WBv12.1_emag_tutorial5_rotating_machine
Techlog软件WBI培训
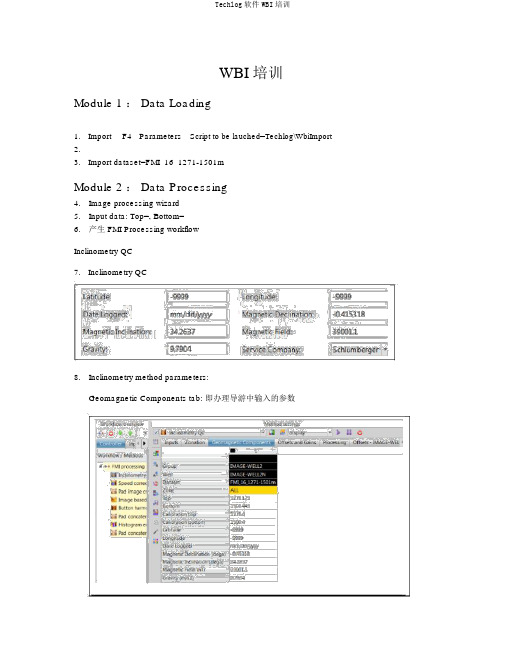
WBI 培训Module 1 : Data Loading1. Import F4 Parameters Script to be lauched=Techlog\WbiImport2.3. Import dataset=FMI_16_1271-1501mModule 2 : Data Processing4.Image processing wizard5.Input data: Top=, Bottom=6.产生 FMI Processing workflowInclinometry QC7. Inclinometry QC8.Inclinometry method parameters:Geomagnetic Components tab: 即办理导游中输入的参数Processing tab: 4 种办理方法。
依据Geomagnetic Components tab 参数对测井获得的magnetometer 磁场和 accelerometer 加快度曲线进行刻度,算出其偏移和增益,并显示在其左边的 Offsets and Gains tab 中。
而后利用这些偏移和增益对丈量的 magnetometer 磁场和 accelerometer 加快度曲线进行校订,既而从头计算新井斜数据。
Offsets and Gains tab:( manual 法时为人工输入)Offsets tab: Measure Point Offset ,Angular Offset :(人工输入)结果:校订前后数据对照,绿色表示合格,红色表示偏差超限。
Ax, Ay, Fx, Fy偏移校订右侧交会图:黑点为校订前点子,绿点为校订后点子。
中间红点为量。
9.Continue without savingSpeed correction10.Cable confidence factor: 同意 0-10,隐含 3,越高加快度变化对测井深度的影响越小。
ANSYS Workbench 12.1官方中文培训教程

Workbench –Mechanical Introduction第一章简介B. ANSYS Workbench 简介Training Manual •什么是ANSYS Workbench?–ANSYS Workbench中提供了与ANSYS系统求解器的强大交互功能的方法。
这个环境提供了一个独特的CAD及设计过程的集成系统。
法这个环境提供了个独特的及设计过程的集成系统•ANSYS Workbench由多种的应用模块组成(例子):–Mechanical:利用ANSYS的求解器进行结构和热分析。
•网格划分也包含在Mechanical应用中。
–Mechanical APDL:采用传统的ANSYS用户界面对高级机械和多物理场进行分析。
–Fluid Flow (CFX):利用CFX进行CFD分析。
–Fluid Flow (FLUENT):使用FLUENT进行CFD分析。
Fl id Fl(FLUENT)–Geometry (DesignModeler):创建几何模型(DesignModeler)和CAD几何模型的修改。
Engineering Data:定义材料性能。
–Engineering Data–Meshing Application:用于生成CFD和显示动态网格。
–Design Exploration:优化分析。
()格行转–Finite Element Modeler (FE Modeler):对NASTRAN和ABAQUS的网格进行转化以进行ansys分析。
–BladeGen (Blade Geometry) :用于创建叶片几何模型。
–Explicit Dynamics:具有非线性动力学特色的模型用于显式动力学模拟。
Training Manual… ANSYS Workbench 简介•Workbench 环境支持两种类型的应用程序:–本地应用(workspaces):目前的本地应用包括工项目管理,工程数据和优化设计本机应用程序的启动完全在窗运行•本机应用程序的启动,完全在Workbench 窗口运行。
AutoDock(英)

Dashboard
3D Viewer
Sequence Viewer Info bar
4
Exercise One: Preprocessing a PDB File
Here’s how 1. In the Dashboard, place the cursor over All Molecules and press Right MouseButton . In the Read Molecule: filebrowser which opens, click on hsg1.pdb and press Open . We will represent those actions like this: DB➞ PMV Molecules ➞ RightMB Read Molecule : ➞ hsg1.pdb ➞ Open 2. In the Dashboard, click on the inverted triangle under Cl to display color choices. Click on By atom type in the drop-down list: DB➞ Cl Ð s ➞ By atom type Click
Contents
Contents .......................................................................................................................................................... 2 Introduction.................................................................................................................................................... 3 Pmv Basics ...................................................................................................................................................... 4 Exercise One: Preprocessing a PDB File ..................................................................................................... 5 Pmv Mouse and Keyboard Bindings............................................................................................................ 6 Exercise Two: Preparing a Ligand for AutoDock. ..................................................................................... 7 Exercise Three: Preparing a Macromolecule............................................................................................ 10 Exercise Four: Setting the Search Space ................................................................................................... 11 AD4 Exercise Five: Preparing the AutoGrid Parameter File ................................................................. 12 AD4 Exercise Six: Starting AutoGrid 4 ..................................................................................................... 13 AD4 Exercise Seven: Preparing the AutoDock4 Parameter File ............................................................ 14 Exercise Eight: Starting AutoDock4 and AutoDock Vina. ...................................................................... 15 AutoDock Vina Exercise Nine: Preparing a Configuration File (optional) ........................................... 16 Exercise Ten: Visualizing AutoDock Vina results…. ............................................................................... 17 Exercise Eleven: Visualizing AD4 results.... .............................................................................................. 18 Exercise Three B (optional): Preparing the flexible residue file ............................................................. 19 Beyond the GUI............................................................................................................................................ 20 Appendix 1: Dashboard Widget ................................................................................................................. 27 Appendix 2: Conformation Player ............................................................................................................. 28
iMosflm tutorial

iMosflm Tutorial1. Introduction1.1 BackgroundMOSFLM can process diffraction images from a wide range of detectors and produces, as output, an MTZ file of reflection indices with their intensities and standard deviations (and other parameters). This MTZ file is passed onto other programs of the CCP4 program suite (POINTLESS, SORTMTZ, AIMLESS, CTRUNCATE) for further data reduction.The MOSFLM program was originally written to process data collected on film. It was then modified to process data collected using the image plate detector developed at the EMBL outstation in Hamburg by Jules Hendrix and Arno Lentfer, and the name was changed to ipmosflm. This is the current version of the program, which will also process data from CCD and pixel detectors.1.2 InstallationThe new GUI (iMosflm) is currently available for Windows, Mac OSX and Linux platforms. For details of installation visit/harry/mosflm and follow the link to the iMosflm version ....1.3 DocumentationThere are two distinct sources of documentation for MOSFLM, although neither of these currently makes any reference to the new GUI. At present, this document is the only documentation available for iMosflm.1. The MOSFLM user guide. This is available as a plain text file (mosflm_user_guide.txt) or on the web (www.mrc-/harry/mosflm/) as a PDF or HTML document. It is a very good idea to look through this guide before starting serious data processing with MOSFLM, although you do not need it for this tutorial.2. The "on-line" help. If you type "help" at the "MOSFLM =>" prompt (after starting the program) all possible keywords arelisted, with information on each keyword. This information is stored in an ASCII file (mosflm.hlp) which can also be read (and searched) with an editor. This relies on having the environment variable "CCP4_HELPDIR" set to the directory containing this file. This is also available on the MOSFLM web pages under "keyword synopses"1.4 Aims of the tutorialYour task is to process 84 images, hg_001.mar1600 to hg_084.mar1600, collected on a Mar345 image plate detector at a synchrotron beamline. These are crystals of a small domain (91 amino acids) that have been soaked in a mercury compound, resulting in a dataset with a strong anomalous signal which can easily be used to solve the structure. These images have kindly been provided by Camillo Rosano.2. Overview of iMosflmStart the program by typing "imosflm". After a brief pause, the following window will appear:The basic operations listed down the left hand side (Images, Indexing, Strategy, Cell Refinement, Integration, History) can be selected by clicking on the appropriate icon, but those that are not appropriate will be greyed-out and cannot be selected.2.1 Drop Down menusClicking on "Session" will result in a drop-down menu that allows you to save the current session or reload a previously savedsession (or add images):Clicking on "Settings" will allow you to see (and modify) Experiment settings, Processing options and Environment variables. Thesewill be described later.The three small icons below "Session" allow you to start a new session, open a saved session or save the current session. Moving themouse over these icons will result in display of a tooltip describing the action taken if the icon is clicked.3. Adding images to a sessionTo add images to a session, use the "Add images..." icon:Select the correct directory from the pop-up Add Images window (the default is the directory in which iMosflm was launched). All files with an appropriate extension (which can be selected) will be displayed. Double-clicking on any file will result in all images with the same template being added to the session. The template is the whole part of the filename prefix, except the number field which specifies the image number. An alternative is to single-click on one image filename and then click on Open.To open one or several images only, check the 'Selected images only' box and then use click followed by Shift+click to select a range of images or Control+click to select individual image files as required.Loaded images will be displayed in the Images window with the start & end phi values displayed (as read from the image header).All images with the same template belong to the same "Sector" of data.Multiple sectors can be read into the same session. Each sector can have a different crystal orientation.Note that a "Warning" has appeared. Click anywhere on the “1 Warning” text to get a brief description of the warning. In this case it is because the direct beam coordinates stored in the MAR image plate images are not 'trusted' by MOSFLM and the direct beam coordinates have been set to the physical centre of the image. Click on the green tick on the right hand side to dismiss this warning; click anywhere outside the warning box to collapse the box; double-click the warning text itself and more details, hints and notes maybe available from MOSFLM.The direct beam coordinates (read from the image header or set by MOSFLM) and crystal to detector distance are displayed. Thesevalues can be edited if they are not correct.Advanced Usage1. The phi values of images can be edited in the Image window. First click on the Image line to highlight it then click with themouse over the phi values to make them editable allowing new values to be given. These new values are propagated for allfollowing images in the same sector. Starting values for the three missetting angles may also be entered following the imagephi values.2. To delete a sector click on the sector name to select it (it turns blue) then use the right mouse button on the sector to bring upa "delete" button. Move the mouse over the delete button (it turns blue) and click to delete the sector and its images.Individual images can be deleted from the Images pane in the same way.3. If one sector is added and used for indexing, and then a second sector from the same crystal is added, the matrix for thesecond sector will not be defined. To define it, double-click on the matrix name for the first sector, save it to a file, double-click on the matrix name (Unknown) for the new sector and read the matrix file written for the first sector.4. Image DisplayWhen images are added to a session, the first image of the sector is displayed in a separate display window.The "Image" drop-down menu allows display of the previous or next image in the series. The "View" drop-down menu allows the image to be displayed in different sizes (related by scale factors of two), based on the image size and the resolution of the monitor. The line below allows selection of different images, either using right and left arrow or selecting one from the drop-down list of all images in that sector. The image being displayed can also be changed by double-clicking on an image name in the "Images" pane of the main window."+" and "-" will zoom the image without changing the centre. The "Fit image" icon will restore the image to its original size (right mouse button will have the same effect). The "Contrast" icon will give a histogram of pixel values. Use the mouse to drag the vertical dotted line, to the right to lighten the image, to the left to darken it. Try adjusting the contrast.4.1 Display IconsThe eight icons on the left, control the display of the direct beam position, spots found for indexing, bad spots, predicted spots, masked areas, spot-finding search area, resolution limits and display of the active mask for Rigaku detectors respectively.These are followed by icons for Zoom, Pan and Selection tools, and tools for adding spots manually (for indexing), editing masks, circle fitting and erasing spots or masks.Lastly on this toolbar are the entry boxes for h, k & l and a button to search for this hkl among the predicted spots displayed on the image. The button resembles a warning sign if the given hkl cannot be found.4.1.1 Masked areas - circular backstop shadowSelect the masked area icon. A green circle will be displayed showing the default position and size of the backstop shadow.Make sure that the Zoom icon (magnifying glass) is selected and use the left-mouse-button (abbreviated to LMB in following text) to drag out a rectangle around the centre of the image. The inner dotted yellow rectangle will show the part of the image that willactually appear in the zoomed area.Choose the Selection Tool. When placed over the perimeter of the circle, the radius of the circular backstop shadow will be displayed. Use the LMB to drag the circle to increase its diameter to that of the actual shadow on the image. The position of the circle can be adjusted with LMB placed on the cross that appears in the centre of the green circle. Adjust the size and position of the circle so that it matches the shadow.4.1.2 Masked areas - general exclusionsChoose the Masking tool. Any existing masked areas will automatically be displayed. Use LMB to define the four corners of the region to be masked. When the fourth position is given, the masked region will be shaded. This region will be excluded from spot finding and integration. This provides a powerful way of dealing with backstop shadows. To edit an existing mask, choose theSelection tool and use the LMB to drag any of the four vertices to a new position.To delete a mask, choose the Spot and mask eraser tool. Place the mouse anywhere within the shaded masked area, and use LMB todelete the mask. Delete any masks that you have created.4.1.3 Spot search areaSelect the Show spotfinding search area icon. The inner and outer radii for the spot search will be displayed as shown below. If theimages are very weak, the spot finding radius will automatically be reduced, but this provides additional control.Either can be changed by dragging with the LMB. Do not change the radii for these images.Advanced UsageThe red rectangle displays the area used to determine an initial estimate of the background of the image. It is important that this does not overlap significant shadows on the image. It can be shifted laterally or changed in orientation (in 90° steps) by dragging with the LMB.4.1.4 Resolution limitsSelect the Show resolution limits icon. The low and high resolution limits will be displayed. The resolution limits can be changed by dragging the perimeter of the circle with LMB (make sure that the Selection Tool has been chosen). The resolution limits will affect Strategy, Cell refinement and Integration, but not spot finding or indexing. The low resolution is not strictly correct (it falls within thebackstop shadow) but does not need to be changed because spots within the backstop shadow will be rejected.4.1.5 Zooming and PanningFirst select a region of the image to be zoomed with the Zoom tool.Select the Pan tool and pan the displayed area by holding down LMB and moving the mouse. This is rapid on a local machine, but may be slow if run on a remote machine over a network.4.1.6 Circle fittingThe circle fitting tool can be used to determine the direct beam position by fitting a circle to a set of points on a powder diffractionring on the image (typically due to icing) or to fit a circular backstop shadow (although using the masking tool is probably easier).Select the circle fitting tool. Three new icons will appear in the image display area. There are two (feint) ice rings visible on theimage at 3.91Å and 3.67Å. Click with LMB on several positions (6-8) on the outer ring (as it is slightly stronger). Then click on thetop circular icon.A circle that best fits the selected points (displayed as yellow crosses) will be drawn, and the direct beam position at the centre of thiscircle will be indicated with a green cross. The direct beam coordinates will be updated to reflect this new position.4.2 Other functionalitiesRight mouse button will return the display to the full size image if it has been zoomed.To get a small zoom window that can be moved over the image, hold down "Shift" with the Zoom tool selected. The area within the dotted square will be zoomed within the solid square.Holding down "Alt" (or "Command" ⌘ on Macs) will display the resolution and the current mouse position on the image in mm and pixels. If positioned over a found spot, the spot coordinates and I/σ(I) will be given. If positioned over a predicted spot position the hkl indices will be shown.5. Spot finding, indexing and mosaicity estimationWhen images have been added, the "Indexing" operation becomes accessible (it is no longer greyed-out).Click on Indexing. This will bring up the major Indexing window in place of the Images window.5.1 Spot FindingBy default, two images 90 degrees apart in phi (or as close to 90 as possible) will be selected and a spot search carried out on both images.Found spots will be displayed as crosses in the Image Display window (red for those above the intensity threshold, yellow for those below). The intensity threshold normally defaults to 20, but will be automatically reduced to 10 or 5 for weak images. The thresholdis determined by the last image to be processed.Images to be searched for spots can be specified in several ways:1. Simply type in the numbers of the images (e.g. 1, 84 above).2. Use the "Pick first image" icon (single blue circle).3. Use the "Pick two images ~90 apart" icon (two blue circles) . This is the default behaviour.4. Use the "Select images ..." icon (multiple circles). If selected, all images in the sector are displayed in a drop-down list. Clickon a image to select it, then double-click on the search icon (resembling a target) for that image to run the spot search. The image will move to the top of the list (together with other images that have been searched).Images to be used for indexing can be selected from those that have been searched by clicking on the "Use" button. If this box was previously checked, then clicking will remove this image from those to be used for indexing. It can be added again by clicking the "Select images ..." icon and clicking on the "Use" box.5.1.1 Difficult imagesParameters affecting the spot search can be modified by selecting the "Settings" drop-down window and selecting "Processing options". The resulting new window contains five tabs relating to Spot finding, Indexing, Processing, Advanced refinement and Advanced integration.The Spot finding window allows the Search area, Spot discrimination parameters, Spot size parameters, Minimum spot separation and Maximum peak separation within spots (to deal with split spots) to be reset. It also allows the choice between a local background determination (preferred) and a radial background determination. The local background method also uses an improved procedure for recognising closely spaced spots. The only parameters commonly changed are:1. Minimum spot separation. This should be the size (in mm) of an average spot (not a very strong spot). Change to valuesestimated by manual inspection of spots if there are difficulties due to badly split spots. This separation parameter is very important when spots are very close, but usually the program will determine a suitable value.2. Minimum pixels per spot. Default value 6, but this will be reduced automatically to 4 if spots are very small.3. Local background box size. Reducing this from 50 to (say) 20 can reduce the number of "false" spots found near any sharpshadow on the image.For data collected in-house spots can be quite large and rather weak. In such cases the spot finding can be greatly improved by the following:1. Reduce the spot finding threshold, e.g. to2.2. Increase the minimum number of pixels per spot to 20-40.3. Reduce the spot rms variation to 1.04. Set the minimum spot separation to a sensible value, e.g. 1.5mm5.2 IndexingProviding there are no errors during spot finding, indexing will be carried out automatically after spot finding. If the image selection or indexing parameters are changed, the "Index" button must be used to carry out the indexing.Autoindexing will be carried out by MOSFLM using spots (above the threshold) from the selected images. The threshold is set by MOSFLM but can be changed using the entry box in the toolbar. The list of solutions, sorted by increasing penalty score, will appear in the lower part of the window. The preferred solution will be highlighted in blue. There will usually be a set of solutions with low penalties (0-20) followed by other solutions with significantly higher penalties. The preferred solution is that with the highest symmetry from the group with low penalty values.Note that all these solutions are really the same P1 solution transformed to the 44 characteristic lattices, with latticesymmetry constraints applied. Therefore, if the P1 solution is wrong, then all the others are wrong as well.For solutions with a penalty less than 50, the refined cell parameters ("ref") will be shown. This can be expanded (click on the + sign) to show the "reg" unrefined but regularised cell (symmetry constraints applied) and the "raw" cell (no symmetry constraints applied). The rms deviation (rmsd) or error in predicted spots positions (σ(x,y) in mm) and the rms error in (σ(φ) in degrees) are given for each solution.Usually the penalty will be less than 20 for the correct solution, although it could be higher if there is an error in the direct beam coordinates (or distance/wavelength). The rmsd (error in spot positions) will typically be 0.1-0.2mm for a correct solution, but if the spots are split or very elongated it can be as high as 1mm or higher.The predicted spot positions for the highlighted solution will be shown on the image display with the following default colour codes: Blue:Fully recorded reflectionYellow:Partially recorded reflectionRed:Spatially overlapped reflection... these will NOT be integratedGreen:Reflection width too large (more than 5 degrees)... not integrated.These colours may be adjusted via the "Tools - Colour predictions" item in the Image display window.Providing there are no errors in the indexing, MOSFLM will automatically estimate the mosaic spread (mosaicity) based on thepreferred solution.Select other solutions with a higher penalty and see how well the predicted patterns match the diffraction image.The rmsd and visual inspection of the predicted pattern are the best ways of checking if a solution is correct. If the agreement is not good, then the autoindexing has probably failed.5.2.1 If the indexing fails - Direct beam searchThe indexing is very sensitive to errors in the direct beam coordinates. For a correct indexing solution, these should be correct to better than half the minimum spot separation. Check, for example, that the current direct beam position is behind the backstop. If there are any ice rings, these can be used to determine the direct beam position (see §4.1.6 Circle fitting).If the accuracy of the direct beam coordinates (generally read from the image header) is uncertain, the program can perform a grid search around the input coordinates. The number and the size of the steps can be set from the Indexing tab of the Processing options menu (Settings - Processing options, see §2.1) but defaults to two steps of 0.5mm on each side of the input coordinates. The direct beam search is started by clicking on the text "Search beam-centre" in the Indexing pane. While in progress, it can be stopped by clicking on the same place, the text of which is now "Abort beam-centre".The indexing will be carried out for each set of starting coordinates (Beam x and Beam y in the table which appears) and, if a solution is found, the refined beam coordinates (Beam x ref, Beam y ref), the unit cell parameters of the triclinic solution and the rmsd error in spot coordinates and in phi will be listed. The correct solution will generally be the one with the smallest rms error in spot positions (σ(x,y)). Note that only the triclinic (P1) solution will be listed. To complete the indexing, double-click on the chosen solution, and the full indexing will carried out for these beam coordinates, with the solutions appearing in the upper window. The results of the direct beam search can be hidden (collapsed) by clicking on the [-] symbol next to "Search beam-centre" and recovered by clicking on [+]5.2.2 If the indexing fails - Other parameters.Errors in other physical parameters (wavelength, crystal to detector distance) can also result in failure. All these parameters should be checked.Several parameters used in autoindexing can also be adjusted using entry fields and buttons that appear in the Indexing toolbar.Weak images1. MOSFLM automatically reduces the I/σ(I) threshold for weak images and it may also reduce the resolution to 4Å but lowervalues can be tried. It is important not to include spots that are not "real" - a small number of false spots can prevent theindexing from working.2. Try changing parameters for spot finding (see 5.1.1)Multiple lattices1. Try increasing the I/σ(I) threshold (default 20), for example to 40 or 60, so that only spots from the stronger lattice are selected.This can be done by entering a new value in the leftmost of the circled icons above (and also via the using the Indexing tab of Processing options).All cases1. Include more images in the indexing.2. In case the crystal orientation has changed, try indexing using only one image.3. If there are ice rings/spots, use the 'Exclude any spot rings during indexing' button.4. If the cell parameters are known, reduce the maximum allowed cell edge to the known maximum cell edge. This can sometimeshelp filter incorrect solutions.5. If the detector distance is uncertain, and the images are high resolution (e.g. 2Å), allow the detector distance to refine duringcell refinement.5.2.2 Space group selectionNote that the indexing is based solely on information about the unit cell parameters. It will therefore be very difficult (or impossible) to determine the correct Laue group in the presence of pseudosymmetry. For example, a monoclinic space group with β ~ 90 will appear to be orthorhombic; an orthorhombic space group with very similar a and b cell parameters will appear to be tetragonal. These can only be distinguished when intensities are available (i.e. after integration) by running POINTLESS.In addition, it is not possible to distinguish between Laue Groups 4/m and 4/mmm, 3 and 3/m, 6/m and 6/mmm, m3 and m3m. This will not affect integration of the images, but it will affect the strategy calculation. In the absence of additional information, the lower symmetry should be chosen to ensure that a complete dataset is collected.The presence of screw axes also cannot be detected, so there is no basis on which to distinguish P21 from P2 etc. This does not affect any aspect of data collection or processing, and can be chosen (on the basis of systematic absences) after integration by running POINTLESS.In this example, there is no way of knowing at this stage if the space group is h3 or h32, so leave it as h3.5.3 Mosaicity estimationThe mosaicity (mosaic spread) will be estimated automatically for the preferredsolution. However, if another solution is chosen, the mosaicity should beestimated again.Click on the "Estimate" button to estimate the mosaicity.A window will appear which plots the total predicted intensity as a function ofmosaic spread, from which an estimate is determined.The predictions displayed on the image will be updated. Entering different valuesof mosaic spread in the box (followed by return) will allow a visual estimate ofthe effect of changing the mosaic spread.The mosaic spread can also be entered in the Images pane by clicking once on theMosaicity line and once again on the value displayed.5.3.1 The mosaic block sizeIn some cases, the apparent mosaic spread is much larger at low resolution than at high resolution, so that if it is set to a value that results in all the low resolution spots being predicted, many more spots are predicted than are visible at high resolution. This effect is best dealt with by adjusting the "Mosaic block size" which is also displayed in the Images pane. This is modelling the size, in microns, of the individual mosaic blocks in the crystal. Decreasing this to values of 0.5-5 microns (say) from the default of 100 microns will result in predicting more spots at low resolution but have virtually no effect at high resolution.6. Saving a sessionThe session can be saved at any time, using the Session drop-down menu referred to in 2.1. The first time that a session is saved, you will be prompted for a filename. The filename convention for saved session files is that the extension is ".mos".Save the current session. Then exit from iMosflm (using the Session drop-down menu), restart iMosflm and read in the saved session. If the program crashes, it should be possible to recover all but the latest actions. Restart iMosflm and it will pop up a "Recover session ..." window.Selecting the Recover button will restore the session as far as possible. This file is written to the directory ".mosflm" in the user's home directory.7. Data collection strategyOnce the crystal orientation and (probable) Laue group have been determined, it is possible to calculate a data collection strategy and the Strategy icon is no longer greyed-out (in fact, all other operations, Strategy, Cell Refinement and Integration become possible at this point).Select the Strategy icon. This will open the Strategy window.The statistics initially presented in the window are based on the assumption that all images between the smallest phi value and the largest phi value in the current sector(s) have been collected (the phi ranges are listed). If processing a dataset that has already been collected (as in this tutorial) this is appropriate.More usually, the Strategy option will be used after two initial images, 90° apart in φ, have been collected. In such cases a complete, shaded segment will not be shown in the lower part of the screen, rather an empty circle will be shown with the matrix label beneath it. In cases where two reference images have been collected and indexed, click the 'Auto-complete' button and, in the pop-up menu that appears, select a rotation angle or 'sweep' of data to collect - or leave the 'Rotation:' setting as 'Auto' for a calculation of maximum completeness for this crystal orientation. Click on Ok and the completeness results will be presented.The orientation of the crystal, expressed as the angles between the a,b,c unit cell axes and the X,Y,Z coordinate frame, is given. X is along the X-ray beam, Z is the rotation axis. A warning will be given if the unique axis is so close to the rotation axis that there will be missing cusp data.7.1 Evaluating completeness manuallyThe completeness of any given segment of data can be determined manually using the circle at the bottom left of the window. Clickon the red segment shown in this circle.。
WB使用技巧集锦
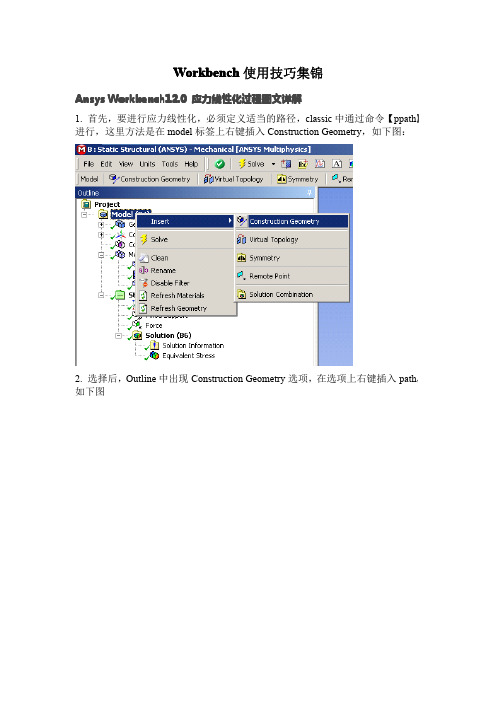
Workbench使用技巧集锦A nsys Workbench12.0应力线性化过程图文详解1.首先,要进行应力线性化,必须定义适当的路径,classic中通过命令【ppath】进行,这里方法是在model标签上右键插入Construction Geometry,如下图:2.选择后,Outline中出现Construction Geometry选项,在选项上右键插入path,如下图3.插入路径后,显示如下图所示路径的Detail选项卡,黄色区域是对路径的定义区域,目前版本只能定义两点的路径,可已通过选择点、线、面或者坐标的方式定义起、止点【默认的,face模式,则取点为面中心,edge模式,取点为其中点,vertex模式,取点为模型上存在的点,坐标模式,取点为鼠标点击的模型表面任一点,选中的点都可以Detail项中的x,y,z坐标值进行调整】4.定义好的路径如下图所示选择方式按钮这里定义路径参照的坐标系,路径取样点数信息在快捷栏选择一种应力线性化,效果是一样的,如下图所示6.插入应力线性化选项后,出现如下图所示的Detail选项卡,黄色为预选的路径定义好的路径会在这里显示,选择一个作为当前线性化路径选择参与线性化的实体选择应力线性化类型,其实就是重新定义线性化结果时间选项,多载荷步求解使用线性化参照的坐标系,可以选择自己定义的坐标系通过subtype 选择的应力类型都会出现在这里,可以看到,这些结果都是可以参数化的,也就是说,可以继续进行基于线性化应力结果的优化7.线性化的结果示例。
查看最大最小值坐标先点击outline 下的solution 以上的图标(如选择Analysis Settings),这时你会发现在菜单Units 下的图标Coordinate 图标激活,点击之,再按住CTRL,左击Outline 下你要的结果云图,鼠标在应力云图上移动,会出现坐标,至于准不准确,研究完了告诉我.......应力线性化选项,做过的朋友都明白,不详细说了用commmandsnsort,..*get,..nx(..)ny(..)nz(..)在后处理上WB比Classic跟为简单明了,但所能进行的后处理极为有限。
蒙特纳科技PG-1275E脉冲发生器用户手册说明书

USER’S MANUALPulse GeneratorPG-1275ERev 21/221.INTRODUCTION (3)2.SAFETY PRECAUTIONS (3)3.DESCRIPTION OF THE GENERATOR PG-1275E (5)3.1G ENERATOR INSTALLATION (5)3.2P ICTURES OF THE EQUIPMENT (5)3.3F RONT PANEL (6)3.3.1NAVIGATION IN MENU : (7)3.3.2MAIN MENU DISPLAY: (7)3.3.3Injected SPIKES configuration menu: (7)3.3.4Injected surge configuration menu: (7)3.3.5ERRORS listing: (8)3.4R EAR PANEL (9)4.HOW TO USE THE TEST EQUIPMENT (10)4.1I NJECTED SPIKES PROCEDURE (10)4.1.1Calibration spikes (10)4.1.2Testing spikes (10)4.2I NJECTED SURGES PROCEDURE (11)4.2.1Calibration surges (11)4.2.2Testing surges (11)4.3P ULSE ENERGY LIMITATION (12)4.3.1Energy for the spikes test (12)4.3.2Energy for the surges test (12)4.4R EMARKS (13)5.SPIKES TEST SETUPS (14)5.1S PIKES CALIBRATION (14)5.2S PIKES TESTING (14)6.SURGES TEST SETUP (15)6.1S URGES CALIBRATION (15)6.2S URGES TESTING WITH INTERNAL DECOUPLING DIODE (16)6.3S URGES TESTING WITH EXTERNAL DIODE MODULE (OPTIONAL) (16)7.SPECIFICATIONS (17)8.GENERATED SIGNALS (18)8.1S PIKES (18)8.2S URGES (19)9.REMOTE CONTROL (20)9.1L IST OF COMMANDS (20)9.2S YNTAX TO SEND A COMMAND (21)9.3Q UERY FORM SYNTAX (21)10.MAINTENANCE (22)11.SERVICING (22)2/22USER’S MANUAL1. IntroductionThis document includes the technical specifications and the instructions for use of the pulsegenerator PG-1275E.This equipment allows performing the following tests according to MIL-STD 1275E.•Injected spikes•Injected surgesNominal operating voltage of electrical device under test (EUT) is 28 V.Surge charging voltage is 200 V max.Spike charging voltage is 2000 V max.Nominal EUT current is limited to 16 A DC (400 A DC with optional external diode module).2. Safety precautionsThe following safety precautions should be observed before using this product and any associatedaccessories. Although some instruments and accessories would normally be used with non-hazardous voltages, there are situations where hazardous conditions may be however present. Exercise extreme caution when a shock hazard is present. Lethal voltages are present on connectors and inside the test equipment.This product is intended for use by qualified personnel only. These should recognize high voltage and be familiar with the safety precautions required to avoid possible injury. Read the operating manualcarefully before using the product.Before operating the instrument, make sure the line cord is connected to a properly grounded power receptacle. Always connect the test equipment to a good earth. Inspect the connecting cables and test leads for possible wear, cracks or breaks before each use. For maximum safety, do not touch theproduct, the test cables or any other accessory while power is applied to the circuit under test. Always remove power from the entire test system before: connecting or disconnecting cables, working on the device or on the equipment under test. Never disconnect conductors carrying current.Be careful while connecting / disconnecting batteries; these contain high level of energy and whenconnected in series, may provide dangerous voltages. Never short the batteries terminal even byaccident (i.e. by leaving a tool falling on it).Don’t work alone. In the event of an emergency, another person’s presence may be essential. Beaware of emergency procedures to follow in case of accident. Workers using a heart pacemaker orsimilar devices should not be present in the vicinity of the test equipment.In case of emergency push the red knob “emergency”.3/22The instrument and accessories must be used in accordance with their specifications and operating instructions otherwise the safety of the equipment may be impaired. Replace fuses with same type and rating for continued protection against fire hazard.If a screw labelled is present, connect it to safety earth using a short conductor having a section of minimum 4 mm2.The symbol on an instrument indicates that the user should refer to the operating instructions located in the manual.The symbol on an instrument shows that it can source or measure 1’000 volts or more, including the combined effect of normal and common mode voltages.High electromagnetic disturbances are produced by the PG-1275E. Sensitive electronic equipment placed in the vicinity may be disturbed.Do not operate the test equipment in an explosive environment or in wet conditions.The access of personnel may be restricted in the test area containing the PG-1275E and the EUT. The access may be controlled by safety barriers or by using a separate room. The doors can be fitted out with safety switches. All safety switches can be connected in series and the resulting safety circuit can be connected to the interlock connector of the test equipment. If a door is opened, the interlock circuit is opened and the generator is put in a safe mode.To maintain protection from electric shock and fire, spare parts in mains circuits, including the power transformer, test leads, and connectors, must be purchased from montena. Standard fuses, with applicable national safety approvals, may be used if the rating and type are the same. Other components that are not safety or high voltage related may be purchased from other suppliers as long as they are equivalent to the original component. Note that selected parts should be purchased only through montena to maintain accuracy and functionality of the product.To clean the test equipment, use a damp cloth or mild, water based cleaner. Clean the exterior of the instrument only. Do not apply cleaner directly to the instrument or allow liquids to enter or spill on the instrument.4/223. Description of the generator PG-1275E3.1 Generator installationKeep clearance on front and rear sides of the generator; this is mandatory to keep space for cooling fan airflow. At least 8 cm should be kept from generator sides to walls or other equipment.3.2 Pictures of the equipmentGenerator (included) 400 A decoupling diode module (optional)3x test cable2 m, Imax 32 A3x test cables1 m, Imax 32 ACalibration resistor 0.5 Ω (included) Cable set (included)Typical LISN (optional) Typical differential voltage probe (optional)5/223.3 Front panelFigure 1 : Front panel of the PG-1275EPULSE:Button to release the defined pulses. Pulse button is enabledonly in READY state, High Voltage being ON.Pulse button is also used to pause the pulses. A short presscontinues the test; a long press resets the counter.HIGH VOLTAGE:Button to switch the high voltage ON and OFF.Red light indicates the high voltage is active. Charging isenabled according to set value.High voltage OFF stops the test when running.High voltage automatically resets to OFF after 10 minutes of noactivity on panel buttons.MENU / LOCAL:Button to display the main menu or to return from remote to localmode.INTERLOCK:Indicator of the interlock circuit state: light is ON if the circuit isopen (safety circuit not OK).EMERGENCY:Emergency knob: when pushing the knob, the PG-1275Eswitches off. Display is blank, high voltage is OFF.To reset, rotate the knob clockwise (see arrows on button).CONTROL:Control knob. Rotate to select the item. Press the button to enterin the menu or to validate the selected value.6/223.3.1 NAVIGATION IN MENU :3.3.2 MAIN MENU DISPLAY:3.3.3 INJECTED SPIKES CONFIGURATION MENU:3.3.4 INJECTED SURGE CONFIGURATION MENU:7/223.3.5 ERRORS listing:8/223.4 Rear panelFigure 2: Rear panel of the PG-1275EPOWER:Mains supply connector with fuse and switchRS-232:SUB-D9 female for RS232 remote operation.USB: USB serial port for remote operation.INTERLOCK:Interlock input.All safety switches must be connected in series between pins 1and 2 to allow the PG-1275E to work. Switch between 1 and 2closed = PG-1275E can be operated.CONTROL 2 Control signals for the optional external diode moduleSPIKES +Positive output, connect to EUT+ for positive spike,respectively to EUT- for negative spike polaritySPIKES -Negative output, connect to EUT- for positive spike,respectively to EUT+ for negative spike polaritySURGES EUT+EUT side output for surges. Connect to the positive terminal ofthe EUT or to the EUT+ plug of the external diode module SURGES POWER+Power supply output for surges. Connect to the positive terminalof the power supply or to the POWER+ plug of the externaldiode moduleCorresponding fuse: F1, 16AT, (6.3x32mm)J1 Jumper (grey colour) for the use of the internal decoupling diodeRemove when an external diode module is used.PROG Button for firmware update, for service only9/224. How to use the test equipment4.1 Injected spikes procedure4.1.1 Calibration spikes•Prepare the setup according to §5.1As the cable length has a negative impact on the rise time, the connections between generatorand probe must kept short.•Set the oscilloscope and the voltage probe with the desired parameters.•Connect the mains supply cable of the PG-1275E.•Make sure the emergency knob is released (pulled).•Switch on the PG-1275E (rear side)•Choose the "SPIKES" generator in the main menu•Set the voltage of the generator to about 1000 V•Press the HIGH VOLTAGE button•Press the PULSE button•Adjust the oscilloscope parameters if needed•Gently increase the Set voltage in order to get the expected +250 V peak value on the measurement•Check the amplitude, rise time and oscillation frequency and compare to the standard•Write down the generator positive "Set" voltage for future testing4.1.2 Testing spikes•Prepare the setup according to §5.2 or Erreur ! Source du renvoi introuvable.•If present set the oscilloscope and the voltage probe with the desired parameters•Connect the mains supply cable of the PG-1275E.•Make sure the emergency knob is released (pulled).•Switch on the PG-1275E (rear side)•Switch on the EUT•Choose the "SPIKES" generator in the main menu•Set the voltage of the generator to about 1000 V•Press the HIGH VOLTAGE button•Press the PULSE button•Adjust the oscilloscope parameters if needed•Gently increase the Set voltage up to value noted during the calibration for positive polarity, but not above the maximum pulse energy (see §4.3).•Apply the required number of pulses with the required repetition period.•Verify the EUT operates as specified while subjected to the voltage spikes. Any deviation from normal operation shall be recognized as a failure of the EUT•Repeat the former steps with negative polarity10/224.2 Injected surges procedure4.2.1 Calibration surges•Prepare the setup according to §6.1 with the calibration resistor 0.5 Ω•Set the oscilloscope and the voltage probe with the desired parameters•Connect the mains supply cable of the PG-1275E.•Make sure the emergency knob is released (pulled).•Switch on the PG-1275E (rear side)•Choose the "SURGE" generator in the main menu•Set the voltage of the generator to about +100 V•Press the HIGH VOLTAGE button•Press the PULSE button•Adjust the oscilloscope parameters if needed•Gently increase the Set voltage in order to get the expected +70 V peak value on the measurement•Check the amplitude, rise time and duration and compare to the standard•Write down the generator "Set" voltage for future testing4.2.2 Testing surges•Prepare the setup according to §6.2 or 6.3•Disable the EUT power switch, if any.•If present, connect the external optional diode module. Cooling fan must start to blow air. It is important to prevent the EUT to draw current from diode module before to have the cooling fan inoperation. There is some risk of overheating the diode module!•Make sure to use conductors with cross-sections according to the EUT maximum power.•Set the oscilloscope and the voltage probe with the desired parameters•Connect the mains supply cable of the PG-1275E.•Make sure the emergency knob is released (pulled).•Switch on the PG-1275E (rear side)•Switch on the EUT•Choose the "SURGE" generator in the main menu•Set the voltage of the generator to about +100 V•Press the HIGH VOLTAGE button•Press the PULSE button•Adjust the oscilloscope parameters if needed•Gently increase the Set voltage up to value noted during the calibration, but not above the maximum pulse energy (see §4.3). Check also the measured voltage as it can be higher thanduring calibration with the 500 mΩ calibration load.•Apply the required number of pulses with the required repetition period.•Verify the EUT operates as specified while subjected to the voltage spikes. Any deviation from normal operation shall be recognized as a failure of the EUT11/224.3 Pulse energy limitationThe MIL-STD-1275 version E defines a limitation of the energy applied to the EUT:2 J for the spikes test60 J for the surges test4.3.1 Energy for the spikes testThe maximum stored energy of the generator at 2000 V charging voltage is about 4 J. In the worstcase, assuming the efficiency of the generator circuit is perfect, 50% of this energy, that is 2 J, could be delivered to the EUT.As this value is not higher than the MIL-STD-1275 limitation of 2 J, there is no need to monitor theinjected energy.4.3.2 Energy for the surges testThe maximum stored energy of the generator at 200 V charging voltage is about 300 J. In the worstcase, 50% of this energy. That is 150 J, could be delivered to the EUT.As this value is higher than the MIL-STD-1275 limitation, the injected energy must be monitored.This energy can be calculated by the oscilloscope, using the differential voltage probe and currentprobe signals. The oscilloscope must be capable of multiplying and integrating the two signals,E(t)=∫u(t)∙i(t)∙dtThe charging voltage of the PG1275E generator is to be adjusted until the calibrated injection level is obtained, while not exceeding the maximum total energy content of a single surge of 60 Joules.12/224.4 Remarks•To interrupt a test, press the pulse button. The test will recover with a short press of the pulse button. A long press will reset the pulses counter.•To cancel a test, press the "HIGH VOLTAGE" button off.•In case of emergency, press the emergency button (note that the cooling fan is always blowing even in emergency to prevent overheating of PG-1275E components).•At the end of the test session, press the button "HIGH VOLTAGE" to set the generator in standby mode. The red light is off. Note that the generator will automatically return to standby mode after10 minutes without activity.•To reset the emergency circuit, rotate the "EMERGENCY" button clockwise.•To return in the local mode, press the "LOCAL" button.•To control the generator with a computer, connect a RS232 or USB cable to the rear side. The connector and the cable must be correctly shielded.•Maximum surge voltage is over 100 V; this voltage level may cause electrical shocks. Do not touch conductors during the surge setup.•Do not shorten the batteries terminals; in general, follow batteries safety instructions.•MIL 1275 tests may create EUT malfunctions that are dangerous for the operators. Always take all required safety precaution to prevent injuries.•To change the spikes polarity, exchange both wires from the generator from the generator output13/225. Spikes test setups5.1 Spikes calibration5.2 Spikes testing14/226. Surges test setup6.1 Surges calibration15/226.2 Surges testing with internal decoupling diodeInsert J1- Insert J16.3 Surges testing with external diode module (optional)Remove J1- Remove J1- Connect CONTROL 216/227. Specifications17/228. Generated signals8.1 SpikesTest conditions :•EUT : no load (only the measurement probe)•EUT supply voltage : 0 V100 V/div 10 us/divTypical spike pulse form, set voltage is 2000 VAt lower levels, due to contact bounce, the waveform might show some discontinuities. Followingfigure shows an example at 200 V set voltage:18/228.2 SurgesTypical surge pulse, 20 V/div 10 ms/divTest conditions :•EUT supply voltage = 0 V•Charging voltage surge generator : 200 V•Test load resistance : 0.5 ΩWith other load impedances than 0.5 Ω, the shape and amplitude of the pulse will vary. Followingfigure shows an example at 100 V set voltage and a load of 1 kΩ:19/229. Remote controlThe remote control can be carried out through the USB or RS 232 connector placed on the rear side of the generator. The RS 232 connector is a Sub-D 9 pins. The port settings are: baud rate 9600, 8data bits, no parity, 1 stop bit, no flow control. The USB is A-type.9.1 List of commandsThe list of the control commands is given below.20/229.2 Syntax to send a commandThe general syntax of a writing command is given as following::<HEAD> < > <ARG> <LF>Example::VLT 0560 sets the voltage to 560 VIn details:9.3 Query form syntaxThe syntax to ask for a parameter from the generator is using the question mark (?):<HEAD>?<LF>Example::VLT?The answer from the generator is given without header and are terminated with <LF>Example:0824<LF>This returned value indicates that the actual set voltage is 824 V21/2210. MaintenanceThis equipment requires no special periodical adjustment or maintenance.11. ServicingMontena Technology SARoute de montena 89CH-1728 RossensSwitzerlandTel. : + 41 26 411 84 84 - Fax : + 41 26 411 17 79Email:********************\MIL1275\PG1275E_V01\4_Dossier_construction\08_Mode_emploi\Users_manual_PG-1275E_R02.docx22/22。
XPSPEAK 说明书

Using XPSPEAK Version 4.1 November 2000Contents Page Number XPS Peak Fitting Program for WIN95/98 XPSPEAK Version 4.1 (1)Program Installation (1)Introduction (1)First Version (1)Version 2.0 (1)Version 3.0 (1)Version 3.1 (2)Version 4.0 (2)Version 4.1 (2)Future Versions (2)General Information (from R. Kwok) (3)Using XPS Peak (3)Overview of Processing (3)Appearance (4)Opening Files (4)Opening a Kratos (*.des) text file (4)Opening Multiple Kratos (*.des) text files (5)Saving Files (6)Region Parameters (6)Loading Region Parameters (6)Saving Parameters (6)Available Backgrounds (6)Averaging (7)Shirley + Linear Background (7)Tougaard (8)Adding/Adjusting the Background (8)Adding/Adjusting Peaks (9)Peak Types: p, d and f (10)Peak Constraints (11)Peak Parameters (11)Peak Function (12)Region Shift (13)Optimisation (14)Print/Export (15)Export (15)Program Options (15)Compatibility (16)File I/O (16)Limitations (17)Cautions for Peak Fitting (17)Sample Files: (17)gaas.xps (17)Cu2p_bg.xps (18)Kratos.des (18)ASCII.prn (18)Other Files (18)XPS Peak Fitting Program for WIN95/98 XPSPEAKVersion 4.1Program InstallationXPS Peak is freeware. Please ask RCSMS lab staff for a copy of the zipped 3.3MB file, if you would like your own copyUnzip the XPSPEA4.ZIP file and run Setup.exe in Win 95 or Win 98.Note: I haven’t successfully installed XPSPEAK on Win 95 machines unless they have been running Windows 95c – CMH.IntroductionRaymond Kwok, the author of XPSPEAK had spent >1000 hours on XPS peak fitting when he was a graduate student. During that time, he dreamed of many features in the XPS peak fitting software that could help obtain more information from the XPS peaks and reduce processing time.Most of the information in this users guide has come directly from the readme.doc file, automatically installed with XPSPEAK4.1First VersionIn 1994, Dr Kwok wrote a program that converted the Kratos XPS spectral files to ASCII data. Once this program was finished, he found that the program could be easily converted to a peak fitting program. Then he added the dreamed features into the program, e.g.∙ A better way to locate a point at a noise baseline for the Shirley background calculations∙Combine the two peaks of 2p3/2 and 2p1/2∙Fit different XPS regions at the same timeVersion 2.0After the first version and Version 2.0, many people emailed Dr Kwok and gave additional suggestions. He also found other features that could be put into the program.Version 3.0The major change in Version 3.0 is the addition of Newton’s Method for optimisation∙Newton’s method can greatly reduce the optimisation time for multiple region peak fitting.Version 3.11. Removed all the run-time errors that were reported2. A Shirley + Linear background was added3. The Export to Clipboard function was added as requested by a user∙Some other minor graphical features were addedVersion 4.0Added:1. The asymmetrical peak function. See note below2. Three additional file formats for importing data∙ A few minor adjustmentsThe addition of the Asymmetrical Peak Function required the peak function to be changed from the Gaussian-Lorentzian product function to the Gaussian-Lorentzian sum function. Calculation of the asymmetrical function using the Gaussian-Lorentzian product function was too difficult to implement. The software of some instruments uses the sum function, while others use the product function, so both functions are available in XPSPEAK.See Peak Function, (Page 12) for details of how to set this up.Note:If the selection is the sum function, when the user opens a *.xps file that was optimised using the Gaussian-Lorentzian product function, you have to re-optimise the spectra using the Gaussian-Lorentzian sum function with a different %Gaussian-Lorentzian value.Version 4.1Version 4.1 has only two changes.1. In version 4.0, the printed characters were inverted, a problem that wasdue to Visual Basic. After about half year, a patch was received from Microsoft, and the problem was solved by simply recompiling the program2. The import of multiple region VAMAS file format was addedFuture VersionsThe author believes the program has some weakness in the background subtraction routines. Extensive literature examination will be required in order to revise them. Dr Kwok intends to do that for the next version.General Information (from R. Kwok)This version of the program was written in Visual Basic 6.0 and uses 32 bit processes. This is freeware. You may ask for the source program if you really want to. I hope this program will be useful for people without modern XPS software. I also hope that the new features in this program can be adopted by the XPS manufacturers in the later versions of their software.If you have any questions/suggestions, please send an email to me.Raymund W.M. KwokDepartment of ChemistryThe Chinese University of Hong KongShatin, Hong KongTel: (852)-2609-6261Fax:(852)-2603-5057email: rmkwok@.hkI would like to thank the comments and suggestions from many people. For the completion of Version 4.0, I would like to think Dr. Bernard J. Flinn for the routine of reading Leybold ascii format, Prof. Igor Bello and Kelvin Dickinson for providing me the VAMAS files VG systems, and my graduate students for testing the program. I hope I will add other features into the program in the near future.R Kwok.Using XPS PeakOverview of Processing1. Open Required Files∙See Opening Files (Page 4)2. Make sure background is there/suitable∙See Adding/Adjusting the Background, (Page 8)3. Add/adjust peaks as necessary∙See Adding/Adjusting Peaks, (Page 9), and Peak Parameters, (Page 11)4. Save file∙See Saving Files, (Page 6)5. Export if necessary∙See Print/Export, (Page 15)AppearanceXPSPEAK opens with two windows, one above the other, which look like this:∙The top window opens and displays the active scan, adds or adjusts a background, adds peaks, and loads and saves parameters.∙The lower window allows peak processing and re-opening and saving dataOpening FilesOpening a Kratos (*.des) text file1. Make sure your data files have been converted to text files. See the backof the Vision Software manual for details of how to do this. Remember, from the original experiment files, each region of each file will now be a separate file.2. From the Data menu of the upper window, choose Import (Kratos)∙Choose directory∙Double click on the file of interest∙The spectra open with all previous processing INCLUDEDOpening Multiple Kratos (*.des) text files∙You can open up a maximum of 10 files together.1. Open the first file as above∙Opens in the first region (1)2. In the XPS Peak Processing (lower) window, left click on 2(secondregion), which makes this region active3. Open the second file as in Step2, Opening a Kratos (*.des) text file,(Page 4)∙Opens in the second region (2)∙You can only have one description for all the files that are open. Edit with a click in the Description box4. Open further files by clicking on the next available region number thenfollowing the above step.∙You can only have one description for all the files that are open. Edit with a click in the Description boxDescriptionBox 2∙To open a file that has already been processed and saved using XPSPEAK, click on the Open XPS button in the lower window. Choose directory and file as normal∙The program can store all the peak information into a *.XPS file for later use. See below.Saving Files1. To save a file click on the Save XPS button in the lower window2. Choose Directory3. Type in a suitable file name4. Click OK∙Everything that is open will be saved in this file∙The program can also store/read the peak parameter files (*.RPA)so that you do not need to re-type all the parameters again for a similar spectrum.Region ParametersRegion Parameters are the boundaries or limits you have used to set up the background and peaks for your files. These values can be saved as a file of the type *.rpa.Note that these Region Parameters are completely different from the mathematical parameters described in Peak Parameters, (Page 11) Loading Region Parameters1. From the Parameters menu in the upper window, click on Load RegionParameters2. Choose directory and file name3. Click on Open buttonSaving Parameters1. From the Parameters menu in the XPS Peak Fit (Upper) window, clickon Save Region Parameters2. Choose directory and file name3. Click on the Save buttonAvailable BackgroundsThis program provides the background choices of∙Shirley∙Linear∙TougaardAveraging∙ Averaging at the end points of the background can reduce the time tofind a point at the middle of a noisy baseline∙ The program includes the choices of None (1 point), 3, 5, 7, and 9point average∙ This will average the intensities around the binding energy youselect.Shirley + Linear Background1. The Shirley + Linear background has been added for slopingbackgrounds∙ The "Shirley + Linear" background is the Shirley background plus astraight line with starting point at the low BE end-point and with a slope value∙ If the slope value is zero , the original Shirley calculation is used∙ If the slope value is positive , the straight line has higher values atthe high BE side, which can be used for spectra with higher background intensities at the high BE side∙ Similarly, a negative slope value can be used for a spectrum withlower background intensities at the high BE side2. The Optimization button may be used when the Shirley background is higher at some point than the signal intensities∙ The program will increase the slope value until the Shirleybackground is below the signal intensities∙ Please see the example below - Cu2p_bg.xps - which showsbackground subtraction using the Shirley method (This spectrum was sent to Dr Kwok by Dr. Roland Schlesinger).∙ A shows the problematic background when the Shirley backgroundis higher than the signal intensities. In the Shirley calculation routine, some negative values were generated and resulted in a non-monotonic increase background∙ B shows a "Shirley + Linear" background. The slope value was inputby trial-and-error until the background was lower than the signal intensities∙ C was obtained using the optimisation routineA slope = 0B slope = 11C slope = 15.17Note: The background subtraction calculation cannot completely remove the background signals. For quantitative studies, the best procedure is "consistency". See Future Versions, (Page 2).TougaardFor a Tougaard background, the program can optimise the B1 parameter by minimising the "square of the difference" of the intensities of ten data points in the high binding energy side of the range with the intensities of the calculated background.Adding/Adjusting the BackgroundNote: The Background MUST be correct before Peaks can be added. As with all backgrounds, the range needs to include as much of your peak as possible and as little of anything else as possible.1. Make sure the file of interest is open and the appropriate region is active2. Click on Background in the upper window∙The Region 0 box comes up, which contains the information about the background3. Adjust the following as necessary. See Note.∙High BE (This value needs to be within the range of your data) ∙Low BE (This value needs to be within the range of your data) NOTE: High and Low BE are not automatically within the range of your data. CHECK CAREFULLY THAT BOTH ENDS OF THE BACKGROUND ARE INSIDE THE EDGE OF YOUR DATA. Nothing will happen otherwise.∙No. of Ave. Pts at end-points. See Averaging, (Page 7)∙Background Type∙Note for Shirley + Linear:To perform the Shirley + Linear Optimisation routine:a) Have the file of interest openb) From the upper window, click on Backgroundc) In the resulting box, change or optimise the Shirley + LinearSlope as desired∙Using Optimize in the Shirley + Linear window can cause problems. Adjust manually if necessary3. Click on Accept when satisfiedAdding/Adjusting PeaksNote: The Background MUST be correct before peaks can be added. Nothing will happen otherwise. See previous section.∙To add a peak, from the Region Window, click on Add Peak ∙The peak window appears∙This may be adjusted as below using the Peak Window which will have opened automaticallyIn the XPS Peak Processing (lower) window, there will be a list of Regions, which are all the open files, and beside each of these will be numbers representing the synthetic peaks included in that region.Regions(files)SyntheticPeaks1. Click on a region number to activate that region∙The active region will be displayed in the upper window2. Click on a peak number to start adjusting the parameters for that peak.∙The Processing window for that peak will open3. Click off Fix to adjust the following using the maximum/minimum arrowkeys provided:∙Peak Type. (i.e. orbital – s, p, d, f)∙S.O.S (Δ eV between the two halves of the peak)∙Position∙FWHM∙Area∙%Lorenzian-Gaussian∙See the notes for explanations of how Asymmetry works.4. Click on Accept when satisfiedPeak Types: p, d and f.1. Each of these peaks combines the two splitting peaks2. The FWHM is the same for both the splitting peaks, e.g. a p-type peakwith FWHM=0.7eV is the combination of a p3/2 with FWHM at 0.7eV anda p1/2 with FWHM at 0.7eV, and with an area ratio of 2 to 13. If the theoretical area ratio is not true for the split peaks, the old way ofsetting two s-type peaks and adding the constraints should be used.∙The S.O.S. stands for spin orbital splitting.Note: The FWHM of the p, d or f peaks are the FWHM of the p3/2,d5/2 or f7/2, respectively. The FWHM of the combined peaks (e.g. combination of p3/2and p1/2) is shown in the actual FWHM in the Peak Parameter Window.Peak Constraints1. Each parameter can be referenced to the same type of parameter inother peaks. For example, for four peaks (Peak #0, 1, 2 and 3) with known relative peak positions (0.5eV between adjacent peaks), the following can be used∙Position: Peak 1 = Peak 0 + 0.5eV∙Position: Peak 2 = Peak 1 + 0.5eV∙Position: Peak 3 = Peak 2 + 0.5eV2. You may reference to any peak except with looped references.3. The optimisation of the %GL value is allowed in this program.∙ A suggestion to use this feature is to find a nice peak for a certain setting of your instrument and optimise the %GL for this peak.∙Fix the %GL in the later peak fitting process when the same instrument settings were used.4. This version also includes the setting of the upper and lower bounds foreach parameter.Peak ParametersThis program uses the following asymmetric Gaussian-Lorentzian sumThe program also uses the following symmetrical Gaussian-Lorentzian product functionPeak FunctionNote:If the selection is the sum function, when the user opens a *.xps file that was optimised using the Gaussian-Lorentzian product function, you have to re-optimise the spectra using the Gaussian-Lorentzian sum function with a different %Gaussian-Lorentzian value.∙You can choose the function type you want1. From the lower window, click on the Options button∙The peak parameters box comes up∙Select GL sum for the Gaussian-Lorentzian sum function∙Select GL product for the Gaussian-Lorentzian product function. 2. For the Gaussian-Lorentzian sum function, each peak can have sixparameters∙Peak Position∙Area∙FWHM∙%Gaussian-Lorentzian∙TS∙TLIf anyone knows what TS or TL might be, please let me know. Thanks, CMH3. Each peak in the Gaussian-Lorentzian product function can have fourparameters∙Peak Position∙Area∙FWHM∙%Gaussian-LorentzianSince peak area relates to the atomic concentration directly, we use it as a peak parameter and the peak height will not be shown to the user.Note:For asymmetric peaks, the FWHM only refers to the half of the peak that is symmetrical. The actual FWHM of the peak is calculated numerically and is shown after the actual FWHM in the Peak Parameter Window. If the asymmetric peak is a doublet (p, d or f type peak), the actual FWHM is the FWHM of the doublet.Region ShiftA Region Shift parameter was added under the Parameters menu∙Use this parameter to compensate for the charging effect, the fermi level shift or any change in the system work function∙This value will be added to all the peak positions in the region for fitting purposes.An example:∙ A polymer surface is positively charged and all the peaks are shifted to the high binding energy by +0.5eV, e.g. aliphatic carbon at 285.0eV shifts to 285.5eV∙When the Region Shift parameter is set to +0.5eV, 0.5eV will be added to all the peak positions in the region during peak fitting, but the listed peak positions are not changed, e.g. 285.0eV for aliphatic carbon. Note: I have tried this without any actual shift taking place. If someone finds out how to perform this operation, please let me know. Thanks, CMH.In the meantime, I suggest you do the shift before converting your files from the Vision Software format.OptimisationYou can optimise:1. A single peak parameter∙Use the Optimize button beside the parameter in the Peak Fitting window2. The peak (the peak position, area, FWHM, and the %GL if the "fix" box isnot ticked)∙Use the Optimize Peak button at the base of the Peak Fitting window3. A single region (all the parameters of all the peaks in that region if the"fix" box is not ticked)∙Use the Optimize Region menu (button) in the upper window4. All the regions∙Use the Optimize All button in the lower window∙During any type of optimisation, you can press the "Stop Fitting" button and the program will stop the process in the next cycle.Print/ExportIn the XPS Peak Fit or Region window, From the Data menu, choose Export or Print options as desiredExport∙The program can export the ASCII file of spectrum (*.DAT) for making high quality figures using other software (e.g. SigmaPlot)∙It can export the parameters (*.PAR) for further calculations (e.g. use Excel for atomic ratio calculations)∙It can also copy the spectral image to the system clipboard so that the spectral image can be pasted into a document (e.g. MS WORD). Program Options1. The %tolerance allows the optimisation routine to stop if the change inthe difference after one loop is less that the %tolerance2. The default setting of the optimisation is Newton's method∙This method requires a delta value for the optimisation calculations ∙You may need to change the value in some cases, but the existing setting is enough for most data.3. For the binary search method, it searches the best fit for each parameterin up to four levels of value ranges∙For example, for a peak position, in first level, it calculates the chi^2 when the peak position is changed by +2eV, +1.5eV, +1eV, +0.5eV,-0.5eV, -1eV, -1.5eV, and -2eV (range 2eV, step 0.5eV) ∙Then, it selects the position value that gives the lowest chi^2∙In the second level, it searches the best values in the range +0.4eV, +0.3eV, +0.2eV, +0.1eV, -0.1eV, -0.2eV, -0.3eV, and -0.4eV (range0.4eV, step 0.1eV)∙In the third level, it selects the best value in +0.09eV, +0.08eV, ...+0.01eV, -0.01eV, ...-0.09eV∙This will give the best value with two digits after decimal∙Level 4 is not used in the default setting∙The range setting and the number of levels in the option window can be changed if needed.4. The Newton's Method or Binary Search Method can be selected byclicking the "use" selection box of that method.5. The selection of the peak function is also in the Options window.6. The user can save/read the option parameters with the file extension*.opa∙The program reads the default.opa file at start up. Therefore, the user can customize the program options by saving the selectionsinto the default.opa file.CompatibilityThe program can read:∙Kratos text (*.des) files together with the peak fitting parameters in the file∙The ASCII files exported from Phi's Multiplex software∙The ASCII files of Leybold's software∙The VAMAS file format∙For the Phi, Leybold and VAMAS formats, multiple regions can be read∙For the Phi format, if the description contains a comma ",", the program will give an error. (If you get the error, you may use any texteditor to remove the comma)The program can also import ASCII files in the following format:Binding Energy Value 1 Intensity Value 1Binding Energy Value 2 Intensity Value 2etc etc∙The B.E. list must be in ascending or descending order, and the separation of adjacent B.E.s must be the same∙The file cannot have other lines before and after the data∙Sometimes, TAB may cause a reading error.File I/OThe file format of XPSPEAK 4.1 is different from XPSPEAK 3.1, 3.0 and 2.0 ∙XPSPEAK 4.1 can read the file format of XPSPEAK 3.1, 3.0 and 2.0, but not the reverse∙File format of 4.1 is the same as that of 4.0.LimitationsThis program limits the:∙Maximum number of points for each spectrum to 5000∙Maximum of peaks for all the regions to 51∙For each region, the maximum number of peaks is 10. Cautions for Peak FittingSome graduate students believe that the fitting parameters for the best fitted spectrum is the "final answer". This is definitely not true. Adding enough peaks can always fit a spectrum∙Peak fitting only assists the verification of a model∙The user must have a model in mind before adding peaks to the spectrum!Sample Files:gaas.xpsThis file contains 10 spectra1. Use Open XPS to retrieve the file. It includes ten regions∙1-4 for Ga 3d∙5-8 for Ga 3d∙9-10 for S 2p2. For the Ga 3d and As 3d, the peaks are d-type with s.o.s. = 0.3 and 0.9respectively3. Regions 4 and 8 are the sample just after S-treatment4. Other regions are after annealing5. Peak width of Ga 3d and As 3d are constrained to those in regions 1 and56. The fermi level shift of each region was determined using the As 3d5/2peak and the value was put into the "Region Shift" of each region7. Since the region shift takes into account the Fermi level shift, the peakpositions can be easily referenced for the same chemical components in different regions, i.e.∙Peak#1, 3, 5 of Ga 3d are set equal to Peak#0∙Peak#8, 9, 10 of As 3d are set equal to Peak#78. Note that the %GL value of the peaks is 27% using the GL sum functionin Version 4.0, while it is 80% using the GL product function in previous versions.18 Cu2p_bg.xpsThis spectrum was sent to me by Dr. Roland Schlesinger. It shows a background subtraction using the Shirley + Linear method∙See Shirley + Linear Background, (Page 7)Kratos.des∙This file shows a Kratos *.des file∙This is the format your files should be in if they have come from the Kratos instrument∙Use import Kratos to retrieve the file. See Opening Files, (Page 4)∙Note that the four peaks are all s-type∙You may delete peak 2, 4 and change the peak 1,3 to d-type with s.o.s. = 0.7. You may also read in the parameter file: as3d.rpa. ASCII.prn∙This shows an ASCII file∙Use import ASCII to retrieve the file∙It is a As 3d spectrum of GaAs∙In order to fit the spectrum, you need to first add the background and then add two d-type peaks with s.o.s.=0.7∙You may also read in the parameter file: as3d.rpa.Other Files(We don’t have an instrument that produces these files at Auckland University., but you may wish to look at them anyway. See the readme.doc file for more info.)1. Phi.asc2. Leybold.asc3. VAMAS.txt4. VAMASmult.txtHave Fun! July 1, 1999.。
ANSYS_Workbench12.0培训教程

• 例子:
相邻体上共用节 点
3-8
General Preprocessing
… 材料属性
• 为体添加材料属性,从目录树中选取体,然后在 下拉菜单中选取 “Material”
– 新的材料数据可以在“Engineering Data”下添加 和输入。然后新的材料就可以从下拉菜单中得到。 – 对于 surface bodies,如上所讲,定义一个厚度 是必要的。
• 基本网格划分控制在“Mesh” 分支下的“Defaults” 中是可用的。
– 用户控制单个网格大小的选项
• “Relevance” 设置在 –100 与 +100之间
Training Manual
- Relevance = 粗划分
+ Relevance = 细划分
3-25
General Preprocessing
Training Manual
3-9
General Preprocessing
… 几何体表格
• 提供体素和已经定义的材料的总表
– 选择 “Geometry”分支和 “Worksheet”
Training Manual
3-10
General Preprocessing
B. 接触
• 存在多个部件时 ,需要确定部分之间的相互关系。
• 初始网格大小将由激活的部件(未抑制的)决定。
Training Manual
– Full Assembly(整个组件):
• 初始网格大小不会受部件的状态(抑制或活动)的影响。
– Parts(部件):
• 初始种子独立地建立在每个部件大小基础上,且网格不会因为部件受抑制而改变。一般给与一个 细化的网格。
ebs R12.1.1-AIX安装手册

汉得信息技术有限公司EBS 系统安装手册Oracle Applications R12 Based On AIX-64System作者: 郭磊创建日期: 2009年2月23日 最后更新: 2009年2月23日 控制码: 版本: 1.0审批签字:客户项目负责人Copy Number _____文档控制修改记录日期作者版本修改参考2009-02-23 郭磊 1.0审阅姓名职位分发拷贝. 姓名地点1234目录EBS系统安装手册1作者:郭磊 (1)审批签字: 1文档控制 (2)修改记录2审阅2目录3安装概述 (4)安装前的系统准备 (5)安装12.1.1PROD环境 (7)已解决的问题 (17)安装概述安装环境:AIX-Based Systems AIX 6.1安装模块:ERP安装过程:1、安装 Applications R12 PROD环境* 安装是基于一台AIX P550主机, 单节点安装。
安装前的系统准备安装介质安装R12 for AIX-64,需用到R12安装介质中的11张DVD设置和检查主机网络和X Window系统检查主机名 hostname ( ebs ) 【vi /etc/hosts】检查IP地址 ifconfig (192.168.101.243)检查域名服务器 /etc/hosts文件/etc/hosts 设置如下127.0.0.1 localhost.localdomain localhost192.168.101.243 erpprodORACLE 的Rapid Install工具是图形化安装工具,主机Linux系统必须安装X Windows。
在终端运行 xhost +命令,以免OUI无法连接X Server。
检查操作系统版本和安装补丁检查操作系统版本AIX6(6.1),需要的补丁如下:AIX 6 (6.1) IZ10223 (TL 0)系统资源限制参数设置操作系统对用户能够使用的资源加以限制,在文件/etc/security/limits 修改资源信息增加如下内容:applprod:fsize = -1core = -1cpu = -1data = -1rss = -1stack = -1nofiles = -1oraprod:fsize = -1core = -1cpu = -1data = -1rss = -1stack = -1nofiles = -1安装JdkR12中的Apache需要使用Jdk1.5.0(以包含在EBS按照介质中,无需单独按照)查看java版本命令Java -versionIBM XL C/C++ Runtime UtilitiesThe XL C/C++ Enterprise Edition V8.0 for AIX, Runtime Environment and Utilities is available from the IBM Web site. Download and install the XL C/C++ runtime utilities and any prerequisites安装需要在xl c/cpp完成后,才能进行。
WB-Mech_Ch-_Results(精品)

Workbench -Mechanical Introduction Introduction第八章结果后处理Training Manual•在本章里,将介绍后处理部分的内容:A.查看结果B.区域结果显示域结果显C.输出结果D.坐标系和指定方向的结果分量标系和指定方向的结果分E.组合求解F.应力奇异性•本章本节描述的应用都能在所有ANSYS版本中使用,除了注明不可以使用本章本节描的应用都能在所有版本中使用除了注明不可以使用的版本。
Training Manual •当选择了一个结果项后,上下文工具栏显示了查看结果的方式:最大/最小Probe显示比例显示方法云图设置纲要显示矢量显示控制•另外,Timeline也有一个动画工具栏,它允许用户设置动画控制分布输出播放暂停图标帧速率控制Training Manual•对于结构分析(静态、模态、屈曲分析),可以改变变形形状:–默认的是一个标量(multiplies)乘上实际的位移。
–用户可以改为真实比例或显示未变形的模型图用户以改为真实比例或显未变的模型图自动选择的位移比例真实比例Training Manual •在图形区的图例上点击鼠标右键,可以调整图例控制。
值编辑输出、输入或保存图例设置增加或减少等高线带水平或垂直放置图例显示日期和时间在图例中显示最大最小图标选择对数比例切换到科学计数法有效数字位数•然后……Training Manual利用图例边界可以更清楚地显示出等高线图的结果分布。
没有改变最大最点击并拖放等高线分配器(或键盘输小值入一个值)来指定等高线范围。
也可以使用非均匀分布的等高线。
Training Manual•Independent Bands (独立带)使用中性色来代表高于或低于指定图例限制的模型区域。
图例等高线范围Training Manual•Geometry 按钮控制着等高线显示方式。
有四种可选选择:IsoSurfacesExterior“Exterior” (外观图)是默认的显示选项,而且是最常用的最常用的。
XTMR_TutoriaL

TutorialTMRTool, ISE 10.1 and Plan Ahead on the Virtex - 4 board-IntroductionThis lab will be an introduction to both the fundamentals of TMRTool and the overall design flow. TMRTool 10.1 is the tool provided by Xilinx for this purpose. The board will be Virtex - 4.-ObjectivesThe objective is to describe the basic design flow of the TMRTool, use the basic features of the TMRTool and verify a design was triplicated in a proper way by observing the netlist with plan ahead.-Prerequisites•Basic knowledge about digital design and FPGAs.•Acquaintance with Xilinx ISE 10.1 and Plan Ahead tools.•TMRTool is not included in ISE but is a program available through the Xilinx university program.-ApplicationThis document is designed for use by individuals who are learning FPGA design and verification.- Design OverviewFor the purpose of demonstrating the TMR processing, we are using a simple AND gate with two inputs A and B and an output Y. It is better to have this ISE project done before starting the Lab. In the TMR design we will be able to see the triplication on inputs, outputs, combinatorial logic and insertion of voter circuitry.-General Flow1. Opening an Existing Project in ISE that is to be triplicated or XTMRed.2. Synthesizing the source code so as to get a .ngc or a .ngo file which serve as an input to the TMRTool.3. Triplicating the netlist with TMRTool.4. Analyze and examine the triplicated netlist or design in Plan Ahead.5. Importing the triplicated netlist into ISE.-ImplementationStep 1: Open an Existing Project using Project Navigator in ISE. For the purpose of this lab we are using an AND gate with the project named And_tmra)In project Navigator, Select File Æ Open Project.b)Browse to the project location. Select the projectc)Click openThe selected project opens with the associated HDL file as shownFigure: 1: Opening ISE ExampleStep 2: Compile the HDL source file, translate the design to create the necessary netlistFile format.Run MAP to quantify the resource utilization.a)In sources window, select the Top-Level source fileb)In the process window, right-click Synthesize-XST and open theproperties menu.Figure: 2: Setting properties for synthesis in ‘processes for source’ tab Under Xilinx specific options tab, set the pack I/O Registers into IOBs option to NO.Figure: 3: Disabling Pack I/O Registers into IOBs option under Xilinx Specific options Note: - Due to the addition of Voter circuits on the outputs, an XTMR design generally does not permit I/O-based registersc)Click OKd)Within the processes for source window, double-click Synthesize-XST tocompile and produce a Xilinx specific netlist.e)Next, expand the menu options under Implement Design and double-clickTranslate.f)Now Right-Click Map and Invoke the properties Menu.Figure: 4: Setting properties for Map in ‘processes for source’ tabg)Turn off the option to pack I/O registers/latches into IOBsFigure: 5: Disabling Pack I/O Registers into IOBs option under map propertiesh)Click OKi)In the processes window Double-Click on Map. Note the map report(orcheck design summary) which can be used to compare the resourceutilization before and after triplication.Figure: 6: Design Summary for Pre XTMR Design ProjectStep 3: Import the .ngc/.ngo file into the TMRTool. Use the TMRTool features to do a specific type of triplication or just the default triplication of the design.a)Open TMRTool from windows menu by clicking on the XTMR icon.b)Create a new TMRTool project, Name it and specify an appropriatelocation (preferably in a new folder in the C drive).c)Check to see if the macro location setting is correct, this is done byselecting EDIT Æ Preferences and point to the location of the macrolibfolder copied during the installation of the TMRTool.Figure: 7: Setting the location for macrolib directoryd)Select Project Æ specify top-level sourcee)Browse to the .ngc file just created in the ISE tool and Click Open.Figure: 8: Opening the .NGC file for the project that is to be XTMRedf)In the Processes for Source window, double-click on Import. The greencheck appears when the process is successfully completed.Figure: 9: Importing the Top-level sourceg)From the processes for source window, double-click EDIT XTMR Types.The resulting display shows each node in the design. For each node shown you can specify the extent of TMR processing. Leave all options at default however for this Tutorial. For further explanation on the topic and the differences between these choices, view the online video on the website.Figure: 10: Setting XTMR typesh)Before clicking Implement XTMR, right-click on implement XTMR andgo to properties.i)Go to process settings and select “Replace SRL 16 with flip-flops” thiswill incorporate the extraction of half latches and replace SRL 16s.Figure: 11: Implement XTMR process settingj)Double-click Implement XTMR.k)After implementing XTMR, the design is exported into ISE environment for standard Place and Route Processing. This is done by double-clicking Export design in the “processes for source” window. The green check appears when the process is completed successfully.Figure: 12: Exporting XTMR NetlistStep 4: Examining the triplicated netlist produced in TMRTool in Plan Ahead software.a)Open Xilinx Plan Ahead 9.2.5 to launch the Plan Ahead Toolb)Select File Æ New Project.c)Create a New Project and Name it.d)Select “Yes, I want to import a synthesized (EDIF or NGC) Netlist.”Figure: 13: Creating new project in Plan Aheade)Browse for the .edf of the project just created in TMRTool.f)Select Next to import Netlist.Figure: 14: Import the netlist just Created in TMRTool(.edf file) g)Select the Virtex 4 family.Figure: 15: Choosing the Product Familyh)Select the Part as xc4vfx60ff1152-10 and Click OK.Figure: 16: Choosing the Devicei)Click NEXT on the import constraints window. We will be Examining theresulting netlist and will not need constraints.j)Click Finish.k)Examine the Netlist under the Netlist window. Select the Top module, Right-click and select Schematic.Figure: 17: Netlist window with the schematic option to examine the modulel)Click on the ‘+’ , to look at different aspects of the primitive. Right click on each primitive and Click Schematic, and observe the schematicrepresentation of the resulting post-TMRTool netlist.Figure: 18: Window with Plan Ahead Schematic for the moduleStep 5: Create a New Project in ISE, Specifying the Triplicated EDIF file as the Top-level Source.a)Create a New ISE Project labeled as And_post_tmr in appropriatelocation. Ensure that the Top-Level Module Type selection is EDIF asshown.Figure: 19: Creating new ISE project with top level EDIF source for Post XTMRb)Click Next.c)On the following screen, specify the input EDIF design file. Browse forthe .edf of the project just created in TMRTool.d)Uncheck copy the input design to the project directory.Figure: 20: Importing Input design file (.edf file created in TMRTool)e)Click Next. Ensure that the Target FPGA device is correct, as shown.Click NEXT and FINISHFigure: 21: Specifying Target FPGA devicef)ISE opens showing the And _tmr as the top-level design file.g)Within the “processes for source” window, right-click MAP and selectproperties. Turn off the pack I/O Registers/Latches in IOBs option by selecting off from the pull-down menu, then Click OK.Figure: 22: Setting properties for Map in ‘processes for source’ tabFigure: 23: Disabling Pack I/O Registers into IOBs option under mappropertiesh)In the sources window, select the top level design file.i)Double click place and route under processes window.Figure: 24: Implementing Place and Routej)In the processes window Double-Click on Map and note the map report or check design summary which can be used to compare the resourceutilization before and after triplication..Figure: 25: Design Summary for Post XTMR Design Project-Author BioName: Naveen Nischal PurushothamGraduate student at UNM in ECE department.E-mail: nischal@.。
gromacs tutorial

GROMACS TutorialStep One: Prepare the Protein TopologyWe must download the protein structure file we will be working with. For this tutorial, we will utilize T4 lysozyme L99A/M102Q (PDB code 3HTB). Go to the RCSB website and download the PDB text for the crystal structure.Once you have downloaded the structure, you can visualize it using a viewing program such as VMD, Chimera, PyMOL, etc. Once you've had a look at the molecule, you are going to want to strip out the crystal waters, PO4, and BME. Note that such a procedure is not universally appropriate (i.e., the case of a bound active site water molecule). For our intentions here, we do not need crystal water or other ligands. We will instead focus on the ligand called "JZ4."If you want a cleaned version of the .pdb file to check your work, you can download it here. The problem we now face is that the JZ4 ligand is not a recognized entity in any of the force fields provided with GROMACS, so pdb2gmx will give a fatal error if you were try to pass this file through it. Topologies can only be assembled automatically if an entry for a building block is present in the .rtp file for the force field. Since this is not the case, we will prepare our system topology in two steps:1.Prepare the protein topology with pdb2gmx2.Prepare the ligand topology using external toolsSince we will be preparing these two topologies separately, we must move the protein and JZ4 into separate coordinate files. Save the JZ4 coordinates like so:grep JZ4 3HTB_clean.pdb > JZ4.pdbThen simply delete the JZ4 lines from 3HTB_clean.pdb. At this point, preparing the protein topology is trivial. There are no missing atoms or residues in the 3HTB structure, so simply run pdb2gmx:pdb2gmx -f 3HTB_clean.pdb -o 3HTB_processed.gro -water spcThe structure will be processed by pdb2gmx, and you will be prompted to choose a force field:Select the Force Field:From '/usr/local/gromacs/share/gromacs/top':1: AMBER03 force field (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)3: AMBER96 force field (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)4: AMBER99 force field (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)5: AMBER99SB force field (Hornak et al., Proteins 65, 712-725, 2006)6: AMBER99SB-ILDN force field (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)8: CHARMM27 all-atom force field (with CMAP) - version 2.09: GROMOS96 43a1 force field10: GROMOS96 43a2 force field (improved alkane dihedrals)11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)14: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)15: [DEPRECATED] Encad all-atom force field, using full solvent charges16: [DEPRECATED] Encad all-atom force field, using scaled-down vacuum charges17: [DEPRECATED] Gromacs force field (see manual)18: [DEPRECATED] Gromacs force field with hydrogens for NMRFor this tutorial, we will use the united-atom GROMOS96 43A1 force field, so type 9 at the command prompt, followed by 'Enter'Step Two: Prepare the Ligand TopologyFor this tutorial, we will use PRODRG to generate a starting topology for our ligand, JZ4. Go to the PRODRG site and upload your JZ4.pdb file. The server presents you with several options for how to treat your ligand:∙Chirality (yes/no): maintain chiral centers in the input molecule (yes), or reconstruct them (no). In our case, we have no chirality, so leave this option set to its default.∙Charges (full/reduced): full charges are for condensed-phase systems (43A1 force field);reduced are for "vacuum" simulations (43B1 force field). Use full charges for nearly allapplications. The accuracy of 43B1 is debatable, anyway.∙EM (yes/no): perform energy minimization (yes), or leave coordinates alone (no). In our case, we want to construct our protein-ligand complex from existing coordinates, so wedo not want PRODRG to minimize our molecule in vacuo. Choose "no" for this option.∙Force field (GROMOS96.1/GROMOS87): "GROMOS96.1" refers to the first version of the GROMOS96 force field, 43A1. "GROMOS87" refers to the (outdated!) GROMOS87 force field. Choose "GROMOS96.1" to get 43A1 parameters for our ligand.We will make use of two files that PRODRG gives us. Save the output of the field "The GROMOS87/GROMACS coordinate file (polar/aromatic hydrogens)" into a text file called"jz4.gro" and "The GROMACS topology" into a file called "drg.itp."Let's take a look at the [ atoms ] section of our JZ4 topology:[ atoms ]; nr type resnr resid atom cgnr charge mass1 CH3 1 JZ4 C4 1 0.000 15.03502 CH2 1 JZ4 C14 2 0.059 14.02703 CH2 1 JZ4 C13 2 0.060 14.02704 C 1 JZ4 C12 2 -0.041 12.01105 CR1 1 JZ4 C11 2 -0.026 12.01106 HC 1 JZ4 H11 2 0.006 1.00807 CR1 1 JZ4 C7 2 -0.026 12.01108 HC 1 JZ4 H7 2 0.006 1.00809 CR1 1 JZ4 C8 2 -0.026 12.011010 HC 1 JZ4 H8 2 0.007 1.008011 CR1 1 JZ4 C9 2 -0.026 12.011012 HC 1 JZ4 H9 2 0.007 1.008013 C 1 JZ4 C10 3 0.137 12.011014 OA 1 JZ4 OAB 3 -0.172 15.999415 H 1 JZ4 HAB 3 0.035 1.0080I immediately notice three things that are wrong with this topology:1.Charge group 2 encompasses the entire aromatic ring, nearly all of the atoms in thismolecule.2.Charges of equivalent functional groups (C-H) are not uniform. In some cases, H atomsare +0.006e, and in other cases they are +0.007e.3.Charges on these atoms bear no resemblance whatsoever to the expected chargesprescribed by the force field.So what do we do? If you read the paper linked above, you would know what I'm going to recommend already. Assign charges and charge groups from equivalent functional groups in a building block-style manner. For any unknown groups, perform the charge calculations suggested in the paper. Since JZ4 involves only functional groups found in proteins (hydrophobic chain, aromatic ring, and an alcohol), life is pretty easy. We can re-assign charges and charge groups from these moieties. I will not go into proper topology validation in this tutorial; it would be far too time-consuming. Suffice it to say that you must satisfy reviewers that the parameters applied to your molecule are reliable. They should reproduce some well-known observable, such as logP or ΔG hydr, as in the GROMOS96 literature. If you cannot satisfy these requirements for your own system, you may want to reconsider your choice of force field.I would construct a topology that has something like this for an [ atoms ] directive:[ atoms ]; nr type resnr resid atom cgnr charge mass1 CH3 1 JZ4 C4 1 0.000 15.03502 CH2 1 JZ4 C14 2 0.000 14.02703 CH2 1 JZ4 C13 2 0.000 14.02704 C 1 JZ4 C12 2 0.000 12.01105 CR1 1 JZ4 C11 3 -0.100 12.01106 HC 1 JZ4 H11 3 0.100 1.00807 CR1 1 JZ4 C7 4 -0.100 12.01108 HC 1 JZ4 H7 4 0.100 1.00809 CR1 1 JZ4 C8 5 -0.100 12.011010 HC 1 JZ4 H8 5 0.100 1.008011 CR1 1 JZ4 C9 6 -0.100 12.011012 HC 1 JZ4 H9 6 0.100 1.008013 C 1 JZ4 C10 7 0.150 12.011014 OA 1 JZ4 OAB 7 -0.548 15.999415 H 1 JZ4 HAB 7 0.398 1.0080Note that equivalent functional groups have equivalent charges, hydrophobic groups are completely uncharged, and the charge groups are of a sensible size (2-3 atoms). The other elements of the topology are sufficiently accurate. Bonded parameters assigned by PRODRG are generally reliable. We are now ready to construct the system topology and reconstruct our protein-ligand complex.Build the ComplexFrom pdb2gmx, we have a file called "conf.gro" that contains the processed, force field-compliant structure of our protein. We also have "jz4.gro" from PRODRG that has included all of the necessary H atoms. Simply copy the coordinate section of jz4.gro and paste it into conf.gro, below the last line of the protein atoms, and before the box vectors, like so:163ASN C 1691 0.621 -0.740 -0.126163ASN O1 1692 0.624 -0.616 -0.140163ASN O2 1693 0.683 -0.703 -0.0115.99500 5.19182 9.66100 0.00000 0.00000 -2.99750 0.00000 0.00000 0.00000becomes (added text in bold green)...163ASN C 1691 0.621 -0.740 -0.126163ASN O1 1692 0.624 -0.616 -0.140163ASN O2 1693 0.683 -0.703 -0.0111JZ4 C4 1 2.429 -2.412 -0.0071JZ4 C14 2 2.392 -2.470 -0.1391JZ4 C13 3 2.246 -2.441 -0.1811JZ4 C12 4 2.229 -2.519 -0.3081JZ4 C11 5 2.169 -2.646 -0.2951JZ4 H11 6 2.135 -2.683 -0.1991JZ4 C7 7 2.155 -2.721 -0.4111JZ4 H7 8 2.104 -2.817 -0.4071JZ4 C8 9 2.207 -2.675 -0.5331JZ4 H8 10 2.199 -2.738 -0.6211JZ4 C9 11 2.267 -2.551 -0.5451JZ4 H9 12 2.306 -2.516 -0.6401JZ4 C10 13 2.277 -2.473 -0.4301JZ4 OAB 14 2.341 -2.354 -0.4341JZ4 HAB 15 2.369 -2.334 -0.5285.99500 5.19182 9.66100 0.00000 0.00000 -2.99750 0.00000 0.00000 0.00000Since we have added 15 more atoms into the .gro file, increment the second line of conf.gro to reflect this change. There should be 1708 atoms in the coordinate file now.Build the TopologyIncluding the parameters for the JZ4 ligand in the system topology is very easy. Just insert a line that says #include "drg.itp" into topol.top after the position restraint file is included. The inclusion of position restraints indicates the end of the "Protein" moleculetype section.; Include Position restraint file#ifdef POSRES#include "posre.itp"#endif; Include water topology#include "gromos43a1.ff/spc.itp"becomes...; Include Position restraint file#ifdef POSRES#include "posre.itp"#endif; Include ligand topology#include "drg.itp"; Include water topology#include "gromos43a1.ff/spc.itp"The last adjustment to be made is in the [ molecules ] directive. To account for the fact that there is a new molecule in conf.gro, we have to add it here, like so:[ molecules ]; Compound #molsProtein_chain_A 1JZ4 1The topology and coordinate file are now in agreement with respect to the contents of the system. Step Three: Defining the Unit Cell & Adding SolventAt this point, the workflow is just like any other MD simulation. We will define the unit cell andfill it with water.editconf -f conf.gro -o newbox.gro -bt dodecahedron -d 1.0genbox -cp newbox.gro -cs spc216.gro -p topol.top -o solv.groStep Four: Adding IonsWe now have a solvated system that contains a charged protein. The output of pdb2gmx told us that the protein has a net charge of +6e (based on its amino acid composition). If you missed this information in the pdb2gmx output, look at the last line of your [ atoms ] directive in topol.top; it should read (in part) qtot。
AIMCGE指南

sector_AC.map map for CGE’s sector and original sector sector_EB.map map of original BC and OC sector , and Energy balance flow
CO2_nonorg_source.set CO2 non organic emission source set
fossil fuel extraction cost in excel file TFP growth rate in excel file Non-energy use in data Renewable energy capacity GDP and population scenario data (GEO4) in excel file a directory for set and map files a set file for activities a set file for commodities a set file for countries a set file for economic industrial classification used in EEDD a set file for economic indicators used in EEDD a set file for energy flow used in EEDD a map file of sales tax set to commodities set a set file for sales tax a set file for energy products used in EEDD a map file from the original SAM classification to that of CGE iv
双向电泳图像分析软件_贾宇峰

基金项目 :国家自然科学基金资助项目 , N o .39928015 收稿日期 :2000 -05 -15 作者简介 :贾宇峰(1971 -), 硕士 , 毕业于华西医科大学公共卫生学院
双向电泳图像分析软件
贾宇峰 刘少君
郭尧君
(军事医学科学院基础医学研究所 北京 100850) (中国科学院生物物理研究所 北京 100101)
摘要 :本文分别使用 I mageM aster 2D Elite 3.01 和 PDQuest 6.0 对 PC12 细胞蛋 白样品双向电泳结果 进行了初步 分析 , 讨论了 软件的图像 扫描 、点检测 、背景消 除 、匹 配 、结 果报告和数 据分析功 能 。 并且对新 一代的 2D 图像分析软 件 , ImageM aster 2D Elite 3.01、PDQuest 6.0 和 M elanie II & 3 的主 要特性进行了比较 。
发布 。 为了有效地评价 2D 图像分析系统 , 我们对 ImageMas-
ter 2D 和 PDQuest 作了初步的分析研究比较 , 并且就 ImageMaster 2D Elite 3.01 、PDQuest 6.0 和 M elanie II &3 的 主要特性进行了比较 。
1 设备与材料
在人类基因组计划即将完成之时 , 越来越多的实验室 开始涉足后基因组 (post_genome)领域 , 蛋白质组研究正在 升温 。 在一些科研机构和生物技术公司的努力下 , 2-DE 技 术也不断出现突破性进展 。 正是在这种情况下 , 第三代图 像分析软件 ImageMaster 2D Elite 、PDQuest 、Melanie II 等应 运而生 , 其共同特点是集成化 、模块化和易于使用 。另外 , 为了处理日渐复杂的 2D 图像数据 , 其算法也更先进 。目前 正在应用的 2D 图像分析软件还有 Gellab_II 、Kepler 、Bio Image 等 , 既有第二代也有第三代软件 , 最近 Melanie 3 也已经
IDL 图像处理第4讲

DIP Lecture 4
Morphological Algorithms
• Boundary Extraction • Region Filling • Connected Components • Hit-or-Miss Transform • Convex Hull • Thinning and Thickening Boundary extraction was done in Lecture 3.
DIP Lecture 4
2
Region Filling Program
Function Region Fill,A,start,filter=B,max iterations=max iterations
IF n params() LT 2 THEN Message,’Too Few Arguments to Function’ IF NOT KEYWORD SET(B) THEN B=[[0b,1b,0b],[1b,1b,1b],[0b,1b,0b]] IF NOT KEYWORD SET(max iterations) THEN max iterations=10000L
1 B=R 1 1
3. Let X0 = p and perform the iteration
P
1 1 1
1 1 S 1
Q
Xk = Xk−1 ⊕ B ∩ A for k = 1, 2, 3, . . .
until Xk = Xk−1
¡
DIP Lecture 4
6
Example: G&W Figure 9.17
Image with three shapes
Structuring elements B1 and B2
MAKER使用指南说明书

Exercise 1. Using MAKER for Genome AnnotationIf you are following this guide for your own research project, please make the following modifications:1. In this exercise, SNAP was used for gene prediction. When you are working on your owngenome, we recommend that you use Augustus. The instructions for using Augustus is in appendix.2. In the exercise, you will be using 2 CPU cores. When you are working on your own genome,you should use all CPU cores on your machine. When you run the command:"/usr/local/mpich/bin/mpiexec -n 2", replace 2 with number of cores available on yourmachine.3. The steps for Repeatmodeler and Repeatmasker are optional in the exercise, but requiredwhen you work on your own genome.The example here is from a workshop by Mark Yandell Lab (/ ) Further readings:1. Yandel Lab Workshop. /MAKER/wiki/index.php/MAKER_Tutorial_for_WGS_Assembly_and_Annotation_Winter_School_2018 .2. MAKER protocol from Yandell Lab. It is good reference. https:///pmc/articles/PMC4286374/3. Tutorial for training Augustus https:///simonlab/bioinformatics/programs/augustus/docs/tutorial2015/training.html4. Maker control file explained: /MAKER/wiki/index.php/The_MAKER_control_files_explainedPart 1. Prepare working directory.1. Copy the data file from /shared_data/annotation2018/ into /workdir/$USER, and de-compress the file. You will also copy the maker software directory to /workdir/USER. The maker software directory including a large sequence repeats database. It would be good to put it under /workdir which is on local hard drive.mkdir /workdir/$USERmkdir /workdir/$USER/tmpcd /workdir/$USERcp /shared_data/annotation2019/maker_tutorial.tgz ./cp -rH /programs/maker/ ./cp -rH /programs/RepeatMasker ./tar -zxf maker_tutorial.tgzcd maker_tutorialls -1Part 2. Maker round 1 - Map known genes to the genomeRun everything in "screen".Round 1 includes two steps:Repeat masking;Align known transcriptome/protein sequences to the genome;1. [Optional] Build a custom repeat database. This step is optional for this exercise, as it is avery small genome, it is ok without repeat masking. When you work on a real project, you can either download a database from RepBase (https:///repbase/, license required), or you can build a custom repeat database with your genome sequence.RepeatModeler is a software for building custom databases. The commands for building a repeat database are provided here.cd example_02_abinitioexport PATH=/programs/RepeatModeler-2.0:$PATHBuildDatabase -name pyu pyu_contig.fastaRepeatModeler -pa 4 -database pyu -LTRStruct >& repeatmodeler.logAt the end of run, you would find a file "pyu-families.fa". This is the file you can supply to "rmlib=" in the control file.2. Set environment to run Maker and create MAKER control files.Every steps in Maker are specified by the Maker control files. The command "maker -CTL" will create three control files: maker_bopts.ctl, maker_exe.ctl, maker_opts.ctl.by.exportPATH=/workdir/$USER/maker/bin:/workdir/$USER/RepeatMasker:/programs/snap:$PATH export ZOE=/programs/snap/Zoeexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libcd /workdir/$USER/maker_tutorial/example_02_abinitiomaker -CTL3. Modify the control file maker_opts.ctl.Open the maker_opts.ctl file in a text editor (e.g. Notepad++ on Windows, BBEdit on Mac, or vi on Linux). Modify the following values. Put the modified file in the same directory“example_02_abinitio”.genome=pyu_contig.fastaest=pyu_est.fastaprotein=sp_protein.fastamodel_org=simplermlib= #fasta file of your repeat sequence from RepeatModeler. Leave blank to skip.softmask=1est2genome=1protein2genome=1TMP=/workdir/$USER/tmp #important for big genome, as the default /tmp is too smallThe modified maker_opts.ctl file instructs MAKER to do two things.a) Run RepeatMasker.The line “model_org=simple” tells RepeatMasker to mask the low complexity sequence (e.g.“AAAAAAAAAAAAA”.The line “rmlib=” sets "rmlib" to null, which tells RepeatMasker not to mask repeatsequences like transposon elements. If you have a repeat fasta file (e.g. output fromRepeatModeler) that you need to mask, put the fasta file name next to “rmlib=”The line “softmask=1” tells RepeatMasker to do soft-masking which converts repeats tolower case, instead of hard-masking which converts repeats to “N”. "Soft-masking" isimportant so that short repeat sequences within genes can still be annotated as part of gene.If you run RepeatMasker separately, as described in https:///darencard/bb10 01ac1532dd4225b030cf0cd61ce2 , you should leave rmlib to null, but set rm_gff to a repeat gff file.b) Align the transcript sequences from the pyu_est.fasta file and protein sequences from thesp_protein.fasta file to the genome and infer evidence supported gene model.The lines “est2genome=1” and “protein2genome=1” tell MAKER to align the transcriptsequences from the pyu_est.fasta file and protein sequences from the sp_protein.fasta file to the genome. These two files are used to define evidence supported gene model.The lines “est=pyu_est.fasta" and "protein=sp_protein.fasta" specify the fasta file names of the EST and protein sequences. In general, the EST sequence file contains the assembled transcriptome from RNA-seq data. The protein sequence file include proteins from closely related species or swiss-prot. If you have multiple protein or EST files, separate file names with ",".4. [Do it at home] Execute repeat masking and alignments. This step takes an hour. Run it in"screen". In the command: "mpiexec -n 2 " means that you will parallelize Maker using MPI, and use two threads at a time. When you work on a real project, it will take much longer, and you should increase this "-n" setting to the number of cores.Set Maker environment if it is new session:exportPATH=/workdir/$USER/maker/bin:/workdir/$USER/RepeatMasker:/programs/snap:$PATH export ZOE=/programs/snap/Zoeexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libExecute the commands:cd /workdir/qisun/maker_tutorial/example_02_abinitio/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd1 >& log1 &After it is done, you can check the log1 file. You should see a sentence: Maker is now finished!!!Part 3. Maker round 2 - Gene prediction using SNAP1. Train a SNAP gene model.SNAP is software to do ab initio gene prediction from a genome. In order to do gene prediction with SNAP, you will first train a SNAP model with alignment results produced in the previous step.If you skipped the step "4. [Do it at home] Execute Maker round 1", you can copy the result files from this directory: /shared_data/annotation2019/cd /workdir/qisun/maker_tutorial/example_02_abinitiocp /shared_data/annotation2019/pyu_rnd1.maker.output.tgz ./tar xvfz pyu_rnd1.maker.output.tgzSet Maker environment if it is new session:exportPATH=/workdir/$USER/maker/bin:/workdir/$USER/RepeatMasker:/programs/snap:$PATH export ZOE=/programs/snap/Zoeexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libThe following commands will convert the MAKER round 1 results to input files for building a SNAP mode.mkdir snap1cd snap1gff3_merge -d ../pyu_rnd1.maker.output/pyu_rnd1_master_datastore_index.logmaker2zff -l 50 -x 0.5 pyu_rnd1.all.gffThe “-l 50 -x 0.5” parameter in maker2zff commands specify that only gene models with AED score>0.5 and protein length>50 are used for building models. You will find two new files: genome.ann and genome.dna.Now you will run the following commands to train SNAP. The basic steps for training SNAP are first to filter the input gene models, then capture genomic sequence immediately surrounding each model locus, and finally uses those captured segments to produce the HMM. You can explore the internal SNAP documentation for more details if you wish.fathom -categorize 1000 genome.ann genome.dnafathom -export 1000 -plus uni.ann uni.dnaforge export.ann export.dnahmm-assembler.pl pyu . > ../pyu1.hmmmv pyu_rnd1.all.gff ../cd ..After this, you will find two new files in the directory example_02_abinitio:pyu_rnd1.all.gff: A gff file from round 1, which is evidence based genes.pyu1.hmm: A hidden markov model trained from evidence based genes.2. Use SNAP to predict genes.Modify directly on the maker_opts.ctl file that you have modified previously.Before doing that, you might want to save a backup copy of maker_opts.ctl for round 1.cp maker_opts.ctl maker_opts.ctl_backup_rnd1Now modify the following values in the file: maker_opts.ctlRun maker with the new control file. This step takes a few minutes. (A real project could take hours to finish). You will use the option “-base pyu_rnd2” so that the results will be written into a new directory "pyu_rnd2".Again, make sure the log2 file ends with "Maker is now finished!!!".Part 4. Maker round 3 - Retrain SNAP model and do another round of SNAP gene predictionYou might need to run two or three rounds of SNAP . So you will repeat Part 2 again. Make sure you will replace snap1 to snap2, so that you would not over-write previous round.1. First train a new SNAP model.2. Use SNAP to predict genes.Modify directly on the maker_opts.ctl file that you have modified previously.Before doing that, you might want to save a backup copy of maker_opts.ctl for round 2.Now modify the following values in the file: maker_opts.ctlmaker_gff= pyu_rnd1.all.gffest_pass=1 # use est alignment from round 1protein_pass=1 #use protein alignment from round 1rm_pass=1 # use repeats in the gff filesnaphmm=pyu1.hmmest= # remove est file, do not run EST blast againprotein= # remove protein file, do not run blast againmodel_org= #remove repeat mask model, so not running RM againrmlib= # not running repeat masking againrepeat_protein= #not running repeat masking againest2genome=0 # do not do EST evidence based gene modelprotein2genome=0 # do not do protein based gene model.pred_stats=1 #report AED statsalt_splice=0 # 0: keep one isoform per gene; 1: identify splicing variants of the same genekeep_preds=1 # keep genes even without evidence support, set to 0 if no/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd2 >& log2 &mkdir snap2cd snap2gff3_merge -d ../pyu_rnd2.maker.output/pyu_rnd2_master_datastore_index.logmaker2zff -l 50 -x 0.5 pyu_rnd2.all.gfffathom -categorize 1000 genome.ann genome.dnafathom -export 1000 -plus uni.ann uni.dnaforge export.ann export.dnahmm-assembler.pl pyu . > ../pyu2.hmmmv pyu_rnd2.all.gff ..cd ..cp maker_opts.ctl maker_opts.ctl_backup_rnd2maker_gff=pyu_rnd2.all.gffsnaphmm=pyu2.hmmRun Maker:/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd3 >& log3 &Use the following command to create the final merged gff file. The “-n” option would produce a gff file without genome sequences:gff3_merge -n -dpyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.log>pyu_rnd3.noseq.gff fasta_merge -d pyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.logAfter this, you will get a new gff3 file: pyu_rnd3.noseq.gff, and protein and transcript fasta files. 3. Generate AED plots./programs/maker/AED_cdf_generator.pl -b 0.025 pyu_rnd2.all.gff > AED_rnd2/programs/maker/AED_cdf_generator.pl -b 0.025 pyu_rnd3.noseq.gff > AED_rnd3You can use Excel or R to plot the second column of the AED_rnd2 and AED_rnd3 files, and use the first column as the X-axis value. The X-axis label is "AED", and Y-axis label is "Cumulative Fraction of Annotations "Part 5. Visualize the gff file in IGVYou can load the gff file into IGV or JBrowse, together with RNA-seq read alignment bam files. For instructions of running IGV and loading the annotation gff file, you can read under "part 4" of this document:/doc/RNA-Seq-2019-exercise1.pdfAppendix: Training Augustus modelRun Part 1 & 2.In the same screen session, set up Augustus environment.cp -r /programs/Augustus-3.3.3/config/ /workdir/$USER/augustus_configexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libexport AUGUSTUS_CONFIG_PATH=/workdir/$USER/augustus_config/export LD_LIBRARY_PATH=/programs/boost_1_62_0/libexport LC_ALL=en_US.utf-8export LANG=en_US.utf-8export PATH=/programs/augustus/bin:/programs/augustus/scripts:$PATHThe following commands will convert the MAKER round 1 results to input files for building a SNAP mode.mkdir augustus1cd augustus1gff3_merge -d ../pyu_rnd1.maker.output/pyu_rnd1_master_datastore_index.logAfter this step, you will see a new gff file pyu_rnd1.all.gff from round 1.## filter gff file, only keep maker annotation in the filtered gff fileawk '{if ($2=="maker") print }' pyu_rnd1.all.gff > maker_rnd1.gff##convert the maker gff and fasta file into a Genbank formated file named pyu.gb ##We keep 2000 bp up- and down-stream of each gene for training the modelsgff2gbSmallDNA.pl maker_rnd1.gff pyu_contig.fasta 2000 pyu.gb## check number of genes in training setgrep -c LOCUS pyu.gb## train model## first create a new Augustus species namednew_species.pl --species=pyu## initial trainingetraining --species=pyu pyu.gb## the initial model should be in the directoryls -ort $AUGUSTUS_CONFIG_PATH/species/pyu##create a smaller test set for evaluation before and after optimization. Name the evaluation set pyu.gb.evaluation.randomSplit.pl pyu.gb 200mv pyu.gb.test pyu.gb.evaluation# use the first model to predict the genes in the test set, and check theresultsaugustus --species=pyu pyu.gb.evaluation >& first_evaluate.outgrep -A 22 Evaluation first_evaluate.out# optimize the model. this step is very time consuming. It could take days. To speed things up, you can create a smaller test set# the following step will create a test and training sets. the test set has 1000 genes. This test set will be splitted into 24 kfolds for optimization (the kfold can be set up to 48, with processed with one cpu core per kfold. Kfold must be same number as as cpus). The training, prediction and evaluation will beperformed on each bucket in parallel (training on hh.gb.train+each bucket, then comparing each bucket with the union of the rest). By default, 5 rounds of optimization. As optimization for large genome could take days, I changed it to3 here.randomSplit.pl pyu.gb 1000optimize_augustus.pl --species=hh --kfold=24 --cpus=24 --rounds=3 --onlytrain=pyu.gb.train pyu.gb.test >& log &#train again after optimizationetraining --species=pyu pyu.gb# use the optionized model to evaluate again, and check the resultsaugustus --species=pyu pyu.gb.evaluation >& second_evaluate.outgrep -A 22 Evaluation second_evaluate.outAfter these steps, the species model is in the directory/workdir/$USER/augustus_config/species/pyu.Now modify the following values in the file: maker_opts.ctlmaker_gff= pyu_rnd1.all.gffest_pass=1 # use est alignment from round 1protein_pass=1 #use protein alignment from round 1rm_pass=1 # use repeats in the gff fileaugustus_species=pyu # augustus species model you just builtest= # remove est file, do not run EST blast againprotein= # remove protein file, do not run blast againmodel_org= #remove repeat mask model, so not running RM againrmlib= # not running repeat masking againrepeat_protein= #not running repeat masking againest2genome=0 # do not do EST evidence based gene modelprotein2genome=0 # do not do protein based gene model.pred_stats=1 #report AED statsalt_splice=0 # 0: keep one isoform per gene; 1: identify splicing variants of the same genekeep_preds=1 # keep genes even without evidence support, set to 0 if noRun maker with the new augustus model/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd3 >& log3 &Create gff and fasta output files:Use the following command to create the final merged gff file. The “-n” option would produce a gff file without genome sequences:gff3_merge -n -dpyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.log>pyu_rnd3.noseq.gff fasta_merge -d pyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.logAfter this, you will get a new gff3 file: pyu_rnd3.noseq.gff, and protein and transcript fasta files. To make the gene names shorter, use the following commands:maker_map_ids --prefix pyu_ --justify 8 --iterate 1 pyu_rnd3.all.gff > id_map map_gff_ids id_map pyu_rnd3.all.gffmap_fasta_ids id_map pyu_rnd3.all.maker.proteins.fastamap_fasta_ids id_map pyu_rnd3.all.maker.transcripts.fasta。
【精品】amber12install(学习资料)

amber12 GNU&intel 编译安装2012-12-18 22:26:07| 分类:默认分类|举报|字号订阅一、amber12 gnu编译安装方法:1. 换源:cd /etc/aptsed '/universe$/ s/^.//' sources.list > /tmp/sources.listsudo mv sources.list sources.list.backupsudo mv /tmp/sources.list sources.listcd ~sudo apt-get update2. 安装必要程序:sudo apt-get install gcc g++ libxt-dev libxext-dev flex gfortran tcshlibnetcdf-dev patch csh bison xorg-dev zlib1g-dev libbz2-devmpi我们选择安装openmpi-dev,即sudo apt-get install openmpi-dev openmpi-bin3. 解压ambertools12和amber12:cd /home/zheng/softwares/ambertar xfj AmberTools12.tar.bz2tar xfj Amber12.tar.bz24. 设置环境变量:cd amber12echo "export AMBERHOME=$PWD" >> ~/.bashrcecho "export PATH=$PATH:$AMBERHOME/bin" >> ~/.bashrcecho "export DO_PARALLEL="mpirun -np 24"" >> ~/.bashrc (N是核心数。
ANSYSWorkbench12.1官方中文培训教程--WB12.1Mechanical模块实例

Workbench -Mechanical Introduction Introduction作业2.121ANSYS Mechanical基础2.1作业Supplement •第一个作业包含了大量的信息,练习时,可以更加的熟悉基本的Workbench Mechanical功能(菜单位置等),因此后续的作业就包含了较少的细节描述。
较少的细节描述•整个作业的菜单路径将被记录如下:“First pick > Second pick > etc.”.作业Supplement•使用Stress Wizard ,建立求解结构模型的应力、变形和安全因子。
问题描述•问题描述:–模型是由Parasolid 文件得到的一个控制箱盖子(如图所示)。
盖子假设是在一个外压下使用(1 Mpa/145 psi )。
–盖子是由铝合金制成的。
–我们的目标是确定这个部件能在假设的环境下使用。
作业Supplement •在深孔施加约束,接合面及内表面使用无摩擦支撑约束.–无摩擦支撑约束是一种施加在整个面的法无摩擦支撑约束是种施加在整个面的法线方向上的约束.除了支撑面的正、负法线方向, 该约束允许其余各方向的平移. 这是一种保守的方法.种保守的方法作业Supplement •载荷: 载荷为1MPa的压力,作用在外壳的17个外表面上.作业Supplement •打开Project page(项目页)•在Units菜单中确定:–Project 单位设为US Customary (lbm, in, s, F, A, lbf, V).–选择“Display Values in Project Units”作业Supplement1.在Toolbox 中建立一个StaticStructural 系统(通过拖放或点击鼠标右键选择)2.在Geometry 子模块上点击鼠标右键选择Import Geometry,选I G选择导入“Cap_fillets.x_t”文件作业Supplement3.双击Model 打开Mechanical application.4.当Mechanical application 打开模型时,会在图形窗中显示出来而窗口中显示出来,而Mechanical ApplicationWizard 会显示在右侧。
Matlab实现bwlabel函数(区域标记)功能

Matlab实现bwlabel函数(区域标记)功能算法分析1. 图像预处理。
对⼆值图进⾏形态学开操作,开操作能去掉细⼩的块,平滑⽬标区域边界且保持⾯积不变2. 遍历⼆值图矩阵,寻找⽬标区域且未被标记的点,若当前像素未标记且当前位置像素点为1(⼆值图的⽬标区域)3. 将该⽬标点⼊队,并标上区域编号label4. BFS,对步骤1中的求得8邻域的像素点,如果该点未越界,是⽬标点(灰度值为1)并且未被标记过则⼊队,并且标上当前编号label,队列中每个点的8领域判断完则出队,当队列为空时⼀个连通区域标记完成,label+15. 回到步骤1,直到⼆值图遍历完成6. 输出,将标记矩阵以彩⾊标记的形式输出,循环结束时label的值多加了1,退出循环时,label的值减1才是该图中的连通区域的个数伪代码function [conn,num] = mybwlabel( I )% 使⽤BFS对⼆值图区域标记% 输⼊:I是⼆值图矩阵% 输出:tmp是标记矩阵,num是连通区域个数[m n] = 计算⼆值图的⼤⼩conn = 开辟⼀个和⼆值图同样⼤⼩的标记矩阵label = 给区域编号设初值1queue = 定义⼆维数组⽤于存放⼀个连通区域的点的坐标(模拟队列)neighbour = 8邻域(坐标⽆序)遍历⼆值图如果当前点属于⽬标区域且未标记queue = 当前点坐标⼊队conn(i,j) = 给该点赋值区域编号队列不空index = 取出队⾸坐标8邻域搜索cur_index = 当前点邻域坐标如果当前坐标未越界如果当前像素邻域像素为1并且标记图像的这个邻域像素没有被标记标记当前点当前坐标⼊队(矩阵合并)队⾸坐标出label = 区域编号加1num = 连通区域的个数为label-1end代码function [conn,num] = mybwlabel( I )% 对⼆值图区域标记% 输⼊:I是⼆值图矩阵% 输出:tmp是标记矩阵,num是连通区域个数[m n] = size(I);conn = zeros(m,n); %标记矩阵label = 1;queue = []; %⽤⼆维数组模拟队列%和当前像素坐标相加得到⼋个邻域坐标(坐标⽆序)neighbour = [-1 -1;-1 0;-1 1;0 -1;0 1;1 -1;1 0;1 1];for i = 2 : m-1for j = 2 : n-1if I(i,j) == 1 && conn(i,j) == 0 %属于⽬标区域且未标记才处理conn(i,j) = label;queue = [i;j]; %记录当前点坐标while ~isempty(queue) %队列不空,这⾥没有出队操作,队列中的每⼀个元素的8领域都判断⼀遍index = [queue(1,1),queue(2,1)];for k = 1 : 8 %8邻域搜索cur_index = index + neighbour(k,:); %加上每⼀⾏的坐标得到周围8邻域的坐标点if cur_index(1) >= 2 && cur_index(1) <= m - 1 && cur_index(2) >= 2 && cur_index(2) <= n - 1 %防⽌坐标越界if I(cur_index(1),cur_index(2)) == 1 && conn(cur_index(1),cur_index(2)) ==0 %如果当前像素邻域像素为1并且标记图像的这个邻域像素没有被标记,那么标记conn(cur_index(1),cur_index(2)) = label;queue = [queue [cur_index(1);cur_index(2)]];endendendqueue(:,1) = []; %队列中第⼀列的坐标出队endlabel = label+1;endendendnum = label - 1; %连通区域的个数(减去背景这个连通区域)end实验结果% 调⽤⽰例:I = imread('rice.png');Ib = im2bw(I);[B,num] = mybwlabel(Ib);subplot(1,3,1),imshow(I),title('原图');subplot(1,3,2),imshow(Ib),title('⼆值图');subplot(1,3,3),imshow(label2rgb(B)),title('区域标记');连通区域个数:图像预处理去噪,由上图中⼆值化之后,⼆值图中有很多噪声点,可以通过形态学开操作去除这些点SE = strel('disk',3); %定义⼀个圆形结构元Ib = imopen(Ib,SE); %开操作去除噪声开操作之后的效果连通区域个数:开操作之后去除噪声,连通区域个数明显减少实验分析在标记连通区域之前可以通过形态学操作,去除噪声点对⼆值图中每⼀个像素点的8邻域,都遍历⼀遍,将8邻域中属于⽬标区域的点的坐标都⼊队,对于当前满⾜条件的像素点判断结束时,就会标记完⼀个连通区域在遍历8领域时为避免重复标记,需要多次判断当前是否已经标记。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Workb kbench-Si Simulatio on Emag
Click on the “New Plane” icon to initiate creation of plane to define direction of PM polarization
January 24, 2010 © 2010 ANSYS, Inc. All rights reserved. ANSYS, Inc. Proprietary Inventory #002406 1-12
Training Manual
Workb kbench-Si Simulatio on Emag
– One coordinate system will be used to define the direction of polarization of the permanent magnet. It will be most convenient to create this coordinate system in DM. Coordinate system “PM_polarization” will have its x axis pointing in the “average radially outward” direction of the permanent magnet outward magnet. Polarization may only be defined only in the positive or negative x direction, so anytime the direction of polarization is not aligned with the Global x direction, a special properly oriented coordinate system must be created for to d fi polarization. define l i i – Another cylindrical coordinate system will be needed to specify the odd periodic boundary condition. It will be most convenient to create this coordinate system in Mechanical. Mechanical
Electromagnetic Analysis
• Objectives
– Set up a transient analysis of one half of the length of a single periodic sector of a rotating machine. Odd periodic boundary conditions will be used to mimic presence of adjacent periodic sectors. – The rotor contains a single g permanent p magnet which is poled radially outward and attached to a permeable ring. – The stator is magnetically permeable structure containing 3 poles. It has no coils in this exercise; a stranded conductor option that can be used to model coils will not be available until version 13 . – Define 90⁰ rotation of the rotor which occurs over a one second time interval. – The PM will be electrically conductive; eddy currents and Joule heating will be calculated calculated. – Net rotor torque will be calculated.
Workb kbench-Si Simulatio on Emag
Start Workbench and Read in Geometry
January 24, 2010 © 2010 ANSYS, Inc. All rights reserved.
ANSYS, Inc. Proprietary
Inventory #002406 1-4
Empty coil slots
Inventory #002406 1-2
Module 5
Electromagnetic Analysis
• Procedure
– WB supports only magnetostatic analyses. Transient analyses, especially those including motion effects, will require command objects. – Rigid body rotation or translational motion can be simulated with an undocumented (even in the traditional ANSYS environment) e o e t) set o of commands (“ARMINT” feature). – Insertion of command objects that set up motion effects is largely automated with a special wizard (supplied courtesy of CADFEM). – “Ladder mesh” requirement will be necessary to use ARMINT feature.
Double click on “Geometry” to bring up the Design Modeler GUI
January 24, 2010 © 2010 ANSYS, Inc. All rights reserved.
ANSYS, Inc. Proprietary
Inventory #002406 1-10
Electromagnetic Analysis
Training Manual
Workb kbench-Si Simulatio on Emag
User-Defined Coordinate Systems for PM Polarization and Periodic Boundary Conditions
Mesh normal to direction of motion ti must t match (and does)
ANSYS, Inc. Proprietary Inventory #002406 1-3
Module 5
Electromagnetic Analysis
Training Manual
ANSYS, Inc. Proprietary
Inventory #002406 1-5
Module 5
Electromagnetic Analysis The Project Page Loads…
Training Manual
Workb kbench-Si Simulatio on Emag
Hold down LMB to drag a Magnetostatic Analysis System into the Project Schematic
January 24, 2010 © 2010 ANSYS, Inc. All rights reserietary
Inventory #002406 1-9
Module 5
Electromagnetic Analysis
Training Manual
Workb kbench-Si Simulatio on Emag
January 24, 2010 © 2010 ANSYS, Inc. All rights reserved.
ANSYS, Inc. Proprietary
Inventory #002406 1-6
Module 5
Electromagnetic Analysis
Training Manual
Workb kbench-Si Simulatio on Emag
Right click on Geometry to browse for SYS.agdb
January 24, 2010 © 2010 ANSYS, Inc. All rights reserved.
ANSYS, Inc. Proprietary
Inventory #002406 1-7
Module 5
Module 5
Electromagnetic Analysis
Training Manual
Workb kbench-Si Simulatio on Emag
Begin to define coordinate system used to define PM polarization
Right click on “Ring” to hide this body and expose the inner cylindrical surface of the PM
EMAG Tutorial 5: Rotating Machine
Tutorial 5
January 24, 2010 © 2010 ANSYS, Inc. All rights reserved.