gene ontology(GO基因注释)
go基因注释与功能分类汇总

另外一种化学注释的方法是 以小分子化学结构的生物学 意义为特征来实现的。 在 KEGG 数据库中,酶与酶 之间的反应信息以及相关的 化学结构信息分别存储在 KEGG REACTION 数据库和 KEGG REPAIR数据库中。 每个化合物的化学结构都被 转 化 为 RDM (atom type changes at R:reaction center D:diffevent atom M:matched atom )模式。
KEGG数据库的改进与更新
为了满足日益增长的科学研究需求,KEGG数据库在最近 几 年 里 不 断 扩 充 , 新 增 加 的 50 多 个 通 路 使 KEGG PATHWAY数据库更加完善。这50多个新增加的通路包括 信号传导通路、细胞生物过程通路和人类疾病通路等。 KEGG对通路数据新增了两个补充内容:第一个补充是一 张全局通路图,这张全局通路图是通过手工拼接 KEGG的 120 多个现存通路图生成的,存储为 SVG 文件。另一个补 充内容是 KEGG MODULE 数据库,这是一个收集了通路 模块以及其他一些功能单元的新型数据库,功能模块是在 KEGG子通路中被定义为一些小的片段,通常包括几个连 续的反应步骤、操纵子、调控单元,以及通过基因组比对 得到的系统发生单元和分子的复合物等。
点击上图右上方的可视化视图 (graphical view)就更清晰地 显示了分子功能记录之间构成 的复杂网状结构,既有上下隶 属关系,也存在平行关系。
2. 用序列检索GO数据库
对于未知基因名的序列,可 以用序列直接检索GO 数据库。 点 击 AmiGO 首 页 上 方 的 “BLAST”。 界面风格类似于其他数据库 BLAST 搜索的网页,在检索 框中铁如氨基酸或核酸序列, 网页能自动识别并相应地做 BLASTP 或 BLASTX 和数据库 中的序列比对。 这里以检索 RPIA 基因的序列 为例,如图所示。
基因功能注释

基因功能注释
基因功能注释是利用不同策略,来确定一个基因的蛋白质产物。
解析基因的功能有助于理解生物体健康和疾病状况,帮助用户分析和保存基因的功能及其相关联的健康风险。
这里推荐一款基因功能注释的软件——Gene Ontology Annotation。
Gene Ontology Annotation(GOA)是一个基于Web的数据库,用于基因组数据注释,主要用于内在属性的分类和分析。
GOA拥有丰富的信息,如基因和基因组概念、分子功能,以及各种细胞类型和发育阶段的功能注释。
GOA的优点:首先,对于对大规模数据进行基因功能注释,GOA提供了针对这类数据的“网状注释”(Web-based annotation)方法。
它是一种快速的整体搜索策略,使用自定义的网状模式(web-based pattern)进行基因注释。
而且,GOA 拥有丰富的信息以及强大的数据库,它可以在短时间内将大量数据转换为可使用的信息,并可以实现复杂的基因功能注释,便于研究者理解不同基因及其细胞类型和发育阶段的功能。
另外,GOA符合人义,它涵盖了各种功能及注释,不仅数量庞大,而且内容丰富,包括超过17万个有用的条目,涵盖细胞生物学、分子生物学、发育学、免疫学、比较基因组学等研究领域等。
用户可以利用它,快速地看到基因的功能,准确地了解不同基因的作用,保存及分析这些相关的信息,一般来说,GOA的操作比较简单,而且搜索结果也很准确。
总之,Gene Ontology Annotation是一个帮助研究者更好地了解基因功能的非常有用的软件,它强大的数据库与丰富的信息,使用起来较为方便,能够有效地帮助研究者细致地研究基因的功能,为生物学研究提供了重要的参考资料,强烈推荐使用它!。
基因本体论(go)功能注释 gene ontology annotation

基因本体论(Gene Ontology,简称GO)是一个标准化的功能分类体系,用于描述基因和基因产物的属性。
GO注释是将基因或基因产物的功能与GO术语相关联的过程。
在GO注释中,基因或基因产物的功能被归类到三个主要的本体论分支中:生物过程(Biological Process)、细胞组分(Cellular Component)和分子功能(Molecular Function)。
每个分支都包含一系列定义明确的术语,这些术语描述了基因或基因产物在细胞中的不同角色和活动。
生物过程分支涵盖了基因或基因产物参与的生物学过程,例如代谢、细胞周期、信号传导等。
细胞组分分支描述了基因或基因产物在细胞内的位置,如细胞核、细胞膜、细胞器等。
分子功能分支则描述了基因或基因产物在分子水平上的活动,如催化活性、结合活性等。
GO注释是基于实验证据和计算预测进行的。
实验方法包括基因突变分析、基因表达研究、蛋白质互作分析等,而计算预测则利用生物信息学工具和算法对基因或基因产物的功能进行预测。
通过GO注释,我们可以更深入地理解基因和基因产物的功能,以及它们在生物体中的相互作用和调控机制。
这些信息对于研究疾病的发病机理、药物设计和基因治疗等领域具有重要意义。
GO功能注释

GO功能注释⽂章转载于 Original 2017-06-12 liuhui ⽣信百科相似的基因在不同物种中,其功能往往保守的。
显然,需要⼀个统⼀的术语⽤于描述这些跨物种的同源基因及其基因产物的功能,否则,不同的实验室对相同的基因的功能的描述不同,将极⼤限制学术的交流。
⽽ Gene Ontology (GO) 项⽬正是为了能够使对各种数据库中基因获基因产物功能描述相⼀致的努⼒结果。
所谓的 GO,是⽣物学功能注释的⼀个标准词汇表术语(GO term),将基因的功能分为三部分:基因执⾏的分⼦功能(Molecular Function)基因所处的细胞组分(Cellular Component)基因以及参与的⽣物学过程(Biological Process)不同的 GO term 通过有向⽆环图关联起来,如下图所⽰:可以看出,不同的 GO term 间的关系由三类:is_a、part_of和regulates。
如regulation of cell projection assembly是⼀种⽣物学过程,是regulation of cell projection organization中的⼀类(is_a),还调节(regulates)cell projection assembly;⼜如cellular component assembly是celluar component biogenesis的⼀部分(part_of)。
值得注意的是,这些关系都是有⽅向的,即反过来不成⽴,因⽽叫做有向⽆环图。
⽬前,GO 注释主要有两种⽅法:(1)序列相似性⽐对(BLAST)(2)结构域相似性⽐对(InterProScan)这⾥以序列相似性⽐对为例,简单介绍 GO 注释的步骤:将基因序列与 swiss-prot 蛋⽩质数据库进⾏ BLAST (blastp 或者 blastx,这篇⽂章介绍了如何做 BLAST 分析:)⽐对,得到如下结果:c49_g1_i1 RNF13_MOUSE 52.00 50 23 1 17 166 240 288 2e-11 65.5c72_g1_i1 RS25_NEUCR 78.72 94 20 0 375 94 1 94 1e-32 116c75_g1_i1 POLX_TOBAC 45.28 53 29 0 162 4 457 509 1e-08 55.1c86_g2_i1 POLX_TOBAC 46.43 112 60 0 339 4 879 990 2e-30 120c91_g1_i1 BUB1_ARATH 55.71 70 28 2 61 264 289 357 1e-14 73.6c143_g1_i1 STL1_YEAST 31.98 172 85 4 6 518 407 547 6e-17 82.8c150_g1_i1 CST26_YEAST 37.63 93 38 3 223 5 142 234 6e-10 58.2c150_g2_i1 YHOE_SCHPO 42.67 75 41 1 227 3 54 126 5e-16 74.7c156_g2_i1 EXOL2_ARATH 47.17 53 28 0 299 141 229 281 6e-06 47.0c169_g1_i1 SPT5_ASPFU 60.98 82 31 1 20 262 725 806 2e-18 84.0其中,第⼆列 swiss-prot 蛋⽩质数据库序列的 ID(UniProtKB ID)。
gene?Ontology?(基因本体论)

gene Ontology (基因本体论)gene ontology为了查找某个研究领域的相关信息,生物学家往往要花费大量的时间,更糟糕的是,不同的生物学数据库可能会使用不同的术语,好比是一些方言一样,这让信息查找更加麻烦,尤其是使得机器查找无章可循。
Gene Ontology就是为了解决这种问题而发起的一个项目。
Gene Ontology中最基本的概念是term。
GO里面的每一个entry都有一个唯一的数字标记,形如GO:nnnnnnn,还有一个term 名,比如"cell", "fibroblast growth factor receptor binding",或者"signal transduction"。
每个term都属于一个ontology,总共有三个ontology,它们分别是molecular function, cellular component 和biological process。
一个基因product可能会出现在不止一个cellular component里面,也可能会在很多biological process里面起作用,并且在其中发挥不同的molecular function。
比如,基因product "cytochrome c" 用molecular function term描述是"oxidoreductase activity",而用biological process term描述就是"oxidative phosphorylation"和"induction of cell death",最后,它的celluar component term是"mitochondrial matrix"和"mitochondrial inner membrane"。
uniprotid和go条目的对应

UniProt ID和GO(Gene Ontology)条目之间的对应关系通常是通过生物信息学数据库的注释来实现的。
UniProt(Universal Protein Resource)是一个全面的、高质量的、经过手工校对的蛋白质序列和功能的数据库。
GO则是一个用于描述基因和基因产物属性的标准词汇表,它提供了三个主要的本体论:分子功能(Molecular Function)、生物过程(Biological Process)和细胞组分(Cellular Component)。
当研究人员对蛋白质进行功能分析时,他们通常会将UniProt ID与GO条目进行匹配。
这种匹配通常基于实验数据、文献报道以及计算预测等。
在UniProt 数据库中,每个蛋白质条目都可能关联有一个或多个GO条目,这些GO条目描述了该蛋白质的功能和参与的生物过程。
要实现UniProt ID和GO条目之间的对应,可以使用多种生物信息学工具和资源,如UniProt网站、Bioconductor包中的函数等。
通过输入UniProt ID,可以检索到与该蛋白质相关的所有GO条目,从而了解其在生物体中的功能和作用。
总之,UniProt ID和GO条目之间的对应关系是通过生物信息学数据库的注释和实验数据等实现的。
这种对应关系有助于研究人员了解蛋白质的功能和参与的生物过程,为生物医学研究提供重要的参考信息。
gene ontology enrichment analysis

gene ontology enrichment analysisGeneOntology富集分析(GeneOntologyEnrichmentAnalysis)是一种生物信息学分析方法,用于解释基因集中的生物学功能和过程。
在基因表达、蛋白质组学等研究中,通常会得到大量基因或蛋白质列表,这些基因或蛋白质在不同的功能或过程中发挥着不同的作用。
通过进行基因集的富集分析,可以帮助研究人员确定哪些生物学功能或过程在研究中起着关键的作用,进而深入研究相关生物学过程的机制。
Gene Ontology(GO)是一个标准化的生物学术语体系,用于描述基因或蛋白质的功能、过程和细胞定位等信息。
该体系包括三个方面:分子功能(Molecular Function)、细胞组成(Cellular Component)和生物学过程(Biological Process)。
在进行GO富集分析时,通常需要使用一些生物信息学工具,如DAVID、Enrichr等,将基因或蛋白质列表映射到GO注释中,并计算每个GO术语的富集程度。
GO富集分析的结果通常包括了每个GO术语的富集水平、显著性水平、富集基因数等信息。
通过分析这些结果,可以得到一些重要的结论,如哪些GO术语在研究中起着重要的作用,哪些基因或蛋白质可能参与到这些生物学过程中。
此外,还可以得到一些新的假设和问题,如某些GO术语富集程度较低是否说明该生物学过程不重要,或者在富集基因中是否存在一些共同的关键基因等。
GO富集分析在生物信息学研究中应用广泛,可以用于研究基因调控、蛋白质相互作用、信号通路等多个方面。
其中,GO富集分析在基因表达芯片数据分析中应用较为普遍,可以帮助研究人员从大量的基因表达数据中快速发现重要的生物学过程和关键基因。
总之,GO富集分析是一种重要的生物信息学分析方法,通过对基因集中的功能和过程进行分析,可以帮助研究人员深入理解生物学过程和机制,为后续的实验研究提供重要的指导和参考。
几种常用的基因功能分析方法和工具

几种常用的基因功能分析方法和工具(转自新浪博客)一、GO分类法最先出现的芯片数据基因功能分析法是GO分类法。
Gene Ontology(GO,即基因本体论)数据库是一个较大的公开的生物分类学网络资源的一部分,它包含38675 个Entrez Gene 注释基因中的17348个,并把它们的功能分为三类:分子功能,生物学过程和细胞组分。
在每一个分类中,都提供一个描述功能信息的分级结构。
这样,GO中每一个分类术语都以一种被称为定向非循环图表(DAGs)的结构组织起来。
研究者可以通过GO分类号和各种GO 数据库相关分析工具将分类与具体基因联系起来,从而对这个基因的功能进行描述。
在芯片的数据分析中,研究者可以找出哪些变化基因属于一个共同的GO功能分支,并用统计学方法检定结果是否具有统计学意义,从而得出变化基因主要参与了哪些生物功能。
EASE(Expressing Analysis Systematic Explorer)是比较早的用于芯片功能分析的网络平台。
由美国国立卫生研究院(NIH)的研究人员开发。
研究者可以用多种不同的格式将芯片中得到的基因导入EASE 进行分析,EASE会找出这一系列的基因都存在于哪些GO分类中。
其最主要特点是提供了一些统计学选项以判断得到的GO分类是否符合统计学标准。
EASE能进行的统计学检验主要包括Fisher 精确概率检验,或是对Fisher精确概率检验进行了修饰的EASE 得分(EASE score)。
由于进行统计学检验的GO分类的数量很多,所以EASE采取了一系列方法对“多重检验”的结果进行校正。
这些方法包括弗朗尼校正法(Bonferroni),本杰明假阳性率法(Benjamini falsediscovery rate)和靴带法(bootstraping)。
同年出现的基于GO分类的芯片基因功能分析平台还有底特律韦恩大学开发的Onto-Express。
2002年,挪威大学和乌普萨拉大学联合推出的Rosetta 系统将GO分类与基因表达数据相联系,引入了“最小决定法则”(minimal decision rules)的概念。
go基因注释与功能分类

其中排在第一位的是人类 基因“PGM1”的相关信息, 点击该条目进入到详细信 息页面。 该页面以表格的形式列出 了该基因有关的详细信息, 包括基因编号,基因的详 细定义,所编码的酶的编 号,基因所在通路,以及 序列的编码信息。同时, 在页面的右侧还提供了该 基因在其他分子生物学数 据库的链接,如OMIM、 NCBI、GenBank等。
KEGG存储内容
KEGG目前共包含了19个子数据库,它们被分类成系统信息、 基因组信息和化学信息三个类别 。
基因组信息存储在GENES数据库里,包括全部完整的基因组序列和部 分测序的基因组序列,并伴有实时更新的基因相关功能的注释。 KEGG中化学信息的 6个数据库被称为 KEGG LIGAND数据库,包含 化学物质、酶分子、酶化反应等信息。KEGG BRITE数据库是一个包 含多个生物学对象的基于功能进行等级划分的本体论数据库,它包括 分子、细胞、物种、疾病、药物、以及它们之间的关系。
一些小的通路模块被存储在MODULE数据库中,该数据库还存储了其 他的一些相关功能的模块以及化合物信息。
KEGG DRUG数据库存储了目前在日本所有非处方药和美国的大部分 处方药品。 KEGG DISEASE是一个存储疾病基因、通路、药物、以及疾病诊断标 记等信息的新型数据库。
KEGG数据库的注释与检索
一、富集分析算法
富集分析方法通常是分析一组基因在某个功能结点上是否 过出现(over-presentation)。这个原理可以由单个基因的注 释分析发展到大基因集合的成组分析。 由于分析的结论是基于一组相关的基因,而不是根据单个 基因,所以富集分析方法增加了研究的可靠性,同时也能 够识别出与生物现象最相关的生物过程。
GeneOntology(GO)分析
Gene Ontology
现今的生物学家们浪费了太多的时间和精力在搜寻生物信息上。这种情况归结为生物学上定 义混乱的原因:不光是精确的计算机难以搜寻到这些随时间和人为多重因素而随机改变的定 义,即使是完全由人手动处理也无法完成。举个例子来说,如果需要找到一个用于制抗生素 的药物靶点,你可能想找到所有的和细菌蛋白质合成相关的基因产物,特别是那些和人中蛋 白质合成组分显著不同的。但如果一个数据库描述这些基因产物为“翻译类”,而另一个描述 其为“蛋白质合成类”,那么这无疑对于计算机来说是难以区分这两个在字面上相差甚远却在 功能上相一致的定义。 Gene Ontology (GO)项目正是为了能够使对各种数据库中基因产物功能描述相一致的努力结 果。这个项目最初是由 1988 年对三个模式生物数据库的整合开始:: FlyBase (果蝇数据库 Drosophila),t Saccharomyces Genome Database (酵母基因组数据库 SGD) and the Mouse Genome Database (小鼠基因组数据库 MGD)。从那开始,GO 不断发展扩大,现在已包含数 十个动物、植物、微生物的数据库。 GO 的定义法则已经在多个合作的数据库中使用,这使在这些数据库中的查询具有极高的一 致性。这种定义语言具有多重结构,因此在各种程度上都能进行查询。举例来说,GO 可以 被用来在小鼠基因组中查询和信号转导相关的基因产物,也可以进一步找到各种生物地受体 酪氨酸激酶。这种结构允许在各种水平添加对此基因产物特性的认识。 GO 的结构包括三个方面:分子生物学上的功能、生物学途径和在细胞中的组件作用。当然, 它们可能在每一个方面都有多种性质。如细胞色素 C,在分子功能上体现为电子传递活性, 在生物学途径中与氧化磷酸化和细胞凋亡有关,在细胞中存在于线粒体质中和线粒体内膜 上。下面,将进一步的分别说明 GO 的具体定义情况。 基因产物 基因产物和其生物功能常常被我们混淆。例如,“乙醇脱氢酶”既可以指放在 Eppendorf 管里 的基因产物,也表明了它的功能。但是这之间其实是存在差别的,一个基因产物可以拥有多 种分子功能,多种基因产物也可以行使同一种分子功能。比如还是“乙醇脱氢酶”,其实多种 基因产物都具有这种功能,而并不是所有的这些酶都是由乙醇脱氢酶基因编码的。一个基因 产物可以同时具有“乙醇脱氢酶”和“乙醛歧化酶”两种功能,甚至更多。所以,在 GO 中,很 重要的一点在于,当使用“乙醇脱氢酶活性”这种术语时,所指的是功能,并不是基因产物。 许多基因产物会形成复合物后执行功能。这些“基因复合物”有些非常简单(如血红蛋白由血
Gene Ontology分析

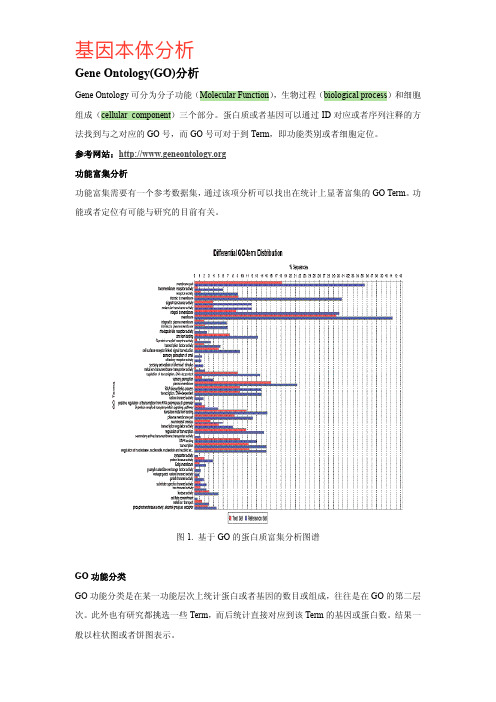
Gene OntologyGO分析Gene Ontology可分为分子功能Molecular Function生物过程biological process和细胞组成cellular component三个部分。
蛋白质或者基因可以通过ID 对应或者序列注释的方法找到与之对应的GO号而GO号可对于到Term即功能类别或者细胞定位。
参考网站 功能富集分析功能富集需要有一个参考数据集通过该项分析可以找出在统计上显著富集的GO Term。
功能或者定位有可能与研究的目前有关。
图1. 基于GO的蛋白质富集分析图谱GO功能分类GO功能分类是在某一功能层次上统计蛋白或者基因的数目或组成往往是在GO的第二层次。
此外也有研究都挑选一些Term 而后统计直接对应到该Term的基因或蛋白数。
结果一般以柱状图或者饼图表示。
1.GO分析根据挑选出的差异基因计算这些差异基因同GO 分类中某几个特定的分支的超几何分布关系GO 分析会对每个有差异基因存在的GO 返回一个p-value小的p 值表示差异基因在该GO 中出现了富集。
GO 分析对实验结果有提示的作用通过差异基因的GO 分析可以找到富集差异基因的GO分类条目寻找不同样品的差异基因可能和哪些基因功能的改变有关。
2.Pathway分析根据挑选出的差异基因计算这些差异基因同Pathway 的超几何分布关系Pathway 分析会对每个有差异基因存在的pathway 返回一个p-value小的p 值表示差异基因在该pathway 中出现了富集。
Pathway 分析对实验结果有提示的作用通过差异基因的Pathway 分析可以找到富集差异基因的Pathway 条目寻找不同样品的差异基因可能和哪些细胞通路的改变有关。
与GO 分析不同pathway 分析的结果更显得间接这是因为pathway 是蛋白质之间的相互作用pathway 的变化可以由参与这条pathway 途径的蛋白的表达量或者蛋白的活性改变而引起。
humann2 genefamilies文件转化为go、kegg注释

humann2 genefamilies文件转化为go、kegg注释摘要:1.介绍humann2 genefamilies 文件2.文件转化为go、kegg 注释的意义3.详细步骤和方法4.结果展示与分析5.总结与展望正文:Humann2 是一种广泛应用于代谢组学数据分析的生物信息学工具,能够对代谢物进行注释和代谢途径分析。
在分析过程中,我们通常会使用humann2 genefamilies 文件,这个文件包含了所有与代谢物相关的基因家族信息。
然而,这个文件通常以一种特定的格式存储,不便于我们直接进行后续的分析。
因此,我们需要将humann2 genefamilies 文件转化为go、kegg 注释格式。
文件转化为go、kegg 注释的意义在于,这两种格式是生物信息学领域广泛接受的标准格式,可以方便地进行后续的功能注释和代谢途径分析。
GO (Gene Ontology)是一种对基因进行功能注释的标准方法,它将基因分为三个层次:生物学过程、细胞组分和分子功能。
KEGG(Kyoto Encyclopedia of Genes and Genomes)则是一种代谢途径数据库,提供了大量的代谢途径信息。
具体的转化步骤如下:1.安装humann2 软件包:首先,我们需要安装humann2 软件包,以便于我们处理humann2 genefamilies 文件。
可以通过运行以下命令进行安装:`conda install -c bioconda humann2`。
2.读取humann2 genefamilies 文件:使用humann2 软件包中的`read_humann2_genefamilies()`函数读取humann2 genefamilies 文件。
例如:`humann2_genefamilies <-read_humann2_genefamilies("path/to/humann2_genefamilies_file.txt")`。
gene_ontology(GO基因注释)

GO(gene ontology)是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的,堆积因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准.GO是多种生物本体语言中的一种,提供了三层结构的系统定义方式,用于描述基因产物的功能.基因本体论(gene ontology)的建立现今的生物学家们浪费了太多的时间和精力在搜寻生物信息上。
这种情况归结为生物学上定义混乱的原因:不光是精确的计算机难以搜寻到这些随时间和人为多重因素而随机改变的定义,即使是完全由人手动处理也无法完成。
举个例子来说,如果需要找到一个用于制抗生素的药物靶点,你可能想找到所有的和细菌蛋白质合成相关的基因产物,特别是那些和人中蛋白质合成组分显著不同的。
但如果一个数据库描述这些基因产物为“翻译类”,而另一个描述其为“蛋白质合成类”,那么这无疑对于计算机来说是难以区分这两个在字面上相差甚远却在功能上相一致的定义。
Gene Ontology (GO)项目正是为了能够使对各种数据库中基因产物功能描述相一致的努力结果。
这个项目最初是由1988年对三个模式生物数据库的整合开始:: FlyBase (果蝇数据库Drosophila),t Saccharomyces Genome Database (酵母基因组数据库SGD) and the Mouse Genome Database(小鼠基因组数据库MGD)。
从那开始,GO不断发展扩大,现在已包含数十个动物、植物、微生物的数据库。
GO的定义法则已经在多个合作的数据库中使用,这使在这些数据库中的查询具有极高的一致性。
这种定义语言具有多重结构,因此在各种程度上都能进行查询。
举例来说,GO可以被用来在小鼠基因组中查询和信号转导相关的基因产物,也可以进一步找到各种生物地受体酪氨酸激酶。
这种结构允许在各种水平添加对此基因产物特性的认识。
go基因注释与功能分类课件

第四节 基因功能预测
Gene Function Prediction
基因功能预测算法
近来已经发展了很多基于GO数据库或KEGG数据库的方 法,利用高通量的基因表达和蛋白质互作数据进行功能预测, 其中一些新开发的方法试图整合多种数据类型,通过构建功 能相关网络的方式预测基因功能。
atom )模式。
点击搜索按钮“GO”进入查询结果页面,该页面会列出针 对基因“PGM1”在KEGG数据库中的搜索结果,除人类外, 包含“PGM1”基因的物种条目也会被列出。
其中排在第一位的是人类 基因“PGM1”的相关信息, 点击该条目进入到详细信 息页面。
该页面以表格的形式列出 了该基因有关的详细信息, 包括基因编号,基因的详 细定义,所编码的酶的编 号,基因所在通路,以及 序列的编码信息。同时, 在页面的右侧还提供了该 基因在其他分子生物学数 据库的链接,如OMIM、 NCBI、GenBank等。
2. 用序列检索GO数据库
对于未知基因名的序列,可 以用序列直接检索GO 数据库。 点 击 AmiGO 首 页 上 方 的 “BLAST”。
界面风格类似于其他数据库 BLAST 搜 索 的 网 页 , 在 检 索 框中铁如氨基酸或核酸序列, 网页能自动识别并相应地做 BLASTP或BLASTX和数据库 中的序列比对。
另外一种化学注释的方法是 以小分子化学结构的生物学 意义为特征来实现的。
在KEGG数据库中,酶与酶 之间的反应信息以及相关的 化学结构信息分别存储在 KEGG REACTION数据库和 KEGG REPAIR数据库中。
每个化合物的化学结构都被 转 化 为 RDM (atom type changes at R:reaction center D:diffevent atom M:matched
go注释原理

go注释原理基因本体论(Gene Ontology, GO)是一种用于描述基因功能的分类系统,其原理主要包括以下几个方面:1. 共享蛋白质的生物作用:基因组测序结果表明,大部分指定核心生物学功能的基因是所有真核生物共有的。
这种共享蛋白质在一个有机体中的生物作用的知识,往往可以转移到其他有机体。
2. 产生动态、可控的词汇表:基因本体论联盟的目标是产生一个动态的、可控的词汇表,可以应用于所有真核生物,即使基因和蛋白质在细胞中作用的知识正在积累和变化。
3. 基因功能注释:当拿到一个非模式生物或者无参考基因组的项目时,经常需要进行基因的功能注释,才能够进行生物信息学的数据分析工作。
4. 统一术语:由于序列相似的基因在不同物种中,其功能往往是保守的,但以前往往会存在不同的实验室对相同的基因的功能描述因为自然语言的模糊性而不尽相同的问题。
因此,需要一个统一的术语用于描述这些跨物种的同源基因及其基因产物的功能,否则这种模糊性将会极大限制不同的科研人员间的学术交流。
5. 分类系统间的直译问题:随着生物信息学数据的积累,出现了不同的应用于描述基因功能的分类数据库。
这些分类系统的目标都是希望能够用于阐述这些跨物种的同源基因的生物学功能。
但是因为分类系统之间的基因功能注释结果可能在自然语言描述上都不尽相同,存在都相互为各自的方言的情况,大部分分类结果都几乎无法在分类系统之间直译。
6. GO注释的定义与分类:GO定义了用于描述基因功能的概念/类以及这些概念之间的关系。
它从三个方面对功能进行分类:细胞组分(cellular component):细胞的每个部分和细胞外环境;分子功能(molecular function):可以描述为分子水平的活性(activity),如催化(catalytic)或结合(binding)活性;生物过程(biological process):生物学过程系指由一个或多个分子功能有序组合而产生的系列事件。
go条目对应的细胞类型

go条目对应的细胞类型介绍细胞是生物体中最基本的结构和功能单位,作为生物学研究的基础,对细胞进行分类和研究对于了解生命的机制至关重要。
Ge ne On t ol og y (G O)是一种常用的细胞分类系统,通过对细胞中的基因功能进行注释来描述不同细胞类型的特征。
本文将介绍一些常见的G O条目对应的细胞类型。
1.神经元细胞神经元细胞是构成神经系统的基本单元,负责传递神经信号。
根据GO 条目,神经元细胞可以被归类为"细胞类型:神经元;细胞过程:神经传导"。
神经元细胞具有高度特化的结构,包括树突、轴突和突触,通过突触传递电化学信号。
它们在神经系统中起着至关重要的作用,参与大脑功能的调控和维持。
2.肌肉细胞肌肉细胞是负责肌肉运动的细胞类型。
根据G O条目,肌肉细胞可以被归类为"细胞类型:肌肉细胞;细胞过程:肌肉收缩"。
肌肉细胞包括骨骼肌细胞、平滑肌细胞和心肌细胞。
它们通过肌纤维的收缩来实现肌肉的运动和功能。
3.血细胞血细胞主要存在于血液中,包括红细胞、白细胞和血小板。
根据G O条目,红细胞对应的细胞类型为"红细胞",细胞过程为"呼吸作用";白细胞对应的细胞类型为"白细胞",细胞过程为"免疫应答";血小板对应的细胞类型为"血小板",细胞过程为"血栓形成"。
血细胞在人体中起着重要的运输、免疫和止血作用。
4.上皮细胞上皮细胞是覆盖人体内外各个组织和器官表面的细胞类型。
根据G O条目,上皮细胞对应的细胞类型为"上皮细胞",细胞过程为"上皮细胞发育"。
上皮细胞具有紧密的排列,形成细胞间连接和组织屏障,起到保护、吸收和分泌等功能。
5.干细胞干细胞是具有自我更新和分化潜能的细胞类型。
根据G O条目,干细胞可以被归类为"细胞类型:干细胞;细胞过程:细胞增殖"。
the go enrichment analysis

the go enrichment analysis
GO enrichment analysis是一种用于分析基因组数据的生物信息学方法,它能够帮助我们理解基因功能和生物过程。
GO代表Gene Ontology(基因本体),是一个标准化的注释系统,用于描述基因和基因产品的功能、进程和定位。
GO enrichment analysis的目标是确定在给定的基因组数据中是否存在显著富集的GO术语。
通常,我们会将基因列表与已知的GO术语进行比较,以确定哪些GO术语在这个基因集中出现的频率高于预期。
这可以帮助我们找到与某个特定生物学研究问题相关的功能或进程。
GO enrichment analysis通常包括以下步骤:
1. 数据预处理:整理基因列表,确保基因标识符的一致性和可用性。
2. GO注释:将基因列表与已知的GO术语进行匹配,以获取每个基因对应的GO注释信息。
3. 统计分析:使用适当的统计方法计算每个GO术语的富集程度,例如超几何检验或Fisher确切检验。
4. 多重检验校正:由于富集分析涉及多个假设检验,需要对得到的P值进行多重校正,以控制错误发现率。
5. 结果解释:根据富集分析的结果,确定哪些GO术语在基因集中富集,并根据需要进行功能注释和生物学解释。
GO enrichment analysis可以应用于各种生物学研究领域,如
基因表达分析、蛋白质组学、转录组学等。
通过富集分析,我们能够深入了解基因集的功能特征,为深入研究提供线索和方向。
go富集分析说明

GO富集分析说明基因本体(Gene Ontology,GO)是一个在生物信息学领域中广泛使用的本体。
1998年由研究三种模式(果蝇、小鼠和酵母)基因组的研究者共同发起组织了一个称为基因本体联盟的专业团队。
创建基因本体的初衷是希望提供一个可具代表性的规范化的基因和基因产物特性的术语描绘或词义解释的工作平台。
现在已包含数十个动物、植物、微生物的数据库。
基因本体涉及的基因和基因产物词汇分为三大类,涵盖生物学的三个方面:细胞组分(cellular component):细胞的每个部分和细胞外环境。
分子功能(molecular function):可以描述为分子水平的活性(activity),如催化(catalytic)或结合(bingding)活性。
生物过程(biological process):生物过程系指由一个或多个分子功能有序组合而产生的系列事件。
其定义有广义和狭义之分,在词义上可以区分为泛指和特指。
一般规律是,一个过程是由多个不同的步骤组成。
注:生物过程与途径或通路(pathway)不是同一回事。
通过将差异基因做GO富集分析,可以把基因按照不同的功能进行归类,达到对基因进行注释和分类的目的。
富集分析方法说明:采取的方法是fisher 精确检验,数据包是clusterProfiler,来自R/bioconductor;挑选的标准是落在某个term/GO 上差异的基因数目>=4,p_value <0.05,画图中取得term/GO 是按照enrich factor 的值从大小降序排列,取前30个结果。
enrich_factor 定义= (某个term中的差异基因数目/总的差异基因数目)/(数据库term中总的基因数目/数据库中总的基因数目)字段说明:上海伯豪生物技术有限公司技术支持部2016年01月。
kegg go fisher检验 方法学描述

kegg go fisher检验方法学描述KEGG(Kyoto Encyclopedia of Genes and Genomes)是一个综合性的数据库,提供了关于基因、蛋白质、代谢途径和疾病等方面的信息。
GO (Gene Ontology)是一种用于描述基因功能和相关生物过程的标准化分类系统。
Fisher检验是一种用于检验两个分类变量之间相关性的统计方法。
在KEGG中,进行GO Fisher检验的方法学描述如下:1. 数据准备:首先,需要从KEGG数据库中获取感兴趣的基因集和相应的GO注释信息。
这些基因可以是在一个生物学实验中鉴定出的差异表达基因集,或者是在特定疾病研究中相关的基因集。
2. 建立基因与GO注释的映射:将基因与其对应的GO注释进行映射,这样可以将基因集转化为对应的功能注释集合。
3. 设定假设:根据研究的目的和问题,设定一个零假设和备择假设。
零假设通常是指基因集在特定GO功能中的分布与全基因组的分布没有显著差异,而备择假设则相反。
4. 进行Fisher检验:使用Fisher精确检验(Fisher's exact test)对基因集与特定GO功能之间的关联性进行检验。
Fisher检验可以计算出一个p值,表示观察到的关联性在零假设下的显著性。
5. 多重检验校正:由于进行多个假设检验可能增加误报的风险,通常需要对得到的p值进行多重检验校正,常用的方法包括Bonferroni 校正和Benjamini-Hochberg校正。
6. 结果解释和可视化:根据得到的p值和校正后的显著性水平,判断哪些GO功能与基因集显著相关。
可以使用图表或其他可视化方式来展示结果。
总而言之,KEGG GO Fisher检验是使用KEGG数据库中的GO注释信息,结合Fisher检验方法来评估基因集与特定功能之间的关联性,并通过统计显著性来判断这种关联性的可信度。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
GO(gene ontology)是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的,堆积因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准.GO是多种生物本体语言中的一种,提供了三层结构的系统定义方式,用于描述基因产物的功能.基因本体论(gene ontology)的建立现今的生物学家们浪费了太多的时间和精力在搜寻生物信息上。
这种情况归结为生物学上定义混乱的原因:不光是精确的计算机难以搜寻到这些随时间和人为多重因素而随机改变的定义,即使是完全由人手动处理也无法完成。
举个例子来说,如果需要找到一个用于制抗生素的药物靶点,你可能想找到所有的和细菌蛋白质合成相关的基因产物,特别是那些和人中蛋白质合成组分显著不同的。
但如果一个数据库描述这些基因产物为“翻译类”,而另一个描述其为“蛋白质合成类”,那么这无疑对于计算机来说是难以区分这两个在字面上相差甚远却在功能上相一致的定义。
Gene Ontology (GO)项目正是为了能够使对各种数据库中基因产物功能描述相一致的努力结果。
这个项目最初是由1988年对三个模式生物数据库的整合开始:: FlyBase (果蝇数据库Drosophila),t Saccharomyces Genome Database (酵母基因组数据库SGD) and the Mouse Genome Database(小鼠基因组数据库MGD)。
从那开始,GO不断发展扩大,现在已包含数十个动物、植物、微生物的数据库。
GO的定义法则已经在多个合作的数据库中使用,这使在这些数据库中的查询具有极高的一致性。
这种定义语言具有多重结构,因此在各种程度上都能进行查询。
举例来说,GO可以被用来在小鼠基因组中查询和信号转导相关的基因产物,也可以进一步找到各种生物地受体酪氨酸激酶。
这种结构允许在各种水平添加对此基因产物特性的认识。
GO发展了具有三级结构的标准语言(ontologies),如表所示。
根据基因产物的相关分子功能,生物学途径,细胞学组件而给予定义,无物种相关性。
本体论内容分子功能本体论基因产物个体的功能,如与碳水化合物结合或ATP 水解酶活性等生物学途径本体论分子功能的有序组合,达成更广的生物功能,如有丝分裂或嘌呤代谢等细胞组件本体论亚细胞结构、位置和大分子复合物,如核仁、端粒和识别起始的复合物等基本来说,GO工作可分为三个不同的部分:第一,给予和维持定义;第二,将位于不同数据库中的本体论语言、基因和基因产物进行联系,形成网络;第三,发展相关工具,使本体论的标准语言的产生和维持更为便捷。
本体论(The ontologies)GO的结构包括三个方面?D?D分子生物学上的功能、生物学途径和在细胞中的组件作用。
当然,它们可能在每一个方面都有多种性质。
如细胞色素C,在分子功能上体现为电子传递活性,在生物学途径中与氧化磷酸化和细胞凋亡有关,在细胞中存在于线粒体质中和线粒体内膜上。
下面,将进一步的分别说明GO的具体定义情况。
基因产物基因产物和其生物功能常常被我们混淆。
例如,“乙醇脱氢酶”既可以指放在Eppendorf管里的基因产物,也表明了它的功能。
但是这之间其实是存在差别的?D?D一个基因产物可以拥有多种分子功能,多种基因产物也可以行使同一种分子功能。
比如还是“乙醇脱氢酶”,其实多种基因产物都具有这种功能,而并不是所有的这些酶都是由乙醇脱氢酶基因编码的。
一个基因产物可以同时具有“乙醇脱氢酶”和“乙醛歧化酶”两种功能,甚至更多。
所以,在GO中,很重要的一点在于,当使用“乙醇脱氢酶活性”这种术语时,所指的是功能,并不是基因产物。
许多基因产物会形成复合物后执行功能。
这些“基因复合物”有些非常简单(如血红蛋白由血红蛋白基因产物α-球蛋白、β-球蛋白和小分子的亚血红素组成),有些非常复杂(如核糖体)。
现在,小分子的描述还没有包括在GO中。
在未来,这个问题可望由和现在的Klotho和LIGAND等小分子数据库联合而解决。
分子功能分子功能描述在分子生物学上的活性,如催化活性或结合活性。
GO分子功能定义功能而不是整体分子,而且不特异性地指出这些功能具体的时空信息。
分子功能大部分指的是单个基因产物的功能,还有一小部分是此基因产物形成的复合物的功能。
定义功能的义项包括催化活性、转运活性、结合活性等,更为狭窄的定义包括腺苷酸环化酶活性或钟形受体结合活性等。
生物学途径生物学途径是由分子功能有序地组成的,具有多个步骤的一个过程。
举例来说,较为宽泛的是细胞生长和维持、信号传导。
一些更为具体的例子包括嘧啶代谢或α-配糖基的运输等。
一个生物学途径并不是完全和一条生物学通路相等。
因此,GO并不涉及到通路中复杂的机制和所依赖的因素。
细胞组件细胞中的位置指基因产物位于何种细胞器或基因产物组中(如糙面内质网,核或核糖体,蛋白酶体等)。
GO的形式GO 定义的术语有着直接非循环式(directed acyclic graphs (DAGs)的特点,而并非是传统的等级制(hierarchy)定义方式(随着代数增加,下一级比上一级更为具体)。
举个例子来说,生物学途径中有一个定义是己糖合成,它的上一级为己糖代谢和单糖合成。
当某个基因被注解为“己糖合成活性”后,它自动地获得了己糖代谢和单糖合成地注解。
因为在GO中,每个术语必须遵循“真途径“法则,即如果下一代的术语可以用于描述此基因产物,其上一代术语也可以适用。
GO的注释(Annotation)那么,GO中的术语如何和相对应的基因产物相联系的呢?这是由参与合作的数据库来完成的,它们使用GO的定义方法,对它们所包含的基因产物进行注解,并且提供支持这种注解的参考和证据。
每个基因或基因产物都会有一个列表,列出与之相关的GO术语。
每个数据库都会给出这些基因产物和GO术语的联系数据库,并且也可以在GO的ftp站点上和WEB方式查询到。
并且,GO联合会提供了简化的本体论术语(GO slim),这样,可以在更高级的层面上研究基因组的功能。
比如,粗略地估计哪一部分的基因组与信号传导、代谢合成或复制有关。
GO对基因和蛋白的注释阐明了基因产物和用于定义他们的GO术语之间的关系。
基因产物指一个基因编码的RNA或蛋白产物。
因为一个基因可能编码多个具有很不相同性质的产物,所以GO推荐的注释是针对基因产物的而不是基因的。
一个基因是和所有适用于它的术语联系在一起的。
一个基因产物可以被一种本体论定义的多种分支或多种水平注释。
注释需要反映在正常情况下此基因产物的功能,生物途径,定位等,而并不包括其在突变或病理状态下的情况。
GO联合会的各个数据库成员采用手动或自动的方式生成注释,这两种方式共有的原理是:一.所有的注释都需要有来源,可以是文字、另一个数据库或是计算机分析结果;二.注释必须提供支持这种基因产物和GO术语之间联系的证据。
GO文件格式GO的所有数据都是免费获得的。
GO数据有三种格式:flat(每日更新)、XML(每月更新)和MySQL(每月更新)。
这些数据格式都可以在GO ftp的站点上下载。
XML 和 MySQL 文件是被储存于独立的GO数据库中。
如果需要找到与某一个GO术语相关的基因或基因产物,可以找到一个相应表格,搜寻到这种注解的编号,并且可以链接到与之对应的位于不同数据库的基因相关文件。
GO浏览器和修改器(browser and editor)GO术语和注释使用了多种不同的工具软件,它们都可以在web方式的“GO 浏览器”下“GO software page”中找到。
大多数GO浏览器都是web模式的,允许你直观的看到术语和其相关信息,如定义、同义词和数据库参考等。
有些GO浏览器如AmiGO和 QuickGO,可以看到每个术语的注释。
而可下载的DAG-Edit编辑器,一样可以离线地显示注释和所有本体论定义的信息。
对于每一个浏览器来说,都可以选择最适用于你要求的工具软件。
常见的三种浏览器AmiGO from BDGP在AmiGO中,可以通过查询一个GO术语而得到所有具有这个注释的基因产物,或查询一个基因产物而得到它所有的注释关系。
还可以浏览本体论,得到术语之间的关系和术语对应的基因产物数目。
AmiGO直接连接GO下的MySQL。
MGI GO Browser MGI GO的功能类似于AmiGO,所不同的在于它所得到的基因为小鼠基因。
MGI GO浏览器直接连接GO下的MGI数据库。
QuickGO at EBI QuickGO,整合在EBI下的InterPro中,可以通过查询一个GO术语而得到它的定义与关系描述、在SWISS-PROT中的定位、在酶分类学(EC)和转运分类学(TC)中的定位和InterPro中的定位等。
其他还有一些特殊的浏览GO的浏览器,其中括号中为建立机构和主要特色:EP GO Browser(EBI,基因表达情况),、GoFish(Harvard,Boolean查询、GenNav (NLM, 图像化展示)、GeneOntology@RZPD(RZPD,UniGene)、ProToGO(Hebrew University,GO的亚图像化)、CGAP GO Browser(癌症基因组解剖工程,癌症)、GOBrowser(Illuminae,perl.、TAIR Keyword Browser (TAIR,拟南芥)、PANDORA(Hebrew University,非一致化蛋白)。
修改器GO 术语和本体论结构可以由任何可以读入GO平板文件的文本修改器进行编辑,但是这需要对平板文件非常熟悉。
因此,DAG-Edit是被推荐使用的,它是为 GO 特别设计的,能够保证文件的句法正确。
GO注释可以被多种数据库特异性的工具所编辑,如TIGR的Manatee和EBI的Talisman tool。
但是GO数据库中写入新的注释是需要通过GO认证的管理员方可进行的,如果想提出新的注释或对本体论的建议,可以联系GO。
主要修改器为DAG-Edit和COBrA。
DAG-Edit基于Java语言,提供了能浏览、查询、编辑具有DAG数据格式的GO数据界面。
在SourceForge可以免费下载,伴随着帮助文件。
COBrA能够编辑和定位GO和OBO本体论。
它一次显示两个本体论,因此可以在不同的水平相应定位。
(如组织和细胞类型水平)优点在于可以综合几种本体论,支持的文件格式多,包括GO平板文件、GO RDF和OWL格式等。
如图为DAG-Edit的界面,可以分为四个部分:1)定义编辑面板(term editor panel)显示当下的本体论。
也是主要的编辑本体论结构的工具,可以通过点击和拖动术语来修改本体论的从属关系。