基因组注释ppt课件
合集下载
细菌全基因组测序 ppt课件
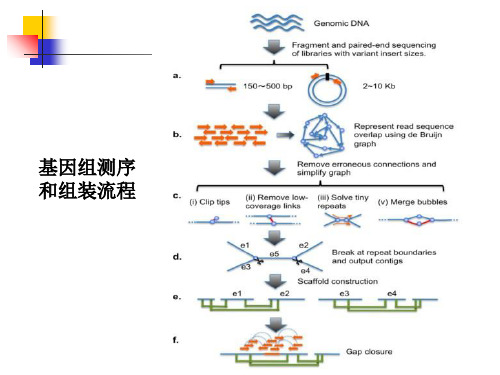
基因家族(gene family) 和基因簇(gene cluster)分析
基因组中来源相同,结构和功能相关的基因 聚集在一起形成基因家族。
基因家族的各个成员紧密成簇排列成大段的串联 重复单位,分布在某一条染色体的特殊区域
genefamily.xls
基因家族聚类结果
genefamily.stat
各基因家族统计信息
培养条件① 培养条件②
或活性较低
测定转录 组mRNA
细菌全基因组测序
比较 新 差异 基因
其他方面的应用研究
❖ 应用NMR、FTIR、UV, 14C标记的木质 素降解机理方面的研究; ❖农药残留物以及其他一些难降解有机物的 降解; ❖ 重金属有机物化合物的降解。
② 木质素降解过程中涉及到的细胞外酶主要有:木质素过氧化物酶
(LiP)和锰过氧化物酶(MnP),以及漆酶(Lac)。此外,一 些附属酶参与过氧化氢的产生,乙二醛氧化酶(glyoxal oxidase, 缩写作GLOX)和芳基醇氧化酶(aryl alcohol oxidase,缩写作 AAO)属于这类酶。
对4株菌的亲缘关系进行分析,确定菌株之间的相互关 系;
通过对4株菌进行进化分析,判定是否为古菌或新的菌 种。
细菌全基因组测序
基因分离
下一步的实验安排
对已注释出的基因进行验证
载体
酶切
酶切
连接
转化
筛选 表达
细菌全基因组测序
未注释出功能的基因鉴定,挖掘新基因
DNA 转录 RNA 翻译 Protein
细菌全基因组测序
“一个物种基因组计划的完成, 就意味着这一物种学科和产业 发展的新开端”
向仲怀院士
谢谢!!
细菌全基因组测序
基因组学PPT课件

9
人类基因组计划的背景-----基因组计划最早始于美国
初衷1945年原子弹事件
1984年12月犹他大学魏特受美国能源部的委托,美国能源部
的广岛之争:突变率调查
资助召开的环境诱变物和致癌物的防护的会议上,
讨论DNA重组技术的发展及测定人类整个基因组
1985年6月,美国加州的会议上, DNA序列的意义,第一次提出测定人体基因和全部DNA序列,
1990年10月1日正式启动实施
目标:完成对人的基因组的30亿个核苷酸对的 全部序列测定工作,阐明人体中全部基因的位置、 功能、结构、表达调控方、德、日、中六国科学家的共同努力下, 2000年6月26日, 国际人类基因组计划与塞莱拉公司联合发布“人类基因组工作草图”。 2001年2月12日 两大科研小组联合发布人类基因组图谱及“基本信息”。宣告人类基因组计划基本完成。10
人类基因组计划是与曼哈顿原子计划、阿波罗登月计划一样伟大宏伟。
人类基因组计划的研究内容
美国的人类基因组计划总体规划是: 拟在15年内至少投入30亿美元,进行对人类全基因组的
分析。 1993年作了修订,其主要内容包括: 人类基因组的基因图构建与序列分析; 人类基因的鉴定; 基因组研究技术的建立; 人类基因组研究的模式生物; 信息系统的建立。 人类基因组研究的社会、法律与伦理问题, 交叉学科的技术训练, 技术的转让, 研究计划的外延等共9方面的内容。
美国能源部正式提出了展开人类
并检测所有的突变,计算真实的突变率。
基因组测序工作,形成了能源部 的“人类基因组计划”初步草案。
1986年6月,新墨西哥州冷泉港吉尔伯特及伯格主持的讨论会上, 进行了可行性讨论。美能源部宣布实施草案。意裔美肿瘤分子生
1987年,美国国家医学研究 院和能源部联合提出了这一 宏伟计划,即HGP),先期
人类基因组计划的背景-----基因组计划最早始于美国
初衷1945年原子弹事件
1984年12月犹他大学魏特受美国能源部的委托,美国能源部
的广岛之争:突变率调查
资助召开的环境诱变物和致癌物的防护的会议上,
讨论DNA重组技术的发展及测定人类整个基因组
1985年6月,美国加州的会议上, DNA序列的意义,第一次提出测定人体基因和全部DNA序列,
1990年10月1日正式启动实施
目标:完成对人的基因组的30亿个核苷酸对的 全部序列测定工作,阐明人体中全部基因的位置、 功能、结构、表达调控方、德、日、中六国科学家的共同努力下, 2000年6月26日, 国际人类基因组计划与塞莱拉公司联合发布“人类基因组工作草图”。 2001年2月12日 两大科研小组联合发布人类基因组图谱及“基本信息”。宣告人类基因组计划基本完成。10
人类基因组计划是与曼哈顿原子计划、阿波罗登月计划一样伟大宏伟。
人类基因组计划的研究内容
美国的人类基因组计划总体规划是: 拟在15年内至少投入30亿美元,进行对人类全基因组的
分析。 1993年作了修订,其主要内容包括: 人类基因组的基因图构建与序列分析; 人类基因的鉴定; 基因组研究技术的建立; 人类基因组研究的模式生物; 信息系统的建立。 人类基因组研究的社会、法律与伦理问题, 交叉学科的技术训练, 技术的转让, 研究计划的外延等共9方面的内容。
美国能源部正式提出了展开人类
并检测所有的突变,计算真实的突变率。
基因组测序工作,形成了能源部 的“人类基因组计划”初步草案。
1986年6月,新墨西哥州冷泉港吉尔伯特及伯格主持的讨论会上, 进行了可行性讨论。美能源部宣布实施草案。意裔美肿瘤分子生
1987年,美国国家医学研究 院和能源部联合提出了这一 宏伟计划,即HGP),先期
基因组信息分析PPT课件

GC含量
碱基G、C相对于A、T的丰度很早就被看作是区分细菌基因组的特征之一 .不同的原核生物中,GC含量(GC content)从25﹪到75﹪,变化非常大。 大部分细菌是通过从其它生物体大规模获得基因(长度为几万甚至几十万个核苷酸)而进化的(水平转移).简而言之,许多细菌基因组表现为具有不同GC含量的区域的组合物,这些区域反映了细菌的进化历史。
G
0.1751306272192
T
0.3248693727808
酵母基因组核苷酸出现频率
在统计过程中,如果同时计算DNA的正反两条链,则根据碱基配对原则,A和T、C和G的出现频率相同。如果仅统计一条链,则虽然A和T、C和G的出现频率不同,但是非常接近。
核苷酸
频率
A
0.344
C
0.155
G
等值区
定义:具有一致碱基组成的长区域 特征 :等值区基因组序列的长度超过1,000,000对碱基虽然不同的等值区其GC含量差别显著,但同一等值区的GC含量始终相对均衡 人类基因组大约可以划分为五个不同类型的等值区:a) L1和L2,平均GC含量分别为39﹪和42﹪(欠GC)) b) H1、H2和H3,GC含量平均值分别为46﹪、49﹪和54﹪ (丰GC)
科学家对这本天书了解最多的部分就是遗传密码 或者说掌握了DNA对蛋白质编码的规律 关于密码子(1)密码子的使用是非随机的 如果密码子的第一、第二位碱基是A、U, 那么第三位将尽可能使用G、C;反之亦然。 如果三位都用G、C,则配对容易,分解难; 三位都用A、U,则相反。 一般地说,高表达的基因,要求翻译速度快, 要求密码子和反密码子配对快、分手也快。
基因结构复杂
基因转录调控方式复杂
真核基因的表达涉及多种RNA聚合酶。与原核生物只使用一种由多个蛋白聚合而成的RNA聚合酶不同,真核生物至少使用由8到12个蛋白组成的三种不同类型的RNA聚合酶。RNA 聚合酶I和III负责转录生成RNA分子,这些分子本身执行重要的功能,在所有的真核细胞中需要始终保持相当恒定的水平。RNA聚合酶II专门负责转录编码蛋白质的基因。 RNA聚合酶II识别的启动子序列的多样性反映了区别基因的复杂程度,即在特定类型的细胞中和在特定的时间,区别哪些基因该表达而哪些基因不该表达。
碱基G、C相对于A、T的丰度很早就被看作是区分细菌基因组的特征之一 .不同的原核生物中,GC含量(GC content)从25﹪到75﹪,变化非常大。 大部分细菌是通过从其它生物体大规模获得基因(长度为几万甚至几十万个核苷酸)而进化的(水平转移).简而言之,许多细菌基因组表现为具有不同GC含量的区域的组合物,这些区域反映了细菌的进化历史。
G
0.1751306272192
T
0.3248693727808
酵母基因组核苷酸出现频率
在统计过程中,如果同时计算DNA的正反两条链,则根据碱基配对原则,A和T、C和G的出现频率相同。如果仅统计一条链,则虽然A和T、C和G的出现频率不同,但是非常接近。
核苷酸
频率
A
0.344
C
0.155
G
等值区
定义:具有一致碱基组成的长区域 特征 :等值区基因组序列的长度超过1,000,000对碱基虽然不同的等值区其GC含量差别显著,但同一等值区的GC含量始终相对均衡 人类基因组大约可以划分为五个不同类型的等值区:a) L1和L2,平均GC含量分别为39﹪和42﹪(欠GC)) b) H1、H2和H3,GC含量平均值分别为46﹪、49﹪和54﹪ (丰GC)
科学家对这本天书了解最多的部分就是遗传密码 或者说掌握了DNA对蛋白质编码的规律 关于密码子(1)密码子的使用是非随机的 如果密码子的第一、第二位碱基是A、U, 那么第三位将尽可能使用G、C;反之亦然。 如果三位都用G、C,则配对容易,分解难; 三位都用A、U,则相反。 一般地说,高表达的基因,要求翻译速度快, 要求密码子和反密码子配对快、分手也快。
基因结构复杂
基因转录调控方式复杂
真核基因的表达涉及多种RNA聚合酶。与原核生物只使用一种由多个蛋白聚合而成的RNA聚合酶不同,真核生物至少使用由8到12个蛋白组成的三种不同类型的RNA聚合酶。RNA 聚合酶I和III负责转录生成RNA分子,这些分子本身执行重要的功能,在所有的真核细胞中需要始终保持相当恒定的水平。RNA聚合酶II专门负责转录编码蛋白质的基因。 RNA聚合酶II识别的启动子序列的多样性反映了区别基因的复杂程度,即在特定类型的细胞中和在特定的时间,区别哪些基因该表达而哪些基因不该表达。
基因组注释详解ppt课件

2000 2002 2003 2005 2006 2007 2008 2009
2010
Hale Waihona Puke In the coming future
Rise of Genbank databases from DNA sequencing
Human Genome Project & Celera
Genomics completes first draft genome
(17)Scenedesmus obliquus
(18)Thraustochytrium Mitochondrial
22
GETORF:Advanced Options
ii.最小的开放阅读框由多少个核甘酸组成,预设值为30,也就 是10个氨基酸。
iii.Type of output:可选择不同的输入结果,包含有: (1)Translation of regions between STOP codons (2)Translation of regions between START and
(1)Standard
(2)Standard (with alternative initiation codons)
(3)Vertebrate Mitochondrial
(4)Yeast Mitochondrial
(5)Mold, Protozoan, Coelenterate Mitochondrial and
FLX 500 Mb / run
Illumina / Solexa/GIIx Genetic Analyzer 50~95GB / run
Illumina / Solexa/HiSeq 200GB / run
基因组学ppt课件

编辑课件
6
染色体带型命名
人类染色体带型最早确定的命名方式是从着丝粒向两侧按数字编号, 短臂以p代表 (p=petit),长臂以q代表. 短臂和长臂又可进一步分区,每个区又分为数个亚区, 亚区又可划分为不同的区带,有的区带又可细分为区亚带。
编辑课件
7
人类染色体核型
编辑课件
8
四、基因组的结构成分
1) SAR和MAR 2) CpG岛 3) 等高线
5) MAR或SAR之间的距离平均为30 kb, 染色体DNA环突
长约25-600 kb, 因此并非所有MAR或SAR均与基质或
骨架结合. 或这说MAR(或SAR)与基质或骨架结合的
位置是动态的, 不固定的.
编辑课件
11
SAR和MAR的应用
由于发现许多功能基因的两侧含有SAR或 MAR的结构,并证实SAR和MAR具有阻止 异染色质位置效应和隔离相邻基因彼此干 扰的功能, 因此为了提高转基因的表达水平, 在构建表达载体时可在基因两侧安装SAR 或MAR顺序, 以减少转基因沉默效应.
编辑课件
12
MAR
的 分 离
编辑课件
13
(2)什么是CpG岛
满足CpG岛的条件为: 1. 连续200 bp的DNA顺序(已修改为500 bp); 2. C+G含量大于50%(已修改为55%); 3. 观测到的CpG双碱基数目与预期的数目
之比大于0.6(已修改为0.65).
(Gardiner-Garden, J.Mol.Bio., 196:261, 1987; Proc Natl Acad Sci USA 99:3740-3745, 2002 )
第6章 真核生物基因组解剖
编辑课件
植物基因组 ppt课件

25000 20000 15000 10000
5000 0
2000年-2014年测序植物基因组大小比较
/bbs/topic/30217258?from=recommend&mode=old
拟南芥 水稻 水稻 杨树 葡萄 衣藻
小立碗藓 番木瓜 百脉根
三角褐指藻 高粱 玉米 黄瓜 大豆
二穗短柄草 褐藻 团藻 蓖麻
小球藻 苹果
森林草莓 可可树
野生大豆 褐潮藻类
麻风树 卷柏
枣椰树 琴叶拟南芥
马铃薯 条叶蓝芥
白菜 印度大麻
木豆 蒺藜苜蓿
蓝载藻 谷子 谷子 番茄 甜瓜 亚麻 盐芥 香蕉
雷蒙德氏棉 大麦 梨 西瓜 甜橙 小麦
棉花(雷蒙德氏棉) 梅花
鹰嘴豆 橡胶树
毛竹 短花药野生稻
小麦A 小麦D
https:///piece/browse.php
2、植物基因组多倍体比较常见,而动物中极其罕见。
1、植物基因组往往比较大染色体数目变化范围大。
世界上现在公认的基因组最大的生物是Paris japonica,中文名 衣笠草,约150Gb,大约是人类(3.2Gb)的50倍,是具有40条 染色体的八倍体。已知的染色体数目最多的一个科叫 Ophioglossum,中文名瓶爾小草科,属于蕨类,该科物种最多 可有1260条染色体。
毛竹 短花药野生稻
小麦A 小麦D
桃树 丝叶狸藻
中国莲 挪威云杉 海洋球石藻
虫黄藻 油棕榈 枣椰树 醉蝶花
莲 桑树 猕猴桃 胡杨 八倍体草莓 康乃馨 甜菜 无油樟(互叶梅) 芝麻 辣椒 火炬松 棉花(亚洲棉) 萝卜 甘蓝 菜豆 野生大豆 普通小麦 野生西红柿 非洲野生稻 油菜 中果咖啡 茄子 多个野生大豆 绿豆 啤酒花 蝴蝶兰
《基因组学》PPT课件

第十章 基因组学 (Genomics)
ppt课件
1
Structural Genomics 结构基因组学 Functional Genomics 功能基因组学
Transcriptomics 转录物组学 Proteomics 蛋白质组学
ppt课件
2
第一节 真核生物基因组组成
Organization of Eukaryotic Genome
ppt课件
16
1. Genetic map 遗传图
The map in which mutant alleles or DNA markers are assigned relative positions along a chromosome on the basis of the recombination frequencies between them
Tetrahymena, GGGGTT Human, GGGATT Telomere-associated sequences: is repetitive and is found both adjacent to and within the telomere. The sequences vary among organisms.
ppt课件
5
Highly repetitive sequences 高度重复序列 5~300bp , 105 copies
Middle-repetitive sequences 中度重复序列 10~1000 copies
Unique sequences 单拷贝序列
ppt课件
6
▪ Gene family(基因家族): a set of genes in one genome all descended from the same ancestral gene.
ppt课件
1
Structural Genomics 结构基因组学 Functional Genomics 功能基因组学
Transcriptomics 转录物组学 Proteomics 蛋白质组学
ppt课件
2
第一节 真核生物基因组组成
Organization of Eukaryotic Genome
ppt课件
16
1. Genetic map 遗传图
The map in which mutant alleles or DNA markers are assigned relative positions along a chromosome on the basis of the recombination frequencies between them
Tetrahymena, GGGGTT Human, GGGATT Telomere-associated sequences: is repetitive and is found both adjacent to and within the telomere. The sequences vary among organisms.
ppt课件
5
Highly repetitive sequences 高度重复序列 5~300bp , 105 copies
Middle-repetitive sequences 中度重复序列 10~1000 copies
Unique sequences 单拷贝序列
ppt课件
6
▪ Gene family(基因家族): a set of genes in one genome all descended from the same ancestral gene.
微生物基因组学_PPT幻灯片

“双脱氧末端终止”的含义
Sanger 双脱氧末端终止法测序原理
自动化测序
荧光染料标记物的发明: 使链终止法用于自动化测序,用不同的荧光 色彩标记ddNTP,如ddATP标记红色荧光, ddCTP标记蓝色荧光,ddGTP标记黄色荧光, ddTTP标记绿色荧光。由于每种ddNTP带有 各自特定的荧光颜色,而简化为由1个泳道同 时判读4种碱基。
(四)影响测序的因素 不管采用随机测序还是定向测序都可碰到下列影响因素。 1.计算机的设备 。 2.靶DNA的性质。 3.完成测序所需的时间 。 4.采用测序策略。
三.微生物基因组的注释 (一)概念:在微生物基因测序的基础上,对其基本结 构和部件进行认定,以进一步研究其功能。
(二)微生物基因组注释的内容 1.碱基组成分析,即G+C Mol%测定。 G+C含量是物种的一个重要特征,在微生物的分类上具有重要意义,是重 要参数之一。 2.开放阅读框的鉴定: 3.编码序列分析
向测序法包括引物测序法和定向缺失测序法。 ⑴引物测序法 即在第一次测序结果的基础上,设计新的寡核苷酸,来充当下一次测
序反应的引物,并依次类推,从而循序渐进获得靶DNA的全部序列。
⑵定向缺失法 定向缺失法是将一个靶DNA变成若干套嵌套的缺失突变体,使靶序列中远
不可测的区段逐渐落入可用通用引物进行测序的方法。
黏粒载体( cosmid )
P1人工染色体载体(PAC)
目前常用的人造染色体载体
23
YAC载体应含有下列元件:
酵母染色体的端粒1 EcoRI CEN4
酵母染色体的着丝粒序列 Apr
pYAC
URA3
4
酵母系统的选择标记
ori
大肠杆菌的复制子标记
基因组注释ppt课件

基因注释软件
1)目前基因注释程序的编写主要依据两种信息内涵:
1.signal terms (信号指令), 如起始密码, 终止密码, 终止信号, 剪接受体位与供体位序列, 多聚嘧啶顺序, 分支点等保守的顺序组成; 2.content terms (内容指令), 如密码子使用偏好.
对结构紧凑的小基因组上述注释软件效果不错,但对大基 因组特别是超长基因的注释有很大困难.在一个长度数十 或数百kb的内含子中, 存在许多可能误判的信号指令. 2) 常 用 的 注 释 软 如 GenScan 主 要 偏 重 于 内 容 指 令 , 而 FgeneSH则着重于信号指令.由于每种生物都有种属专一 性的密码子偏好,也存在某些非保守的信号指令, 因此在 超长基因注释中常出现正向错误(false-positive, 多注 释)或负向错误(false-negetive, 少注释).
3) EBI: 27 462 (2003, nature 423:576) 4) Genscan: 65 452 许多人倾向于不可能知道人类基因组精确的基因数.
几种模式生物注释的基因总数
大肠杆菌(E.coli): 4 800 酵母(yeast): 6 200 线虫(nematode): 19 000 果蝇(fly): 13 600 拟南芥(Arabidopsis): 25 000 水稻(rice): 60 000 玉米(maize): 59 000 老鼠(mouse): 30 000
76??typesdnachipstypesdnachipsexpressionchipsgenomicchipssequencingchipsdnachips77?基因芯片研制的总体蓝图研制方向的确定基因组序列分析与待检基因探针序列的确定检测样品的制备探针阵列的准备检测设备的研制杂交检测与数据分析78?表达芯片的制备检测流程79表达芯片胞cdna未处理的细胞cdna杂交杂交激光共聚焦扫描发现17个差异表达基因11个被热诱导6个被热抑制发现其中3个为未发现的新基因80蛋白质组定义
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
相似性与一致性
249 MFN-MAI PFGAGAYAQALNQQQAALMASVAQGG 232 I LTSL TLPFS AGAYAQALNQQQTTV I S - -T S GS 注: 红色为一致性氨基酸, 蓝色为可取代氨基酸, 白色为趋
异氨基酸. 一致性氨基酸百分比为红色氨基酸所占的 比例, 相似性氨基酸百分比为红色和蓝色氨基酸相加 所占的比例.
GCA,GCC或GCT,而GCG很少使用。 特定种属有特征性的密码子偏爱,这些序列在 编码区常常出现,非编码区只保持平均的碱基 分布水平。
2)外显子-内含子边界
外显子和内含子的边界有一些明显的特征如: 内含子的5‘端或称供体位(donor site)常见的顺序为 5’-AG↓GTTAAGT-3’; 3’端又称受体位(acceptor site), 多为 5‘PyPyPyPyPyPyCAG-3’(“Py”嘧啶核苷酸,T或C);
达基因产物。
5.1.1 根据基因结构特征搜寻基因
基因不是核苷酸的随机排列而是具有明显特征: 基因的编码区是可读框。
可能的六种ORF
1. 根据开放读码框预测基因
a. 起始密码子ATG: 第一个ATG的确定则依据Kozak规则: Kozak规则是基于已知数据的统计结果, 所谓Kozak规则,即第一个ATG侧翼序列的 碱基分布所满足的统计规律。
内含子使ORF扫描复杂化
内含子的出现给计算机判读基因带来不少问题, 对ORF扫描的基本程序的编写要考虑以下几个问 题:
1)密码子偏倚; 2)外显子—内含子边界; 3)上游调控序列。
1)密码子偏爱性
编码同一氨基酸的不同密码子称为同义密码, 其差别仅在密码子的第3位碱基不同。 不同种属间使用同义密码的频率有很大差异, 如人类基因中,丙氨酸(Ale)密码子多为
终止密码子: TAA, TAG,TGA GC% = 50% 终止密码子每 64 bp出现一次; GC% > 50% 终止密码子每100-200 bp 出现 一次;
由于多数基因 ORF 均多于50个密码子,因此 最可能的选择应该是 ORF 不少于100 个密码子。
细菌基因组的ORF阅读DNA效果不佳。
5.1.2 同源基因查询
通过已存入数据库中的基因序列与待查的 基因组序列进行比较,从中查找可与之匹 配的碱基序列及其比例,用于界定基因的 方法称为同源查询。
同源有如下几种情况:
A. DNA序列某些片段完全相同; B. 开放读码框排列类似,如有等长外显子; C. 开放读码框翻译成的氨基酸序列的相同; D. 模拟多肽高级结构相似。
基因注释软件
1)目前基因注释程序的编写主要依据两种信息内涵:
1.signal terms (信号指令), 如起始密码, 终止密码, 终止信号, 剪接受体位与供体位序列, 多聚嘧啶顺序, 分支点等保守的顺序组成; 2.content terms (内容指令), 如密码子使用偏好.
对结构紧凑的小基因组上述注释软件效果不错,但对大基 因组特别是超长基因的注释有很大困难.在一个长度数十 或数百kb的内含子中, 存在许多可能误判的信号指令. 2) 常 用 的 注 释 软 如 GenScan 主 要 偏 重 于 内 容 指 令 , 而 FgeneSH则着重于信号指令.由于每种生物都有种属专一 性的密码子偏好,也存在某些非保守的信号指令, 因此在 超长基因注释中常出现正向错误(false-positive, 多注 释)或负向错误(false-negetive, 少注释).
引自: Nature reviews genetics, 4:741-749,2003.
不同注释软件之间的效率
Performance of three popular gene prediction programs on 42 semiartificial genomic sequences containing 178 known human gene sequences (900 exons). Sensitivity is percentage of exons that are predicted correctly. Selectivity is percentage of predicted exons that are correct. Reproduced with changes from Yada et al., 2002 Cold Spring Harbor Genome Sequencing and Biology Meeting, May 7-11, 2002. FGENESH is by far the most accurate of three programs.
同源查询
当在氨基酸水平进行比较时,两个序列之间缺少同源性 就更明显。
同源性,一致性和相似性
1) 同源性(homology)基因系指起源于同一祖先但序列已经 发生变异的基因成员。 分布在不同物种间的同源基因又称直向同源基因。 同一物种的同源基因则称共生同源基因(水平基因), 水平基因由重复后趋异产生。
基因同源性只有“是”和“非”的区别, 无所谓百 分比.
2) 一致性(identity):指同源DNA顺序的同一碱 基位置的相同的碱基成员, 或者蛋白质的同一 氨基酸位置的相同的氨基酸成员, 可用百分比 表示.
3) 相似性(similarity):指同源蛋白质的氨基酸 序列中一致性氨基酸和可取代氨基酸所占的 比例。可取代氨基酸系指具有相同性质如极 性氨基酸或非极性氨基酸的成员, 它们之间的 代换不影响蛋白质(或酶)的生物学功能。
若将第一个ATG中的碱基A,T,G分别标为1, 2,3位,则Kozak规则可描述如下: (1) 第4位的偏好碱基为G; (2) ATG的5’端约15bp范围的侧翼序列内不含
碱基T; (3) 在-3,-6和-9位置,G是偏好碱基; (4) 除-3,-6和-9位,在整个侧翼序列区,C是
偏好碱基。
b. 终止密码子
上游外显子-内含 子边界的共有序列 在真正基因中发现 的真实序列之间的 关系。
3)上游控制顺序
几乎所有基因(或操纵子)上游都有调控序 列,它们可与DNA结合蛋白作用,控制基因 表达。
另外个别生物的基因组特有组成也可作为判 别依据,如脊椎动物基因组许多基因的上游 都有CpG岛。 大多数CpG岛都位于管家基因和大部分组织 专一性表达基因的5’侧翼区以及基因的第一 个外显子区。
第5章 基因组序列注释
学习重点:
1) 基因注释的方法 2) 基因功能的研究方法
基因组序列所包含的全部遗传信息是什么? 基因组作为一个整体如何行使其功能? 用什么方法寻找基因? 用什么方法研究基因的功能?
计算机分析+实验
5.1 寻找基因
基因组序列 查找基因。有两种常见的方法: 1. 计算机分析寻找与基因有关的序列。 2. 通过对DNA序列进行实验分析,看其能否表