EMA_关于基因毒性杂质限度指南的问答(中文)
ema亚硝胺杂质问答翻译 -回复

ema亚硝胺杂质问答翻译-回复标题:EMA亚硝胺杂质问答:了解亚硝胺及其相关问题导言:在日常生活和食品加工中,亚硝胺杂质是一个重要的关注点。
欧洲药品管理局(EMA)是负责监管和监督药品和相关杂质的机构之一。
本文将以EMA亚硝胺杂质为主题,一步一步回答相关问题,帮助读者更好地了解亚硝胺及其相关问题。
一、什么是亚硝胺?亚硝胺是一类有机化合物,由亚硝化剂和胺类物质反应生成。
它们可以存在于食品、化妆品、水源和环境中。
亚硝胺的形成与温度、pH值和存储条件等因素有关。
二、亚硝胺杂质对健康的影响是什么?亚硝胺类物质与人类健康密切相关。
它们被认为是潜在的致癌物质,尤其是与胃癌和肠癌相关。
此外,长期暴露于含有亚硝胺杂质的食品和环境中,还与其他健康问题如糖尿病和心血管疾病的发生有关。
三、EMA的职责是什么?欧洲药品管理局(EMA)是负责监管和监督医疗产品的机构。
它的任务包括评估药物的安全性、疗效和质量要求,为医疗专业人员和公众提供建议和信息。
四、为什么亚硝胺杂质是一个关注点?亚硝胺杂质因其潜在的致癌性质和存在于许多常见产品中的普遍性而备受关注。
过去几年中,许多产品被检测出含有亚硝胺杂质,包括食品、药品、化妆品和饮用水。
因此,监管机构和消费者对亚硝胺杂质的控制和减少提出了更高的要求。
五、EMA如何监管亚硝胺杂质?EMA对医疗产品中的亚硝胺杂质制定了严格的监管标准。
它要求药品制造商在生产过程中控制和监测亚硝胺杂质的含量,以确保产品质量和安全性。
此外,EMA还与其他监管机构合作,制定共同监管标准和共享信息以加强亚硝胺杂质的风险评估和控制。
六、如何最大程度降低亚硝胺杂质的风险?个人和企业可以采取多种措施来降低亚硝胺杂质的风险。
一方面,消费者可以选择新鲜和天然的食品,避免过度加工的食品。
另一方面,制造商可以优化生产过程,控制生产条件,避免亚硝胺杂质的形成。
此外,科研机构和监管机构可以加强监测和风险评估,及时发布相关信息以指导消费者和制造商。
基因毒性杂质的评估与控制

6. Istituto Superiore di Sanita (Italy) 7. Prous Institute (Spain) 8. Swedish Toxicology Science Research Center(Sweden)
1.临床使用剂量增加 2.用药时长显著增加 3.病情严重或危及生命的病患状态下采用较高可接 受摄入量,变为不那么严重的病患情况后,原有的 杂质可接受摄入量不再适当 4.使用新的给药途径 5.扩大使用患者群包括孕妇和(或)小儿
◆新的杂质被确知属于第一类或第二类诱变性杂
质
THREE
基因毒性杂质的评估
一、评估内容
五、杂质残留风险评估考量
基毒杂质清除因子计算表
代码
GTI
到API 步骤
3 2 1
反应活 性
1-100 X X
溶解性
1-10(100) X X
挥发性
1-10 X X
电离性
5 X X
消除因 子/步
XXX
总消除因 子
清除 原理
Xx,xxx,xxx
参考文献: Teasdale A, Elder D, Chang S-J, et al. Risk assessment of genotoxic impurities in new chemical entities: strategies to demonstrate control[J]. Org Process Res Dev, 2013, 17: 221-230.
FDA
2008 年签发《原料药和成品药中遗传 毒性和致癌性杂质,推荐方法》
内容和EMA指南基本一致,主要包括: ◆ 原料药和制剂中的基因毒性杂质生 成的预防办法
EMA关于API中基因毒性、金属杂质和残留溶剂的标准(中英文对照)

EMA关于API中基因毒性、金属杂质和残留溶剂的标准(中英文对照)EMA关于API中基因毒性,金属杂质和残留溶剂的标准1.What is a reasonable policy forsetting specifications for potentially genotoxic impurities which aretheoreti cal or actual impurities in a drug substance manufacturing process?HJune20121.什么是设定潜在基因毒性杂质(原料药生产工艺中理论或实际的杂质)标准的基本原则?人用药品,2012年6月Different possible scenarios can beidentified and the applicable policies to be applied for each of them arede scribed below:不同的可能情况可被划分,并且不同的适用原则对应于下面描述的各个情况:Example1–A potential genotoxicimpurity举例1—潜在基因毒性杂质The definition for a potentialgenotoxic impurity is derived from the definition for'potential impurity':animp urity that theoretically can arise during manufacture or storage.It may ormay not actually appear in the(new) drug substance(ICH Q3A,glossary).潜在基因毒性杂质的定义起源于“潜在杂质”的定义:理论上可能在生产中或贮藏中出现的杂质,它可能会或不会实际存在于(新)原料药中(ICH Q3A,术语)。
EMA制剂质量问答-12杂质—杂质阈值计算2问201710

EMA制剂质量问答-12杂质—杂质阈值计算2问201710Impurities - Calculation of thresholds for impurities杂质—杂质阈值计算1. What is the basis for the calculation of thresholds to set limits for impurities in the finished product specification? H March 2015制剂质量标准中杂质限度设定的阈值计算基础是什么?H 2015年3月Mono-component products 单组份药品For medicinal products containing only one active substance, the calculated thresholds should be based on the highest maximum daily dose of the respective active substance in authorised medicinal products and the limits in the specification set accordingly. The threshold for impurities should be the same for all strengths. The applicant is responsible to consider the maximum daily dose (MDD) that is approved for a given active substance in those Member States where the medicinal product is to be licensed.对于只含有一种活性物质的药品,计算阈值应基于批准的制剂中对应活性物质的日最高剂量和相应质量标准系列中的限度。
(完整版)基因毒性杂质的评估与控制

序号 1 2 3 4
名称 Toxicological Data Network Toxicology Literature Online U.S.Food and Drug Administration National Instirute for Health
简称 TOXLINE TOXLINE
FDA NIH
1. 美 国 PhRMA 发 布 白 皮书,提出阶段化TTC 概念。
2.EMA 颁 布 《 基 因 毒 性杂质限度指南》。
ICH M7公布,总结构 效 关 系 ( Q ) SAR 和 毒理学关注阈值 (TTC)评估。
国家药典委员会《遗传 毒性杂质控制指导原则
》(征求意见稿)。
二、官方指南:EMA及FDA
◆上市产品已建立其总体控制策略和质量标准后 所获得的新的相关杂质危害数据
◆新的杂质被确知属于第一类或第二类诱变性杂 质
THREE
基因毒性杂质的评估
一、评估内容 存在
判断
分类
来源
评估内容
评估
二、杂质来源
工艺杂质
起始物料及其杂质,溶剂及 其杂质,中间体,副产物,
催化剂,辅料杂质
生产设备及包材 引入杂质
Leadscope
MultiCASE
NTP PAN Pharma Pendium RTECS ToxNet /ChemID Plus TRACE from BIBRA VITIC from Lhasa Limited
特点 公开,毒性物质及疾病登记,包括危害性评价的毒理学研究资料 公开,包括化学致癌物、结构及试验数据,1985—2011 阶段研究 公开,1980—2011,致癌性数据库 公开,可按结构查询的毒性数据库,包括来自CPDB, ISSCAN 等数据库的信息 公开,欧洲化学品信息 商用,包括Gene-Tox 和CCRIS 公开,美国环保局公布、经专家审评过的3 000 种化学物质的致突变性研究结果 公开,美国国立癌症研究所 公开,国际化学品安全性项目总结 公开,美国环保署用以人群健康风险评价,着重在危害确认及剂量反应关系评价 公开,化学致癌物,包括结构及试验数据 公开,日本现有化学品数据库,包括高生产容量化学品( High production volume chemicals)
基因毒性杂质限度指南(转载中英文)

20060628 EMEA/CHMP/QWP/251344/2006 基因毒性杂质限度指南(转载中英文)London, 28 June 2006CPMP/SWP/5199/02EMEA/CHMP/QWP/251344/2006TABLE OF CONTENTS 目录EXECUTIVE SUMMARY 内容摘要 (3)1. INTRODUCTION 介绍 (3)2. SCOPE 范围 (3)3. LEGAL BASIS法律依据 (3)4. TOXICOLOGICAL BACKGROUND 毒理学背景 (4)5. RECOMMENDATIONS 建议 (4)5.1 Genotoxic Compounds With Sufficient Evidence for a Threshold-Related Mechanism具有充分证据证明其阈值相关机理的基因毒性化合物 (4)5.2 Genotoxic Compounds Without Sufficient Evidence for a Threshold-Related Mechanism不具备充分证据支持其阈值相关机理的基因毒性化合物 (5)5.2.1 Pharmaceutical Assessment 药学评价 (5)5.2.2 Toxicological Assessment 毒理学评价 (5)5.2.3 Application of a Threshold of Toxicological Concern 毒理学担忧阈值应用 (5)5.3 Decision Tree for Assessment of Acceptability of Genotoxic Impurities基因毒性杂质可接受性评价决策树 (7)REFERENCES. 参考文献 (8)EXECUTIVE SUMMARY 内容摘要The toxicological assessment of genotoxic impurities and the determination of acceptable limits for such impurities in active substances is a difficult issue and not addressed in sufficient detail in the existing ICH Q3X guidances. The data set usually available for genotoxic impurities is quite variable and is the main factor that dictates the process used for the assessment of acceptable limits. In the absence of data usually needed for the application of one of the established risk assessment methods, i.e. data from carcinogenicity long-term studies or data providing evidence for a threshold mechanism of genotoxicity, implementation of a generally applicable approach as defined by the Threshold of Toxicological Concern (TTC) is proposed. A TTC value of 1.5 μg/day intake of a genotoxic impurity is considered to be associated with an acceptable risk (excess cancer risk of <1 in 100,000 over a lifetime) for most pharmaceuticals. From this threshold value, a permitted level in the active substance can be calculated based on the expected daily dose. Higher limits may be justified under certain conditions such as short-term exposure periods.基因毒性杂质的毒理学评估和这些杂质在活性药物中的可接受标准的测定是一件困难的事情,并且在现有的ICH Q3X指南中也没有详细的规定。
基因毒性杂质(genotoxic

TTC用于计算未做研究的化学物质的接触量,这些 化学物质不会有明显的致癌性或者其他毒性。
ConcentrationLimit ( ppm) TTC (ug / day) dose(g / day)
TTC理论不可以应用于那些毒性数据(长期研究) 充分的致癌物质,也不可以做高风险毒性物质的风 险评价。
TTC是一个风险管理工具,它使用的是概率方法。所以 TTC不能被理解为绝对无风险的保障。
TTC
意思是:假如有一个基因毒性杂质,并且我们对 它的毒性大小不了解,如果它的每日摄入量低于 TTC值,那么,该基因毒性杂质的致癌风险将不 会高于100000分之一的概率。
某些特定情况,TTC值高于1.5μg/day也是可以 接受的。比如药物的短期接触,即治疗某些声明 预期在5年以下的某些严重疾病,或者这种杂质是 一种已知物质,人类在其他方式上对它的摄入量 会更高(比如在食品上)。这个需要根据实际情 况再进行推算。
应该有合理的分析方法去检测和量化这些杂质的 残留量。
毒理学研究
为一个不存在阀值的基因毒性致癌物定义一个安 全的摄入量水平(零风险观点)是不可能的,并 且从活性药物成分中完全的除去基因毒性杂质经 常是很难做到。这样就要求我们建立一个可接受 的风险水平,例如对一个低于可忽略风险的每日 摄入量进行评价。
判断是否为基因毒性杂质
通过Carcinogenic potency database (CPDB) 数据库查询,数据库中现有1574种致癌物质的列 表。链接 /chemnamein dex.html ,还可查询到关于基因毒性方面研究 的出版物。
基因毒性杂质卤代烃的风险评估
有数据表明氯乙烷、氯甲烷为基因毒性杂质,因 此有理由怀疑其他低分子卤代烃类也有类似的作 用。在生产中应该对其进行相应的控制。
EMEA人用药品委员会(CHMP)《遗传毒性杂质限度指导原则》中文译稿

EMEA人用药品委员会(CHMP)《遗传毒性杂质限度指导原则》原文:European Medicines Agency: Guideline on the Limits of Genotoxic Impurities.CPMP/SWP/5199/02。
EMEA/CHMP/QWP/251344/2006。
London, 28 June 2006摘要遗传毒性杂质的毒理学评估和原料药中此类杂质的可接受限度确定是一个难题,现有ICHQ3X指南中未充分说明。
常用遗传毒性杂质数据库差异很大,而数据库是决定可接受限度评估所用方法的主要因素。
当运用已建立的风险评估方法所需资料缺乏时,包括致癌性长期试验资料或提供遗传毒性阈值机制证据的资料等,建议采用毒理学担忧阈值(TTC)所定义的普遍适用方法。
对大部分药物,遗传毒性杂质摄入量为1.5µg/天的TTC值时,认为相关的风险可接受(终身癌症风险<1/100000)。
根据该阈值,原料药中遗传毒性允许水平可根据预计每日剂量计算得到。
短期给药等特定情况下可能有理由提高限度。
1.介绍在原料药(Q3A,新原料药中的杂质)和药物制剂(Q3B,新药物制剂中的杂质)的指导原则中描述了杂质限度确定的一般概念,将限度确定定义为获得和评价特定水平下单个杂质或特定杂质谱的生物学安全性资料。
对于有潜在遗传毒性的杂质,确定可接受剂量水平通常被认为是特别重要的问题,尚未被现有专门指导原则涵盖。
2. 适用范围本指导原则阐述了如何处理新原料药中遗传毒性杂质的一般框架和实践方法。
该指导原则也适用于已有原料药的新申请,如果其合成路线、过程控制和杂质研究尚无法确保不会产生新的或更高含量的遗传毒性杂质(与EU目前批准的相同原料药相比)。
该指导原则同样适用于已上市原料药有关合成方面的变更申请。
不过,除非有特殊原因,本指导原则不适用于已批准药品。
本文中,将化合物(杂质)归类为遗传毒性物质,一般指在主要着重于检测有直接损伤DNA潜力的DNA反应物质的既定体外或体内遗传毒性试验中有阳性结果。
EMA关于 API 中基因毒性、金属杂质和残留溶剂的标准(中英文对照)

EMA关于API中基因毒性,金属杂质和残留溶剂的标准1.What is a reasonable policy forsetting specifications for potentially genotoxic impurities which aretheoreti cal or actual impurities in a drug substance manufacturing process?HJune20121.什么是设定潜在基因毒性杂质(原料药生产工艺中理论或实际的杂质)标准的基本原则?人用药品,2012年6月Different possible scenarios can beidentified and the applicable policies to be applied for each of them arede scribed below:不同的可能情况可被划分,并且不同的适用原则对应于下面描述的各个情况:Example1–A potential genotoxicimpurity举例1—潜在基因毒性杂质The definition for a potentialgenotoxic impurity is derived from the definition for'potential impurity':animp urity that theoretically can arise during manufacture or storage.It may ormay not actually appear in the(new) drug substance(ICH Q3A,glossary).潜在基因毒性杂质的定义起源于“潜在杂质”的定义:理论上可能在生产中或贮藏中出现的杂质,它可能会或不会实际存在于(新)原料药中(ICH Q3A,术语)。
基因毒性杂质限度指南-中文

基因毒性杂质限度指南-中文《基因毒性杂质限度指南》European Medicines Agency: Guideline on the Limits of Genotoxic Impurities.CPMP/SWP/5199/02。
EMEA/CHMP/QWP/251344/2006。
London, 28 June 2006 关键词:杂质;基因毒性;毒理学关注阈值(TTC);构效关系(SAR)摘要进行基因毒性杂质的毒理学评估和这些杂质在原料药中的可接受限度的确定是个难题,并且在现有的ICHQ3X指南中并未详细规定。
现有的关于基因毒性杂质的相关数据是容易变化的,也是对杂质可接受标准如何进行评价的主要影响因素。
如果缺少风险评估方法所需要的数据,比如,致癌作用的长期研究数据或为基因毒性的阈值提供证据的数据,一般建议使用一般通用的被定义为毒理学关注阈值 (TTC)的方法。
一个“1.5ug/day”的TTC值,即相当于每天摄入1.5ug的基因毒性杂质,被认为对于大多数药品来说是可以接受的风险(一生中致癌风险小于十万分之一)。
按照这个阈值,可以根据这个预期的每日摄入量计算出原料药中可接受的杂质水平。
但是在某些情况下制定更高的限度也是合理的,例如:短期暴露。
1.前言在原料药(Q3A,新药物活性物质中的杂质)或药物制剂(Q3B,新药物制的一般概念,其中将“杂质界定”定义“是剂中的杂质)指南中描述了杂质界定获得和评估数据的过程,该数据用于建立单个杂质或在特定水平的已知杂质的生物安全性”。
对于潜在基因毒性的杂质,确定其可接受的剂量水平通常被认为是特别重要的问题,但是这个问题在现有的指南中没有被讨论。
12.适用范围本指南阐述了如何处理新原料药中基因毒性杂质的一般框架和实用方法。
该指南也适用于已有原料药的新申请,如果其合成路线、过程控制和杂质研究尚无法确保不会产生新的或更高含量的遗传毒性杂质(与EU目前批准的相同原料药相比)。
基因毒性杂质

基因毒性杂质什么是基因毒性杂质对于基因毒性杂质的定义主要是指:在以DNA反应物质为主要研究对象的体内/体外试验中,如果发现它们对DNA有潜在的破坏性,那可称之为基因毒性。
对没有进行体内实验的情况下,也可以根据关联系做一些相关的体外实验去评估该物质在体内的毒性。
如果没有关联评估的,体外基因毒性物质经常被考虑为假定的体内诱变剂和致癌剂。
GUIDELINE ON THE LIMITS OF GENOTOXIC IMPURITIES (EMEA/CHMP/QWP/251344/2006)基因毒性杂质的风险按照目前的法规来说,(体内)基因毒性物质在任何摄入量水平上对DNA都有潜在的破坏性,这种破坏可能导致肿瘤的产生。
因此,对于基因毒性致癌物,不能说“不存在明显的阀值,或是任何的摄入水平都具有致癌的风险”。
可接受风险的摄入量对于那些可以与DNA进行反应的化合物,由于在较低的剂量时机体保护机制可以有效的运行,按照摄入量由高到低所造成的影响进行线性推断是很困难的。
目前,对于一个给定诱变剂,我们很难从实验方面证明它的基因毒性存在一个阀值。
特别是对某些化合物,它们可以与非DNA靶点进行反应,或一些潜在的突变剂,在与关键靶位结合之前就迅速失去了毒性。
由于缺乏支持基因毒性阀值存在的有力证据,而使得我们很难界定一个安全的服用量。
所以有必要采取一个新观点:确定一个可接受其风险的摄入量。
可接受其风险的摄入量即毒理学阈值一般通用的被定义为Threshold of Toxicological Concern(TTC) 。
具体含义为:一个“1.5ug/day”的TTC值,即相当于每天摄入1.5ug的基因毒性杂质,被认为对于大多数药品来说是可以接受的风险(一生中致癌的风险小于100000分之1)。
按照这个阀值,可以根据预期的每日摄入量计算出活性药物中可接受的杂质水平。
在特定的条件下一些基因毒性杂质也可以有较高的阈值。
如接触时间比较短等,这个需要根据实际情况再进行推算。
EMA关于基因毒性杂质限度指南的问答(中文)

EMA关于基因毒性杂质限度指南的问答2010-9-23背景:本问答文件的目的是对2006年出版的基因毒性杂质限度指南(EMEA/CHMP/QWP/251344/2006)进行相关内容统一和说明。
问答Q1:该指引并不要求对已批准销售的产品进行基因毒性杂质再评估,除非有一个特定的“重要原因”(cause-for-concern)。
请问什么是“重要原因”?A1:如果原料药的生产过程基本上没有改变,就不需要对基因毒性杂质进行重新评价。
但是,如果新知识表明有新原因时,例如几年前发现的甲磺酸盐药物可能形成甲磺酸烷基的基因毒性杂质,这需要进行基因毒性杂质的再评估,包括EP药典中收载的所有甲磺酸盐类产品,并出示“生产声明”。
Q2:该指引指出:即使按决策树程序其水平低于毒理学关注阈值(threshold of toxicological concern,TTC),也要尽可能地减少已知或未知的诱变杂质(mutagenic impurity)。
如果已知其诱变杂质的水平低于TTC(TTC是一个非常保守的值),为什么要还进一步减少呢?实际上这还涉及定量限在1ppm左右的分析方法,可以这样做但可能没结果,这是否有必要呢?A2:如果一个诱变杂质的水平低于毒理学关注阈值(相当于临床剂量≤1.5微克/天),就没有必要这样做。
除非它具有一个高度关注的风险结构:如N -亚硝基,黄曲霉毒素类和氧化偶氮物就需要这样做。
Q3:该指引规定:“当一个潜在的杂质包含“警示结构”(structural alerts)时,应考虑用细菌突变试验对其杂质进行的基因毒性分析”。
i)如果一个杂质能诱发“警示结构”,该杂质的致突变试验(Ames)结果为阴性时,是否就足以得出结论:该化合物不属于关注的遗传毒性杂质?是否还需要进一步的确认研究?ii)“警示结构”不存在就足以说明不属于关注杂质呢?iii)假设某杂质属于“警报结构”,但只要加以控制确保其杂质水平低于TTC,不进行常规检测是否可以接受?A3回答:i)是的。
基因毒性杂质控制相关文件及指南介绍
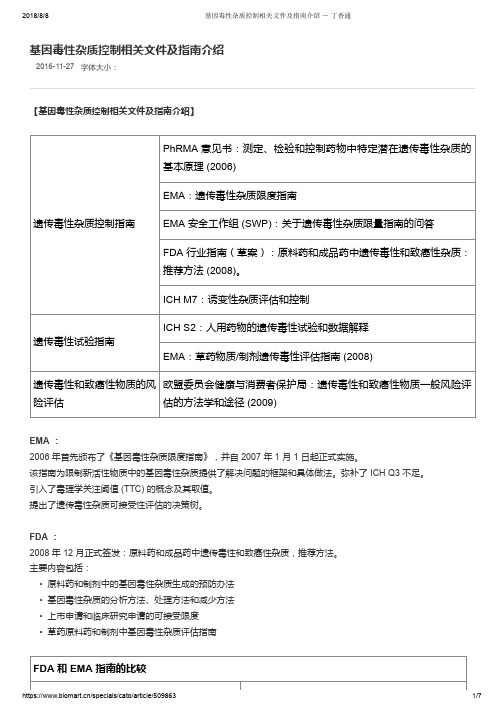
2016-11-27字体大小:基因毒性杂质控制相关文件及指南介绍【基因毒性杂质控制相关文件及指南介绍】遗传毒性杂质控制指南PhRMA 意见书:测定、检验和控制药物中特定潜在遗传毒性杂质的基本原理 (2006)EMA:遗传毒性杂质限度指南EMA 安全工作组 (SWP):关于遗传毒性杂质限量指南的问答FDA 行业指南(草案):原料药和成品药中遗传毒性和致癌性杂质:推荐方法 (2008)。
ICH M7:诱变性杂质评估和控制遗传毒性试验指南ICH S2:人用药物的遗传毒性试验和数据解释EMA:草药物质/制剂遗传毒性评估指南 (2008)遗传毒性和致癌性物质的风险评估欧盟委员会健康与消费者保护局:遗传毒性和致癌性物质一般风险评估的方法学和途径 (2009)EMA :2006 年首先颁布了《基因毒性杂质限度指南》,并自 2007 年 1 月 1 日起正式实施。
该指南为限制新活性物质中的基因毒性杂质提供了解决问题的框架和具体做法。
弥补了 ICH Q3 不足。
引入了毒理学关注阈值 (TTC) 的概念及其取值。
提出了遗传毒性杂质可接受性评估的决策树。
FDA :2008 年 12 月正式签发:原料药和成品药中遗传毒性和致癌性杂质,推荐方法。
主要内容包括:• 原料药和制剂中的基因毒性杂质生成的预防办法• 基因毒性杂质的分析方法、处理方法和减少方法• 上市申请和临床研究申请的可接受限度• 草药原料药和制剂中基因毒性杂质评估指南FDA 和 EMA 指南的比较相似点不同点推荐的鉴定和认证潜在遗传性杂质的方法相同 推荐的处理遗传毒性和致癌性杂质的方法相同FDA 指南包含致癌性杂质TTC 设定为 1.5 μg/天指南允许的 14 天内用药的 TTC 水平为 120 μg , 而非仅针对单次用药临床试验中短期暴露的 TTC 更高FDA 指南不允许根据现售药品的短期暴露情况而 提高 TTC ICH M7【基因毒性杂质的控制策略】具有阳性致癌数据的诱变杂质(第1类)---计算可接受摄入量( AI ): • M7 Addendum 中列出的 15 种化合物中有 10 个为该计算方法计算 • Carcinogenicity Potency Database (CPDB )中列明了 1574 种致癌物质的 TD50 值毒理学关注门槛---TTC 法(第 2/3 类): • ICHM7 主要讨论的方法,主要针对第 2/3 类基因毒性杂质,比如低级磺酸酯类等。
ema亚硝胺杂质问答翻译 -回复

ema亚硝胺杂质问答翻译-回复【EMA亚硝胺杂质问答翻译】引言:近年来,亚硝胺杂质成为了食品和药品行业中备受关注的热点问题。
欧洲药品管理局(EMA)对亚硝胺杂质进行了严格监管,并制定了一系列政策和指南,以确保公众的食品和药品安全。
本文将逐步回答有关EMA亚硝胺杂质的常见问题,帮助读者更好地了解这一问题。
问题一:什么是亚硝胺杂质?亚硝胺是一类有机化合物,通常由亚硝酰胺和胺类化合物反应生成。
它们广泛存在于环境中,包括水、气体和食品中。
一些亚硝胺杂质具有致癌性和致畸性作用,因此对其含量的监管成为了必要的举措。
问题二:为什么亚硝胺杂质会存在于食品和药品中?亚硝胺杂质存在于食品和药品中的原因多种多样。
其中一种主要原因是加工过程中的化学反应。
例如,食品中的亚硝酸盐可以与含有氨基化合物的食材(比如肉类和鱼类)发生反应,形成亚硝胺杂质。
此外,水源和环境因素也可能会引入亚硝胺杂质,进而影响到食品和药品的质量。
问题三:亚硝胺杂质对人体健康有什么影响?一些亚硝胺杂质被证实具有致癌性和致畸性作用。
它们与人体内部的DNA和RNA发生反应,导致基因突变和细胞损伤。
长期摄入含有高含量亚硝胺杂质的食品和药品可能与癌症、肝脏损伤和免疫系统异常等健康问题相关。
问题四:EMA如何监管亚硝胺杂质?欧洲药品管理局(EMA)非常重视亚硝胺杂质的监管,采取了一系列措施,确保食品和药品的安全。
EMA要求制药企业进行亚硝胺杂质的检测和监测,并设定了容许含量标准。
EMA还与欧洲监管机构和国际组织合作,分享信息和最佳实践。
问题五:亚硝胺杂质监管的最新进展是什么?近期,EMA对亚硝胺杂质的监管有了新的进展。
2022年1月1日起,EMA 将会强制要求制药企业对所有非致命(non-lifethreatening)药物的亚硝胺杂质进行检测。
此举旨在进一步降低亚硝胺杂质对人体健康的潜在风险。
结论:亚硝胺杂质作为食品和药品行业中的重要问题,得到了EMA的积极关注和监管。
ema亚硝胺杂质问答20翻译

ema亚硝胺杂质问答20翻译(实用版)目录1.EMA 亚硝胺杂质的定义与来源2.EMA 亚硝胺杂质的危害3.EMA 亚硝胺杂质的检测方法4.如何避免 EMA 亚硝胺杂质的产生5.结论正文EMA 亚硝胺杂质问答 20 翻译EMA 亚硝胺杂质是一种在药品和食品中常见的有害物质,它的存在对人体健康具有很大的危害。
本文将对 EMA 亚硝胺杂质的定义与来源、危害、检测方法以及如何避免其产生的方法进行详细介绍。
一、EMA 亚硝胺杂质的定义与来源EMA 亚硝胺杂质是一类含有亚硝基的化合物,主要来源于食品和药品的合成过程以及储存过程中的化学反应。
此外,食品和药品在加工、加热、腌制等过程中也可能产生亚硝胺杂质。
二、EMA 亚硝胺杂质的危害EMA 亚硝胺杂质具有致癌、致畸、致突变等危害,长期摄入会对人体健康产生严重影响。
因此,对于药品和食品中的 EMA 亚硝胺杂质,我国有严格的限量标准。
三、EMA 亚硝胺杂质的检测方法检测 EMA 亚硝胺杂质的方法有多种,包括薄层色谱法、气相色谱法、高效液相色谱法等。
这些方法各有优缺点,具体选择需要根据实际检测需求和样品特性来确定。
四、如何避免 EMA 亚硝胺杂质的产生为了避免 EMA 亚硝胺杂质的产生,可以从以下几个方面入手:1.在生产过程中,采用合理的工艺参数和设备,减少亚硝酸盐的生成。
2.在储存过程中,注意温度、湿度、氧气和光照等因素,避免药品和食品发生化学反应。
3.在食品加工过程中,减少加热、腌制等可能产生亚硝胺杂质的环节。
4.定期对药品和食品进行检测,确保其符合安全标准。
五、结论总之,EMA 亚硝胺杂质是一种对人体健康具有很大危害的有害物质。
第1页共1页。
基因杂质限度指南

基因杂质限度指南《基因杂质限度指南》嘿,朋友!今天来跟你唠唠基因杂质限度这个事儿哈。
我一开始接触的时候也是两眼一抹黑呢。
一、基本注意事项首先呢,你得搞清楚什么是基因杂质。
简单来说,就像是一锅好汤里不小心混进去的小沙子(比喻可能不太准,但你能理解意思就行),它在基因层面上是些不该有的东西。
在检测基因杂质限度的时候,你一定要严格按照规定的流程来。
比如说取样本这个环节,可不能马虎呀。
我当时就是图快,取样本的时候没太注意规范,结果数据就不对,重新做了好几次。
这就像建房子,地基打不好,上面全白搭。
二、实用建议工具要用对喽。
检测的时候,不同的基因杂质可能需要不同的检测试剂或者仪器。
这就好比做菜,做红烧肉得用酱油来上色,做清蒸鱼就不用,得根据食材(基因杂质)的不同来选择调料(试剂和仪器)。
还有啊,要多做几个平行样呢。
比如你测三个平行样,如果结果差别很大,那就得好好找原因了。
我就有过这样的经历,三个结果差得离谱,后来发现是样本在处理的时候有一个被污染了。
三、容易忽视的点哦,对了。
这个环境因素很容易被忽视啊。
你知道吗,检测的环境如果不干净,很可能就会带来新的杂质,影响你的结果。
我当初没重视这个,后来才知道,就像在灰尘大的地方晾刚洗的衣服,衣服很容易又脏了。
还有记录的细节问题,每一步操作的时间、温度这些都得记录准确了,要不然回头你都不知道问题出在哪。
四、特殊情况有时候会遇到一些很特殊的基因结构,它们对检测试剂有特殊反应,这就需要你有更多的经验或者去查阅更多的资料。
我曾经碰到过一种很罕见的基因杂质,按常规方法检测老是不对头。
我想了好久,后来换了一种检测方法,还特别咨询了几个专家,才把它搞明白。
这就像你走在路上碰到一堵墙,常规的路走不通了,就得想办法找别的路绕过去。
五、总结要点总的来说呢,基因杂质限度这个事儿,从样本的选取到检测的过程,再到对特殊情况的应对,环境的维护和准确的记录,哪一个环节都不能出岔子。
你得要有耐心,像个细心的工匠一样,一点一点把这个活儿做好。
基因毒性杂质(genotoxic..[文字可编辑]
![基因毒性杂质(genotoxic..[文字可编辑]](https://img.taocdn.com/s3/m/1bfdec66a6c30c2258019e42.png)
EMA对基因毒性杂质分类
? EMA对基因毒性杂质的指导原则适用于上市申请 和临床研究。
生物系统的纠错功 能使试验不具备可 行性。
引入一个 新观点: 确定一个 可接受其 风险的摄 入量
TTC
? 可接受其风险的摄入量一般被定义为Threshold of Toxicological Concern (TTC) 。
? 具体含义为:1.5μg/天的TTC值。 ? 相当于人每天摄入1.5μg的基因毒性杂质,被认为对于大
? 甲磺酸烷基酯,如甲磺酸甲酯( MMS)和甲磺酸 乙酯(EMS),是甲磺酸与 甲醇,乙醇 ,或其它 低级醇 形成的酯。特别是在以甲磺酸盐或甲磺酸 酯形式存在的药物活性成分中或其合成过程中用 到了甲磺酸的药物活性成分中,甲磺酸烷基酯会 被视为潜在杂质。
? 在以羟乙基磺酸盐,苯磺酸盐和对甲苯磺酸盐形 式存在的药物活性成分中也会发现类似的磺酸烷 基酯或芳基酯污染。需说明出现这些污染的风Байду номын сангаас。
? 一、有足够实验数据的阈值
? 对于有足够的(实验性的)数据来支持阈值界定 的基因毒性杂质:可参考“ Q3C Note for guidance on impurities: Residual Solvents ” 中2级溶剂的规定,计算出了一个“允许的日摄入 量(PDE—permitted daily exposure )”
判断是否为基因毒性杂质
? 通过Carcinogenic potency database (CPDB) 数据库查询,数据库中现有 1574种致癌物质的列 表。链接 /chemnamein dex.html ,还可查询到关于基因毒性方面研究 的出版物。
基因毒性杂质培训及讨论

M7 Addendum中使用AI计算的10种化合物
.
其他致癌性已知杂质的毒性计算---黄曲霉素
黄曲霉毒素(1402-68-2):基本结构为二呋喃环和香豆素,该类目前已经鉴定出12种。
遗传毒性和致癌性物质一般风险评估的方法学和途径2009有关基因毒性杂质的指南ichm7二ichm7介绍及实例讨论m7适用范围介绍基因毒性杂质控制策略情景原料药制剂备注新原料药和制剂m7基本意图新化合物及制剂的临床申请m7基本意图m7适用范围介绍上市后变更原料药研发生产和控制特别是变更评估要确定其是否会导致任何新的诱变性杂质或已知诱变性对原料药中间体或起始物料的生产场所的变更或变更原料供应商则不需要对诱变性杂质风险重新进行评原料药用于新剂型不需要重新评估上市后变更制剂研发生产和控制上市后申报涉及制剂例如成分生产工艺剂型则应对所有新的诱变性降解产物或已有诱变性降解产物更高的可接受标准进行评估
▪ 原料药和制剂中的基因毒性杂质生成的预防办法 ▪ 基因毒性杂质的分析方法、处理方法和减少方法
.
▪ 上市申请和临床研究申请的可接受限度
有关基因毒性杂质的指南
ICH M7:
.
二、ICH M7介绍及实例讨论
➢M7适用范围介绍 ➢基因毒性杂质评估及分类 ➢基因毒性杂质控制策略
.
M7适用范围介绍
情景 新原料药和制剂
第4类:具有警示结构、与API有关、基因毒性(突变性) 未知的杂质
第5类:没有警示结构,没有基因毒性(突变性)的 杂质
.
基因毒性杂质分类及控制
.
Ames试验(埃姆斯实验)---污染物致突变性检测
➢B.N.Ames等经十余年努力,于1975年建立并完善的沙门氏菌回复突 变试验(亦称Ames试验)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
EMA 关于基因毒性杂质限度指南的问答
2010-9-23
背景:本问答文件的目的是对 2006 年出版的基因毒性杂质限度指南
( EMEA/CHMP/QWP/251344/2006 )进行相关内容统一和说明。
问答
Q1:该指引并不要求对已批准销售的产品进行基因毒性杂质再评估,除非有一个特定的“重要原因”(cause-for-concern )。
请问什么是“重要原因”?
A1:如果原料药的生产过程基本上没有改变,就不需要对基因毒性杂质进行重新评价。
但是,如果新知识表明有新原因时,例如几年前发现的甲磺酸盐药物可能形成甲磺酸烷基的基因毒性杂质,这需要进行基因毒性杂质的再评估,包括EP 药典中收载的所
有甲磺酸盐类产品,并出示“生产声明”。
Q2:该指引指出:即使按决策树程序其水平低于毒理学关注阈值 (threshold of toxicological concern,TTC) ,也要尽可能地减少已知或未知的诱变杂质( mutagenic impurity )。
如果已知其诱变杂质的水平低于 TTC (TTC 是一个非常保守的值),为什么要还进一步减少呢?实际上这还涉及定量限在 1ppm 左右的分析方法,可以这样做但可能没结果,这是否有必要呢?
A2:如果一个诱变杂质的水平低于毒理学关注阈值 (相当于临床剂量≤1.5微克 /天),就没有必要这样做。
除非它具有一个高度关注的风险结构:如N - 亚硝基,黄曲霉毒素
类和氧化偶氮物就需要这样做。
Q3 :该指引规定:“当一个潜在的杂质包含“警示结构”(structural alerts )时,应考虑用细菌突变试验对其杂质进行的基因毒性分析”。
i )如果一个杂质能诱发“警示结构”,该杂质的致突变试验( Ames)结果为阴性时,
是否就足以得出结论:该化合物不属于关注的遗传毒性杂质?是否还需要进一步的确认研究?
ii )“警示结构”不存在就足以说明不属于关注杂质呢?
iii )假设某杂质属于“警报结构”,但只要加以控制确保其杂质水平低于 TTC ,不进行常规检测是否可以接受?
A3 回答:
i)是的。
只要按 Q3A/B 的要求做 Ames 试验显示阴性时,即可认定该杂质不属于“警报
结构”,就不需要进一步确认研究。
ii )是的。
通过仔细评估,如果“警报结构”不存在,即可认定“关注杂质”(concern impurity )不存在。
通常这种评估常用构效关系的评估软件,如 DEREK 或 MCASE 软件。
iii )是的。
当一个潜在的基因毒性杂质水平控制在 TTC 的水平时,就不强制要求进行基因毒性的常规检测,除非它具有非常强的基因毒性物质类 (N -亚硝基化合物及偶氮化合物或黄曲霉毒类化合物)。
Q4:什么是限定新杂质出现的适当战略 (在三期临床阶段或商业批生产阶段) ?,例如,下列战略是否可以接受?当发现一个新未知杂质在 0.05-0.09%的范围就不需采取行
动,在 0.10至0.15%范围,即使它会诱导出“警示结构”,只要对含有这种杂质的成分进行 Ames 试验即可?
A4:出现新的低于鉴别阈值( identification threshold)的未知杂质时, ICH 指南没
有要求有任何行动;但是当高于鉴别阈值,低于确认阈( qualification threshold)并且可
能成为“警示结构”时,并且该原料药中含该杂质的最小浓度为 250 μ板g/( Ames方法的评估检测线,详见 Kenyon et等,Reg Tox & Pharm, 2007, 75-86),就需要对含有这种杂质
的活性成分进行 Ames 实验并成阴性。
Q5:该指引指出,“毒理学关注阈( TTC )值高于 1.5微克/天,在一定条件下是可以接受的,例如短期接触⋯ . ”,阶段性 TTC 与临床接触的持续时间有关,例如抗生素。
如果是这样的话,什么是短期接触的可接受水平?
A5:高于 1.5 μ天g /微克以上的基因毒性杂质的要逐案处理。
对于短期治疗,更高
水平的接受能力原则见问题 6 的回答。
Q6:该指引的措辞意味着它覆盖了 ICH Q3A 没有涉及的问题。
ICH 指南范围不包括NDA 前的临床申请(或备忘录)。
然而,实践表明,临床研发阶段涉及的基因毒性指南同样适合于注册法规要求。
在研发阶段如何控制基因毒性杂质呢?
A6:根据该指南的范围,它主要适用于“新的活性物质”中的基因毒性杂质,包括临床申请和备忘录中提到的新活性物质和临床试验申请中提到的新活性物质。
事实上,临床试验的化学品和药品质量文件要求指( CHMP/QWP/185401/2004)已经对 IMP 中的杂质对志愿者和患者的安全性提出过要求。
要基于安全和毒性数据提供杂质的规格与接受标准。
CHMP 认可在临床研发阶段采用 TTC 分段概念。
在临床研发时段,短期的 TTC 应符合下列要求。
允许的日吸入量与接触时间关系见下表。
面这些值都是考虑了单次校正因子为 2 后通过线性外推的模式来计算偏差得来的
Q7:按指引条款要求,意味着 1.5 微克/天的 TTC 值可适用于每一个原料药中的基因毒性杂质,请问这样的理解是否确切?
A7:在原料药中存在一个以上的基因毒性杂质时, 1.5微克/天 TTC 值对每一个结构不相关的杂质是适用的。
在结构相似的情况下,基因毒性杂质的作用模式一致,并具有相同的分子靶向,在这种情况下,建议基因毒性杂质的总和为 1.5 微克 /天。
这可能不符合实际,特别是最大日剂量很高而且又要求按低限要求申报时。
此时应根据下列因素逐案考虑:
原料药的每日最高剂量;
治疗适应症 ;
产生基因毒性杂质的合成步骤;
消除这些杂质生产纯化能力;
控制这些杂质的分析方法能力。
在这种情况下将这种检测方法能力强的分析方法作为常规检测将是困难的,人们可以在开发过程或首次商业批生产中考虑使用这种分析方法,以充分证明其实际值低于 TTC 值。
在这种情况下,使用风险评估的方法,定期检测而不是常规检测是可以接受的。
Q8.:2008年 3月欧洲药典委员会对具有潜在基因毒性杂质颁发过一个政策。
这项政策将在制定和修订药典中得到应用。
它提供了非常务实的一个政策来指导如何处理现行药典中收载的活性物质中存在的潜在基因毒性杂质。
这些政策是否适用于没有收载到药典中的 API 吗?
A8 :是的。
在这个指南执行前已批准的 API 是适用的。
在注册申报文件中应递交相关的规格要求。
只有当研究数据表明存在遗传毒性杂质时才需要采取行动,仅存在“警
示结构”时不足以引发的后续措施,除非在出现一个高度关注的风险结构,例如:N -亚
硝基,黄曲霉毒素类和氧化偶氮化合物。
如果似使用新的合成路线,该合成路线可能引起潜在基因遗传毒性杂质出现或先前确认的潜在基因毒性杂质水平提高,这种情况应与主管机关
进行讨论。
Q9:在原料药生产过程中,对于理论推测存在或实际存在的潜在基因毒性杂质质量标准的设定依据是什么?
A9 回答:各种不同情形时的潜在基因毒性杂质质量标准可以依据下面的情况来进行设定:
例 1:一个潜在基因毒性杂质
潜在基因毒性杂质的定义是基于“潜在杂质”的定义得来的。
潜在杂质是指按照理论推测在生产或储存过程中可能产生的杂质。
它可能在(新)原料药中存在,也可能不存在(ICH Q3A ,术语)。
如果一个潜在基因毒性杂质仅仅是理论上推测存在,也就是说基于理论推测存在但生产中并未实际检测到(由生产工艺开发阶段的研究证实),则不需要将其列入到原料药的质量标准中。
例 2:合成最后一步前,实际生成或引入了一个(潜在)基因毒性杂质
如果在最后一步合成前,实际生成或引入了一个(潜在)基因毒性杂质,可以不将其纳入原料药质量标准中,但是必须保证该杂质在合成中间体中有合理的控制限度并且在原料药检测结果中明确其含量不超过来源于 TTC 或其他认可标准的 30%(推荐使用标准加入法检测原料药)。
但是如果不能满足这些要求,就必须在原料药质量标准中进行日常检测。
如果一个基因毒性杂质不可在中间阶段控制,那么适用例 3 情况。
例 3:最后一步合成中,实际生成或引入了一个(潜在)基因毒性杂质
如果在最后一步合成中实际生成或引入了一个(潜在)基因毒性杂质,必须在原料药质量标准中控制该杂质。
如果在原料药中该杂质的含量不超过 TTC 或其他认可标准的 30%,可
实行定期检测。
至少要提交 6 批中试批或 3 批大生产的数据。
如果不能满足这个要求,就
必须在原料药质量标准中进行日常检测。
以下为文件中涉及的定义
基因毒性杂质:某种杂质在适当的基因毒性试验模式中表示为基因毒性,例如细菌基因诱变( Ames)试验
潜在基因毒性杂质:某种杂质其结构警示可能具有基因毒性,但未经过试验测试模式。
这里潜在意指可能具有基因毒性,而不是指杂质的存在与否。
参考文献
M.O. Kenyon, J.R. Cheung, K.L. Dobo, W.W. Ku: An evaluation of the sensitivity of the
Ames assay to discern low-level mutagenic impurities. Reg Tox & Pharm, 2007, 75-86.。