高中生物“伴性遗传”知识点总结
高三伴性遗传知识点

高三伴性遗传知识点遗传学是生物学的重要分支之一,研究个体之间遗传信息的传递和变化规律。
而伴性遗传是遗传学中的一个重要概念,主要描述基因在染色体上的位置与该基因表型表达之间的关系。
本文将对高三伴性遗传的知识点进行论述,帮助读者更好地理解这一概念以及其在遗传学中的应用。
一、伴性遗传的基本原理伴性遗传最早由著名的遗传学家摩尔根(Thomas Hunt Morgan)发现,他通过对果蝇的研究,发现有些性状是与性别相联络的,也就是与染色体的性别决定区域相同。
二、伴性遗传的特点与规律1. 伴性遗传通常由一个位点上的基因影响;2. 伴性遗传的现象通常在雄性表现出来;3. 伴性遗传是因为伴随着雌雄性染色体的差异,导致不同性别个体的表达差异。
三、伴性遗传的机制伴性遗传的机制主要涉及到两个方面:基因与染色体的关系以及遗传物质的性别决定。
1. 基因与染色体的关系伴性遗传的基因通常位于性染色体的非交叉区域上,因此sex-linkage也称为染色体连锁。
2. 遗传物质的性别决定在人类中,女性拥有两个X染色体,而男性则拥有一个X染色体和一个Y染色体。
当X染色体上携带有伴性遗传的基因时,只要男性携带该基因,即可在表型上表现出来。
因为男性只有一个X染色体,其所携带伴性遗传基因无法与另一个X染色体上的基因进行抵消。
四、伴性遗传的疾病和案例伴性遗传的特点使得一些疾病主要发生在男性,并且往往具有家族性遗传。
1. 血友病血友病是由于凝血因子基因突变导致的出血性疾病,通常伴随族内成员之间传递。
男性因只有一个X染色体,即使携带一个突变的凝血因子基因也会表现出疾病。
2. 红绿色盲红绿色盲是一种常见的色觉缺陷疾病,主要影响男性。
该疾病由于X染色体上的视蛋白基因突变导致。
五、伴性遗传的研究方法与应用1. 连锁图通过连锁图可以画出染色体上各个基因的相对位置,进而研究基因之间的共真和交换频率。
2. 疾病的预防和诊断了解伴性遗传的规律和机制,可以为一些伴性遗传疾病的预防和诊断提供依据。
高中生物“伴性遗传”知识点总结

高中生物“伴性遗传”知识点总结高中生物“伴性遗传”知识点总结伴性遗传的最大特点就是性状与性别的关联,这部分常考题目主要有伴性遗传的判断和相关计算。
判断是伴性遗传还是常染色体遗传,常用同型的隐形个体与异型的显性个体杂交,根据后代的表现型进行判断。
以XY型性别决定的生物为例,如果为伴X隐性遗传,雌性隐性个体与雄性显性个体杂交,如果后代雄性个体中出现了显性性状,即为常染色体遗传,否则即为伴X遗传。
一.常见遗传病的遗传方式:(1) 单基因遗传:常染色体显性遗传:并指、多指; 常染色体隐性遗传:白化病、失天性聋哑 X连锁隐性遗传:血友病、红绿色盲; X连锁显性遗传:抗维生素D佝偻病; Y连锁遗传:外耳道多毛症;(2)多基因遗传:唇裂、先天性幽门狭窄、先天性畸形足、脊柱裂、无脑儿; (3 )染色体病:染色体数目异常:先天性愚型病; 染色体结构畸变:猫叫综合症。
二、单基因遗传病单基因遗传病是指受一对等位基因控制的遗传病, 较常见的有红绿色盲、血友病、白化病等。
根据致病基因所在染色体的种类,通常又可分四类:1、常染色体显性遗传病致病基因为显性并且位于常染色体上,等位基因之一突变,杂合状态下即可发病。
致病基因可以是生殖细胞发生突变而新产生,也可以是由双亲任何一方遗传而来的。
此种患者的子女发病的概率相同,均为1/2。
此种患者的异常性状表达程度可不尽相同。
在某些情况下,显性基因性状表达极其轻微,甚至临床不能查出,种情况称为失显。
由于外显不完全,在家系分析时可见到中间一代人未患病的隔代遗传系谱,这种现象又称不规则外显。
还有一些常染色体显性遗传病,在病情表现上可有明显的轻重差异,纯合子患者病情严重,杂合子患者病情轻,这种情况称不完全外显。
2、常见常染色体显性遗传病的病因和临床表现1)、多指(趾)、并指(趾)。
临床表现:5指(趾)之外多生1~2指(趾),有的仅为一团软组织,无关节及韧带,也有的有骨组织。
2)、珠蛋白生成障碍性贫血。
高一必修二伴性遗传知识点

高一必修二伴性遗传知识点高中生物课程中,必修二部分涵盖了一系列的遗传学知识,其中之一是伴性遗传。
伴性遗传是一种基因传递方式,与常染色体遗传有所不同。
下面将对高一必修二中与伴性遗传相关的知识点进行详细介绍。
一、伴性遗传的定义与特点伴性遗传是指遗传性状不仅与性别有关,而且此类遗传物质定位于性染色体上的基因遗传方式。
一般来说,伴性遗传主要与X染色体相关,因为Y染色体上的基因较少。
伴性遗传通常表现为一种基因在同一性别中出现的频率较高,而在另一性别中出现的频率较低的现象。
二、伴性遗传的经典案例——血友病血友病是一种由于凝血因子遗传异常导致的出血性疾病,它是伴性遗传的典型案例之一。
正常情况下,凝血因子位于X染色体上的基因负责血液凝固的过程。
而当这个基因发生突变时,就会导致凝血因子缺乏,从而引发血友病。
由于该基因位于X染色体上,所以男性很容易受到该病的影响,而女性则常常是健康的基因携带者。
三、伴性遗传的概率计算要了解伴性遗传的概率,我们需要掌握一些重要的概念:正常基因、突变基因、杂合子和纯合子等。
正常基因是指没有突变的基因,而突变基因则是发生了突变的基因。
杂合子指在同一基因位点上有一对不同的等位基因,而纯合子则是在同一基因位点上有一对相同的等位基因。
在伴性遗传中,女性通常是正常基因和突变基因的杂合子,因为她们有两个X染色体。
而男性只有一个X染色体,所以如果他们携带突变基因,就会表现出相应的遗传疾病。
四、伴性遗传的其他案例和知识点除了血友病外,伴性遗传还涉及到其他疾病,如色盲、肌肉萎缩症和多发性鳞状疣等。
这些疾病的发生也与突变基因在X染色体上的位置有关。
此外,在伴性遗传中,女性有时也可能表现出某些遗传疾病,这是因为突变基因发生在她们两个X染色体的相同位置上,从而导致疾病的发生。
五、伴性遗传的临床意义和研究进展伴性遗传对于临床医学有着重要的意义,不仅可以帮助医生诊断与伴性遗传相关的疾病,还能指导患者的治疗方案。
同时,科学家们也在不断深入研究伴性遗传的机制,希望能找到更好的治疗方法,从而改善患者的生活质量。
伴性遗传总结归纳

伴性遗传总结归纳伴性遗传是指染色体上的基因与性别染色体连锁在一起遗传的现象。
它与常染色体遗传略有不同,常染色体遗传是指遗传物质的传输与配对无关,而伴性遗传则与性别染色体的传输密切相关。
本文将对伴性遗传作一个综述,总结并归纳其中的重要内容。
一、伴性遗传的特点伴性遗传主要有以下几个特点:1. 伴性遗传常见于X染色体上的基因,因为Y染色体上较少存在基因,故较少参与伴性遗传。
2. 伴性遗传一般表现为在一代中雄性突变的现象,而雌性则通常是正常表达基因。
3. 由于男性只有一个X染色体,一旦携带了有问题的伴性基因,就会表现出相应的遗传疾病。
4. 雌性因为拥有两个X染色体,所以即使一个染色体携带了不正常的基因,另一个染色体上正常的基因可以起到补偿作用。
二、常见的伴性遗传疾病1. 血友病:血友病是一种常见的伴性遗传疾病,主要表现为凝血因子Ⅷ或Ⅸ的缺乏,导致患者出血时间延长及出血量增加。
2. 肌萎缩侧索硬化症:肌萎缩侧索硬化症是一种运动神经元退行性疾病,主要表现为进行性肌肉萎缩和肌力减退。
3. 艾尔综合征:艾尔综合征又称间质性精索结节性纤维化,主要表现为内脏器官异常、生长发育异常以及智力低下等症状。
4. 艾迪森病:艾迪森病是一种自身免疫性疾病,主要表现为肾上腺皮质功能不全,全身各种代谢功能受到影响。
5. 艾文氏综合征:艾文氏综合征是一种神经发育异常疾病,最常见的症状为眼球震颤、共济失调和智力低下。
三、伴性遗传的诊断和治疗伴性遗传疾病的诊断通常通过患者的家族史以及对相关基因的检测来确定。
对于携带伴性遗传基因的人群,可以通过遗传咨询和基因咨询进行相关的预防和治疗。
预防措施主要包括:1. 遗传咨询:对于容易遗传伴性基因的夫妻,应该在结婚前进行遗传咨询,了解相关的风险和预后,做出明智的决策。
2. 孕前筛查:对怀孕计划的夫妻,可以通过孕前筛查来了解是否携带有伴性遗传疾病的基因,避免遗传给下一代。
3. 人工辅助生殖:对于携带伴性遗传基因的夫妻,可以选择通过人工受精、胚胎筛查等方法来降低下一代患病的风险。
“伴性遗传”知识梳理及考点详析

“伴性遗传”知识梳理及考点详析“伴性遗传”是高考高频考点,侧重于考查学生综合分析和应用知识的能力。
一、生物的性别决定特别提醒:①并不是所有的生物都有性别之分,如大部分的雌雄同株植物,没有性染色体,没有性别之分。
②性染色体决定性别是性别决定的主要方式,此外还有其它方式,如蜜蜂是由染色体组数目决定性别的。
例1 下列关于人类性别决定与伴性遗传的叙述,正确的是()A.性染色体上的基因都与性别决定有关B.性染色体上的基因都伴随性染色体遗传C.生殖细胞中只表达性染色体上的基因D.初级精母细胞和次级精母细胞中都含Y染色体解析:本题主要考查性别决定和伴性遗传的相关知识。
性染色体上的基因不都与性别决定有关,如色盲基因,故A错;染色体上的基因都随染色体的遗传而遗传,故B对;生殖细胞既有常染色体上的基因也有性染色体上的基因,它们都选择性地表达,故C错;X和Y 染色体是特殊的同源染色体,在减Ⅰ时X和Y染色体分离,次级精母细胞中有的只含X染色体,有的只含Y染色体,故D错。
答案:B二、伴性遗传的类型及规律1. X、Y染色体的形态特点及其基因分布高等生物如人类的Y染色体比X染色体小得多,大部分不是同源区段。
下图为X、Y不同区段上分布的基因遗传都与性别相联系,都是伴性遗传。
又具体分为以下几种情况:①基因只存在于Y染色体的非同源区段中,则遗传为伴Y遗传(也叫限雄遗传)。
这种情况判断起来很简单,即,相关性状只出现在雄性个体中。
在基因型的书写中,常染色体上的基因不需标明其位于常染色体上(即只须写基因符号),性染色体上的基因则需将性染色体及其上基因一同写出,基因符号写在“Y”的右上角,“X”的右上角不需要写,一般常染色体上的基因写在前,性染色体及其上基因写在后,如AAXY b。
②基因只存在于X染色体上的非同源区段,则遗传为伴X遗传。
这种情况就是教材上介绍的诸如人类红绿色盲等性状的遗传,属于考试考查得最多的一种情形。
相关性状在雌、雄性个体中均有出现。
高一生物伴性遗传知识点

高一生物伴性遗传知识点第一节:伴性遗传的概述伴性遗传是指某些特定特征在遗传过程中与性别有关。
一般来说,这些特征主要表现在X染色体上。
本节将介绍一些与伴性遗传相关的基本概念。
第二节:男性染色体在人类细胞中,男性拥有一对性染色体,分别为X染色体和Y染色体。
在生殖过程中,男性可以将X或Y染色体传递给下一代。
第三节:女性染色体相比之下,女性拥有两对X染色体,即XX。
在生殖过程中,女性只能将X染色体传递给下一代。
第四节:伴性遗传的性别差异由于男性只有一个X染色体,当其携带的某个基因发生突变时,后代表现出该特征的概率会相对较高。
而女性有两个X染色体,因此她们需要两个突变的X染色体才会表现出某一特征。
第五节:伴性遗传的示例1. 红绿色盲:红绿色盲是一种与伴性遗传相关的疾病。
该疾病主要发生在男性身上,因为只需一个突变的X染色体就能引发该病。
2. 血友病:血友病也是一种与伴性遗传相关的遗传疾病。
该病主要表现在男性身上,因为只需一个突变的X染色体就能引发该病。
女性只有在两个X染色体都突变时才会表现出血友病的症状。
第六节:伴性遗传的机制伴性遗传中的突变基因位于X染色体上。
当一个母亲携带这样的突变基因时,她的男性后代有50%的几率携带该基因,而女性后代有50%的几率成为携带者。
第七节:携带者的特点通常情况下,女性携带者不表现出伴性遗传疾病的症状,因为她们拥有两个X染色体,其中一个正常的X染色体可以抵消突变基因的影响。
然而,她们仍承担着将该基因传递给下一代的风险。
第八节:诊断和预防基因检测可以用于确定一个人是否携带伴性遗传疾病的突变基因。
在诊断出携带者后,家庭成员可以进行遗传咨询,以了解如何降低突变基因传递给下一代的风险。
结论伴性遗传是一种与性别有关的遗传现象,可导致一些特定特征和疾病在男性中更常见,并对携带者的后代有一定的遗传风险。
通过基因检测和遗传咨询,我们可以更好地了解伴性遗传的机制,并为预防和治疗提供指导。
关于高一生物伴性遗传知识点

关于高一生物伴性遗传知识点伴性遗传是指与一对常染色体相关的性染色体上的一对等位基因所具有的性别差异相关的一种遗传方式。
在伴性遗传中,一般男性是表现型而雌性是隐性携带者。
下面是高一生物伴性遗传的知识点。
1.性染色体的组成与性别决定-人类的性染色体由一对X和Y染色体组成。
-男性的性别决定是由于他们有一个X和一个Y染色体,被称为XY型。
-女性有两个X染色体,被称为XX型。
-此外,Y染色体上有少数决定男性性别的基因。
2.X连锁遗传-X连锁遗传是指那些位于X染色体上的基因的遗传,因为X染色体中还有一部分基因位点,可以与Y染色体的对应基因位点匹配。
-男性只有一个X染色体,所以位于这个X染色体上的基因会直接影响物种表现型。
-女性有两个X染色体,如果一些位点上的两个等位基因中有一个是突变的,则另一个正常的等位基因可以补偿这一不良效应,女性通常是携带者。
3.伴性基因和常见的性连锁遗传疾病-葡萄糖-6-磷酸脱氢酶缺乏症:主要影响男性,女性多数是隐性携带者。
-血友病:男性患者由于X染色体携带有突变基因而出现凝血功能障碍,大多数女性为隐性携带者。
-迪伦菲尔氏综合症:男性患者由于X染色体上的突变基因,而在骨骼和其他器官中出现异常,女性通常是携带者。
4.X连锁显性遗传疾病-一些X连锁遗传疾病可以以显性方式表现,并且常常影响男性。
-这些疾病的特征是,患者的母亲通常是受影响的,而患者的父亲通常是正常的,因为男性只有一个X染色体。
5.良性X连锁顶叶癫痫-一种X连锁显性遗传疾病,主要影响男性。
-主要特征是部分性发作和全面性发作。
-患者通常在婴儿期或儿童早期开始出现症状。
总之,高一生物伴性遗传的知识点有性染色体的组成与性别决定、X 连锁遗传、伴性基因和常见的性连锁遗传疾病、X连锁显性遗传疾病以及良性X连锁顶叶癫痫等。
理解这些知识点对于深入理解遗传学和了解遗传疾病的发生机制都非常重要。
高中生物 必修二 伴性遗传总结

必修二_伴性遗传总结在正式开始伴性遗传的部分之前,先简述一下生物性别决定的几种类型1.性染色体决定(1)XY型和XO型对于XY型生物,XX表现为雌性,XY表现为雄性,代表生物有人类、哺乳类、部分鱼类、两栖类、双翅目昆虫对于XO型生物,XX表现为雌性,X表现为雄性(雄性的性染色体只有一条),代表生物有蝗虫、蟑螂、蟋蟀等(2)ZW型和ZO型对于ZW型生物,ZZ和WW表现为雄性,ZW表现为雌性,代表生物有鸟类、部分鱼类、两栖类、爬行类和鳞翅目昆虫对于ZO型生物,Z表现为雌性(雌性只有一条性染色体且没有W染色体),ZZ表现为雄性,代表生物有家禽(鸡、鸭)、部分鱼类和鳞翅目昆虫2.染色体数量决定部分生物的性别是由单倍体、二倍体决定的,代表生物蚂蚁和蜜蜂3.温度决定部分生物的性别是由卵孵化时的温度决定的,代表生物为大部分爬行类动物(鳄鱼、蜥蜴)伴性遗传的基本概念1.定义:伴性遗传是指性染色体上的基因所控制的形状总是伴随性别而遗传的现象,又称为性连锁遗传。
2.遵循规律:分离定律和自由组合定律3.分类(暂不考虑XY同源区段问题,但在最后会进行补充):(1)伴X隐形遗传,如红绿色盲、果蝇的红白眼、血友病(2)伴X显性遗传,如抗维生素D佝偻病、遗传性脑智力超常型孤独症(3)伴Y遗传(无显隐性区别),如外耳道多毛症伴X隐性遗传1.特点:(1)人群中男性患者多于女性患者(2)存在“无中生有”的情况(隔代遗传)(3)无法在男性间传递(交叉遗传)(4)正常男性的女孩一定正常2.配婚方式(以伴性遗传病为例)(1)X B X B× X B Y正常女性:正常男性 = 1:1X B X B : X B Y = 1:1(2)X B X B× X b Y女性携带者:正常男性 = 1:1X B X b : X B Y = 1:1(3)X B X b× X B Y正常女性:女性携带者:正常男性:男性患者1 : 1 : 1 : 1X B X B:X B X b:X B Y :X b Y = 1:1:1:1(4) X B X b× X b Y正常女性:女性患者:正常男性:男性患者1 : 1 : 1 : 1X B X B:X b X b:X B Y :X b Y = 1:1:1:1(5)X b X b× X B Y女性携带者:男性患者 = 1:1X B X b : X b Y = 1:1(6)X b X b× X b Y女性患者:男性患者 = 1:1X b X b : X b Y = 1:12.交叉遗传致病基因只能由母本传给子一代中的雄性个体,再传给子二代中的雌性个体3.隔代遗传父本和子二代中雄性个体患病,但子一代中雌性个体正常伴X显性遗传1.特点:(1)人群中女性患者多于男性患者(2)不存在“无中生有”的情况(无法隔代遗传)(3)患病男性的女儿一定患病,儿子一定正常2.配婚方式(以伴性遗传病为例)(1)X B X B× X B Y女性患者:男性患者 = 1:1X B X B : X B Y = 1:1(2)X B X B× X b Y女性患者:男性患者 = 1:1X B X b : X B Y = 1:1(3)X B X b× X B Y女性患者:男性患者:正常男性2 : 1 : 1X B X B:X B X b:X B Y :X b Y = 1:1:1:1(4) X B X b× X b Y正常女性:女性患者:正常男性:男性患者1 : 1 : 1 : 1X B X B:X b X b:X B Y :X b Y = 1:1:1:1(5)X b X b× X B Y女性患者:正常男性 = 1:1X B X b : X b Y = 1:1(6)X b X b× X b Y正常女性:正常男性 = 1:1X b X b : X b Y = 1:1伴Y遗传1.特点:(1)只在男性间传递且不存在患病女性(2)只要父本患病,则子代雄性个体一定都患病2.配婚方式介于伴Y遗传情况较为单一且简单,在此不做展开3.由于只存在Y染色体上的基因很少,故伴Y遗传现象极为少见,同时由于情况简单,在题目中几乎没有考察XY同源区段问题1.特点:(1)人群中女性患者多于男性患者(2)不存在“无中生有”的情况(无法隔代遗传)(3)患病男性的女儿一定患病,儿子一定正常2.配婚方式(以伴性遗传病为例且默认为隐性基因致病)介于情况较多,这里只列举杂合子交配情况,其它情况可以参考常染色体遗传(1)X B X b × X B Y b正常女性:女性携带者:男性携带者:男性患者1 : 1 : 1 : 1X B X B:X B X b:X B Y b:X b Y b = 1:1:1:1(2)X B X b × X b Y B正常女性:女性患者:正常男性:男性携带者1 : 1 : 1 : 1X B X B:X b X b:X B Y B:X b Y B = 1:1:1:13.在题目中没有出现同源区段这一关键词时,考试时不必从同源区段这一角度进行考虑总体上来讲,只要熟练掌握几种遗传病的常见类型,能够正推子代类型,反推亲本基因型,伴性遗传问题还是易于解决的。
(完整版)伴性遗传知识点小结

伴性遗传知识点总结1、伴X隐性遗传病特点:①患者中男性多于女性(女性中同事含有两个患病基因才患病,男性中只要有一个致病基因就患病)②具有隔代交叉遗传现象。
母亲→儿子→孙女,父亲→女儿→外孙③女性患者的父亲和儿子一定是患者,即“母病子必病,女病父必病”。
④男性正常,其母亲、女儿全都正常。
例如:色盲、血友病、另外还有果蝇的红眼与白眼。
2、伴X显性遗传病特点:①女性患者多于男性患者。
②表现为代代相传,具有世代连续性。
③男患者的母亲和女儿一定患病。
(父患女必患,子患母必患)④女性正常,其父亲、儿子全都正常。
例如:抗维生素D佝偻病3、伴Y遗传特点:致病基因位于Y染色体上,只能在男性中表现出来,即:父传子、子传孙,传男不传女。
例如:外耳道多毛症4、常染色体遗传病常染色体隐性遗传病特点:女病,其父亲和儿子中有正常。
例如:白化病常染色体显性遗传病特点:男病,其母亲和女儿中有正常。
例如:多指症5、细胞质遗传(母系遗传)病特点:母正常则子女均正常,母患病则子女均患病例如:人线粒体肌病【遗传系谱图判定口诀】(1)子女同母为母系;父子相传为伴Y(2)无中生有为隐性,隐性遗传看女病,母患子必患,女患父必患为伴X隐;男(父子)有正为常隐(3)有中生无为显性;显性遗传看男病,父患女必患,子患母必患为伴X显;女(母女)有正为常显(4)伴性与常染色体遗传都有可能时,无性别差异,男女患者数相同,最可能为常染色体遗传;有性别差异,男比女多,最可能为伴X隐性;女比男多,最可能为伴X显性。
患者全部是男性,最有可能是伴Y【练一练】1、如下图为甲病(A—a)和乙病(B—b)的遗传系谱图,其中乙病为伴性遗传病,请回答下列问题:(1)甲病属于________,乙病属于________。
A.常染色体显性遗传病B.常染色体隐性遗传病C.伴X染色体显性遗传病D.伴X染色体隐性遗传病E.伴Y染色体遗传病(2)Ⅱ5为纯合子的概率是________,Ⅱ6的基因型为________,Ⅲ13的致病基因来自于________。
高一生物伴性遗传知识点

高一生物伴性遗传知识点伴性遗传可归纳为下列规律:1. 当同配性别的性染色体(如哺乳类等为XX为雌性,鸟类ZZ为雄性)传递纯合显性基因时,F1雌、雄个体都为显性性状。
F2性状的分离呈3显性:1隐性;性别的分离呈1雌:1雄。
其中隐性个体的性别与祖代隐性体一样,即1/2的外孙与其外祖父具有相同的表型特征。
2.当同配性别的性染色体传递纯合体隐性基因时,F1表现为交叉遗传,即母亲的性状传递给儿子,父亲的性状传递给女儿,F2中,性状与性别的比例均表现为1:1。
3.存在于Y染色体差别区段上的基因(特指人类或哺乳类)所决定的性状,或由W染色体所携带的基因所决定的性状,仅仅由父亲(或母禽、母鸟)传递给其儿子(或雌禽、母鸟)。
表现为特殊的Y连锁(或W连锁)遗传。
高一生物伴性遗传知识点二:怎么判断一种性状的准确遗传方式呢?常染色体显性常染色体隐性X性染色体显性X性染色体隐性Y染色体遗传细胞质遗传1.首先确定显隐性:①“无中生有为隐性”②“有中生无为显性”2、再确定致病基因的位置:①“无中生有为隐性,女儿患病为常隐”②“有中生无为显性,女儿正常为常显”③“母病子病,女病父病,男性患者多于女性”――最可能为“X隐”④“父病女病,子病母病,女性患者多于男性”――最可能为“X显”3.常染色体与性染色体同时存在的处理方法:当既有性染色体又有常染色体上的基因控制的两对及以上的性状遗传时: 由性染色体上的基因控制的性状按伴性遗传处理;由常染色体上的基因控制的性状按分离规律处理,整体上则按基因的自由组合定律来处理.高一生物伴性遗传知识点三:遗传性疾病通常可分为三类:1、染色体疾病主要是染色体数目的异常,又分为常染色体异常和性染色体异常。
前者如21-三体综合征,即比正常人多了第21条染色体;后者如先天性卵巢发育不全,即正常女性的染色体应该为XX,而这种病人是XO,缺少了一个X 染色体。
2、单基因遗传病指同源染色体上的等位基因,其中的1个或2个发生异常,根据遗传方式又可分为三种:⑴常染色体显性遗传:如果父母双方之一带有常染色体的病理性基因是显性的,那么只要有一个这样的病理性基因传给子女,子女就会出现和父母同一种的疾病。
高二伴性遗传生物知识点

高二伴性遗传生物知识点
高二伴性遗传生物知识点
伴性遗传生物知识点概念:伴性遗传此类性状的遗传控制基因位于性染色体上,因而总是与性别相关联。
类型:X染色体显性遗传:抗维生素D佝偻病等
X染色体隐性遗传:人类红绿色盲、血友病
Y染色体遗传:人类毛耳现象
一、X染色体隐性遗传:如人类红绿色盲
1、致病基因Xa 正常基因:XA
2、患者:男性XaY 女性XaXa
正常:男性XAY 女性 XAXA XAXa(携带者)
3、遗传特点:
(1)人群中发病人数男性大于女性
(2)隔代遗传现象
(3)交叉遗传现象:男性女性男性
二、X染色体显性遗传:如抗维生素D佝偻病
1、致病基因XA 正常基因:Xa
2、患者:男性XAY 女性XAXA XAXa
正常:男性XaY 女性XaXa
3、遗传特点:
(1)人群中发病人数女性大于男性
(2)连续遗传现象
(3)交叉遗传现象:男性女性男性。
高中生物伴性遗传知识要点归纳

高中生物伴性遗传知识要点归纳伴性遗传是指通过性染色体携带的基因在非性染色体上表现出来的遗传现象。
在高中生物学课程中,学生需要掌握伴性遗传的相关知识,以便更好地理解遗传规律以及相关生物现象的产生。
下面是对高中生物伴性遗传知识的要点归纳:一、伴性遗传的概念伴性遗传是遗传学中的一个概念,指的是某些特定基因的表达与性别染色体的携带有关。
这些基因位于非性染色体上,但在性染色体上也有一部分相应的区域。
二、伴随现象1. 男性与女性的发病率不同:由于男性只有一个X染色体,所以他们具有更高的可能患上与基因相关的疾病。
2. 女性携带者的存在:女性通常有两个X染色体,所以她们可以是携带者,即携带有相应基因但并不表现出疾病症状的个体。
三、伴性遗传的传递方式1. 父母的遗传:男性与女性有不同的性染色体组合,所以伴性遗传通常是由父母传递给子女的。
2. 母亲到儿子的传递:若女性是携带者(heterozygote),则她有50%的概率将其携带的基因传递给儿子,使其成为患者。
3. 父亲到女儿的传递:若男性是患者,则他的所有女儿都将成为携带者。
4. 母亲到女儿的传递:若母亲是携带者,则她的所有女儿都有50%的概率成为携带者。
四、伴性遗传的例子1. 色盲:红绿色盲是一种常见的伴性遗传疾病,男性患者较多,女性通常是携带者。
2. 血友病:血友病也是一种常见的伴性遗传疾病,男性患者很多,女性通常是携带者。
3. 杜氏肌营养不良症:杜氏肌营养不良症是一种严重的伴性遗传疾病,主要影响男性患者,女性很少发病。
五、伴性遗传的分子机制伴性遗传是由于非性染色体上的基因与性染色体上的相应区域之间发生重组导致的。
重组使得非性染色体上的基因与性染色体上的相应区域产生连锁,从而影响到基因的表达。
六、伴性遗传的重要意义伴性遗传研究揭示了性染色体的特殊性质以及基因与表型之间的关系。
通过对伴性遗传的研究,人们能更好地理解遗传规律,提高对遗传疾病的认识,并为相关疾病的预防与治疗提供理论依据。
伴性遗传必背知识点

伴性遗传必背知识点一、概念:遗传控制基因位于性染色体上,因而总是与性别相关联。
记忆点:1.生物体细胞中的染色体可以分为两类:常染色体和性染色体。
2.性别类型:XY型:XX雌性 XY雄性————大多数高等生物:人类、动物、高等植物ZW型:ZZ雄性 ZW雌性————鸟类、蚕、蛾蝶类二、XY型性别决定方式:XY型的性别决定方式:雌性体内具有一对同型的性染色体XX,雄性体内具有一对异型的性染色体XY。
减数分裂形成精子时,产生了含有X染色体的精子和含有Y染色体的精子。
雌性只产生了一种含X染色体的卵细胞。
受精作用发生时,X精子和Y精子与卵细胞结合的机会均等,所以后代中出生雄性和雌性的机会均等,比例为1:1。
染色体组成n对:雄性:n-1对常染色体 + XY 雌性:n-1对常染色体 + XX性比:一般 1 : 1常见生物:全部哺乳动物、大多雌雄异体的植物,多数昆虫、一些鱼类和两栖类。
三、三种伴性遗传的特点:1伴X隐性遗传的特点:① 男 > 女② 隔代遗传交叉遗传即外公→女儿→外孙③ 女患,父必患。
母患,子必患。
2伴X显性遗传的特点:① 女>男② 连续发病③ 子患,母必患父患,女必患3伴Y遗传的特点:传男不传女附:常见遗传病类型要记住:伴X隐:色盲、血友病、果蝇眼色、女娄菜伴X显:抗维生素D佝偻病、钟摆型眼球震颤常隐:先天性聋哑、白化病常显:多并指Y染色体上遗传如外耳道多毛症4伴性遗传与基因的分离定律之间的关系:伴性遗传的基因在性染色体上,性染色体也是一对同源染色体,伴性遗传从本质上说符合基因的分离定律。
四、遗传病类型的鉴别:一先判断显性、隐性遗传:无中生有,为隐性有中生无,为显性二再判断常、性染色体遗传:1、父母无病,女儿有病——常、隐性遗传2、已知隐性遗传,母病儿子正常——常、隐性遗传3、已知显性遗传,父病女儿正常——常、显性遗传4、如果家系图中患者全为男性女全正常,且具有世代连续性,应首先考虑伴Y遗传,无显隐之分。
必修二生物伴性遗传知识点

必修二生物伴性遗传知识点
1. 伴性遗传是指某种性状或基因在性染色体上遗传的现象。
2. 性染色体决定了一个个体的性别,通常由一个X染色体和一个Y染色体组成。
3. X染色体上的基因遗传给下一代的方式与常染色体上的基因不同,因为男性只有一
个X染色体。
4. 如果一个基因位于X染色体上,那么这个基因就是由母亲传给子女的。
这意味着男
性只能从母亲处继承这个基因。
5. 由于男性只有一个X染色体,所以男性患上一个伴性遗传疾病的概率比女性高。
女
性通常需要从父亲和母亲那里都继承这个基因才会患病。
6. 常见的伴性遗传疾病包括血友病和色盲。
7. 在血友病中,血液凝血过程中的一种蛋白质缺乏或功能异常。
男性可能患上血友病,而女性通常是携带者。
8. 色盲是一种视觉缺陷,使得人们难以区分某些颜色。
很多女性是色盲基因的携带者,而男性则更容易受到影响。
9. 伴性遗传还可以表现为一些其他的特征,例如在一些昆虫种群中,雄性具有不同颜
色的翅膀或眼睛等。
10. 伴性遗传可以通过遗传学研究和家族历史来确定。
一般来说,如果一个家族有伴性遗传疾病的历史,那么父母需要做额外的检查和咨询,以了解他们携带伴性遗传病的风险。
必修二生物伴性遗传知识点

必修二生物伴性遗传知识点
必修二生物的伴性遗传知识点主要包括以下内容:
1. 伴性遗传的概念:伴性遗传是指某些基因位于性染色体上,且这些基因的表达与性
别有关。
因此,这些基因的遗传方式与性染色体的遗传方式相似。
2. 性染色体和性别的确定:人类的性别由性染色体决定,女性有两条X染色体(XX),而男性有一条X染色体和一条Y染色体(XY)。
3. 伴性遗传的特点:伴性遗传通常表现为在一性别中的某些性状或疾病的出现率较高,而在另一性别中几乎不出现。
4. 雌雄同体:某些生物如果蝇等雄性个体和雌性个体在形态上几乎相同,而只有配子
形成的方式不同。
5. 伴性遗传的例子:经典的例子是红绿色盲。
这是一种在视网膜感光细胞中某种色素
缺乏导致的视觉缺陷,通常由X染色体上的突变基因引起。
因为女性有两条X染色体,所以女性携带一个致病基因时,通常还有一条正常的基因,因此只有一部分女性表现
出色盲症状。
而男性只有一条X染色体,如果这条染色体上的基因突变,就会导致男
性患病。
6. 伴性遗传的遗传模式:伴性遗传是通过性染色体传递的,所以遗传模式主要涉及到
X染色体。
一般来说,如果母亲是携带致病基因的,她的子女中男性有一半有可能患病,女性有一半有可能成为携带者;如果父亲是携带致病基因的,他的女儿有一半有
可能成为携带者,而他的儿子则不会患病。
7. 伴性遗传的重要性:伴性遗传的研究对于了解性别差异以及一些与性别相关的性状
和疾病具有重要意义,也对于表明基因的遗传方式提供了有益的信息。
以上是必修二生物的伴性遗传的一些知识点,希望能对你有所帮助!。
高中生物伴性遗传知识要点归纳

高中生物伴性遗传知识要点归纳伴性遗传是生物必修二的知识,这部分知识是高考比考察的内容之一,很多学生觉得这部分知识过难,但其实是由于对基本知识理解的不够扎实造成的。
下面是店铺为大家整理的高中生物必备的知识,希望对大家有用!高中生物伴性遗传知识要点归纳篇1一、名词:1、染色体组型:也叫核型,是指一种生物体细胞中全部染色体的数目、大小和形态特征。
观察染色体组型最好的时期是有丝分裂的中期。
2、性别决定:一般是指雌雄异体的生物决定性别的方式。
3、性染色体:决定性别的染色体叫做~。
4、常染色体:与决定性别无关的染色体叫做~。
5、伴性遗传:性染色体上的基因,它的遗传方式是与性别相联系的,这种遗传方式叫做~。
二、语句:1、染色体的四种类型:中着丝粒染色体,亚中着丝粒染色体,近端着丝粒染色体,端着丝粒染色体。
2、性别决定的类型:(1)XY型:雄性个体的体细胞中含有两个异型的性染色体(XY),雌性个体含有两个同型的性染色体(XX)的性别决定类型。
(2)ZW型:与XY型相反,同型性染色体的个体是雄性,而异型性染色体的个体是雌性。
蛾类、蝶类、鸟类(鸡、鸭、鹅)的性别决定属于“ZW”型。
3、色盲病是一种先天性色觉障碍病,不能分辨各种颜色或两种颜色。
其中,常见的色盲是红绿色盲,患者对红色、绿色分不清,全色盲极个别。
色盲基因(b)以及它的等位基因——正常人的B就位于X染色体上,而Y染色体的相应位置上没有什么色觉的基因。
4、人的正常色觉和红绿色盲的基因型(在写色觉基因型时,为了与常染色体的基因相区别,一定要先写出性染色体,再在右上角标明基因型。
):色盲女性(XbXb),正常(携带者)女性(XBXb),正常女性(XBXB),色盲男性(XbY),正常男性(XBY)。
由此可见,色盲是伴X隐性遗传病,男性只要他的X上有 b基因就会色盲,而女性必须同时具有双重的b才会患病,所以,患男>患女。
5、色盲的遗传特点:男性多于女性一般地说,色盲这种病是由男性通过他的女儿(不病)遗传给他的外孙子(隔代遗传、交叉遗传)。
伴性遗传知识点总结

伴性遗传知识点总结1. 什么是伴性遗传?伴性遗传是指一种特定的遗传方式,其中基因突变发生在X染色体上。
由于X 染色体在男性中只有一条,在女性中有两条,这导致伴性遗传通常在男性中表现出来,并且女性成为携带者。
2. 伴性遗传的主要特点伴性遗传具有以下主要特点:•受影响的基因位于X染色体上,通常不受Y染色体控制。
•男性更容易显露出遗传病的症状,因为他们只有一条X染色体。
•女性成为携带者,因为她们有两条X染色体,但只有一条突变的X 染色体是不足以导致症状的。
•女性成为携带者后,她们的子女有50%的概率携带遗传病。
3. 伴性遗传的常见疾病伴性遗传引起的疾病有很多,以下是一些常见的例子:色盲色盲是一种视觉缺陷,主要表现为无法区分红色和绿色。
该疾病由X染色体上突变的基因引起,通常只在男性中显露。
血友病血友病是一种由凝血因子缺乏引起的遗传性出血性疾病。
这种疾病主要发生在男性中,女性通常成为携带者。
肌营养不良症肌营养不良症是一组肌肉退化疾病,导致肌肉无法正常发育和运作。
这种疾病主要影响男性,而女性成为携带者。
4. 伴性遗传的诊断和治疗诊断伴性遗传的疾病通常需要进行基因测序和分析。
这可以通过检测特定基因的突变来确定是否存在遗传病。
治疗伴性遗传的疾病目前仍然是一个挑战,因为无法根治这些疾病。
然而,一些疾病可以通过症状管理和康复治疗来改善患者的生活质量。
5. 伴性遗传的咨询和预防对于携带伴性遗传疾病的女性来说,咨询和预防措施非常重要。
他们可以通过咨询遗传专家来了解他们携带疾病的风险以及可能的遗传后果。
此外,他们可以通过避免与有患病家庭成员进行婚姻或进行辅助生殖技术来减少遗传疾病的概率。
6. 伴性遗传的研究进展随着基因测序和生物技术的进步,我们对伴性遗传的理解和研究也在不断深入。
新的筛查方法和治疗方案正在研究和开发中,以帮助患者和他们的家庭更好地应对这些疾病。
结论伴性遗传涉及到一系列基因突变引起的疾病,这些疾病通常在X染色体上。
高中生物知识点总结——人类遗传病及伴性遗传!
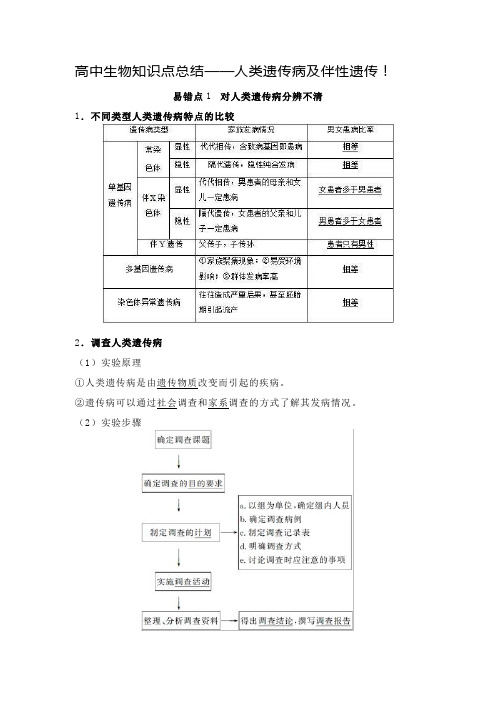
高中生物知识点总结——人类遗传病及伴性遗传!易错点1 对人类遗传病分辨不清1.不同类型人类遗传病特点的比较2.调查人类遗传病(1)实验原理①人类遗传病是由遗传物质改变而引起的疾病。
②遗传病可以通过社会调查和家系调查的方式了解其发病情况。
(2)实验步骤(3)遗传病发病率与遗传方式调查的区别易错点2 混淆“男孩患病”与“患病男孩”的概率问题1.携带致病基因≠患病,如隐性遗传病中的携带者Aa不患病。
2.不携带致病基因≠不患遗传病,如21三体综合征。
3.速判遗传病的方法4.患病概率计算技巧(1)由常染色体上的基因控制的遗传病①男孩患病概率=女孩患病概率=患病孩子概率。
②患病男孩概率=患病女孩概率=患病孩子概率×1/2。
(2)由性染色体上的基因控制的遗传病①若病名在前、性别在后,则从全部后代中找出患病男(女),即可求得患病男(女)的概率。
②若性别在前、病名在后,求概率时只考虑相应性别中的发病情况,如男孩患病概率则是指在所有男孩中患病的男孩所占的比例。
5.快速突破遗传系谱题思维模板6.判断基因是位于X、Y染色体的同源区段还是仅位于X染色体上(1)方法:隐性雌性×纯合显性雄性。
(2)结果预测及结论①若子代全表现为显性性状,则相应的控制基因位于X、Y染色体的同源区段。
②若子代中雌性个体全表现为显性性状,雄性个体全表现为隐性性状,则相应的控制基因仅位于X 染色体上。
易错点3 不能正确判断遗传系谱图中遗传病的遗传方式1.判定是否为伴Y染色体遗传若系谱中,遗传具有“父传子,子传孙”的特点,即患者都是男性且有“父→子→孙”的传递规律,则为伴Y染色体遗传;否则不是伴Y染色体遗传。
2.判定是显性遗传还是隐性遗传(1)若双亲正常,其子代中有患者,则此单基因遗传病一定为隐性遗传(即“无中生有为隐性”)。
(2)若患病的双亲生有正常后代,则此单基因遗传病一定为显性遗传(即“有中生无为显性”)。
如果没有上述明显特征,则采用假设法,即先假设为显性或隐性遗传,如果与遗传图谱相符,则假设成立,否则假设不成立。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
高中生物“伴性遗传”知识点总结高中生物“伴性遗传”知识点总结
伴性遗传的最大特点就是性状与性别的关联,这部分常考题目主要有伴性遗传的判断和相关计算。
判断是伴性遗传还是常染色体遗传,常用同型的隐形个体与异型的显性个体杂交,根据后代的表现型进行判断。
以XY型性别决定的生物为例,如果为伴X隐性遗传,雌性隐性个体与雄性显性个体杂交,如果后代雄性个体中出现了显性性状,即为常染色体遗传,否则即为伴X遗传。
一.常见遗传病的遗传方式:
(1) 单基因遗传:
常染色体显性遗传:并指、多指; 常染色体隐性遗传:白化病、失天性聋哑 X连锁隐性遗传:血友病、红绿色盲; X连锁显性遗传:抗维生素D佝偻病; Y连锁遗传:外耳道多毛症;
(2)多基因遗传:唇裂、先天性幽门狭窄、先天性畸形足、脊柱裂、无脑儿; (3 )染色体病:染色体数目异常:先天性愚型病; 染色体结构畸变:猫叫综合症。
二、单基因遗传病
单基因遗传病是指受一对等位基因控制的遗传病, 较常见的有红绿色盲、血友病、白化病等。
根据致病基因所在染色体的种类,通常又可分四类:
1、常染色体显性遗传病
致病基因为显性并且位于常染色体上,等位基因之一突变,杂合状态下即可发病。
致病基因可以是生殖细胞发生突变而新产生,也可以是由双亲任何一方遗传而来的。
此种患者的子女发病的概率相同,均为1/2。
此种患者的异常性状表达程度可不尽相同。
在某些情况下,显性基因性状表达极其轻微,甚至临床不能查出,种情况称为失显。
由于外显不完全,在家系分析时可见到中间一代人未患病的隔代遗传系谱,这种现象又称不规则外显。
还有一些常染色体显性遗传病,在病情表现上可有明显的轻重差异,纯合子患者病情严重,杂合子患者病情轻,这种情况称不完全外显。
2、常见常染色体显性遗传病的病因和临床表现
1)、多指(趾)、并指(趾)。
临床表现:5指(趾)之外多生1~2指(趾),有的仅为一团软组织,无关节及韧带,也有的有骨组织。
2)、珠蛋白生成障碍性贫血。
病因:珠蛋白肽链合成不足或缺失。
临床表现:贫血。
3)、多发性家族性结肠息肉。
病因:息肉大小不等,可有蒂,也可以是广底的,分布在下段结肠或全部结肠。
临床表现:便血,常有腹痛、腹泻。
4)、多囊肾。
病因:肾实质形成大小不等的囊泡,多为双侧。
临床表现:腹痛,血尿,腹部有肿块,高血压和肾功能衰竭。
5)、先天性软骨发育不全。
病因:长骨干骺端软骨细胞形成障碍,软骨内成骨变粗,影响骨的长度,但骨膜下成骨不受影响。
临床表现:四肢粗短,躯干相对长,垂手不过髋关节,手指短粗,各指平齐,头围较大,前额前突出,马鞍型鼻梁,下颏前突,腰椎明显前突,臀部后凸。
6)、先天性成骨发育不全。
临床表现:以骨骼易折、巩膜蓝色、耳聋为主要特点。
7)、视网膜母细胞瘤。
临床表现:视力消失,瞳孔呈黄白色,发展可引起青光眼,眼球突出。
二、常染色体隐性遗传病致病基因为隐性并且位于常染色体上,基因性状是隐性的,即只有纯合子时才显示病状。
此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女。
子代有1/4的概率患病,子女患病概率均等。
许多遗传代谢异常的疾病,属常染色体隐性遗传病。
按照“一基因、一个酶”或“一个顺反子、一个多肽”(one 的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常。
3、常见常染色体隐性遗传病的病因和临床表现
1)、白化病。
病因:黑色素细胞缺乏酪氨酸酶,不能使酪氨酸变成黑色素。
临床表现:毛发银白色或淡黄色,虹膜或脉络膜不含色素,因而虹膜和瞳孔呈蓝或浅红色,且畏光,部分有曲光不正、斜视及眼球震颤,少数患者智力低下。
2)、苯丙酮尿症。
肝脏中缺乏苯丙氨酸羟化酶,使苯丙氨酸
不能氧化成酪氨酸,只能变成苯丙酮酸,大量苯丙氨酸及苯丙酮酸累积在血和脑积液中,并随尿排出,对婴儿神经系统造成不同程度的伤害,并抑制产生黑色素的酪氨酸酶,致使患儿皮肤毛发色素浅。
临床表现:不同程度的智力低下,皮肤毛发色浅,尿有发霉臭味,发育迟缓。
3)、半乳糖血症。
病因:由于α1-磷酸半乳糖尿苷转移酶缺乏,使半乳糖代谢被阻断,而积聚在血、尿、组织内,对细胞有损害,主要侵害肝、肾、脑及晶状体。
临床表现:婴儿出生数周后出现体重不增、呕吐、腹泻、腹水等症状,可出现低血糖性惊厥、白内障、智力低下等。
4)、粘多糖病。
病因:粘多糖类代谢的先天性障碍,各种组织细胞内积存大量的粘多糖,形成大泡。
临床表现:出生时正常,6个月到2岁时开始发育迟缓,可有智力及语言落后,表情呆板,皮肤略厚,似粘液水肿,可有骨关节多处畸形。
5)、先天性肾上腺皮质增生症。
病因:肾上腺皮质合成过程中的各种酶缺乏。
临床表现:女性患者男性化,严重者可呈两性畸形;男性患者外生殖器畸形,假性性早熟,可合并高血压、低血钾等症状。
三、X连锁显性遗传病
1、X连锁显性遗传
一些性状或遗传病的基因位于X染色体上,其性质是显性的,这种遗传方式称为X连锁显性遗传(X-linked dominant
inheritance),这种疾病称为X连锁显性遗传病。
目前所知X连锁显性遗传病不足20种。
由于致病基因是显性的,并位于X染色体上,因此,不论男性(XAY)和女性(XAXa)只要有一个这种致病基因XA就会发病。
与常染色体显性遗传不同之处是,女性患者既可将致病基因传给生子,又可以传给女儿,且机会均等;而男性患者只能将致病基因传给女儿,不传给儿子。
由此可见,女性患者多于男性,大约为男性的1倍。
另外,从临床上看,女性患者大多数是杂合子,病情一般较男性轻,而男患者病情较重。
抗维生素D佝偻病(vitamin D resistant rickets, VDRR)可以作为X连锁显性遗传病的实例。
VDRR是一种以低磷酸血症导致骨发育障碍为特征的遗传性骨病。
患者主要是肾远曲小管对磷的转运机制有某种障碍,困而尿排磷酸盐增多,血磷酸盐降低而影响骨质钙化。
患者身体矮小,有时伴有佝偻病等各种表现。
患者用常规剂量的维生素D治疗不能奏效,故有抗维生素D佝偻病之称。
从临床观察,女性患者的病情较男性患者轻,多数只有低血磷,佝偻症状不太明显,表现为不完全显性,这可能是女性患者多为杂合子,其中正常X 染色体的基因还发挥一定的作用。
男性患者(XHY)与正常女性(XhXh)结婚,所生子女中,儿子全部正常,女儿全部发病;女性患者(XHXh)与正常男性(XhX)
结婚,子女中正常与患者各占1/2. X连锁显性遗传病病种较少,有抗维生素D性佝偻病等。
这类病女性发病率高,这是由于女性有两条X染色体,获得这一显性致病基因的概率高之故,但病情较男性轻。
男性患者病情重,他的全部女儿都将患病。
2、常见X伴性显性遗传病的病因和临床表现
1)、抗维生素D佝偻病。
病因:甲状腺功能不足,影响体内磷、血钙的代谢过程,致使血磷降低,且维生素D治疗效果不好。
临床表现为:身材矮小,可伴佝偻病和骨质疏松症的各种表现。
2)、家族性遗传性肾炎。
病因:肾小管发育异常,集合管比常人分支少,呈囊状,远曲小管薄,但近曲小管变化轻。
临床表现为:慢性进行性肾炎,反复发作性血尿,1/3~1/2患者伴神经性耳聋。
四、X连锁隐性遗传病
致病基因在X染色体上,性状是隐性的,女性只是携带者,这类女性携带者与正常男性婚配,子代中的男性有1/2是概率患病,女性不发病,但有1/2的概率是携带者。
男性患者与正常女性婚配,子代中男性正常,女性都是携带者。
因此X连锁隐性遗传在患病系中常表现为女性携带,男性患病。
男性的致病基因只能随着X染色体传给女儿,不能传给儿子,称为交叉遗传。
常见X伴性隐性遗传病的病因和临床表现
1、血友病A。
病因:血浆中抗血友病球蛋白减少,AHG即第Ⅷ因子凝血时间延长。
临床表现:轻微创伤即出血不止,不出血时与常人无异。
2、血友病B。
病因:血浆中缺乏凝血酶成份PTC,即第Ⅸ因子。
临床表现同血友病A。
3、色盲。
临床表现:全色盲对所有颜色看成无色,红绿色盲为不能区别红色和绿色。
4、进行性肌营养不良。
病因:为原发性横纹肌变性并进行性发展。
临床表现:初为行走笨拙,易跌到,登梯及起立时有困难,从仰卧到起立必须先俯卧,双手撑地,再用两手扶小腿、大腿才能站起。
进行性肌肉萎缩,但一般不累及面部及手部肌肉。
五、隔代遗传
从遗传学的角度看,致病基因的传递是代代相传的,一个个体一旦没有从亲代继承到某个特定的致病基因,那么,其后代一般也不必担忧此种致病基因所带来的遗传病。