fda发布咀嚼片关键质量属性指导原则(1)
FDA药-械组合产品桥接指导原则介绍

【审评规范】FDA药-械组合产品桥接指导原则介绍萧惠来国家药品监督管理局药品审评中心,北京100022摘要:美国食品药品管理局(F D A)的“供企业用药-械和生物制品-器械组合产品的桥接指导原则(草案)”详细描述 了药-械组合产品注册申请人,利用另外的开发方案产生的信息,作为拟开发产品的注册资料(即桥接),替代试验研究资 料的方法,支持拟申报产品的批准;推荐渐进式5步骤法,确定桥接策略和信息需求并列举了3个示例予以解读。
详细介绍 该指导原则的内容,期望为中国药-械组合产品研发和监管开辟新思路,建议在合适的条件下可考虑利用“桥接”方法减少 试验研究,加速研宄进程,缩短研宄周期,节省研宄经费,促进药-械组合产品的开发。
关键词:美国食品药品管理局:药-械组合产品:桥接:指导原则中图分类号:R951 文献标志码: A 文章编号:1674-6376 (2021) 03-0461-07DOI :10.7501/j.issn. 1674-6376.2021.03.001Introduction to FDA's bridging for drug-device combination products guidanceXIAO HuilaiCenter for Drug Evaluation, National Medical Products Administration, Beijing 100022, ChinaAbstract: The FDA's Bridging for Drug-Device and Biologic-Device Combination Products Guidance for Industry (Draft) described in detail that the applicant for registration of drug-device combination products can use the information developed by another development program as the registration information of the product to be developed (i. e. bridging) and replace the experimental research data to support the approval of the product to be applied for. That is to say, the stepwise five step approach is recommended to determine the bridging strategy and information requirements are determined, and three examples are given to interpret. This paper introduces the guidance in detail, hoping to open up new ideas for the research and development as well as supervision of drug-device combination products in China. In suitable conditions, it can be considered to use "bridging" method to reduce experimental research, speed up the research process, shorten the research cycle, save research funds, and promote the development of drug-device combination products.Key words: FDA; drug-device combination product; bridging; guidance美国食品药品管理局(FDA)于2019年12月发 布了“供企业用药-械和生物制品-器械组合产品的 桥接指导原则(草案)”[1]。
FDA数据完整性法规解读与初步认知

》第9节:良好文件规范中说明:只要能够达到GMP的要求,纸质的追踪记录也 13 可以被接受。(任何删改都经过申请、审核、批准)(切记与报警系统一致) 13
指南解读
问题
FDA 回答 及要 求
Q7:审计追踪应多长时间审查一次?
FDA建议,采集关键数据变更的审计追踪应在每次记录和最终批准记录前审查。需要定 期审查的审计追踪应包括但不限于以下内容:最终产品检验结果的更改历史、样品运行序 列的更改、样品标识的更改、关键工艺参数的更改。
解读与 讨论
1)按文件要求,复核检验记录的复核人应需要对电子记录和审计追踪进 行进行核实。(做不到怎么办?) 2)部门负责人、QA专人定期审核,审核期限根据参数影响评估。
15
15
指南解读
问题
Q1:术语定义——“静态记录”和“动态记录”定义
静态用于表示固定数据文件,例如纸质记录或电子图像;
FDA 回答 及要 求
7
7
数据完整性基础定义
原始性(original)
原始数据应当被审核; 应当留存原始数据和/或认证的真实、准确副本,副本 保存了原始数据的内容及含义; 在记录留存期内,原始记录应当完整、持久而且容易 获得、易读;
原始记录应当被审核; 数据审核应当被记录(纸质记录前面)
电子数据的源记录应当被审核
解读 与讨 论
14
14
指南解读
问题
Q8.应由谁来审查审计追踪?
审计追踪被认为是相关记录的一部分。根据CGMP负责记录审查的人员, 应审查采集与记录相关的对关键数据更改的审计追踪,如他们审查其它记
FDA回 答及要 求
录一样(例如,§§ 211.22(a),211.101(c),211.194(a)(8) 例如,所有 生产和控制记录,包括审计追踪,必须由质量部门审核和批准(§ 211.192)。这与FDA对企业在审核数据时在纸上划痕标注的预期是一样 的。
美国食品药品监督管理局(FDA)发布3D打印医疗产品技术指导意见

3D打印医疗产品迅速发展,个性化的医疗器械和能够显著改善健康状况的创新药物使患者从中受益。
美国食品药品监督管理局(FDA)已经审查了目前市面上100多个由3D打印技术生产的医疗产品。
为跟上技术革新,FDA正努力提供更全面的监管途径,帮助促进基于增材制造技术的安全有效地创新。
一、增材制造背景及视角FDA所制定的技术指导意见代表其对增材制造技术应用于医疗产品生产领域的初步意见。
(一)增材制造相关定义增材制造设备:“增材制造流水线的一部分,包括硬件、机器控制软件、数据处理软件和完成零件生产所必需的外围附件”。
制造周期:“一个或多个组件在增材制造系统内分层堆积的单次过程循环。
”制造准备软件:“用于将制造产品三维模型数据转换为可用于增材制造装备加工格式的处理软件。
”设计处理软件:“允许针对特定情况(例如患者匹配)修改医疗产品设计的计算机程序。
”(二)增材制造医疗产品介绍医疗产品制造行业最常用的相关技术有:粉体熔化成型:依靠激光或电子束选择性地熔化或烧结金属或聚合物粉末,逐渐成型。
立体光固化成型:通过特定波长与强度的激光选择性聚焦到光固化材料表面使之凝固。
熔融挤出成型:通过熔化位于热熔喷头的固体长丝,按照零件预定轨迹以固定的速率进行熔体凝固。
液态挤出成型:通过喷射液体使之固化(方法包括曝光、溶剂蒸发或其他化学过程)。
(三)增材制造技术指导意见概述本指导意见主要涉及设计和制造注意事项和产品测试注意事项两部分内容。
“设计和制造注意事项”涉及的环节应遵循产品质量体系(Quality System,QS)的要求。
此类要求依照产品的法定类别及适用于产品的相关法规决定。
“产品测试注意事项”描述了产品上市前应向FDA递交的上市前审批(Premarket Approval,PMA)申请、人道主义产品豁免(Humanitarian Device Exemption HDE)申请、新创请求和增材制造研究性产品豁免(Investigational Device Exemption,IDE)申请等信息。
FDA发布行业指南草案《ANDA提交质量管理规范》

FDA发布行业指南草案《ANDA提交质量管理规范》2018年伊始,FDA 即一并发布了两份文件,旨在简化和改善仿制药申请(ANDA)的提交和审评。
第一份文件是行业指南草案《ANDA 提交质量管理规范》(Good ANDA Submission Practices),强调了我们在仿制药申请中看到的可能导致审批延迟的常见缺陷。
第二份文件是《ANDA 评估质量管理规范》(Good ANDA Assessment Practices),其中概述了FDA 工作人员的ANDA 评估实践。
其中,行业指南草案《ANDA 提交质量管理规范》(Good ANDA Submission Practices)中,从专利和排他性缺陷、标签缺陷、产品质量缺陷和生物等效性缺陷四个角度对ANDA提交中的常见缺陷进行了系统的整理,用于指导制药公司及其代理人的ANDA提交工作,其中大体内容如下:Ⅰ专利和排他性缺陷A. 法律诉讼的文件和通知B. 解决或提起法律诉讼C. P IV证明通知D. 橙皮书中新增或修订的信息E. 对未经批准的ANDA进行的修改F. 商业营销通知Ⅱ标签缺陷A. 容器标签和纸盒标签样稿B. 容器标签和纸盒标签的颜色差别C. 标签格式D. 注射剂1. 包装类型2. 产品规格3. 套圈和瓶盖顶封Ⅲ产品质量缺陷A. 原料药1. 活性药物成分起始原料2. API 生产工艺3. 杂质a API表征信息b 实际和潜在杂质的致突变能力的安全性评估4. 可分离中间体的质量标准5. 特定关键质量属性的检测B. 制剂1. 建立关键质量属性2. 杂质的鉴别、控制和质量a 鉴别和控制杂质b 原料药或制剂中杂质超过相关限度阈值的安全限定3. 非活性成分a 引用非活性成分数据库进行说明b 仿制药制剂中非活性成分超出IID最大值的安全性说明4. 分析方法的验证C. 体外溶出度(生物药剂学)1. 当溶出测试不能标准化时,开发和验证内部溶出度测试方法2. 溶出度接受标准D. 设施1. 生产设备的标识2. 核查的准备3. 合同生产设备和CGMP的选择E. 商业生产工艺F. 微生物学考虑1. 过程中的生物负载测试和验收标准2. 细菌内毒素测试方法的描述和验证3. 支持延长存储时间的微生物数据Ⅳ生物等效性缺陷A. 生物分析研究数据B. 临床概要C. 特定产品指导原则的偏离D. 有关BE的信息和体内BE研究的安全性E. 配方和非活性成分的差异F. 依据21 CFR 314.99(b)的豁免要求本文转自:制剂汇网,转发仅为学习交流,如有侵权,请联系小编删除。
咀嚼片(化学药品)质量属性研究技术指导原则(征求意见)
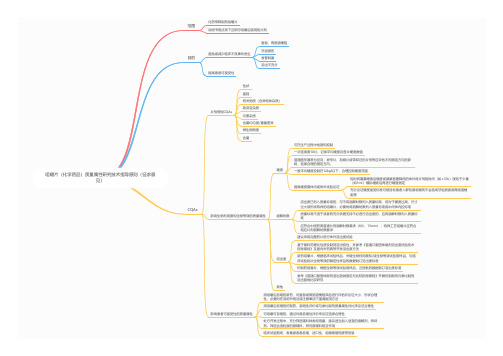
咀嚼片(化学药品)质量属性研究技术指导原则(征求意见)范围化药常释制剂咀嚼片说明书用法项下注明可咀嚼后服用的片剂目的避免或减少临床不良事件发生窒息、胃肠道梗阻牙齿损伤食管刺激溶出不充分CQAs提高患者可接受性片剂常规CQAs性状鉴别有关物质(含异构体杂质)致突变杂质元素杂质含量均匀度/重量差异微生物限度含量影响生物利用度和生物等效的质量属性硬度崩解时限溶出度其他影响患者可接受性的质量属性须咀嚼后吞咽的新药,对窒息或胃肠道梗阻风险进行评估并论证大小、形状合理性,必要时在说明书用法或注意事项下强调服用方法须咀嚼后吞咽的仿制药,吞咽性评价或与参比制剂质量属性对比来论证合理性可咀嚼可吞咽的,通过对易吞咽性评价来论证选择合理性处方开发过程中,充分筛选辅料种类和用量,建议适当加入适宜的崩解剂,矫味剂,筛选合适粒度的原辅料,研究原辅料相互作用临床试验期间,收集患者易吞咽、适口性、咀嚼难易程度等信息可在生产过程中检测和控制一次连续测10片,记录平均硬度及各片硬度数值普通圆形通常为径向;异形片、刻痕片或带印记的片剂等应评估不同测定方向的影响,选择合理的测定方向。
一般平均硬度控制在12kgf以下,合理控制硬度范围提高硬度需体内或体外试验论证短时间暴露唾液后硬度或崩解显著降低的体外将片剂短时间(如≤30s)浸泡于少量(如1ml)模拟唾液后再进行硬度测定充分论证硬度接受标准可被目标患者人群轻易咀嚼而不会造成牙齿损害或胃肠道梗阻等溶出度已列入质量标准的,可不将崩解时限列入质量标准,但对于硬度过高、尺寸过大或形状特异的咀嚼片,必要时将崩解时限列入质量标准或中间体内控标准质量标准不适于或者有充分依据支持不必进行溶出度的,应将崩解时限列入质量标准应符合中国药典普通片剂崩解时限要求(6片,15min);特殊工艺咀嚼片应符合相应片剂崩解时限要求建议采用完整药片进行体外溶出度试验基于原料药理化性质及制剂溶出特性,并参考《普通口服固体制剂溶出度试验技术指导原则》及国内外药典等开发溶出度方法新药咀嚼片,根据临床试验样品、关键生物利用度和/或生物等效试验用样品、与临床试验批次生物等效的稳定性样品的数据制订溶出度标准仿制药咀嚼片,根据生物等效试验用样品、注册批的数据制订溶出度标准参考《普通口服固体制剂溶出曲线测定与比较指导原则》开展仿制制剂与参比制剂溶出曲线比较研究。
美国FDA指导原则CPGSEC555500所有食品卫生检查分类英文原版

美国FDA指导原则CPGSEC555500所有食品卫生检查分类英文原版The U.S. Food and Drug Administration (FDA) provides guidance for food establishments on how to maintain food safety. One such important guidance document is the Code of Federal Regulations (CFR) Title 21, Part 110, which outlines the Current Good Manufacturing Practice (CGMP) regulations for food processors.Under the CGMP regulations, food establishments are required to follow specific guidelines to ensure that the food producedis safe for consumption. These guidelines cover a wide range of areas, including personnel training, sanitation practices, equipment maintenance, and recordkeeping.Here is a breakdown of the different food safety inspection categories outlined in the FDA's guidance document, CPG Sec. 555.500:1. Personnel: This section focuses on the training and health requirements for individuals who handle food. It includes guidelines on cleanliness, handwashing, and the use ofprotective clothing.2. Plant and grounds: This section covers the maintenance and cleanliness of the food processing facility, including building construction, water supply, plumbing, lighting, and ventilation.3. Sanitary operations: This section outlines the practices necessary to prevent contamination during food processing. It includes guidelines on cleaning and sanitizing equipment and utensils, preventing cross-contamination, and controlling pests.4. Sanitary facilities and controls: This section addresses the design and maintenance of facilities like restrooms, handwashing stations, and employee break areas. It also includes guidance on controlling physical hazards, such as glass or metal fragments.5. Equipment and utensils: This section provides guidelines for the design, construction, and maintenance of equipment used in food processing. It also covers the proper cleaning and storage of utensils.6. Processes and controls: This section focuses on the control measures necessary to ensure the safety of specific food products. It includes guidelines for pasteurization, heat treatment, and other processing methods to eliminate or reduce pathogens.7. Warehousing and distribution: This section covers the storage and transport of food products. It includes guidelines on temperature control, pest control, and preventing contamination during distribution.8. Defect action levels: This section provides guidance on acceptable levels of quality defects in certain food products.It helps to ensure that products with significant defects are not sold to consumers.9. Food additives: This section outlines the requirements for the safe use of food additives, including labeling and documentation.10. Color additives: This section provides guidelines for the use of color additives, including permitted colors and labeling requirements.。
实施“质量源于设计”的五个关键因素

实施“质量源于设计”的五个关键因素在药品⽣产⾏业,对药品质量的控制经历了从“检验决定质量”模式到“⽣产决定质量”模式并逐渐向“质量源于设计”模式的发展。
质量源于设计的相关理念始源于20世纪70年代Toyota为提⾼汽车质量⽽提出的创造性的概念,并经过在通信、航空等领域的发展逐渐形成。
美国⾷品药品监督管理局(FDA)已逐渐将“质量源于设计”的理念使⽤贯穿于药品诞⽣周期的全过程的各个阶段,其⽬的在于更好地控制药品质量及维护患者⽤药的安全性。
1、 “质量源于设计”简介质量源于设计(Quality by Design,QbD)这⼀理念⾸先出现在⼈⽤药品注册技术规定国际协调会议(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)发布的Q8中,其定义为“在可靠的科学和质量风险管理基础之上的,预先定义好⽬标并强调对产品与⼯艺的理解及⼯艺控制的⼀个系统的研发⽅法”。
ICH Q8指出,质量不是通过检验注⼊到产品中,⽽是通过设计赋予的。
要获得良好的设计,必须增加对产品的认知和对⽣产的全过程控制。
实施QbD是将过程分析技术与风险管理综合应⽤于药品⼯艺开发的过程,它的⽬的不是消灭⽣产过程中的偏差,⽽是建⽴⼀种可以在⼀定范围内调节偏差来保证产品质量稳定性的⽣产⼯艺。
通过QbD可以找出这个范围,并建⽴设计空间(Design Space)。
ICH Q8对设计空间的定义为“已被证明有质量保障作⽤的物料变量和⼯艺参数的多维组合和相互作⽤”,就是各种影响产品质量的关键因素和参数的组合。
设计空间允许企业在研究的基础上确定⼀个可以保证产品质量的操作空间。
⼀旦确定产品⽣产的设计空间,则在此设计空间中的各种⼯艺等⽅⾯的变化,⽆需进⾏进⼀步的注册审批,减少上市后的变更申报。
咀嚼片化学药品质量属性研究技术指导原则试行溶瘤病毒产品药学研究与评价技术指导原则试行(一)

咀嚼片化学药品质量属性研究技术指导原则试行溶瘤病毒产品药学研究与评价技术指导原则试行(一)咀嚼片化学药品质量属性研究技术指导原则试行咀嚼片是一种常见的口服制剂形式,其可以直接在口腔中咀嚼,释放药物成分,更容易被机体吸收,因此得到广泛应用。
然而,咀嚼片化学药品的质量属性研究一直是制药研发和质量控制的关键问题之一。
为了保证制剂的有效性和安全性,需要对咀嚼片药品的质量属性进行全面、精细的研究,制定科学的技术指导原则。
一、咀嚼片的性质及其对药品质量属性的影响咀嚼片是一种特殊的制剂形式,它比普通口服制剂在药物释放、药物转运和药效表现等方面具有独特的优势。
因此,在咀嚼片的研发和质量控制中,需要考虑以下因素:1.咀嚼片的组成和结构2.咀嚼片的口感和咀嚼性3.咀嚼片的溶解度和离子强度4.咀嚼片的药物释放速度和药效持续时间以上因素都会对咀嚼片的药物质量属性产生影响。
因此,在研究和评价咀嚼片化学药品的质量属性时,需要全面考虑这些影响因素。
二、咀嚼片化学药品质量属性研究技术指导原则试行为了提高咀嚼片化学药品的质量和有效性,需要制定科学合理的研究技术指导原则。
以下是一些可能的技术指导原则:1.药品质量属性研究应注重口感和咀嚼性口感和咀嚼性是咀嚼片区别于其他制剂的重要特点。
因此,在研究药品质量属性时,需要注重这些特点的评价。
例如,可以采用口感评估仪、颗粒度分析仪等工具,对咀嚼片的口感和咀嚼性进行精细化评估。
2.药品质量属性研究应注重药物释放速度和药效持续时间咀嚼片的药物释放速度和药效持续时间是咀嚼片制剂性能的重要指标。
在研究药品质量属性时,需要注重这些指标的评价。
可采用离体、原位等不同方法,对药物释放速度和药效持续时间进行研究和评价。
3.药品质量属性研究应注重溶解度和离子强度咀嚼片的溶解度和离子强度是药物在体内吸收和转运的重要影响因素。
因此,在研究药品质量属性时,需要注重这些指标的评价。
应注意选取合适的试验条件和测定方法,提高测试的准确性和可重复性。
美国FDA药物分析程序及方法验证指导原则(中文版)

药品及生物制品的分析方法和方法验证指导原则目录1.介绍...................... (1)2.背景..................... .. (2)3.分析方法开发. ..................... . (3)4.分析程序内容.............................................. ......... ..................................... .. 3A.原则/范围 (4)B.仪器/设备............................................. . (4)C.操作参数.............................................. .. (4)D.试剂/标准............................................. . (4)E.样品制备.............................................. .. (4)F.标准对照品溶液的制备............................................ .. (5)G.步骤......... ....................................... (5)H.系统适应性..... (5)I.计算 (5)J.数据报告 (5)5.参考标准和教材............................................ (6)6分析方法验证用于新药,仿制药,生物制品和DMF (6)A.非药典分析方法............................................. (6)B.验证特征 (7)C.药典分析方法............................................. .. (8)7.统计分析和模型 (8)A.统计 (8)B.模型 (8)8.生命周期管理分析程序 (9)A.重新验证 (9)B.分析方法的可比性研究............................................ . (10)1.另一种分析方法............................................... .. (10)2.分析方法转移的研究 (11)C.报告上市后变更已批准的新药,仿制药,或生物制品 (11)9.美国FDA方法验证............................................... . (12)10.参考文献前言本指导原则草案,定稿后,将代表美国食品和药物管理局(FDA)目前关于这个话题目前的想法。
高效液相色谱法检测德拉昔布咀嚼片含量方法的建立

高效液相色谱法检测德拉昔布咀嚼片含量方法的建立于泓潇,李思鸿,肖天石,张艺馨,杨雨齐,李佳瑞,杨洪亮,张秀英(东北农业大学动物医学学院,黑龙江哈尔滨150030)摘要:为建立检测德拉昔布咀嚼片含量的方法,本试验采用高效液相色谱法对德拉昔布咀嚼片和Deoms咀嚼片进行含量测定。
样品经粉碎与甲醇溶解,超声处理20min,0.22%m滤膜滤过后进样测定。
色谱条件:色谱柱Wasters C18 (4.6mm x250mm,5%m);流动相为磷酸盐缓冲液(pH=4.5):乙G(52:48,a/");检测波长252nm;流速1mLmin&柱温30e。
结果显示,拉昔布标以甲醇为溶剂,进样浓度在0.5-8%g/mL,标,样的线性关系良好(m=0.9992)。
仪器精密度为0.56%,日内差异为0.94%,日间差异为1.85%,溶液稳定性为0.57%,平均加样回收率为(99.68±0.53)%$所的高效量法、、专属性强,可于德拉昔布咀嚼量的$关键词:德拉昔布咀嚼片;高效液相色谱法&方法学研究中图分类号:S859.1文献标志码:A文章编号:0529—6005(2021)01—0092—04Establishment of the HPLC Method for Determination of Deracoxib Chewable TabletrYU Hong-xiaa,LI Si-hong,XIAO Tian-shi,ZHANG Yi-xin,YANG Yu-qi,LI Jin-pi,YANG Hong-Fang,ZHANG Xiu-ying(Co e geoeVeieeinaeyMedicine,NoeiheasiAgeicueiueaeUnieeesiiy,Haebin150030,China) Abstract:To establish a method to determine the content of deracoxib chewable tablets,this study determined the contents of deracoxib chewable tablets and deomaxx chewable tablets by high perormanco liquid chromatography(HPLC).The samples were crushed,and then dissolved by methanol.After the ultrasonic treatment for20min,the solution was filtrated by0.22%m fi/er membrane then analyzed by HPLC.The chromatographic conditions were as follows:the HPLC column was Wasters C18(4.6mm X250 mm,5%m);the mobile phase was phosphate buffer(pH=4.5) :acetonitrile(52:48)at a low rate1mL/min;the detection wavelength was252nm;the column Wmperatuo was kept at30e.Results showed that the deocoxin standards were dissolved by methanol,the linear range of the calibration curve for deocoxin was0.5-8%g/mL,and the linear relationship between sample concentration and peak area was good(r2=0.9992).The instrument precision was0.56%,inWr-day coefficient of variation was 0.94%and iniea-daycoe e i cienioeeaeiaiion was1.85%,ihesoeeenisiabieiiywas0.57%,and iheaeeeageeecoeeeywas(99.68±0.53)%.Theeesueisindicaieihaiihemeihod issimpeeand accueaiewiih high sensiiieiiyand specieiciiy,which coued beused iode-ieemineiheconienioedeeacoiib chewabeeiabeeis.Key word(:deeacoiib chewabeeiabeeis;high peeeoemanceeiquid cheomaiogeaphy;meihod eaeidaiion eeseaechCorre(pondnng aureor:ZHANGXiu-ying,E-maie:*********************.cn德拉昔布属于非笛体类解热镇痛抗炎药物,化学名为4内5-(3-氟内-甲)R-二氟甲基-1H-咪卩坐-1-基*,分子式为C17H14F3N3O3S,分子量为397.38,是环-2选择性抑制剂中的一员,也是第1款国及药物管理局(Fed and Dog Administration, FDA)批准用于犬的环氧酶内选收稿日期:2018—12—11作者简介:于泓潇(1995-),男,硕士生,研究方向为基础兽医学,E-mail:593200864@通信作者:张秀英,E-mail:zhanyxiuying@ 择性抑制剂[1],临床上用于治疗犬的骨关节炎症及疼痛,并可预防犬牙科与骨科手的疼痛[2]。
FDA发布咀嚼片关键质量属性指导原则(中英文对照)

FDA发布咀嚼片关键质量属性指导原则(中英文对照)This draft guidance, when finalized, will represent the current thinking of the Food and Drug Administration (FDA or Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the FDA staff responsible for this guidance as listed on the title page.该指南草案定稿后,代表FDA对这一主题当前的想法,并未赋予任何人权利,且对FDA或公众不具有任何约束力。
如果满足适用的法规和条例要求,也可以使用另外一种方法。
如需讨论另外一种方法,请与本指南封面中列出的FDA工作人员联系。
I. INTRODUCTIONI.引言Thisguidance provides manufacturers of chewable tablets for human use with theCenter for Drug Evaluation and Research’s (CDER) current thinking on thecritical quality attributes that should be assessed during the development ofthese drug products.2 This guidance also provides recommendationsabout submitting developmental, manufacturing, and labeling information forchewable tablets that must be approved by CDER before they can be distributed.The recommendations in this guidance apply mainly to new drug applications(NDAs), abbreviated new drug applications (ANDAs),3and certainchemistry, manufacturing, and controls (CMC) supplements to these applications.4some of therecommendations about the submission of developmental information may alsoapply to investigational new drugapplications (INDs). The recommendationsabout assessing critical quality attributes apply to all chewable tablets forhuman use, including non-application products.本指南向生产者提供了药品审评研究中心(CDER)对人用咀嚼片在研发过程中应评估的关键质量属性的当前想法2。
仿制药外形指导原则FDA

Size, Shape, and Other Physical Attributes ofGeneric Tablets and Capsules Guidance for IndustryU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)June 2015Pharmaceutical Quality/CMCSize, Shape, and Other Physical Attributes of Generic Tablets andCapsulesGuidance for IndustryAdditional copies are available from:Office of Communications, Division of Drug InformationCenter for Drug Evaluation and ResearchFood and Drug Administration10001 New Hampshire Ave., Hillandale Bldg., 4th FloorSilver Spring, MD 20993Phone: 855-543-3784 or 301-796-3400; Fax: 301-431-6353druginfo@/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htmU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)June 2015Pharmaceutical Quality/CMCContains Nonbinding RecommendationsTABLE OF CONTENTSI.INTRODUCTION (1)II.BACKGROUND (2)A.Differences in Size and Shape of Tablets and Capsules between a Reference Listed Drugand a Drug Product Subject to an Abbreviated New Drug Application (2)1.Size (2)2.Shape (3)3.Patient Factors (4)B.Other Physical Attribute Considerations (4)III.RECOMMENDATIONS (4)A.Size (4)B.Shape (5)C.Other Physical Attributes (6)D.Biowaivers (6)Size, Shape, and Other Physical Attributes of GenericTablets and CapsulesGuidance for Industry1This guidance represents the current thinking of the Food and Drug Administration (FDA or Agency) onthis topic. It does not create any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. Todiscuss an alternative approach, contact the FDA staff responsible for this guidance as listed on the titlepage.I. INTRODUCTION 简介Tablets and capsules are widely manufactured and prescribed and may provide a number of advantages over other dosage forms, including ease of storage, portability, ease of administration, and accuracy in dosing.片剂和胶囊剂是被广泛生产和使用的剂型,相对其它剂型,片剂和胶囊剂具有诸多优势,如存储方便,易于携带,方便服用,剂量精确等。
CAR-T细胞产品的质量控制和非临床研究——一般原则和关键问题-

一、引言嵌合抗原受体T(CAR-T)细胞是一种经基因工程化的T细胞,通常表达能识别特定肿瘤抗原的嵌合抗原受体(chimeric antigen receptor,CAR),进而激活免疫系统以消灭肿瘤。
CAR通常包含三个结构域:识别肿瘤相关抗原的胞外域(如scFv)、信号转导结构域(如CD3ζ)和一个或多个胞内共刺激结构域(如可源自CD28、4-1BB、OX40等)。
CAR-T细胞免疫疗法通常是从采集的患者血液中分离出T细胞,然后在GMP生产车间对其进行基因改造,通过逆转录病毒和慢病毒载体、转座系统(如SB转座系统)或直接将mRNA转导到T细胞内,使T细胞表面表达CAR。
在生产车间对这些T细胞进行扩增后将其回输到患者体内,这种经过修饰的T细胞能够特异、高效地识别和杀死肿瘤细胞,从而达到在治疗肿瘤的同时又避免对正常组织的损伤。
在靶向CD19分子治疗B细胞恶性肿瘤(急性B淋巴细胞白血病及B细胞淋巴瘤等)的临床试验中,CAR-T细胞显示出令人振奋的疗效及较少的副作用。
2017年8月31日,诺华(Novartis)公司宣布,美国食品药品监督管理局(Food and Drug Administration,FDA)已批准其开发的靶向CD19的CAR-T产品Tisagenlecleucel(曾用名为CTL019,商品名为Kymriah)上市,用于治疗B细胞前体急性淋巴性白血病(acute lymphoblastic leukemia,ALL)。
不到两个月后,FDA又批准了第二个CAR-T药物上市,商品名为Yescarta(axicabtagene ciloleucel),是Kite Pharma公司开发的同样靶向CD19的CAR-T疗法,用于治疗某些类型的大B细胞淋巴瘤的成年患者。
针对其他恶性肿瘤的CAR-T细胞治疗也在不断进展。
目前已有500多项关于CAR-T的临床试验正在进行。
对于恶性血液病,需要继续开展临床试验,以确定新靶点和新组合。
美国FDA发布了关于口腔崩解片及指导原则

美国FDA发布了关于口腔崩解片的指导原则(草案)Guidance for Industry OrallyDisintegrating Tablets一、FDA发布的口腔崩解片指导原则内容介绍1、背景介绍为提高药物使用的方便性,解决特定适应症和特殊患者人群的用药顺应性,制药企业开发了一类只需放在舌面上就可以咽下的产品。
该类产品被设计为当接触唾液后能够迅速崩解和溶解,而无需咀嚼、整片吞咽或用水服用。
这种给药方式最初希望能对儿童患者、老年患者、吞咽困难患者以及服药顺应性差的患者(如精神失常患者)带来益处。
1998年,FDA的CDER命名标准委员会根据对早期该类产品的审评情况,首次将该类产品定义为一种新的剂型——口腔崩解片(Orally disintegrating tablet,ODT)。
其定义如下:口腔崩解片是一种含药的固体制剂,当放在舌面上时,可以迅速地、通常在几秒钟内崩解。
早期开发的该类产品的特点包括片重低、尺寸小、所含组分溶解性好以及崩解迅速,这些特点支持这类产品的使用目的。
然而,当后续的制药企业使用不同的生产技术和方法生产出另外的产品时,许多后续产品显示出与早期产品较大的变异性。
由于这些产品特性的变化可能会对其特定用途的适用性产生影响,因而FDA提出了该针对制药企业的指导原则。
2、讨论该指导原则中给出的建议是基于该类产品最初定义的目的以及CDER对采用该剂型的NDAs和ANDAs申请的经验做出的。
FDA针对申报单位递交的申报资料进行了调研,完成了一篇文献综述,并收集了实验室研究的信息,结果显示:尽管这些产品的崩解时间在几秒钟到1分钟以上,但绝大多数产品的崩解时间皆在大约30秒钟或更少。
代表了不同生产技术、不同尺寸和片重以及不同崩解方法的多种产品表明相对快速的崩解时间较易实现。
标称为口腔崩解片的产品必须与该类产品的特征相匹配(在唾液中迅速崩解,无需咀嚼或服用时使用液体)。
基于最初产品的基本原则和CDER的经验,FDA建议:除最初的定义外,口腔崩解片应被看作一种能在口腔中迅速崩解的固体口服制剂,当使用USP崩解时限实验方法或其他方法时,体外崩解时间应在大约30秒钟或更短。
孟鲁司特钠咀嚼片制剂学研究

整粒参数
整粒的目的是使颗粒粒度分布均 匀,控制好整粒的筛网目数和筛 网振幅等参数。
压片参数
压片时控制好压力和片重等参数, 以保证片剂的硬度和含量均匀性。
工艺验证与优化
验证方案
制定详细的工艺验证方案,包括验证目的、方法、 评价标准等。
验证结果分析
对验证数据进行统计分析,评估工艺的可行性和 可靠性。
ABCD
临床需求
针对不同疾病和患者情况,需选择合适的剂 型以满足临床需求。
市场因素
剂型选择还需考虑市场接受度和竞争情况, 以实现良好的市场表现。
03
孟鲁司特钠咀嚼片处方设计
主药成分的选择与剂量确定
主药成分
孟鲁司特钠,是一种口服的白三烯受 体拮抗剂,用于治疗哮喘和慢性阻塞 性肺病。
剂量确定
根据临床试验和文献资料,确定孟鲁 司特钠的剂量范围,以满足治疗需要 并确保安全性。
安全性分析
对使用孟鲁司特钠咀嚼片过程中出现的不良反应进行监测和分析,评估其安全性。
感谢您的观看
THANKS
用途
主要用于预防和治疗哮喘、慢性阻塞性 肺疾病(COPD)等呼吸道疾病,以及 减轻过敏性鼻炎症状。
药物发现与研发历程
孟鲁司特钠由默克公司研发,最初是作为一种新型非甾体抗炎药进行研发。
在研发过程中,科学家发现其具有强大的白三烯受体拮抗作用,因此被重新定位并开发为抗哮喘药物。
经过临床试验验证其疗效和安全性后,孟鲁司特钠于1998年获得美国食品药品监督管理局(FDA)批准 上市。
包装材料
选用符合规定的包装材料,确保 包装密封性、阻光性、防潮性等 符合要求。
质量检测方法研究
含量测定
建立准确可靠的含量测定方法,确保制剂中孟鲁司 特钠的含量符合规定。
咀嚼片化学药品质量属性研究技术指导原则试行(一)

咀嚼片化学药品质量属性研究技术指导原则试行(一)随着科技的不断进步,医药行业得到了空前的发展和迅速的增长,呈现出多样化的形态。
化学药品的研发和生产水平也日益提高,其中咀嚼片是一类较为特殊的药品,它的品质属性研究也成为了医药品质研究的重要方向。
本文将重点讨论咀嚼片化学药品质量属性研究技术指导原则试行。
一、咀嚼片化学药品的特点咀嚼片是一种具有明显特点的药品。
首先,它的口感、呈味、口感和口感因素更丰富;其次,它需要兼顾咀嚼功能、膳食特点和给药剂量准确性;还有它具有更高的毒副作用和过敏反应的危险性,因此,对于咀嚼片的质量属性研究是非常重要的。
二、咀嚼片药品质量研究的技术指导原则1、选用合适的分析方法及技术在咀嚼片的质量研究中,分析方法的选择是非常关键的。
因为其物理、化学及药剂学特性差异,需要在质量研究中采取不同的分析方法来进行测试。
包括的常用方法有:高效液相色谱法、气相色谱法、储存稳定性试验等。
2、注意对环境因素及样品的存储咀嚼片药物对环境和存储条件的敏感度较大,因此,进行质量属性研究需要注意对环境温湿度、光照和放置位置的控制。
同时,对于样品的存储也有着严格的要求,需要保证样品隔绝空气和湿度,避免受潮、变质、失效等。
3、进行质控计划的制定咀嚼片药物的质控计划的制定是质量属性研究中不可缺少的一步。
主要内容包括:制定药品质量控制标准、培训检验员以及质量记录和质量分析报告的管理等。
4、建立质量评价标准咀嚼片药品的质量评价标准包括以下方面:外观和理化性质、保质期和稳定性、病人使用的舒适性、药品的有效性和安全性、毒副作用和不良反应的监测等。
三、小结本文重点讨论了咀嚼片化学药品质量属性研究技术指导原则。
通过选用合适的分析方法、注意环境因素及样品的存储、进行质控计划的制定、建立质量评价标准等多方面的方法,可以达到有效提升咀嚼片药品的质量属性和适用性,对于其进一步的研究和生产也有着积极的意义和作用。
咀嚼片硬度要求标准 -回复

咀嚼片硬度要求标准-回复咀嚼片硬度是指咀嚼片在一定条件下对压力的抵抗能力。
它是制定咀嚼片质量标准以及确保患者正常使用的重要因素之一。
本文将从咀嚼片硬度的定义、测试方法、影响因素以及相关标准要求等方面进行详细阐述。
一、咀嚼片硬度的定义咀嚼片硬度可以简单地理解为咀嚼片在咀嚼过程中需要施加的力量。
它反映了咀嚼片在口腔中的稳定性和耐咀嚼性能,直接影响患者的用药体验和治疗效果。
咀嚼片硬度的测试通常采用硬度计或者牙套等工具来进行。
二、咀嚼片硬度的测试方法咀嚼片硬度的测试方法有多种,包括压痕法、牙印法和切割法等。
其中,最常用的是压痕法。
压痕法是通过将一定药片厚度的咀嚼片,放置在硬度计平台上,然后用固定的力量进行压痕,测量所需的压力。
根据药典标准,常用的测试仪器有洛氏硬度计、杜氏硬度计和微出指等。
三、影响咀嚼片硬度的因素咀嚼片硬度受多种因素的影响,主要包括药物成分、配方比例、制剂工艺和贮存条件等。
1. 药物成分:咀嚼片的活性成分类型、含量和药材处理方式等都会对硬度产生影响。
一般来说,活性成分较多或者药材处理不当会导致咀嚼片硬度过大。
2. 配方比例:药物配方中的各个成分比例不同,会对咀嚼片的硬度产生重要影响。
例如,填充剂和粘结剂的比例过大,会使咀嚼片硬度升高。
3. 制剂工艺:制剂过程中的干燥时间、研磨方法和成型压力等也会直接影响咀嚼片的硬度。
4. 贮存条件:咀嚼片在贮存过程中的温度和湿度等环境因素,对硬度也有一定的影响。
一般来说,较高的温度和湿度会导致咀嚼片变软。
四、相关标准要求咀嚼片硬度的相关标准主要包括国家药典、欧洲药典和美国药典等。
这些标准规定了测试方法以及最低咀嚼片硬度要求。
例如,国家药典规定了咀嚼片硬度应该在20-50 N之间。
在临床应用中,咀嚼片的硬度要求根据患者的需求和用药目的而定。
一般来说,儿童和老年患者对咀嚼片的硬度要求较低,以防止对口腔组织造成伤害。
而对于一般成人患者,较高的咀嚼片硬度则能更好地满足口腔清洁和药物释放的要求。
碳酸镧咀嚼片质量标准

碳酸镧咀嚼片质量标准碳酸镧咀嚼片是一种用于治疗高磷血症的药物,其主要成分为镧离子。
这种药物通过与食物中的磷结合,形成不溶性磷酸镧,从而降低胃肠道对磷的吸收,达到降磷的效果。
以下是对碳酸镧咀嚼片质量标准的详细说明:一、质量标准概述碳酸镧咀嚼片的质量应符合国家药品监管部门制定的相关标准。
为了确保药品的安全性和有效性,质量标准应包括以下方面:药物的成分、纯度、稳定性、崩解性、处方和制备工艺的合理性、包装材料的适宜性、无菌性、微生物限度、药理毒理试验、临床试验等。
二、成分碳酸镧咀嚼片的主要成分是镧离子(La3+),以碳酸盐的形式存在。
此外,药品中可能还含有其他辅料,如赋形剂、甜味剂等,以改善药物的口感和外观。
三、纯度碳酸镧咀嚼片应具有高纯度,其中La3+的含量应符合相关规定。
杂质的存在可能会影响药物的疗效和安全性,因此应进行严格控制。
四、稳定性碳酸镧咀嚼片在贮存期间应保持稳定,以确保药物的疗效和安全性。
因此,质量标准应对药品的稳定性进行评估,包括加速试验和长期试验等。
五、崩解性崩解性是指药物在口腔内迅速释放出有效成分的能力。
碳酸镧咀嚼片应具有良好的崩解性,以便于患者服用后迅速发挥作用。
因此,质量标准应对药品的崩解性进行评估。
六、处方和制备工艺的合理性碳酸镧咀嚼片的处方和制备工艺应经过严格的审核和验证,以确保其安全性和有效性。
质量标准应对处方和制备工艺的合理性进行评估,包括对原料药的质量控制、生产环境的卫生条件、设备的清洁与消毒等方面。
七、包装材料的适宜性碳酸镧咀嚼片应采用适宜的包装材料进行包装,以确保药品的安全性和稳定性。
质量标准应对包装材料的适宜性进行评估,包括对包装材料的材质、密封性、阻隔性等方面的考察。
八、无菌性碳酸镧咀嚼片应具有无菌性,以确保患者的安全。
质量标准应对药品的无菌性进行严格控制,包括对生产过程中的卫生条件、灭菌工艺等方面的考察。
九、微生物限度微生物限度是指药品中微生物的数量和种类应符合相关规定,以确保药品的安全性和有效性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
FDA发布咀嚼片关键质量属性指导原则(中英文对照)I. INTRODUCTIONI.引言This guidance provides manufacturers of chewable tablets for human use with the Center for Drug Evaluation and Research’s (CDER) current thinking on the critical quality attributes that should be assessed during the development of these drug This guidance also provides recommendations about submitting developmental, manufacturing, and labeling information for chewable tablets that must be approved by CDER before they can be distributed. The recommendations in this guidance apply mainly to new drug applications(NDAs), abbreviated new drug applications (ANDAs),3 and certain chemistry, manufacturing, and controls(CMC) supplements to these some of there commendations about the submission of developmental information may also apply to investigational new drug applications (INDs). The recommendations about assessing critical quality attributes apply to all chewable tablets for human use, including non-application products.本指南向生产者提供了药品审评研究中心(CDER)对人用咀嚼片在研发过程中应评估的关键质量属性的当前想法2。
该指南也提供了必须向CDER提交并被其批准的咀嚼片的研发、生产及说明书信息的建议。
该指南的这些建议主要针对新药申请(NDAs)、仿制药申请(ANDAs)3和一些化学、生产和质控(CMC)补充申请4。
某些建议同样适合于研究性新药申请(即新药临床申请,INDs)。
关于评估关键质量属性的建议适用于所有人用咀嚼片,包括非申请产品。
Ingeneral, FDA’s guidance documents do not establish legally enforceableresponsibi lities. Instead, guidances describe the Agency’s current thinking ona topic and should be viewed only as recommendations, unless specificregulatory or statutory requirements are cited. The use of the word should inAgency guidances means that something is suggested or recommended, but notrequired.通常,FDA的指导文件不具有法律强制性,指南中描述的主题仅代表FDA机构目前的看法,只作为建议,除非是引用具体的法规或条例要求。
建议或推荐使用该指导原则,但不是必须的。
II. BACKGROUNDII.背景Chewabletablets are an immediate release (IR) oral dosage form intended to be chewedand then swallowed by the patient rather than swallowed whole. They should be designed to have a pleasanttaste and be easily chewed and swallowed. Chewable tablets should be safe and easyto use in a diverse patientpopulation, pediatric, adult, or elderly patients, who are unable or unwillingto swallow intact tablets due to the size of the tablet or difficulty withswallowing. The availability of safe, easy-to-use dosage forms is important inclinical practice. Chewable tablets are available for many over-the-counter(OTC) and prescription drug products.咀嚼片是患者经咀嚼后立即释放的口服剂型,而不是整个吞咽。
其应被设计为可口的味道且易于咀嚼和吞咽。
咀嚼片应是安全的,易于那些因片子大小或吞咽困难导致不能或不愿吞服的特殊人群、儿童、成年、或老年患者服用。
能获得安全的、易于服用的剂型在临床实践中非常重要。
在许多OTC和处方药中均有咀嚼片。
TheUnited States Pharmacopeia (USP) recognizes and differentiates between twotypes of chewable tablets: (1) thosethat may be chewed for ease of administration, and (2) those that must bechewed or crushed before swallowing to avoid choking and/or to ensure therelease of theactive The concepts in this guidance are applicable to both types of chewabletablets.USP药典中识别和区分两种类型的咀嚼片:(1)可以咀嚼以方便服用的咀嚼片;(2)必须咀嚼或压碎以避免吞咽窒息和/或确保活性成分充分释放的咀嚼片5。
本指南中的概念适用于这两种类型的咀嚼片。
Adverseevents for chewable tablets can include gastrointestinal (GI) obstructionresulting from patients swallowing whole or incompletely chewed tablets, as wellas tooth damage and denture breakage resulting from excessive tablet A related potential adverse event thatsponsors should also consider is esophageal irritation from chewabletablets. A review of numerous approveddrug product applications for chewable tablets revealed that in certain casescritical quality attributes such as hardness, disintegration, and dissolutionwere not given as much consideration as may have been warranted. This was evidenced by instances of incompletemonitoring of all relevant critical quality attributes or the use of widelyranging values that were not justified as acceptance criteria. In addition, a wide variation in analyticalprocedures has been ,8,9咀嚼片的不良反应包括患者整片吞咽或不完全咀嚼导致的胃肠道(GI)阻塞,以及片剂过硬导致牙齿损伤和假牙破损6。
也应考虑咀嚼片引起的食道刺激这一潜在不良事件。
从过去批准的很多咀嚼片来看,许多产品对硬度、崩解时限、溶出度等关键质量属性的考察仍不充分,例如,对所有相关的关键质量属性监管不完全,或质量指标范围很宽泛但未证明其在可接受的标准之内。
此外,据报道,分析方法也存在很大差异7,8,9。
Thisguidance describes the critical quality attributes that should be consideredwhen developing chewable tablets and recommends that the selected acceptancecriteria be appropriate and meaningful indicators of product performancethroughout the shelf life of the product.本指南建议了开发咀嚼片时应考虑的关键质量属性、可选择的合适的可接受标准、产品有效期内的有意义的产品性能指标。
III.讨论Avariety of physical characteristics should be considered in the manufacturingprocess for chewable tablets. An idealchewable tablet should be:•Easy to chew•Palatable (taste masked or of acceptable taste)•Of appropriate size and shape10•Able to disintegrate readily to minimize aspiration and facilitate dissolution.在咀嚼片剂生产工艺中,应考虑各种物理特性。