,转移至200ml量瓶中,加水至刻度,取出5.00ml,置于50ml 量瓶中,加6mol/L HCl 4ml,加水至刻度。
吹扫捕集—气相色谱法测定地表水中的乙醛

吹扫捕集—气相色谱法测定地表水中的乙醛文章建立了吹扫捕集样品管在35℃下进样管中富集、气相色谱仪(具FID 检测器)检测生活饮用水中乙醛的测定方法。
乙醛在5μg/L~500μg/L范围内有良好的线性,方法的检出限为0.007μg/L,相对标准偏差为3.64~8.90,加标回收率在89.6%~107%范围内。
该方法快速、简单、没有二次污染,适用于地表水生活饮用水及水源水中乙醛的测定。
标签:乙醛;气相色谱法;吹扫捕集乙醛是一种微毒性极性有机物[1],水溶性强且,是丙氨酸、吡啶衍生物、乙酸、乙醇、乙酸乙酯和农药DDT等的合成原料。
高浓度乙醛可引起急性中毒,肺水肿致死,亚急性和慢性中毒引起神经中枢系统中毒的现象[2],乙醛还具有致畸性。
随着检测仪器的不断更新,关于水中乙醛测定方法的报道有顶空气相色谱法,吹扫捕集-气相色谱法/气相色谱法-质谱法[3],和大体积直接进样气相色谱法。
但由于乙醛的水溶性很强,吹扫温度对乙醛检出的峰面积[4]有很大影响,本文在35℃吹扫条件下对水中乙醛进行测定,检出限、精密度和准确度的结果很理想,完全满足地表水水源中的乙醛的测定。
1 实验部分1.1 主要仪器和试剂气相色谱仪(Agilent 7890A),具FID检测器,自动进样器;30m ×0.25mm×0.25μm BR-Swax石英毛细管色谱柱(BRUKER公司);吹扫捕集浓缩仪(CDS7000,北京博赛德科技有限公司),5mL石英吹扫管;实验室超纯水机(型号ELGA,Veolia Water公司);10μL~100μL、100μL~1000μL移液器(德国Brand公司);乙醛标准溶液(美国百灵威公司,100mg/L,水)氮气:纯度≥99.999%1.2 色谱条件色谱柱温度:45℃(1min)65℃(1min);进样口温度:180℃;分流进样,分流比15:1;检测器温度:250℃;载气流量:0.8mL/min;尾吹流量:25mL/min;空气:400mL/min;氢气35:mL/min;1.3 吹扫捕集条件采用氮气为吹扫的载气,吹扫温度为35℃、流量45mL/min、时间20min;解析温度为150℃、时间5min;烘烤温度为220℃、时间11min。
气相色谱法的内标与外标

气相色谱中内标法与外标法的应用”一、内标法1.什么叫内标法?怎样选择内标物?内标法是一种间接或相对的校准方法。
在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。
内标法在气相色谱定量分析中是一种重要的技术。
使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。
采用内标法定量时,内标物的选择是一项十分重要的工作。
理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。
当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。
需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。
2.在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值?影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。
由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。
化学方面的因素包括:1、内标物在样品里混合不好;2、内标物和样品组分之间发生反应,3、内标物纯度可变等。
对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。
气相色谱基础知识
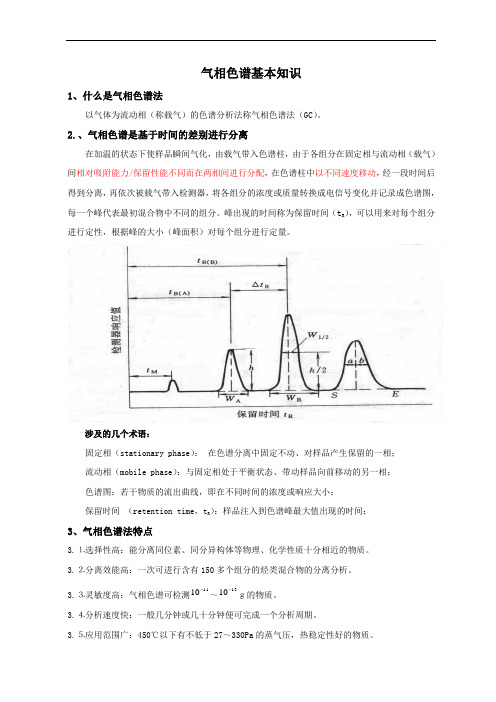
气相色谱基本知识1、什么是气相色谱法以气体为流动相(称载气)的色谱分析法称气相色谱法(GC )。
2.、气相色谱是基于时间的差别进行分离在加温的状态下使样品瞬间气化,由载气带入色谱柱,由于各组分在固定相与流动相(载气)间相对吸附能力/保留性能不同而在两相间进行分配,在色谱柱中以不同速度移动,经一段时间后得到分离,再依次被载气带入检测器,将各组分的浓度或质量转换成电信号变化并记录成色谱图,每一个峰代表最初混合物中不同的组分。
峰出现的时间称为保留时间(t R ),可以用来对每个组分进行定性,根据峰的大小(峰面积)对每个组分进行定量。
涉及的几个术语:固定相(stationary phase ): 在色谱分离中固定不动、对样品产生保留的一相; 流动相(mobile phase ):与固定相处于平衡状态、带动样品向前移动的另一相; 色谱图:若干物质的流出曲线,即在不同时间的浓度或响应大小;保留时间 (retention time ,t R ):样品注入到色谱峰最大值出现的时间;3、气相色谱法特点3.⒈选择性高:能分离同位素、同分异构体等物理、化学性质十分相近的物质。
3.⒉分离效能高:一次可进行含有150多个组分的烃类混合物的分离分析。
3.⒊灵敏度高:气相色谱可检测1110-~1310-g的物质。
3.⒋分析速度快:一般几分钟或几十分钟便可完成一个分析周期。
3.⒌应用范围广:450℃以下有不低于27~330Pa 的蒸气压,热稳定性好的物质。
3.⒍缺点:不适应于大部分沸点高的和热不稳定的化合物;需要有已知标准物作对照。
4、气相色谱系统主要包括五大系统:载气系统、进样系统、分离系统、检测系统和记录系统。
基本流程如下脱水管限流器4.1、载气系统:可控而纯净的载气源。
载气从起源钢瓶/气体发生器出来后依次经过减压阀、净化器、气化室、色谱柱、检测器,然后放空。
载气必须是纯洁的(99.999%),要求化学惰性,不与有关物质反应。
中国药科大学-分析化学课件-第17色谱分析

峰宽和之半
tR2 W1
tR1 W2
2
R 2(tR2 tR1) 1.177(tR2 tR1)
W1 W2
W1 2(1) W1 2(2)
讨论
• 设色谱峰为正常峰,W1≈W2= 4σ
R 1.0 tR 4 基本分离 R 1.5 tR 6 完全分离(定量分析前提)
R 1.0 完全未分开
调整保留体积VR’:保留体积与死体积之差,即组分 停留在固定相时所消耗流动相的体积
VR'
VR
V0
t
' R
FC
注:VR' 与Fc无关;t
' R
1 Fc
V0 和 Vm、t0 和 tm 的区别
• V0 :由进样器至检测器的流路中未被固定相占有的空 间体积 ; 流定相充满死体积所需的时间为t0 。
• Vm :平衡时流动相在色谱柱中占有的体积,流动相经 过色谱柱所需时间用tm 表示。
线性:对称峰 凸形:拖尾峰
• 对称因子(symmetry factor)
——衡量色谱峰对称性
色谱峰
正常峰(对称)——fs在0.95~1.05之间
非正常峰 前沿峰 ——fs小于0.95 拖尾峰 ——fs大于1.05
对称因子:(拖尾因子)
fs
W0.05h 2A
A B 2A
8.分离因子和分离度:—分离参数
➢吸附色谱:利用物理吸附性能的差异(固定相固体)
( absorption chromatography)
➢离子交换色谱:利用离子交换原理(固定相离子交换树脂)
(ion exchange chromatography )
➢空间排阻色谱:利用排阻作用力的不同(固定相凝胶)
第十七章 气相色谱法

第十七章气相色谱法思考题和习题1.名词解释:噪音检测限死体积分离度程序升温保留温度分流进样分流比线性分流相对重量校正因子麦氏常数2.说出下列缩写的中文名称:TCD FID ECD TID FPD WCOT柱PLOT柱SCOT柱FSOT柱3.简述范氏方程在气相色谱中的表达式以及在分离条件选择中的应用。
4.某色谱柱理论塔板数很大,是否任何两种难分离的组分一定能在该柱上分离?为什么?5.气相色谱仪主要包括哪几部分?简述各部分的作用。
6.在气相色谱中,如何选择固定液?7.说明氢焰、热导以及电子捕获检测器各属于哪种类型的检测器,它们的优缺点以及应用范围。
8.在气相色谱分析中,应如何选择载气流速与柱温?9.气相色谱定量分析的依据是什么?为什么要引入定量校正因子?常用的定量方法有哪几种?各在何种情况下应用?10.毛细管柱气相色谱有什么特点?毛细管柱为什么比填充柱有更高的柱效?11.当出现下列三种情况时,Van Deemter曲线是什么形状?(1)B/u=Cu=0;(2)A=Cu=0;(3)A=B/u=012.用气相色谱法分离某二元混合物时,当分别改变下列操作条件之一时,推测一下对t R、H、R的影响(忽略检测器、气化室、连接管道等柱外死体积)。
(a)流速加倍,(b)柱长加倍,(c)固定液液膜厚度加倍,(d)色谱柱柱温增加。
13.当色谱峰的半峰宽为2mm,保留时间为4.5min,死时间为1min,色谱柱长为2m,记录仪纸速为2cm/min,计算色谱柱的理论塔板数,塔板高度以及有效理论塔板数,有效塔板高度。
(11200 ,0.18mm;6790,0.29mm)14.在某色谱分析中得到如下数据:保留时间t R =5.0min ,死时间t 0=1.0min ,固定液体积V s =2.0ml ,载气流速F =50ml/min 。
计算:(1)容量因子;(2)分配系数;(3)死体积;(4)保留体积。
(4.0,100,50ml ,250ml )15.用一根2米长色谱柱将两种药物A 和B 分离,实验结果如下:空气保留时间30秒,A 与B 的保留时间分别为230秒和250秒,B 峰峰宽为25秒。
气相色谱法检测食品中反式脂肪酸的研究

气相色谱法检测食品中反式脂肪酸的研究作者:杨丹青来源:《山东工业技术》2016年第24期摘要:反式脂肪酸是食品中一种非常重要的成分,检测这种成分的化学方法也有许多种,气相色谱法是检测反式脂肪酸成分的一种常用的方法,其检测相比其他方法更加准确、方便。
如果食品中反式脂肪酸含量太多时,会对人们的身体健康产生一定程度的影响,含量不同产生的伤害程度也不尽相同,然而目前我国检测反式脂肪酸技术还不是太先进,食物中含量也没有规定具体标准,所以我国还需在这方面有巨大的研究空间。
本文在反式脂肪酸概念基础上,论述了气相色谱法检测方法以及反式脂肪酸的来源。
关键词:气相色谱法;反式脂肪酸;来源DOI:10.16640/ki.37-1222/t.2016.24.0381 反式脂肪酸的概念以及危害1.1 反式脂肪酸的概念脂肪酸是在化学中非常常见的羧酸化合物,它是由碳氢组成的烃类基团链接的羧基构成的,是一些食品中非常重要的部分,其中最常见的脂肪就是由甘油和脂肪酸两部分构成的三酰甘油酯。
同时,根据不同依据可以化成不同分类,比如可以根据脂肪酸空间结构不同,分成顺式脂肪酸和反式脂肪酸两种;还可以根据其饱和程度分成单不饱和脂肪酸、多不饱和脂肪酸、饱和脂肪酸三种。
反式脂肪酸属于非共轭不饱和脂肪酸,化学成分组合是由一个或者多个反式双键,通常反式脂肪酸比顺式脂肪酸的熔点较高,顺式油酸熔点一般大约为13℃,在室温下呈现液体,然而反式脂肪酸的熔点大约为46℃,在室温情况下呈现固体状态。
反式脂肪酸有天然存在和人工制造两种形式。
在人乳和牛乳中都存在天然的反式脂肪酸。
同时,人工也可以制造反式脂肪酸,它是在对植物油进行氢化改性过程中产生的,成为氢化油,它可以防止脂肪酸变质而改变风味。
1.2 反式脂肪酸对人体的危害虽然反式脂肪酸可以改变事物的口感,延长食物的保质期,但是反式脂肪酸的摄入会对人体有所伤害,尤其是对心血管的健康有着非常明显的影响,这一点已经被科学家通过对动物的试验证实,被广为接受。
气相色谱法的建立

气相色谱法的建立第一章前言1,气相色谱法气相色谱法是根据气-固、气-液、气-液-固之间的相平衡,借溶质分配系数的不同而进行分离的方法。
建立相平衡的“界面”最好是无穷大,气相色谱法能满足在这个极大的表面上瞬间建立相平衡的条件。
由于一般用惰性气体作载气,故可认为溶质和载气分子之间基本上没有相互作用。
为减少色谱柱中的纵向扩散,流动相最好用分子量大的载气。
另外,还存在一个使理论塔板高度(H)最小的最佳线性流速。
但气相色谱法在选择色谱柱时基本上可以忽略这些。
研究固定相液体、载体表面、吸附剂以及溶质在液相中或固体表面上的分子间相互作用,才是选择色谱柱的必要事项。
2,哪些样品可以作为分析对象分析样品的物性(如沸点、官能团、反应性、溶解的溶剂系统等)与选择气相色谱的分离条件密切相关。
保留体积(Vg)与样品沸点(TB)之间的关系:式中:M1—液相的分子量,γ—溶质的活度系数(同系物溶质的γ基本相同,则保留体积的对数与TB成直线关系),T—色谱柱温。
(1)溶质之间沸点相差20℃时:容易用标准色谱柱分离;(2)溶质之间沸点相差10℃时:若选择与溶质有相似极性的固定液,很容易分离;(3)溶质之间沸点相差5℃时:用较长的色谱柱或用结构与溶质很类似的固定液;(4)溶质之间沸点相差0~2℃时:当两者的沸点相近时,若每种溶质的官能团不同,选用与其中一种溶质的极性相近的固定液就容易进行分离。
但对具有相同官能团的同系物要选择可以利用结构差异的固定相(如分离o-,m-,p-位取代苯可用FFAP/Carbopak C等气-液-固体系);(5)溶质沸点在-50℃以下:用强吸附剂作填料,而且柱温要置于低温;(6)溶质沸点在-50~20℃:用吸附剂或以吸附剂为载体,且在担体上涂渍极性固定液的填料;(7)溶质沸点在20~300℃:几乎所有的填料均可使用;(8)溶质沸点在300℃以上:用高沸点固定液或根据情况将样品衍生化后再供分析用,或者用液相色谱法测定。
第十七章 气相色谱法

1、名词解释相对极性:Px色谱中的相对极性与化学上的极性不同,它指固定液与被测组分之间相互作用力的强弱。
因此,固定液相对极性不仅与固定液本身有关,而且与被测组分有关。
麦氏常数:某组分在被测固定液和角鲨烷柱上的保留指数之差,用于表示固定液与某类化合物相互作用力的大小。
色谱手册上列出的麦氏常数有5个数据,分别表示与苯、正丁醇、戊彤-2、硝基苯烷、吡啶的作用力大小。
各麦氏常数的总和可作为固定液的相对极性,小于300的为非极性固定液。
检测限:某组分的峰高恰为噪声2倍时,单位时间内由载气引入检测器中该组分的质量或单位体积载气中所含该组分的量。
浓度型检测器:响应值与载气中组分的浓度成正比。
质量型检测器:响应值与单位时间内进入检测器的组分质量成正比。
灵敏度(S):浓度型检测器时Sc为1ml载气携带一毫克的某组分通过检测器时产生的电压。
质量型检测器时Sm为每秒钟有1g的某组分被载气携带通过检测器时产生的电压。
分流比:进入毛细管柱的物质量与被分流的物质量之比,通常为进入色谱柱的流量与分流流量之比。
漂移:基线在单位时间内单方向缓慢变化的幅值。
噪声:由于仪器本身和工作条件等的偶然因素引起的基线起伏。
相对校正因子:被测物质与标准物质的绝对校正因子之比。
程序升温:在一个分析周期中,按照既定程序改变色谱柱温度,以使沸点差距较大的各组分均得到良好分离。
涂壁毛细管柱:这种毛细管柱把固定液涂在毛细管内壁上。
2、TCD热导检测器FID氢焰离子化检测器ECD电子捕获检测器NPD氮磷检测器TID热离子化检测器FPD火焰光度检测器WCOT柱涂壁毛细管柱PLOT柱多孔层毛细管柱SCOT柱载体涂层毛细管柱FSOT柱融融石英毛细管柱3、见16章第2题4、简述范式方程中各项的含义,他们的改变将如何影响柱效?5、范式方程对选择色谱分离条件有何指导意义?H = A + B/u + Cudp和填充物的填充不规则因子有关。
填充柱色谱中,A=2λdp 所以,采用均匀、较细粒径的载体,并且填充均匀可减小涡流扩散项,提高柱效。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第十七章气相色谱法
思考题和习题
1.名词解释:噪音检测限死体积分离度程序升温保留温度分流进样分流比线性分流相对重量校正因子麦氏常数
2.简述范氏方程在气相色谱中的表达式以及在分离条件选择中的应用。
3.气相色谱仪主要包括哪几部分?简述各部分的作用。
4.说明氢焰、热导以及电子捕获检测器各属于哪种类型的检测器,它们的优缺点以及应用范围。
5.在气相色谱分析中,应如何选择载气流速与柱温?
6.气相色谱定量分析的依据是什么?为什么要引入定量校正因子?常用的定量方法有哪几种?各在何种情况下应用?
7.毛细管柱气相色谱有什么特点?毛细管柱为什么比填充柱有更高的柱效?
8.用气相色谱法分离某二元混合物时,当分别改变下列操作条件之一时,推测一下对t R、H、R的影响(忽略检测器、气化室、连接管道等柱外死体积)。
(a)流速加倍,(b)柱长加倍,(c)固定液液膜厚度加倍,(d)色谱柱柱温增加。
9.在某色谱分析中得到如下数据:保留时间t R=5.0min,死时间t0=1.0min,固定液体积V s=2.0ml,载气流速F=50ml/min。
计算:(1)容量因子;(2)分配系数;(3)死体积;(4)保留体积。
(4.0,100,50ml,250ml)10.用一色谱柱分离A、B两组分,此柱的理论塔板数为4200,测得A、B 的保留时间分别为15.05min及14.82min。
(1)求分离度;(2)若分离度为1.0时,理论塔板数为多少?
(0.25,67200)11.用气相色谱法测定正丙醇中的微量水分,精密称取正丙醇50.00g及无水甲醇(内标物)0.4000g,混合均匀,进样5µl,在401有机担体柱上进行测量,测得水:h=5.00cm,W1/2=0.15cm,甲醇h=4.00cm,W1/2=0.10cm,求正丙醇中微
量水的重量百分含量。
(相对重量校正因子水f =0.55,甲醇f =0.58) (1.42%)。