实验2 序列比对
基因组测序中的序列比对使用教程

基因组测序中的序列比对使用教程序列比对在基因组测序中扮演着重要的角色,它是将测序得到的短序列与已知基因组进行比对,以确定这些短序列在基因组中的位置和功能。
本文将为您提供一份基因组测序中序列比对的详细使用教程。
一、理解序列比对的基本概念序列比对的基本概念是将测序得到的短序列与已知基因组进行匹配。
测序通常会产生大量的短序列,这些短序列需要通过比对才能确定其在基因组中的位置和功能。
在序列比对中,通常会引入一个参考基因组,该参考基因组是一个已知的基因组序列,可以是某个物种的基因组或某个特定区域的基因组。
二、选择合适的序列比对工具选择合适的序列比对工具对于准确地比对测序数据非常重要。
常见的序列比对工具包括Bowtie、BWA、BLAST等。
以下是这些工具的简介:1. Bowtie:Bowtie是一款非常快速的短序列比对工具,适合于比对长度较短的序列。
2. BWA:BWA适用于比对长度较长的序列,比如全基因组测序。
3. BLAST:BLAST是一款广泛应用于序列比对的工具,可以根据序列的相似性进行比对。
根据实际需求和数据类型选择合适的比对工具,以确保比对的准确性和效率。
三、准备比对所需的参考基因组和测序数据在进行序列比对之前,需要准备比对所需的参考基因组和测序数据。
参考基因组可以从公共数据库(如NCBI)下载,也可以使用自己的实验室已有的基因组数据。
测序数据通常是以FASTQ文件格式存储的,包括了测序reads的序列和对应的质量分数。
在比对之前,需要先将FASTQ文件进行质量控制和预处理,例如使用Trimmomatic工具去除低质量reads和适配体序列。
四、进行序列比对选择合适的比对工具后,可以开始进行序列比对。
以下是比对的一般流程:1. 将参考基因组索引化:大部分比对工具都需要将参考基因组进行索引化,以加快比对速度。
通过运行工具提供的索引化命令将参考基因组转换为索引文件。
2. 进行比对:根据选择的比对工具和参数设置,将准备好的测序数据与参考基因组进行比对。
生物信息学-序列比对-实验报告

姓名
学号
专业年级
基础学院生物信息学教研室
题目
序列比对
日期
实验者
一、实验目的
掌握BLAST 2的使用和功能
了解点阵法进行双序列比对的优点
二、实验器材
电脑
三、方法与步骤
见下文
四、结果与讨论
1,例题中其它的最佳比对结果
2,用动态规划法找出两序列的所有最佳比对,要求写出详细过程。打分矩阵采用{(4,-3,-4},即匹配得4分,不匹配得-3分,空位得-4分。序列1:AAAG,序列2:ACG。
61 GCCCCGGCTCAGGGCCAAGAACAGATGAGACAGCTGAGTGATGGGCCAAACAGGATATCT
121 GTGGTAAGCAGTTCCTGCCCCGGCTCGGGGCCAAGAACAGATGGTCCCCAGATGCGGTCC
序列2与序列3比对:两者为反向互补序列,可以发现可能的发夹状结构。
(1)给动态规划矩阵赋初值
0
A
A
A
Gபைடு நூலகம்
0
0
-4
-8
-12
-16
A
-4
C
-8
G
-12
(2)按照最优分的递归算法填充动态规划矩阵
0
A
A
A
G
0
0
-4
-8
-12
-16
A
-4
4
0
-4
-8
C
-8
0
1
-3
-7
G
-12
-4
-3
2
1
(3)从最后一个单元格开始,回溯最优化比对路径
生物信息学中的序列比对算法使用方法解析

生物信息学中的序列比对算法使用方法解析序列比对在生物信息学中是一项重要的技术,用于寻找DNA、RNA或蛋白质序列之间的相似性和差异性。
它是理解生物学结构和功能的基石之一。
在本文中,我们将解析生物信息学中常用的序列比对算法的使用方法。
序列比对算法主要分为全局比对和局部比对。
全局比对用于比较完整的序列,而局部比对则更适用于在序列中查找相似区域。
在这两个主要类别中,有几种经典的序列比对算法,包括Pairwise Sequence Alignment、BLAST、Smith-Waterman算法和Needleman-Wunsch算法等。
首先,我们来看Pairwise Sequence Alignment(两两序列比对)算法。
这个算法是基本的序列比对方法,通过比较两个序列中的每一个碱基、氨基酸或核苷酸,并根据其相似性和差异性对它们进行排列。
Pairwise Sequence Alignment算法使用动态规划的思想,通过计算匹配、替代和插入/删除的分数,来确定两个序列的最佳匹配方案。
在生物信息学中,常用的实现包括Needleman-Wunsch算法和Smith-Waterman算法。
Needleman-Wunsch算法是一种全局比对算法,用于比较两个序列的整个长度。
它是通过填充一个二维矩阵来计算最佳匹配路径的。
算法的核心思想是,通过评估每个格子的分数,根据路径选择的最佳分数进行全局比对。
这个算法不仅可以计算序列的相似性,还可以计算每个位置的分数,从而获得两个序列的对应二面的对应关系。
Smith-Waterman算法是一种局部比对算法,用于寻找两个序列中的最佳匹配片段(子序列)。
它与Needleman-Wunsch算法的计算思路相同,但不同之处在于允许负分数,这使得算法能够确定具有高分数的局部匹配片段。
通过动态规划计算,Smith-Waterman算法可以寻找到两个序列中的相似片段,并生成比对的结果。
另一种常用的序列比对算法是基本本地搜索工具(BLAST)。
生物信息学中的序列比对与分析教程

生物信息学中的序列比对与分析教程序列比对与分析在生物信息学中扮演着非常重要的角色。
通过对不同生物体的DNA、RNA或蛋白质序列进行比较和分析,我们可以揭示它们之间的相似性和差异性,从而推断它们的功能和进化关系。
本教程将介绍序列比对的基本概念、工具和方法,并探讨如何进行常见的序列分析。
1. 序列比对的基本概念序列比对是用于比较两个或多个生物序列之间的相似性和差异性的过程。
在序列比对中,我们会使用特定的算法和方法,将不同序列中的相似区域进行匹配,以找到它们之间的共同点。
常用的序列比对算法包括全局比对(如Needleman-Wunsch算法)和局部比对(如 Smith-Waterman算法)等。
2. 序列比对的工具现在有许多序列比对工具可供选择,其中一些是免费提供的。
其中最常用的工具之一是BLAST(Basic LocalAlignment Search Tool)。
BLAST可以快速找到一个或多个与给定序列相似的其他序列,并给出相似性得分。
除了BLAST,还有一些其他的序列比对工具,比如ClustalW、MUSCLE和T-Coffee等。
3. DNA序列比对DNA序列比对是研究生物体间遗传关系和进化关系的重要工具。
DNA序列之间的相似性可以用来确定物种的亲缘关系、寻找共同的进化起源以及研究基因的功能。
在DNA序列比对中,常用的方法是使用BLAST等工具,通过将查询序列与数据库中的已知序列进行比对来找到相似的区域。
4. RNA序列比对RNA序列比对主要用于研究基因表达和功能相关的RNA分子。
与DNA序列比对相似,RNA序列比对也可以通过BLAST等工具进行。
此外,对于非编码RNA序列的比对,可以使用RAPSearch和PIRCH等专门的工具。
5. 蛋白序列比对蛋白序列比对是分析蛋白质结构和功能的关键步骤。
蛋白质序列比对可以通过BLAST等工具进行,还可以使用更高级的算法和方法,如Smith-Waterman算法和多序列比对算法,来找到更为精确的比对结果。
生物信息学中的序列比对算法综述

生物信息学中的序列比对算法综述序列比对(sequence alignment)是生物信息学中一项重要的任务,其目的是找出两个或多个生物序列中的相似性和差异性。
在生物信息的研究和应用中,序列比对算法起到了至关重要的作用。
本文将对生物信息学中的序列比对算法进行综述。
1. 引言序列比对是生物信息学中的一个基本问题,它在基因组学、蛋白质学、进化生物学等领域都得到了广泛的应用。
通过比对不同生物序列之间的相似性和差异性,可以进一步研究基因功能、蛋白质结构以及物种进化等重要问题。
因此,序列比对算法的研究具有重要的理论价值和实际意义。
2. 序列比对的基本概念在进行序列比对之前,首先需要了解序列之间的相似性和差异性的度量方法。
常用的序列相似性度量方法包括编辑距离、相似度百分比、贝叶斯统计等。
其中,编辑距离是一种常见的度量方式,它衡量了两个序列之间的差异程度。
3. 序列比对算法分类序列比对算法可以分为全局比对和局部比对两类。
全局比对算法着重于找出整个序列的相似性和差异性,常用的算法包括Needleman-Wunsch算法和Smith-Waterman算法。
而局部比对算法则注重于找出序列中的局部相似性和差异性,常用的算法有BLAST和FASTA。
4. 全局比对算法全局比对算法的核心思想是将两个序列通过插入、删除和替换等操作转化为相同长度的序列,然后计算它们的相似性得分。
Needleman-Wunsch算法是一种经典的全局比对算法,通过动态规划的方式找到序列之间的最佳比对方式。
Smith-Waterman算法是基于Needleman-Wunsch算法的改进,它将负得分和局部比对引入到全局比对中,提高了比对的准确性。
5. 局部比对算法局部比对算法主要用于序列中的片段比对,其核心思想是通过寻找序列中的相似片段来找出序列的结构和功能区域。
BLAST算法是一种常用的局部比对算法,它通过生成字典和索引的方式实现快速比对。
FASTA算法则是一种早期的局部比对算法,其基本原理是通过序列片段之间的kmer匹配来寻找相似性。
实验2 序列比对

实验二:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析。
实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlign用dotplot方法能够直观地认识两条序列比对,但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍由DNASTAR公司开发的一个比较全面的生物信息学软件包--Lasergene,它包含了7个模块,其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 2.3),选择完毕点击Done回到程序页面。
实验二 双序列比对分析

实验三双序列比对分析一.实验目的Tay-Sachs是一种常染色体隐性遗传疾病,它的起因是第15号染色体的等位基因HEXA突变。
人类的HEXA基因在GenBank中的编号为“NM_000520”,小鼠的HEXA 基因在GenBank中的编号为“AK080777”,它们是核苷酸序列,以这两条序列为例,学习双序列比对分析。
1.学习和掌握在MATLAB平台上应用Bioinformatics工具包有关核苷酸和蛋白质双序列比对的命令和功能。
2.学习和掌握在MATLAB平台上应用Bioinformatics工具包访问GenBank,并提取核苷酸和蛋白质序列数据的方法。
3.学习和掌握在MATLAB平台上应用Bioinformatics工具包制作核苷酸或蛋白质两条序列比对的点阵图的方法。
4.学习和掌握在MATLAB平台上应用Bioinformatics工具包进行核苷酸或蛋白质双序列的局部比对和全局比对的方法。
二.实验内容1.在MATLAB平台上应用Bioinformatics工具包访问GenBank,提取核苷酸序列并转换为蛋白质序列。
①用“web”命令在MATLAB平台上打开NCBI网页。
web('/')web('/books/bv.fcgi?call=bv.View..ShowSection&rid=gnd')②用“getgenbank”功能从GenBank中读序列信息到MARLABhumanHEXA = getgenbank('NM_000520')mouseHEXA = getgenbank('AK080777')在MATLAB的workshop打开humanHEXA 和mouseHEXA查看其内容。
③从GenBank中提取2条核苷酸序列后,首先要做的是用全局比对来寻找两条序列中的相似序列。
因为进行蛋白质序列的比对更能体现其生物学本质,所以常常进行蛋白质序列的比对。
实验二_数据库相似性搜索与序列比对

实验二_数据库相似性搜索与序列比对实验二数据库相似性搜索与序列比对实验原理:数据库相似性搜索以两两序列比对为基础,将感兴趣的基因序列与序列数据库中的每个序列进行比较,鉴别出相似的序列。
搜索结果显示出与最佳匹配序列的对位排列及匹配记分。
序列数据库搜索对发现基因的功能非常有效。
fasta和blast是两个著名的用于数据库相似性搜索的软件包。
其中blast(basiclocala1ignmentsearchtool)基于局部比对的搜索工具,是一种启发式搜索算法服务软件,包括blastp,blastn,blastx,tblastn 和tblastx程序。
实验目的和要求:学习数据库相似性检索和序列比对的程序的使用,能够理解程序给出的结果,从中获取有关功能和结构的信息。
(1)要求学生使用所学的数据库检索方法检索数据库中的特定基因(2)掌握数据库相似性搜索工具blast的基本比对方法,参数设置及结果分析(3)掌握核酸和蛋白质序列的比对方法、参数设置和结果分析实验材料:未知核酸序列;未知氨基酸序列;SOD基因工具软件:(1)数据库检索工具Entrez一、利用blast中的special类下的aligntwosequences(bl2seq)比较人与老鼠的sod基因蛋白质序列的相似性程度(1)人类aab27818是通过NCBI 1的ntrez和小鼠3gtt_E的SOD基因氨基酸序列或登录号(SOD分为SOD1或SOD2等,检索时注意选择完全相同的SOD基因)搜索蛋白质数据库获得的。
(2)进入NCBI的blast网页,然后选择specializedlast下的align two sequences(bl2seq)程序来比较这两个序列(3)选择blastp子程序,将序列或登录号分别粘贴到序列框中(4)其他选项采用默认的设置,运行程序(5)分析结果,并回答以下问题NCBI的Entrez搜索中使用了哪些关键词?humanandsodmouseandsod人和小鼠SOD基因蛋白质序列的注册号是多少?人aab27818 1和鼠标3gtt_e两序列比对得到的一致性百分比和相似性百分比分别为多少?识别127/153(83%)阳性135/153(88%)两序列比对结果中哪些区域出现了gap?差距0/153(0%)二、利用specielizedblast的conserveddomain进行蛋白质保守结构域分析(1)进入ncbi的blast网页(2)选择specialize last to enter下的保守域超链接(3)在cazy数据库查找一个糖苷水解酶glycosidehydrolases(gh+学号),获得其蛋白质序列或蛋白质序列的genbank登录号aek59386.1(4)在保守域页面的输入框中输入糖苷水解酶的登录号或蛋白质(5),选择默认参数,点击提交进行提交分析(6)阅读得到的结果,点击各hit的超链接了解找到的结构域的功能(7)将结构域图形和表格记录在实验报告中三、利用blast在数据库中搜索不同物种的同源基因(1)利用文献检索工具检索clostridiumthermocellum嗜热梭菌与其纤维素降解功能相关的基因,例如糖苷水解酶glycosidehydrolases(gh+学号)或多糖裂解酶polysaccharidelyases(pls)或碳水化合物酯酶carbohydrateesterases(ces)等(2)利用ncbi的entrez检索该基因获得其核酸序列ab125373或者使用(2)中的蛋白质注册号通过NCBI数据库中的相关信息链接到核酸数据库,以获得基因的核酸注册号或序列(3)利用blastn进行数据库相似性搜索搜索其他微生物中的同源基因(4)分析blast结果,并回答以下问题检索获得基因名称是?chi19-1该基因的登录号是多少?ab125373进行blastn搜索的数据库选项为?nr请列出其他3-5种具有该基因及其同源基因的微生物的注册号?ap009493.1。
序列比对——生物信息学实习报告

序列⽐对——⽣物信息学实习报告实习⼆:序列⽐对学号20090***** 姓名**** 专业年级⽣技技术****实验时间2012.6.13 提交报告时间2012.6.14实验⽬的:1.学会使⽤EMBOSS软件包的NEEDLE和WATER进⾏两条序列⽐对2.学会使⽤MegAlign进⾏两条和多条序列⽐对3.学会使⽤ClustalX和MUSCLE进⾏多条序列⽐对分析实验内容:1.两条序列⽐对EMBOSS全称是The European Biology Open Software Suite,是⼀个开放源代码的分⼦⽣物学分析软件包。
本次实习利⽤分别利⽤全局⽐对软件Needle和局部⽐对软件Water的在线版本进⾏。
1.1动态规划算法全局⽐对(Needle):在线软件⽹址为/doc/cb8211483.html/Tools/psa,利⽤⽕狐浏览器进⼊,选择Needle⼯具对⾃⼰选择的序列进⾏序列全局⽐对。
1.2动态规划算法局部⽐对(Water):在线软件⽹址为/doc/cb8211483.html/Tools/psa,利⽤⽕狐浏览器进⼊,选择Water⼯具对⾃⼰选择的序列进⾏序列局部⽐对,并和Needle⽐对结果进⾏⽐较,分析差异产⽣的原因。
2.利⽤MegAlign软件分别对核酸序列和蛋⽩质序列进⾏两条和多条序列⽐对,并分析⽐对结果。
3.利⽤Clustalx软件分别对核酸序列和蛋⽩质序列进⾏多条序列⽐对,并分析⽐对结果。
4.利⽤MUSCLE在线⼯具分别对核酸序列和蛋⽩质序列进⾏多条序列⽐对,并分析⽐对结果。
5.⽐较MegAlign,Clustalx,MUSCLE⽐对结果的异同。
作业:1.从上节课搜索到的同源核酸和蛋⽩质序列中各任意选两条,分别使⽤Needle和water进⾏⽐对,分析对⽐结果是否存在差异,为什么?答:选⽤核酸序列为NM_002964.4和XM_001137986.1;选⽤蛋⽩质序列为:XP_001110530.1和NP_001139616.1。
第2讲-序列比对

T -5 1 -1 -5
C -5 -1 1 -5
G -1 -5 -5 1
AGTCGA
?
AATCGT
-2
21/ 77
2、蛋白质打分矩阵
• (i)等价矩阵
1 i j Rij 0 i j
• • • •
其中Rij代表打分矩阵元素 i、j分别代表字母表第i和第j个字符。
(ii) 遗传密码矩阵GCM (iii)疏水矩阵 (iv)PAM矩阵(Point Accepted Mutation) (v) BLOSUM矩阵 (Blocks Amino Acid Substitution Matrices)
两序列有90%的相似性 两序列有90%的同源性
6/ 77
• 直系同源(orthology):不同物种内的同源序列。
• 旁系同源(paralogy):同一物种内的同源序列。
7/ 77
人类与模式生物——小鼠
因为他们各自的 kit基因都存在缺陷
8/ 77
2、序列比对的概念
基本概念:
• 序列:由一些字母组成的字符串,包括核酸和蛋白质序列。
26/ 77
c s t p
27/ 77
针对不同的进化距离采用不同的PAM 矩阵
序列相似度 = 40% | 打分矩阵 = PAM120
50% | PAM80
60% | PAM 60
PAM250
→ 14% - 27%
28/ 77
模块氨基酸替换矩阵
BLOSUM 62
29/ 77
BLOSUM90
BLOSUM80
AGTCGATAGTCGAT AGT---TAGTCGAT
AGTCGATAGTCGAT A-TCGAT-GTC-AT
05_双序列比对

• Successive shifts would be represented as adjacent diagonal lines:
简单的序列比对
• 将两条序列左端对齐,放在两行中。 • 如果某列中两条序列的字符相同,则用竖线(|)将
它们连起来。 • 这是最佳比对结果吗?
是否有更好的比对结果?
• 如果我们仅仅将它们相对移动两个位置, 就可得到更好的比对结果:
结论
• We can not simply put both sequences one alongside the other, but we must compare them in all possible shiftings looking for the best alignment.
1. 为什么要进行序列比对? 2. 序列比对相关术语 3. 最佳比对 4. 利用计算机进行序列比对 5. 序列的点阵作图比对 6. 双序列比对工具
5.1 点阵作图中的一些特征图形
• It is easier to visualize how this works if you imagine a two dimensional chart, where you compare each residue on one of the sequences against every other in the other sequence:
Is it the end of the story?
生物采用多种机制产生变化
实验二 核酸序列分析

实验二核酸序列分析【实验目的】1、掌握已知或未知序列接受号的核酸序列检索的基本步骤;2、掌握使用BioEdit软件进行核酸序列的基本分析;1、熟悉基于核酸序列比对分析的真核基因结构分析(内含子/外显子分析);2、了解基因的电子表达谱分析。
【实验原理】针对核酸序列的分析就是在核酸序列中寻找基因,找出基因的位置和功能位点的位置,以及标记已知的序列模式等过程。
在此过程中,确认一段DNA序列是一个基因需要有多个证据的支持。
一般而言,在重复片段频繁出现的区域里,基因编码区和调控区不太可能出现;如果某段DNA片段的假想产物与某个已知的蛋白质或其它基因的产物具有较高序列相似性的话,那么这个DNA片段就非常可能属于外显子片段;在一段DNA序列上出现统计上的规律性,即所谓的“密码子偏好性”,也是说明这段DNA是蛋白质编码区的有力证据;其它的证据包括与“模板”序列的模式相匹配、简单序列模式如TATA Box等相匹配等。
一般而言,确定基因的位置和结构需要多个方法综合运用,而且需要遵循一定的规则:对于真核生物序列,在进行预测之前先要进行重复序列分析,把重复序列标记出来并除去;选用预测程序时要注意程序的物种特异性;要弄清程序适用的是基因组序列还是cDNA序列;很多程序对序列长度也有要求,有的程序只适用于长序列,而对EST这类残缺的序列则不适用。
1. 重复序列分析对于真核生物的核酸序列而言,在进行基因辨识之前都应该把简单的大量的重复序列标记出来并除去,因为很多情况下重复序列会对预测程序产生很大的扰乱,尤其是涉及数据库搜索的程序。
2. 数据库搜索把未知核酸序列作为查询序列,在数据库里搜索与之相似的已有序列是序列分析预测的有效手段。
在理论课中已经专门介绍了序列比对和搜索的原理和技术。
但值得注意的是,由相似性分析作出的结论可能导致错误的流传;有一定比例的序列很难在数据库里找到合适的同源伙伴。
对于EST序列而言,序列搜索将是非常有效的预测手段。
生物信息学实验报告

生物信息学实验报告班级::学号:日期:实验一核酸和蛋白质序列数据的使用实验目的了解常用的序列数据库,掌握基本的序列数据信息的查询方法。
教学基本要求了解和熟悉NCBI 核酸和蛋白质序列数据库,可以使用BLAST进行序列搜索,解读BLAST 搜索结果,可以利用PHI-BLAST 等工具进行蛋白质序列的结构域搜索,解读蛋白质序列信息,可以在蛋白质三维数据库中查询相关结构信息并进行显示。
实验容提要在序列数据库中查找某条基因序列(BRCA1),通过相关一系列数据库的搜索、比对与结果解释,回答以下问题:1. 该基因的基本功能?2. 编码的蛋白质序列是怎样的?3. 该蛋白质有没有保守的功能结构域 (NCBI CD-search)?4. 该蛋白质的功能是怎样的?5. 该蛋白质的三级结构是什么?如果没有的话,和它最相似的同源物的结构是什么样子的?给出示意图。
实验结果及结论1. 该基因的基本功能?This gene encodes a nuclear phosphoprotein that plays a role in maintaining genomic stability, and it also acts as a tumor suppressor. The encoded protein combines with other tumor suppressors, DNA damagesensors, and signal transducers to form a large multi-subunit protein complex known as the BRCA1-associated genome surveillance complex (BASC). This gene product associates with RNA polymerase II, and through the C-terminal domain, also interacts with histone deacetylase complexes. This protein thus plays a role in transcription, DNA repair of double-stranded breaks, and recombination. Mutations in this gene are responsible for approximately 40% of inherited breast cancers and more than 80% of inherited breast and ovarian cancers. Alternative splicing plays a role in modulating the subcellular localization and physiological function of this gene. Many alternatively spliced transcript variants, some of which are disease-associated mutations, have been described for this gene, but the full-length natures of only some of these variants has been described. A related pseudogene, which is also located on chromosome 17, has been identified. [provided by RefSeq, May 2009]2. 编码的蛋白质序列是怎样的?[Homo sapiens]1 mdlsalrvee vqnvinamqk ilecpiclel ikepvstkcd hifckfcmlk llnqkkgpsq61 cplcknditk rslqestrfs qlveellkii cafqldtgle yansynfakk ennspehlkd121 evsiiqsmgy rnrakrllqs epenpslqet slsvqlsnlg tvrtlrtkqr iqpqktsvyi181 elgsdssedt vnkatycsvg dqellqitpq gtrdeislds akkaacefse tdvtntehhq241 psnndlntte kraaerhpek yqgssvsnlh vepcgtntha sslqhenssl lltkdrmnve301 kaefcnkskq pglarsqhnr wagsketcnd rrtpstekkv dlnadplcer kewnkqklpc361 senprdtedv pwitlnssiq kvnewfsrsd ellgsddshd gesesnakva dvldvlnevd421 eysgssekid llasdpheal ickservhsk svesniedki fgktyrkkas lpnlshvten481 liigafvtep qiiqerpltn klkrkrrpts glhpedfikk adlavqktpe minqgtnqte541 qngqvmnitn sghenktkgd siqneknpnp ieslekesaf ktkaepisss isnmelelni601 hnskapkknr lrrksstrhi halelvvsrn lsppnctelq idscssseei kkkkynqmpv661 rhsrnlqlme gkepatgakk snkpneqtsk rhdsdtfpel kltnapgsft kcsntselke721 fvnpslpree keekletvkv snnaedpkdl mlsgervlqt ersvesssis lvpgtdygtq781 esisllevst lgkaktepnk cvsqcaafen pkglihgcsk dnrndtegfk yplghevnhs 841 retsiemees eldaqylqnt fkvskrqsfa pfsnpgnaee ecatfsahsg slkkqspkvt 901 feceqkeenq gknesnikpv qtvnitagfp vvgqkdkpvd nakcsikggs rfclssqfrg 961 netglitpnk hgllqnpyri pplfpiksfv ktkckknlle enfeehsmsp eremgnenip 1021 stvstisrnn irenvfkeas ssninevgss tnevgssine igssdeniqa elgrnrgpkl 1081 namlrlgvlq pevykqslpg snckhpeikk qeyeevvqtv ntdfspylis dnleqpmgss 1141 hasqvcsetp ddllddgeik edtsfaendi kessavfsks vqkgelsrsp spfththlaq 1201 gyrrgakkle sseenlssed eelpcfqhll fgkvnnipsq strhstvate clsknteenl 1261 lslknslndc snqvilakas qehhlseetk csaslfssqc seledltant ntqdpfligs 1321 skqmrhqses qgvglsdkel vsddeergtg leennqeeqs mdsnlgeaas gcesetsvse 1381 dcsglssqsd ilttqqrdtm qhnliklqqe maeleavleq hgsqpsnsyp siisdssale 1441 dlrnpeqsts ekavltsqks seypisqnpe glsadkfevs adsstsknke pgversspsk 1501 cpslddrwym hscsgslqnr nypsqeelik vvdveeqqle esgphdltet sylprqdleg 1561 tpylesgisl fsddpesdps edrapesarv gnipsstsal kvpqlkvaes aqspaaahtt 1621 dtagynamee svsrekpelt astervnkrm smvvsgltpe efmlvykfar khhitltnli 1681 teetthvvmk tdaefvcert lkyflgiagg kwvvsyfwvt qsikerkmln ehdfevrgdv 1741 vngrnhqgpk raresqdrki frgleiccyg pftnmptdql ewmvqlcgas vvkelssftl 1801 gtgvhpivvv qpdawtedng fhaigqmcea pvvtrewvld svalyqcqel dtylipqiph 1861 shy3. 该蛋白质有没有保守的功能结构域 (NCBI CD-search)?有保守的供能结构域。
序列比对,构建进化树
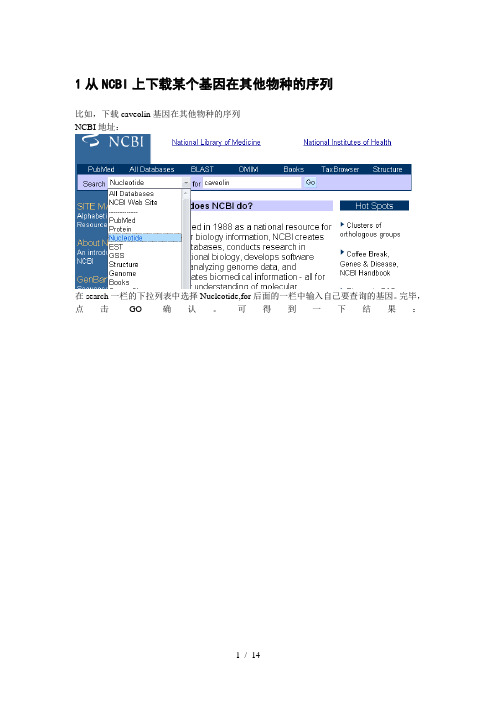
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
两条序列比对与多序列比对
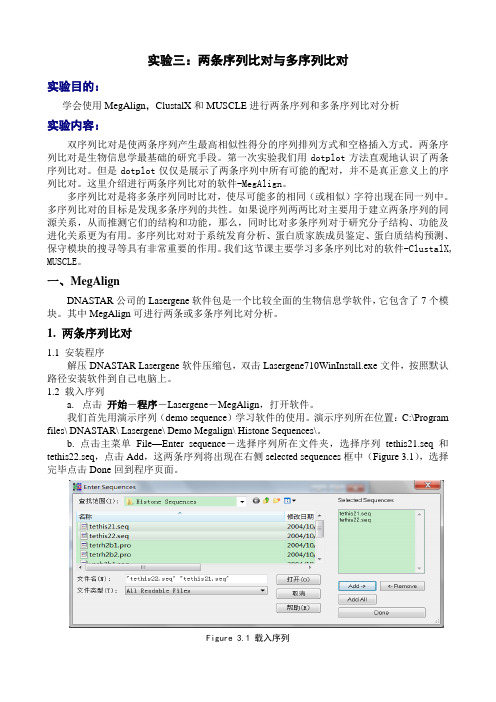
实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。
ncbi序列比对方法与操作实例

NCBI序列比对方法与操作实例一、序列比对方法概述1. 序列比对的概念序列比对是指通过对两个或多个生物序列进行比较分析,找到它们之间的相似性和差异性。
序列比对是生物信息学中的重要工具之一,可以帮助研究人员理解DNA、RNA、蛋白质等生物分子的结构和功能,进而推动生物医药和生物科学领域的发展。
2. 序列比对的意义在生物学研究中,通过对不同生物序列进行比对分析,可以揭示它们之间的进化关系、基因结构、功能和调控机制等重要信息,有助于揭示生物系统的内在规律。
序列比对还可以在分子生物学实验设计、基因工程、疾病诊断、新药开发等方面发挥重要作用。
3. 序列比对的方法常用的序列比对方法包括全局比对、局部比对和多序列比对等,其中全局比对适用于寻找整个序列间的相似段,局部比对适用于寻找两个序列中的部分匹配段,多序列比对则适用于比较多个序列之间的相似性和差异性。
二、NCBI序列比对工具介绍1. NCBI数据库NCBI(National Center for Biotechnology Information)是美国国家生物技术信息中心,是全球生物学信息资源的重要提供者之一。
NCBI数据库中包含大量生物信息数据,包括基因组序列、蛋白质序列、原始文献、生物信息学工具等。
2. NCBI序列比对工具NCBI提供了一系列用于序列比对的工具,其中包括BLAST(Basic Local Alignment Search Tool)、BLAT(BLAST-Like Alignment Tool)、ClustalW、MAFFT等。
这些工具可以帮助研究人员进行序列比对分析,找到感兴趣的生物序列在数据库中的同源序列或相似序列。
三、NCBI序列比对操作实例以BLAST工具为例,介绍NCBI序列比对的操作步骤。
1. 打开NCBI全球信息湾打开NCBI全球信息湾(),在全球信息湾首页的搜索栏中输入“BLAST”,进入BLAST工具的页面。
2. 输入查询序列在BLAST工具的页面中,选择适当的数据库,粘贴或上传待比对的查询序列,可以选择标准蛋白数据库、EST数据库、基因组数据库等作为比对的对象。
2双序列比对

哈尔滨医科大学 生物信息学院
李霞教授
第一节
引言
同源(homology)- 具有共同的祖先
直向同源(Orthologous ) 共生同源(paralogous )
相似(similarity)
同源序列一般是相似的,相似序列不 一定是同源的
2
3
4
通过点矩阵进行序列比较
5
6
7
Normalize Total Mutation Rate to 1%
This defines an evolutionary period: the period during which the 1% of all sequences are mutated (accepted of course)
1
2 1 2 2 1 0 1 2 2 2 1 2 1 2 2
1
1 1 2 2 2 1 0 2 1 1 2 2 2 1 2
1
2 2 1 1 2 2 2 0 1 2 2 2 1 2 2
1
2 1 1 2 2 2 1 1 0 2 2 1 1 1 1
2
1 2 2 2 2 2 1 2 2 0 1 1 2 2 2
但这并不意味100次PAM后,每个氨基酸都发生变化,因为其中一些位置可能 会经过多次突变,甚至可能会变回到原来的氨基酸。
PAM矩阵的制作步骤 •构建序列相似(大于85%)的比对 •计算氨基酸 j 的相对突变率mj(j被其它氨基酸替换的次数) •针对每个氨基酸对 i 和 j , 计算 j 被 i 替换次数 •替换次数除以相对突变率(mj)
41
取最小值!
计算过程:
42
计算过程:
•按行计算
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实验二:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析。
实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlign用dotplot方法能够直观地认识两条序列比对,但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍由DNASTAR公司开发的一个比较全面的生物信息学软件包--Lasergene,它包含了7个模块,其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 2.3),选择完毕点击Done回到程序页面。
Figure 2.3 载入序列此时程序窗口分为三部分,最左侧较窄的是sequence name,中间显示的是序列起始位置,最右侧显示序列末尾部分,可以通过拖动窗口底部滚动条,查看序列其它部分(Figure 2.4)。
若想改变字体显示方式,点击主菜单OPTIONS,选择Font改变字体,选择Size改变字号大小。
若要移除序列,选中sequence name的序列名,右击,选clear。
Figure 2.4 载入序列后(注意标注的绿色箭头,即为坐标位置)1.3 进行两条序列比对按住Shift选择序列tethis21和tethis22,然后点击主菜单Align-One pair,由于目前输入的是核酸序列,此时有三个选项,Wilbur-Lipman Method、Martiner NW Method和Dotplot。
如果输入的是蛋白质序列,前两个选项将是灰色,只能用Lipman-Pearson Method和Dotplot进行比对。
Wilbur-Lipman Method是一种以word为单位的(word-based)启发式局部比对方法;Martiner NW Method是一种改进了的全局动态规划算法。
Lipman-Pearson Method是序列相似度搜索软件Fasta的比对算法,也是一种以word为单位的快速启发式算法。
选择其中一个,出现比对参数设定窗口(Figure 2.5),选择默认参数不做更改,直接点击OK即可。
Figure 2.5 Wilbur-Lipman比对方法参数设定这时出现一个新窗口,即为比对结果。
可以选择OPTION-size,放大字号观察比对结果。
可以看到在窗口上部显示的是比对方法名称,所用参数,两条序列各自的起止位置,相似度值,比对结果中空位数目,长度和一致序列的长度。
随后就是比对结果部分,其中第一行是第一条序列,它上面的v70是标尺,其中的“V”的位置对应的是第一条序列的第70个核苷酸所在位置;第三行是第二条序列,它下方的数字同样对应该序列位置坐标;中间那行是根据两条序列比对结果中匹配部分推断出来的一致序列(consensus sequence),错配或空位显示为空白(Figure 2.6)。
Figure 2.6 Wilbur-Lipman方法比对结果设置比对结果显示方式:点击比对结果窗口最左侧的按钮,出现Alignment View Options窗口,可以选择匹配,错配和一致序列的字符颜色和其它显示选项。
推荐使用设置:选择match为红色,mismatch为绿色,consensus为蓝色,并选择show identities as vertical bars (一致序列显示为竖线),则得到Figure 2.7。
还可以尝试选中或不选show header, show ruler,show names,show contest四个选项,看看显示结果有何变化。
Figure 2.7 Alignment View OptionsTIP:MegAlign分析自己下载的序列时要注意序列扩展名如果是直接下载的fasta格式文件,可以象上面一样,用enter sequence直接将序列读入程序。
但是如果序列文件是复制粘贴到txt文档中的,MegAlign程序无法识别扩展名为txt 的文件,此时可将每条序列文件(fasta或genbank格式皆可)扩展名改为MegAlign可以识别的类型(核酸序列为seq,蛋白质序列为pro),即可从File-Enter sequence载入。
1.4 设定序列比对位置MegAlign允许使用者选择序列的一部分进行比对分析,例如,可以根据GenBank格式的序列中Features部分关于编码区(CDS)位置的描述,设定只对此编码区进行分析。
a. 点击最左侧Sequence Name框中的第一条序列tethis,然后选择主菜单OPTIONS-Set sequence limits-from feature table(Fig 2.8)。
此时根据feature内容,出现四个可以选择的片段,第一个为全长,从序列起始到末尾(1-906),其它三个则只包括序列的一部分,选择最后一个Histone H2B-1—CDS,点击Change the Reset,点击OK,同样对第二条序列进行上述操作,回到主界面工作区,此时窗口中的序列起始和终止位置已经发生了变化(Fig 2.9)。
Figure 2.8 利用Feature Table选择序列特定部分Figure 2.9 选择序列特定部分b. 我们还可以通过设定序列坐标进行部分序列比对,首先选定序列,选择主菜单OPTIONS-Set sequence limits-by coordinates,输入起始和终止位置坐标来选择部分序列进行分析。
注意:只有genbank格式的序列才可以Set sequence limits from feature table,fasta格式的序列因为没有feature那一项内容,只可以Set sequence limits by coordinates。
2. 多序列比对2.1 载入序列进行多条序列比对的演示序列(demo sequence)在c:\program files\ dnastar\ lasergene\ demo megalign\ Calmodulin Sequences\ 文件夹里。
点击主菜单File-Enter Sequence-根据路径到达Calmodulin Sequences文件夹,点击Add All,此时14条序列全都出现在右侧的selected sequences框中,点击Done,回到主程序工作区。
(Figure 2.10)这是来自14个物种的钙调蛋白。
Figure 2.10载入14条序列2.2 序列比对第一步,选择比对所用的打分矩阵。
点击主菜单Align-Set residue Weight Table,由于钙调蛋白比较保守,我们选择PAM100作为打分矩阵,点击OK结束设定(Figure 2.11)。
Figure 2.11 选择打分矩阵此时还可以通过点击Align-Method Parameters设定比对所用的其它参数。
打开的新窗口中包含三个选项卡,Jotun Hein、Clustal V和Clustal W,对应程序中多条序列比对可用的三种算法。
推荐大家不做修改,使用默认参数即可。
第二步,比对。
点击Align-by Clustal V Method,此时出现窗口显示比对进度,比对结束后,回到原来工作窗口,显示比对结果。
注意序列上方彩色条块,颜色代表对应列中相似程度,相似度由低到高,依次以黑、深蓝、浅蓝、绿、桔、红几种颜色代表(Figure 2.12)。
Figure 2.12 比对后结果2.3 查看比对结果此时可以通过几种方式观察比对结果。
a. 点击View-Alignment Reports出现新窗口,显示比对结果报告。
点击OPTIONS-Alignment report contents,选中show consensus strength,其它不变,点击OK。
在序列上方出现条块,显示每一列序列的相似程度(Fig 2.13)。
Figure 2.13选择show consensus strength显示结果设置比对结果显示方式:突出显示匹配或错配的氨基酸。
点击OPTIONS-New Decorations,在alignment decoration name框里输入shade disagreements(自己定义名字),选择decoration parameters为shade—residues differing from—the consensus,此时下方出现新的选项,选择对选定字符突出显示的颜色,选择完毕,点击OK,则与majority序列不同的字符将突出显示。
(Figure 2.14)Figure 2.14 修改alignment report显示模式b.点击View-Sequence Distances出现新窗口(Fig 2.15),显示两两序列percent identity(上半部分)和divergence(下半部分)。
Figure 2.15比对结果-一致度(identity)c.点击View-Residue Substitutions出现新窗口,显示比对中所有替换的类型和数目(Fig 2.16)。
Figure 2.16 比对结果-替换情况d.点击View-Phylogenetic Tree出现新窗口,显示根据14条序列比对结果构建出的进化树(Fig2.17)。