omim对表型系列的分类
Omenn综合征临床表型及分子诊断

Omenn综合征 临床表型及分子诊断
袁建 涛 雷 婷 咸 宁市第 一人 民医院检验科 (湖北咸 宁 4370nn综 合 征 的临 床特 点及 其致 病 基 因。方 法 回顾 分 析 2例 Omenn综合 征 患儿 的临 床资 料 和 基 因检测结果。结果 男女患儿各 1例,分别为6个月、8个月 ;均有反复感染、皮疹病史 ;免疫球蛋 白水平均降低 ,CD8 T 淋 巴细胞 降 低 ,CD 19 B淋 巴细 胞严 重 降低 ,自然杀 伤 细胞数 量 升高 。利 用高 通量 测序 方法 对 2例 患儿 及其 家庭 成员 进行 免 疫 缺 陷相关 基 因检 测 ,并利 用 sanger测序 法 验证 ,例 1的 RAG1基 因外显 子 2存在 c.1211G>A(p.R404Q)纯合 突变 ,分 别 遗传 自其 父 母 ,患儿 叔叔 亦为 携带 者 。例 2的 RAG2存 在母 源 C.830A>G(p.Y277C)突变 以及父 源 c.104G>C(p.G35A) 突变 ,2个 突变 位点 均未 见报 道 ,生物 信 息学 预测 2个 突变均 为可 能致 病性 异 常 。结 论 Omenn综合 征患 者 发病年 龄较 早 , 免 疫功 能低 下 。基 因检测 有利 于早 期诊 断 及遗传 咨 询 。确认 了 2个 可 引起 Omeu n 综 合征 的新 突变 。
infection and skin rash.The level of IgA ,IgM and IgG was decreased.Both of them have a lower level of CD 8 T lym phocytes
汗孔角化症的临床表型及基因型研究进展

汗孔角化症的临床表型及基因型研究进展朱培秋;姜薇【摘要】汗孔角化症是较少见的慢性遗传性皮肤病,依据临床表现的不同,可分为6种经典类型及多种新类型.家系遗传性汗孔角化症较散发病例更为常见.目前本病的研究热点为甲羟戊酸代谢通路相关基因的突变.本文就汗孔角化症的临床分型、遗传学研究进展进行了综述.【期刊名称】《中国麻风皮肤病杂志》【年(卷),期】2018(034)009【总页数】5页(P564-568)【关键词】汗孔角化症;临床表型;基因型【作者】朱培秋;姜薇【作者单位】北京大学第三医院皮肤科,北京,100191;北京大学第三医院皮肤科,北京,100191【正文语种】中文1 汗孔角化症的概述汗孔角化症(porokeratosis,PK)是较少见的慢性遗传性皮肤病,多数汗孔角化症表现为常染色体显性遗传,仍有一部分为无家族遗传史的散发病例。
遗传因素、日光暴露及机体免疫抑制状态可能均参与疾病的发生。
本病主要临床特点为边缘隆起呈环状的角化性丘疹或斑块,在组织病理学中有特异的排列紧密的角化不全细胞柱。
经典分型包括:Mibelli型、浅表播散型、光化性浅表播散型、线状型、点状型、掌跖播散型,其中Mibelli型、线状型及点状型皮损较局限,浅表播散型、光化性浅表播散型及掌跖播散型则为泛发性表现。
疣状型、面部型、巨大型、肥厚型、网状型、瘙痒性发疹型汗孔角化症均为较少见的类型[1]。
各种类型可同时存在表现为混合型汗孔角化症[2]。
在遗传学方面,甲羟戊酸代谢通路相关基因突变已日渐成为PK发病机制的研究热点。
2 汗孔角化症的临床经典分型2.1 Mibelli型汗孔角化症(porokeratosis of Mibelli,PM) PM是最经典的临床类型,由Vittorio Mibelli在1893年首次命名,此类型较少见,病情进展缓慢,可为常染色体显性家族遗传,但更多表现为散发病例[3]。
PM常于儿童期发病,多见于有家族史者,且男性好发(男女比例为2.17∶1)[4]。
在线人类孟德尔遗传数据库(OMIM)

在线人类孟德尔遗传数据库(OMIM)中国分子心脏病学杂志2001年12月第一卷第一期(总第l期)在线人类孟德尔遗传数据库(OMIM)甄一松1丰谢攀2史海波1欧国斌1潘月亮1综述张利2审校l中国医学科学院心血管病研究所中围协和医科大学阜外心血管病医院2清华大学电子工程系『摘要』在线人类盂德尔遗传数据库足H前分遗传学中最苇要的生物信息学数据库之一.本文简单地介绍了该数据库所包含的内容,以及如何使用数据库进行盘询和搜索.同时也简单地讨论了这类数据库目前仔任的缺陷.『关键词』人类盂德尔遗传数据库数据内存询方法‟在线人类孟德尔遗传数据库…(On-lineMencle—lianInheritanceinMan,OMIM)是涵慨关r人类遗传病和基因座位等相关信息和文献的中心级数据库【.该数据库的数据内容采用一定的格式以文本形式存贮,总文件大小约为30M,可以从美国国牛物技术信息中心(NCBI)的ftp服务器【下载所有内容,以供本地研究使用.在查询使用上,数据库可以允许使用布尔算子(booleanoperatorS),可以实现固定格式的,比较复杂的查询.一,OMIM的功能【OMIM是在McKusick博士编撰的人类孟德尔遗传一书的基础上发展起来的,前数据库的整理和注释仟务均由JohnHopkins人学医学院的一部分专职从事r科学写作和评论的(医学)博士们承担,他(她)们都具有良好的遗传学基础,町以保证数据注释的质量.对于临J未工作者,通过体现病人临床特征的关键浏,可以从OMIM数据库中寻找最近的临床检测标准和发展趋势.在教学研究方而,OMIM可以迅速,简单地提供给学者们关1:基因和遗传病办面最关键的信息和综评,并且实现表型到基因型的分析,而这些是联机医学检索系统(Medline)无法比拟的.当然,OMIM最具魅力之处是它能够提供给遗传学家关十基因序列,图谱,文献等其它数据库关于该类注释的详尽信息.因此,这样的无缝集成能够提供和lI:我们了解疾病基因组领域的前沿性进展和以及该领域活跃的研究T作.+作者通讯地址:甄一松阜外医院中德实验室北京l00037电了.信箱:********************同时,基于OMIM的知识发现(也就是所谓的数据挖掘, data—mining)也有助于我们掌握生化途径及疾病的分子致病机理I._二,OMIM的特点]1.文本记录(entry)OMIM每一条记录内容都有一个唯一的编号(MIM登录号),对应一个基因或者是某种疾病,这一点体现了它与”序列”为核心的数据库的不同.它不包含EST,假基因和遗传标记,而且所有的cDNA序列存在对应的功能注释.一般而言,每?个基因座位和记录内容存在一一映射的关系. 此外,许多疾病现孟德尔遗传特征,虽然没有在基因和生化水平I:捕述,但仍然包括在记录内容中.OMIM的另一个主要LI标足实现已定位的表型和对应基因序列的关联. MIM登录的六位数字系统的含义见表一.如果存在等位基,则可以MIM录号后添加四位数的小数数字用以区分不同的变异体.例如友病因了.IV的所有变异体(或突变)可以分别命名为306900,000l到306900.0l0l.此外,MIM登录号前的星()指该表型的遗传方式已经得到证明或确认,而且该基因座位的基因对应的表与其它带星号的记录表型可以区分.而没有星号则可以认为目前该疾病的遗传模式没有被确队.MIM登录号前的(#)表明这类疾病的表型可以由两个或两个以上基因的任何一个基因突变引起.MIM登录号前的()表明该条记录已经删除或者赋予了新的MIM号.OMIM在临床疾病的记录,包括等位变异(allelic variation),检测方法,处理方案,都是经过仔细挑选,文献来源可靠,能够保证数据注解的准确性.文本记录的内容按照时间顺序的发展进行数据滓释.对于较大的文本*MCC*VOL.115DECEMBER200146T刀丁‟Ilr~7网于>-R面二uu1十l二月昂一仕一删思表…?MIM登录号的含义MIM登录号遗传特征创建时间100000一常染色体显性建立于19945.15以前200000-常染色体隐性建于l9945.15以前300000一X连锁(或表型呈x一连锁)400000一Y一连锁(戒表曼Y一连锁)500000一线粒体遗传(或表型为母系遗传)600000一常染色体(或表为常染色体遗传)建下l9945.15以后注释记录,又分为几个专题区域.这里应当包括以几个方面:2.使用评论(usercomments)OMIM鼓励使用者对现有的记录内容进行评论和注释,同时也可以提供改进意见和辅助材料.评论1人】容和注释通过NCBI转交给JohnHopkins大学的专职数据沣释员.3.临床大纲(clinicalsynopsis)采用有限词汇(controlledvocabulary)描述医学上的表型特征.包括病征,实验室检测方法和遗传学特征等.这样的主要优势是在文本奄询时能够准确地定位所要的信息.4.等位变异体(alleliCvariant)主要收集序列的突变信息,有时也包括不引起疾病的中性遗传多态.但是对f体细胞突变,如癌症,则不在OMIM收录之内.此外,OMIM并不是搜集所有的粜个基因座位的变异体的数据,相反,它丰要搜集相对常见的,对疾病机制起到一定揭示作用的突变或有一定历史重要性的突变数据.对此,许多疾病基冈座位专一(1ocus—specific)的数据库可以起到有益的补充作用.5.OMIM基因图(OMIMgenemap)基以染色体位置为排序标准,和疾病图谱(mot—bidmap)+起,通过图表的方法描述了OMIM掌握的关于疾病基因的信息.这里的内容包含染色体位置,基因命名(简称),全称,MIM录号,疾病名称,注释,定能方法和模式动物的有关信息.6.文献引用(citation)所有参考文献列于数据记录1人】容的最后.每-?条记录通过PubMed的文献ID与摘要相连.7.编辑历史(edit.例如,我们查询”与心肌病有关的定位于11号染色体的常染色体显性遗传疾病基因”,nr以在查询栏内填入“cardiomyopathy1Iautosomaldominant”,得到6条结果(杏徇时『白】2001/1O/12).2.高级查询可以通过历史(hiSt0rY),索引(index)和限制(1imit)三个菜单的组合方式灾现比较多样的查询历史菜单可以迅速提供先前的查询结果,支持布尔算子(AND, OR,NOT)的组合,修改先前的查询.查向时,尔算子必须为火写,支持”“通配符和括号”()”限制.限制菜的功能包括指定诸如标题,MIM登录号,参考文献等查询范围,以及染色体位置,登录号前缀,如#,+等,记录建立和修改的时间,其它属性诸如等位变体和皋因图座何. 索引包括有关数语(terms)的字母列表,包含对应术语的记录文件数,可以提供和选择多个术语用j:查徇.这里,我们采取与简单查询同样的例子,来说明高级查询的使用方法.第一步,我f『J往杏询栏内填入”cardiomyopathy”,选中”限制菜单(1imit)”,选中复选框”Text”和”Chromosomes1l”,点击”go”Ⅵ兀*MCC*VOL.1}15DECEMBER200147中国分子心脏病学杂志2001年l2月第一卷第一期(总第l期) ■■—■l■哺l■….…,.■‟■l-….…—蛆墨:.:‟蜘I习I:”嘲》璺I…-姆岍蛐.囊州#坩f‟??-_¨;”{nl…-£…IrlIj.-t_--m?‟rzISga~chi_,t-I●:fb-?●阻Ikmb~r●‟E-:”p.rlItriLltrt(-i~JI$lr?:=::=::.l_rt-}.…广II^I娜广-tll…Lr:_●”…rrItnf”●…I~~……‟”.._i…I…~,…m--t-C.)£抽-F0-1№舶r_L1k:d-4?T-I广i广广Z-5广广广年1广】£:1i一:1i*:1-厂1『1rl;广l{广l:r:l¨Jlr‟I:”…;?rl广】0广:广l『lr:2rrf『iI{},I■厂tjn士1,l广,_…E…,I……………一-_fI?‟Ilr-}-mI.1‟r,iOL,t(~¨j…l”I‟l图2高级查询的表单(见图2).第二步,在查啕栏内填入”aut0s0mal dominant”,选中”限制菜单”,选中查询范围(SearchinField(s))复选框内的”clinicalsynopsis”,点击”go”.第三步,选中”历史菜单(history)”,敲入上两步的历史代码,如”#lAND#2”,得到4条结果.3.复杂布尔查询这种查询方式实际上是高级查询的命令行方式,£要优势是一步即可完成高级查询的任务例如我们前面提到的例子可以改写为布尔算式cardiomyopathy[txt】ANDll【chrom】ANDautosomaldominant[clin】.我们可以看到,所有域限定词(fieldqualifiers)均封闭在方括号1人J([】).如果没有指定一个域限制词,则系统默认为所有领域(AllFields).限定同和搜索同可以不必用空格格开,但算了_的左右两端则必须是空格.运算符的演算顺序是自左到右,遵循括号优先原!j!IJ.四,OMIM的的缺陷OMIM数据库模式(databaseschema)和数据模型(datamode1)不透明,所以无法实现即席查询(adhoc),也没有提供相应的查询界面.也就意味着使用者无法利用SQL查询语句自行编写查询语句进行数据库的知识发现.对于复杂性疾病,例如哮喘,由于所需分析的数据类型异常复杂,OMIM目前提供的解决方案似乎无法满足日益增长的研究需要.而且OMIM包含的内容显然没有座位专一数据库(1ocus—specificdatabase)那样丰富:突变数据没有完全收集,缺乏引物设计的信息,基表达谱等等.数据注解仅限于遗传学方面.值得一提的是,中国医学科学院心血管病研究所生物信息中心正在筹建分子心脏病学数据库,其主要形式将与OMIM和SwissPro相似,是以文本注解为特征,采用专家方式对文献中的数据进行整理,将更多的语义信息以知识表示形式存入系统的一种知识库(knowledgebase).我们借此也希望全国的同道加入到我们的队伍中来,建立一个具有国际水平的关于心血管疾病方面的生物信息平台, 从而推动我国在分子心脏病学领域的研究和协作,以及在世界范围内该领域的研究互动I81.『参考文献』l_/entrez/query.fc~?db=OMIM2BrownAF,McKieMA.MuStaRandothersoftwareforl0cus—specificmutationdatabases.HumMutat.(2000)15(1):76—85.3.ftp:///mlmsitory/OMIM/4.LetovskyS.In:Bio/nformatics:Databaseand Systems.Pp77—84.KluwerAcademicPublishers5.Boyad0ievSA&JabsEW.OnlineMendelianInherit—anceinMan(OMM)asaknowledgebaseforhumandevel—opmentaldisorders.ClinGenet.[标签:快照]。
OMIM数据库:大神私藏的数据库,99.9%的人都不知道!

OMIM数据库:⼤神私藏的数据库,99.9%的⼈都不知道!“GEO、NCDB、TCGA、SEER数据库这些我都知道,但OMIM是什么⿁?OMIM(Online Mendelian Inheritance in Man)数据库,中⽂称在线⼈类孟德尔遗传数据库。
OMIM包括了现在所有已知的遗传病和超过15000个基因的信息。
OMIM侧重于疾病表型与其致病基因之间的关联。
”也就是说当你知道某个病的时候,但不知道它受什么基因影响——选OMIM数据库!当你知道某个基因的时候,但不知道它与什么疾病有关——选OMIM数据库!当你想了解某个基因与某个疾病之间的关系——选OMIM数据库!OMIM数据库包括gene entry基因条⽬;allelic variations 等位基因变异;gene map 基因图谱;phenotypic series 表型系列;phenotype entry 表型条⽬;clinical synopsis 临床提要;externallinks 外部链接。
接下来将从两个⾓度来⼿把⼿教你如何使⽤OMIM壹使⽤OMIM搜索表型(包括病症)或者临床特征以肺癌为例,在搜索条中输⼊:lung cancer。
双击进⼊#211980 LUNG CANCER词条在这个主页你可以查询到:1表型与基因的关系解读:location代表相关基因在染⾊体中的位置;phenotype代表基因相关的表型;phenotypeMIM number代表表型的MIM编号;inheritance代表遗传,是指该基因的遗传类型,如AD是指常染⾊体显性遗传,SMu是指体细胞突变,⿏标点击缩写符号就会出现不同缩写代表的具体含义;phenotype mapping key代表表型映射关键,3代表该疾病的分⼦基础是已知的;Gene/Locus代表对应的基因或位点;Gene/Locus MIM number代表对应的基因或基因座MIM编号。
利用生物信息学分析疾病基因

利用生物信息学分析疾病基因人类疾病是人类健康的威胁,其复杂性让我们需要了解更多关于基因与疾病之间的关系。
基因意味着生物个体的遗传信息,而疾病则是个体健康状态的一种异常现象。
事实上,基因变异与遗传学因素经常会导致疾病进一步的发展,这就需要我们利用生物信息学的方法进行分析。
本文将简要介绍生物信息学的应用,并探究如何使用生物信息学工具来分析疾病基因。
一、生物信息学的应用生物信息学是一门研究大型分子生物信息的交叉学科,涉及计算机科学、数学和生物学。
生物信息学主要研究生物大分子的结构、功能、进化以及调控,其目的是利用计算机和其他技术手段处理和解析大量的生物数据。
这些数据可以包括基因组、蛋白质组、代谢组、转录组、蛋白质结构及其相互作用等方面的信息。
生物信息学的应用非常广泛,涉及到很多生命科学领域,包括:1.基因组学:研究基因组的结构、功能及遗传变异对生物进化和表型的影响;2.蛋白质组学:研究蛋白质在不同环境中的表达、结构、功能和相互作用;3.代谢组学:研究细胞代谢通路以及细胞内代谢产物的定量和定性分析;4.转录组学:研究基因转录和RNA的稳定性、结构和功能;5.结构生物学:研究蛋白质分子的三维结构和它与其他化合物的相互作用;6.系统生物学:综合应用多种数据集,研究复杂生物系统之间的关系和相互作用。
生物信息学的应用可以帮助我们更好地理解和分析生物学现象,并为疾病研究带来了全新的机遇。
二、利用生物信息学分析疾病基因生物信息学的应用已经在疾病研究中得到了广泛的应用,其中最重要的应用之一是利用生物信息学的方法分析疾病基因。
疾病基因是指导致疾病的基因或某个基因突变的变种。
下面我们将介绍如何使用生物信息学工具来分析疾病基因。
1.数据库相关数据库是分析疾病基因的关键,这里我们介绍一些重要的生物信息学数据库:1.1 OMIM (Online Mendelian Inheritance in Man)OMIM数据库包含了所有已知人类遗传疾病的基因、表型及其影响的信息。
关于遗传基因检测中基因变异临床意义分级的建议

561
建议与共识
关于遗传基因检测中基因变异临床意义分级的建议
天津市医学会医学遗传学分会,天津市医学会遗传咨询分会
摘要:目前基因检测报告主要对基因变异的致病性及临床含义进行描述,实验室人员撰写报告时往往把焦点放 在变异本身的性质,而临床医生的关注点主要在案例的临床情况。这种关注点的差异时常造成临床医生对检测报 告的误解。本建议提出基因变异的临床分级方案,即在基因检测报告中不仅应对基因变异的致病性进行分类,还应 增加临床意义的分级。推荐将基因变异的临床指导意义分为 5 个级别:具有明确的临床指导意义、具有潜在的临床 指导意义、临床指导意义不明确、具有意外发现的临床指导意义和没有明显的临床指导意义。本方案强调了临床表 型的准确性、全面性,以及实验室-临床沟通的重要性,并且提倡表型描述的标准化,这将有助于临床医生与实验室 人员之间的相互理解,有利于基因检测报告的解读和遗传咨询。
基金项目:国家重点研发计划项目(2017YFC1001900,2020YFC2008100);国家自然科学基金资助项目(81771589);京津冀专项项目 (19JCZDJC65400);天津市卫生行业重点攻关项目(16KG166);天津市重大疾病防治科技重大专项(18ZXDBSY00170,18ZXDBSY00230);天津 市卫生健康科技项目(ZC20120,KJ20166)
在完成了基因检测之后,还可以根据基因检测 报告所提示的疑似诊断再次进行表型采集,以便发 现较为次要的表型,进一步确认或者排除诊断。尤 其是针对携带可疑基因变异的家庭成员,也应尽量 采集其表型,帮助对基因型-表型之间的相关性进行 评估及确认。 1.2 辅助进行表型描述以及查询的数据库和网站 1.2.1 人 类 表 型 标 准 用 语 联 盟(Human Phenotype Ontology,HPO)和 中 文 人 类 表 型 标 准 用 语 联 盟 (CHPO)表型标准化数据库 随着对人类疾病研究 的逐渐深入,科研工作者们越来越意识到临床表型 数据的重要性,对基因型与表型数据进行联合分析 成为众多疾病研究的一个方向。HPO 旨在提供人类 疾病中用于描述表型异常的标准词汇,目前包含约 11 000 个名词和超过 115 000 条关于遗传性疾病的 注 释 ,还 提 供 了 一 套 针 对 约 4 000 种 疾 病 的 注 释
omim筛选条件

omim筛选条件标题:使用OMIM筛选条件提高基因研究效率导语:基因研究在现代医学领域中起着重要的作用。
为了提高基因研究的效率和准确性,研究人员常常使用OMIM(Online Mendelian Inheritance in Man)数据库来筛选目标基因。
本文将介绍如何利用OMIM筛选条件来优化基因研究,并提供相关案例来说明其重要性。
第一部分:OMIM数据库的介绍OMIM数据库是一个全球公认的基因研究工具,其中包含了大量与人类遗传疾病和基因相关的信息。
它提供了疾病描述、基因变异、表型特征和遗传模式等详细信息,为研究人员提供了宝贵的参考资料。
第二部分:利用OMIM筛选条件提高基因研究效率1. 根据遗传模式筛选:OMIM数据库提供了丰富的遗传模式信息,包括常染色体显性、常染色体隐性、X连锁隐性等。
研究人员可以根据疾病的遗传模式来选择合适的筛选条件,从而缩小研究范围,提高效率。
2. 根据表型特征筛选:OMIM数据库中详细描述了各种疾病的表型特征,研究人员可以根据患者的表型特征来筛选相关基因。
例如,对于某一具有特定表型特征的疾病,研究人员可以利用OMIM数据库提供的表型信息进行筛选,找出可能相关的基因。
3. 根据基因变异筛选:OMIM数据库提供了大量的基因变异信息,包括突变类型、变异位置等。
研究人员可以根据已知的基因变异信息来筛选目标基因,从而减少研究的盲目性,提高准确性。
第三部分:OMIM筛选条件的重要性案例分析通过分析一些基因研究的案例,我们可以看到使用OMIM筛选条件的重要性。
例如,在一项研究中,研究人员利用OMIM数据库的遗传模式筛选功能成功找到了一种罕见的遗传性疾病的致病基因。
另外,在另一项研究中,研究人员利用OMIM数据库的表型特征筛选功能发现了与某种特定表型相关的基因变异。
结尾:总结一下,利用OMIM筛选条件可以大大提高基因研究的效率和准确性。
通过根据遗传模式、表型特征和基因变异等筛选条件,研究人员可以缩小研究范围,减少盲目性,找到可能相关的基因,为基因研究提供宝贵的参考资料。
OMIM使用简要说明

OMIM使用简要说明
OMIM (Mendelian Inheritance in Man)是一个全球公认的遗传性疾病基因数据库,它记录了人类遗传疾病与基因之间的关联关系。
OMIM的建立旨在提供临床医生、生物学家、遗传学家和研究人员等专业人士一个可以快速查询、获取遗传疾病基因信息的有用工具。
OMIM的主要功能包括:
1.提供疾病描述:OMIM中的每个疾病都有详细的描述,包括临床表现、遗传模式、遗传缺陷等等。
这些信息可以帮助医生更好地理解疾病的本质以及其基因突变的影响。
2.提供基因信息:OMIM收录了与遗传疾病相关的基因,包括基因的命名、位置、功能描述以及相关的突变。
这些信息对于研究人员来说非常有用,可以帮助他们深入研究这些基因的作用机制。
3.提供基因突变信息:OMIM中的每个疾病都有详细的突变信息,包括已知的突变类型、突变区域以及突变对疾病表型的影响等。
这些信息对研究人员来说非常有价值,可以帮助他们更好地了解特定突变与疾病之间的关联。
4.提供表型信息:OMIM还收录了与遗传疾病相关的表型信息,包括临床特征、实验室检查等。
这些信息可以帮助医生更好地诊断疾病并制定治疗方案。
总之,OMIM是一个非常有用的工具,给医生、生物学家、遗传学家和研究人员等专业人士提供了一个方便快捷的查询遗传疾病基因信息的平台。
它不仅收录了丰富的遗传疾病和相关基因信息,还提供了一系列实用工具和资源,帮助用户更好地利用和理解这些数据。
目前,OMIM已经成
为遗传学领域的权威数据库,对于促进疾病诊断、研究和治疗有重要的意义。
2021糖原贮积病Ⅱ型GAA基因变异特点及基因型与表型的关系(全文)

2021糖原贮积病Ⅱ型GAA基因变异特点及基因型与表型的关系(全文)糖原贮积病Ⅱ型(OMIM号:232300)又称庞贝病,因α-葡萄糖苷酶(acid α-glucosidase,GAA)基因(OMIM号:606800)缺陷导致溶酶体酸性GAA缺乏所致的一种罕见溶酶体贮积病,呈常染色体隐性遗传。
随着重组人GAA替代药物的应用,该病成为可治疗罕见病,早期诊断和早期干预治疗可显著改善预后。
GAA基因变异具有种族及地域差异,且部分基因型与临床表型有一定关系。
中国尚缺乏大样本的有关该病的临床谱及分子流行病学资料。
本研究回顾性分析2010年1月至2020年5月广州市妇女儿童医疗中心确诊的18岁前起病的57例糖原贮积病Ⅱ型患者的临床表现及GAA基因检测结果,探索GAA基因变异特点及基因型与表型的关系,并以广州市2 395名健康儿童GAA基因变异谱根据人群致病变异携带率推断该病的理论发病率。
对象和方法一、对象回顾性病例研究,广州市妇女儿童医疗中心于2010年1月至2020年5月通过GAA活性测定及GAA基因变异分析确诊糖原贮积病Ⅱ型患者共63例,选择18岁前起病的57例患者为研究对象,其诊断年龄为1月龄至27岁。
同期广州市妇女儿童医疗中心儿科研究所提供2 395名健康儿童全外显子测序中GAA基因检测结果。
本研究经广州市妇女儿童医疗中心医学伦理委员会批准(2012[024])并获儿童(监护人)或患者知情同意。
二、方法1.临床资料收集及整理:通过医院电子病例系统收集患者病史、临床表现、实验室及影像学检查结果,根据发病年龄、心脏是否受累分为婴儿型、晚发型、非典型婴儿型3种临床表型。
2.淋巴细胞GAA活性测定:采集外周静脉血4 ml,乙二胺四乙酸抗凝,分离淋巴细胞,超声破碎细胞,制备细胞匀浆,蛋白定量,采用荧光底物法测定GAA活性。
取6 mmol/L的4-甲基伞酮-ɑ-D-葡萄糖苷(美国Sigma公司)20 μl,加80 μmol/L阿卡波糖(美国Sigma公司)5 μl,加细胞混悬液20 μl(30 μg蛋白),37 ℃恒温震荡孵育1 h,加150 mmol/L乙二胺四乙酸缓冲液(pH=11.3)200 μl终止反应,Bio-Tek FLx 800 荧光分析仪测定4-甲基伞酮荧光强度,激发光波长350 nm,发射光波长460 nm,以4-甲基伞酮(美国Sigma公司)制定标准曲线。
omim数据库

OMIM数据库OMIM,全称为Online Mendelian Inheritance in Man(在线人类遗传性疾病目录),是一个记录遗传性疾病和相关基因信息的数据库。
该数据库由美国约翰·霍普金斯大学的儿童中心医学专家维护和更新,旨在为科研人员、医生和患者提供有关遗传疾病的详尽信息。
数据来源OMIM数据库收集、整理和提供了来自世界各地的研究成果、临床报告和遗传测试结果。
数据来源主要包括科研文献、基因测序数据库、临床试验报告等。
OMIM的更新速度相对较快,能够及时反映最新的遗传疾病研究成果。
数据结构OMIM数据库中的信息主要分为两大类:遗传性疾病和相关基因信息。
每种遗传疾病都有一个唯一的编号,其条目包括疾病的名称、症状描述、遗传方式、流行病学数据等。
同时,每个基因也有一个独特的标识符,相关信息包括基因的名称、功能、变异类型等。
数据应用科研人员可以利用OMIM数据库深入研究遗传疾病的发病机制、诊断方法和治疗策略。
医生可以通过OMIM查找特定疾病的症状及遗传信息,帮助患者进行更精准的诊断。
患者及家属也可以通过OMIM了解疾病的遗传风险及病情发展情况,从而更好地管理和处理疾病。
未来展望随着遗传学研究的深入和技术的进步,OMIM数据库的内容和功能将不断扩展和完善。
未来,可以预见OMIM将成为遗传疾病研究、临床诊断和个体化治疗的重要工具,为人类健康事业做出更大的贡献。
在数字时代,OMIM数据库作为一个重要的遗传疾病信息库,为遗传疾病研究和临床诊断提供了便利和支持,有望在全球范围内推动遗传医学领域的发展。
钙敏感受体激活性和失活性突变所致骨矿代谢异常疾病及治疗

DOI:10.3969/j.issn.l674-2591.2021.01.011•综述・钙敏感受体激活性和失活性突变所致骨矿代谢异常疾病及治疗董冰子,李成乾,孙晓方[摘要]钙敏感受体(calcium-sensing receptor,CaSR)是感知细胞外钙离子浓度,调节甲状旁腺素分泌及尿钙重吸收,维持钙稳态的关键受体。
CaSR激活性和失活性突变导致钙调定点的移动,引起相应的钙矿物质代谢异常疾病和骨代谢异常表现。
本文综述CaSR突变所致疾病的发病机制、临床表现、治疗策略,通过动物实验模型阐述CaSR的病理生理功能,并探讨针对上述疾病的治疗及CaSR配体药物的研究进展。
[关键词]钙敏感受体;家族遗传性低尿钙性高钙血症;常染色体显性遗传性低钙血症;溶钙素;拟钙剂中图分类号:R589文献标志码:AGain and loss-of-function mutations of calcium-sensing receptor associated boneand mineral metabolic disorders and the therapiesDONG Bing-zi,LI Cheng-qian,SUN Xiao-fangDepartment of Endocrinology and Metabolism,The Affiliated Hospital of Qingdao University,Qingdao266003,Shandong,China[Abstract]Calcium-sensing receptor(CaSR)senses the extracellular calcium concentration,regulates the parathyroid hormone(PTH)secretion,and renal calcium reabsorption,plays an important roles in maintaining the calcium homeostasis.Gain and loss-of-function mutations of CaSR lead to the shift of calcium set-point,and result in the mineral and bone diseases.We reviewed the pathophysiology of diseases associated with activating and inactivating mutations of CaSR,and their clinical characteristics,therapeutic strategy,and animal models.To achieve better understanding of CaSR,the allosteric modulators of CaSR may become promising therapeutic options.[Key words]calcium-sensing receptor;familial hypocalciuric hypercalcemia;autosomal dominant hypocalcemia;calcilytics;calcimimetics钙敏感受体(calcium-sensing receptor,CaSR)主要表达于甲状旁腺、肾脏、骨组织、肠道等,通过感受细胞外钙离子浓度,调节甲状旁腺主细胞对甲状旁腺素(parathyroid hormone,PTH)的合成和释放,同时调节肾小管对钙的重吸收,是维持机体钙稳态的关键受体[1]。
NCBI、OMIM、HGMD数据库使用教程

➢ 基因表达、结构、定位 ➢ 基因功能 ➢ 分子遗传学:报道案例
➢ 基因型与表型的关系
1.空隙位置的突变更易导致婴儿期严重肌阵挛性癫痫,发病年龄早。 2.癫痫相关的突变主要聚集在C端和蛋白质前3个结构域的第5段和第6段之间的环。 3.并不是所有的基因都有该项描述
➢ 突变位点
报道的突变的位点及家系的基本情况: Baulac等人报道了一个家系,三代存在 全身癫痫伴发热性癫痫加2型,检测发现ARG1648HIS位点突变,其中家系中一位 成员存在突变确无表型,提示外显不全 发病年龄、性别发病情况(XL)、外显不全
获e
8.保守结构域查询(protein-Conserved Domains)
NCBI-Genetic-ClinVar数据(类似HGMD数据库)
基因的缺失重复
点突变
NCBI-Genetic-dbvar数据(类似decipher数据库)
Decipher数据库
疾病种类有限
National Center for Biotechnology Information https:///
NCBI-Gene-gene
1.别名查询 3.基因定位 4.外显子数目
2.基因功能查询 5.基因长度、转录本、核苷酸、氨基酸数目
National Center for Biotechnology Information https:///
OMIM-疾病
➢ 疾病描述总结
报告中的疾病描述: Dravet综合征又称早期发作癫痫性脑病(Eiee),其特点是全身强直,阵挛和强 直阵挛发作,最初是由发烧引起的,开始于生命的第一年。 癫痫发作通常难以 治疗。 后来,患者还表现出其他发作类型,包括无意识、肌阵挛和部分发作。 脑电图最初通常是正常的,但后来表现为广义尖峰波活动。精神运动发育停滞 在生命的第二年左右,受影响的个体表现出随后的精神衰退和其他神经学表现。
共济失调毛细血管扩张症四例临床表型及基因突变分析

共济失调毛细血管扩张症四例临床表型及基因突变分析郑岚;刘晓黎;曹立【摘要】Objective To report 4 cases of ataxia-telangiectasia (AT) with ATM genetic mutation and to summarize the clinical and genetic characteristics of AT by literatures review.Methods Clinical data of 4 patients from 3 AT families was collected in detail and genomic DNA of the patients and family members was extracted from peripheral blood.Whole exon sequencing (WES) and polymerase chain reaction (PCR) of Sanger sequencing was used to analyse ATM genic mutation.Results Four patients were characterized by progressive cerebellar ataxia with onset in childhood,oculocutaneous telangiectasia,recurrent infection caused by immunodeficiency,α-fetoprotein (AFP) elevation and cerebellar atrophy shown in brain MRI were presented.Sequence analysis of ATM gene revealed two known compound heterozygous mutations c.8287C > T (p.Arg2763X) and c.9139C > T (p.Arg3047X) which were nonsense mutation in Case 1 and Case 2.In Case 3,there were two compound heterozygous mutations,including nonsence mutation c.8911C > T(p.Gln2971X) and deficit mutation c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18) both of which were not reported previously.Case 4 carried homozygotic mutation c.1402_1403delAA(p.Lys468GlufsX18).Conclusions Four patients were diagnosed as AT with typical clinical manifestations.Patients with variant AT present mild nervous system symptom,normal head MRI and less involvement other thannervous systemt.Definite diagnosis should be dependant on ATM genetic testing.%目的报道4例经基因检测明确诊断的共济失调毛细血管扩张症患者,结合文献总结该病临床表型和基因突变特点.方法采集3个共济失调毛细血管扩张症家系共4例患者临床资料,并提取患者及其父母外周静脉血,采用全外显子测序和Sanger测序进行ATM基因突变分析.结果 4例共济失调毛细血管扩张症患者均表现为儿童期发病的进行性进展的小脑共济失调、球结膜和皮肤毛细血管扩张、免疫缺陷导致反复感染,血清甲胎蛋白水平升高,头部MRI显示小脑萎缩.ATM基因检测显示,例1和例2存在已知复合杂合突变c.8287C> T(p.Arg2763X)和c.9139C>T(p.Arg3047X),均为无义突变;例3存在2种未报道的复合杂合突变,包括无义突变c.8911C> T(p.Gln2971X)和缺失突变c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18);例4存在纯合突变c.1402_1403delAA (p.Lys468 GlufsX 18).结论 4例患者具有典型共济失调毛细血管扩张症临床表现.变异型共济失调毛细血管扩张症患者神经系统受累较轻,头部MRI通常正常,神经系统以外受累少见,明确诊断仍依靠ATM基因检测.【期刊名称】《中国现代神经疾病杂志》【年(卷),期】2017(017)007【总页数】7页(P519-525)【关键词】共济失调性毛细血管扩张;表型;基因;突变【作者】郑岚;刘晓黎;曹立【作者单位】201199 上海市闵行区中心医院神经内科;200025 上海交通大学医学院附属瑞金医院神经科上海交通大学医学院神经病学研究所;200025 上海交通大学医学院附属瑞金医院神经科上海交通大学医学院神经病学研究所【正文语种】中文共济失调毛细血管扩张症[AT,在线人类孟德尔遗传数据库(OMIM)编号:208900]亦称Louis⁃Bar综合征,是常染色体隐性遗传性多系统疾病,系ATM基因突变(OMIM 编号:607585)所致,累及神经系统、免疫系统、内分泌系统和皮肤等[1⁃2].该病全球发病率1/10万~1/4万,通常于婴幼儿期(1~5岁)发病,临床主要表现为进行性小脑共济失调,球结膜和面部毛细血管扩张,反复发作的呼吸道感染[3],其中,神经系统受累还可以表现为吟诗样语言、肌阵挛、肌张力障碍、眼运动性共济失调等;实验室检查血清甲胎蛋白(AFP)水平升高和染色体不稳定;头部MRI显示小脑萎缩;疾病晚期易合并恶性肿瘤.本研究报道4例经基因检测明确诊断的共济失调毛细血管扩张症患者,并复习相关文献,总结该病临床表型和基因突变特点.一、研究对象本研究纳入2014年9月6日-2015年9月10日在上海交通大学医学院附属瑞金医院神经科门诊就诊的3个共济失调毛细血管扩张症家系共4例患者,临床诊断均符合Harding标准[4];均为汉族,男性3例,女性1例;年龄10~15岁,平均14.75岁;发病年龄14~18个月,平均15.75个月;其中例1和例2为同胞姊弟;所有患者父母(6例)均身体健康,无相似临床症状,否认近亲婚配;遗传方式符合常染色体隐性遗传.选择我院既往进行流行病学调查的200名具有相同遗传背景的正常人作为对照,均为汉族.本研究经上海交通大学医学院附属瑞金医院道德伦理委员会审核批准,所有患者及其父母均知情同意并签署知情同意书.二、研究方法1.样本采集采集4例患者及其父母外周静脉血各2 ml,予乙二胺四乙酸(EDTA,国药集团化学试剂有限公司)抗凝.采用标准酚氯仿法提取患者及其父母基因组DNA,操作步骤严格按照试剂盒说明书进行.采用多功能酶标仪(美国Biotek公司)检测DNA纯度.2.基因检测采用全外显子测序(WES)对先证者外周静脉血DNA进行检测并进行生物信息学分析,发现存在ATM基因突变.根据测序结果采用聚合酶链反应(PCR)扩增ATM基因相应外显子编码区,再行Sanger测序验证.针对ATM基因第10、49、57、62、63外显子及侧翼序列设计引物,由英潍捷基(上海)贸易有限公司合成,正向引物序列分别为ATM⁃10F:5'⁃CTCCCAAATGCTGGGAAAC⁃3'、ATM⁃49F:5'⁃AATTTGTGTTGGGCCACATT⁃3'、ATM⁃57F:5'⁃GGGCAGGCTCTCAAACATC ⁃3'、ATM⁃62F:5'⁃AAGTGTGCATGATGTTTGTTCC ⁃3'、ATM⁃63F:5'⁃GACCAAGAATGCAAACGAAA⁃3',反向引物序列分别为ATM⁃10R:5'⁃CTTCTCGGCCAAACAAGAAA⁃3'、ATM⁃49R:5'⁃CGAAAAGAACCCAAAAGACC⁃3'、ATM⁃57R:5'⁃TTTTCACTCACACACTTTCATTCT⁃3'、ATM⁃62R:5'⁃CAGTGCCTTCTTCCACTCC⁃3'、ATM⁃63R:5'⁃CTGGGATTACAGGCGTGAG⁃3'.PCR 反应体系25 μl,依次加入dNTPs 2.50 mmol、2XGC 缓冲液Ⅰ12.50 μl、各引物序列 5 pmol、模板 DNA 100 ng、r⁃Taq 1U,再加水补充至25 μl;反应条件为95 ℃预变性5 min,95℃ 90 s、62 ℃ 1 min、72℃ 1 min,共循环35次,72℃延伸10 min.然后将Sanger测序结果与人类基因组ATM基因序列(GenBank序列号:NM_000051)进行比对,采用DNAStar软件包中SeqMan软件(美国DNAStar公司)进行基因突变分析.针对未报道的基因突变,对200例具有相同遗传背景的正常对照者进行相应基因检测,并采用Mutation Taster软件[5]预测其致病性.一、临床特征例1 女性,25岁,主因行走不稳25年、扭转性痉挛反复发作11年,于2014年9月6日至我院神经科门诊就诊.患者刚会行走(1岁)时即出现行走不稳,呈醉酒步态;16个月时出现球结膜充血;4~5岁时可见眼睛有明显血丝;5岁时出现扭转性痉挛,表现为头向两侧扭转痉挛,发作频率逐渐增加,逐渐出现四肢痉挛,头部MRI未见明显异常;6岁时头部抖动更加明显,经肌肉注射A型肉毒毒素后症状缓解;8岁时再行MRI 检查显示小脑萎缩;9岁时不能独立行走,需搀扶,进行性加重;24岁时完全不能站立,困于轮椅中,同时伴言语障碍、吞咽困难和饮水呛咳,其中言语障碍自学习说话时即有体现,表现为言语含糊不清,目前言语难以理解.患者足月剖宫产,出生时3.60 kg,无产伤和窒息史,2个月会俯卧抬头,3个月会翻身,5个月可独坐片刻,7个月会爬行,14个月可独立行走但常跌倒,此后逐渐不能生活自理,需喂养,17个月时会说话;8岁时有反复低热史(37.5~38.0℃),原因不明,自行痊愈;上小学后学习成绩中等,小学六年级(11岁)后辍学,反应较同龄正常儿童稍慢;14岁时曾出现一次阴道流血,后未再出现,第二性征已发育;咀嚼稍慢,饮水呛咳.父母身体健康,无相似症状,否认近亲婚配;其弟表现出相似临床症状.门诊神经系统检查:神志清楚,言语欠清晰;双眼球结膜毛细血管扩张,双眼水平眼震,眼球各向活动可,双侧瞳孔等大、等圆,直径约3 mm,余脑神经检查未见明显异常;发作性头部向后仰,四肢不自主抖动与肌阵挛,双足内翻,尤以右侧显著;四肢肌力检查不配合,肌张力增高;不能独自站立,双侧快复轮替动作、指鼻试验差,跟⁃膝⁃胫试验和Romberg征不配合;四肢腱反射对称,病理反射未引出.实验室检查血清甲胎蛋白318.61 μg/L(<7 μg/L).头部 MRI显示小脑萎缩.例2 例1之弟,男性,15岁,因进行性行走不稳伴言语不清13年,于2014年9月6日至我院神经科门诊就诊.患儿临床症状与其姊(例1)相似,包括进行性共济失调、构音障碍和球结膜毛细血管扩张,不同之处为持续性流涎和左眼向外斜视.患儿足月剖宫产,出生时3.20 kg,无产伤和窒息史;16个月时会独立行走,但行走不稳,易跌倒,出现球结膜充血;18个月时会说话,言语含糊不清;3.50岁时仍有流涎;10岁时无法独自站立,搀扶可行走;尚可生活自理,智力发育与同龄正常儿童无差异.门诊神经系统检查:神志清楚,言语欠清晰;双眼球结膜毛细血管扩张,双眼水平眼震,左眼向外斜视,双眼各向活动充分,双侧瞳孔等大、等圆,直径约3 mm,余脑神经检查未见异常;无法独自站立,困于轮椅中,无高弓足;四肢肌力5级,肌张力正常;双侧快复轮替动作、指鼻试验、跟⁃膝⁃胫试验差;腱反射减弱,病理反射未引出.实验室检查血清甲胎蛋白水平为426.33 μg/L.头部MRI显示小脑萎缩.例3 男性,14岁,主因进行性行走不稳伴言语欠流利12年,于2014年6月3日至我院神经科门诊就诊.患儿生长发育里程碑正常,12个月会说话,言语欠流利;18个月可独立行走,呈阔基步态,易跌倒,可见足内翻畸形;行走不稳和言语障碍进行性加重;9岁时出现平衡障碍,伴四肢和躯干不自主抖动;11岁时需助行器辅助行走;目前只能依靠轮椅,言语难以理解;幼儿期反复呼吸道感染史.父母身体健康,无相似症状,否认近亲婚配.门诊神经系统检查:神志清楚,言语含糊不清;双眼球结膜明显毛细血管扩张,双眼水平眼震,眼球各向活动充分,双侧瞳孔等大、等圆,直径约3.50 mm,余脑神经检查未见异常;四肢意向性震颤,紧张时加重;四肢肌力和肌张力均正常,双侧快复轮替动作和指鼻试验差,跟⁃膝⁃胫试验和Romberg征不配合,深浅感觉和腱反射正常,病理征阴性.实验室检查血清甲胎蛋白511 μg/L.头部MRI显示小脑萎缩.例4 男性,10岁,主因进行性行走不稳9年,于2015年7月7日至我院神经科门诊就诊.患儿15个月时可独立行走,易跌倒;5岁时出现四肢不自主抖动,明显行走不稳,进行性加重;目前上小学3年级,学习成绩中等偏下;病程中可见肢体抽搐.患儿足月剖宫产,出生时3.60 kg,无产伤和窒息史;12个月时会说话.父母身体健康,无相似症状,否认近亲婚配.门诊神经系统检查:神志清楚,言语尚清晰;双眼球结膜明显毛细血管扩张,无明显眼震,眼球各向活动尚可,双侧瞳孔等大、等圆,直径约3 mm,余脑神经检查未见异常;可见共济失调步态,一字步不能;四肢肌力5级,肌张力正常;双侧快复轮替动作、指鼻试验和跟⁃膝⁃胫试验差,Romberg征阳性;双上肢腱反射减弱、双下肢正常,病理反射未引出.头部MRI显示小脑萎缩.二、基因检测对4例患者及其父母进行ATM基因检测,共发现5种突变:例1和例2(图1)为ATM基因复合杂合突变c.8287C>T(p.Arg2763X)和c.9139C>T(p.Arg3047X),均为无义突变,其中,c.8287C>T(p.Arg2763X)突变源自其父(图2a),c.9139C>T(p.Arg3047X)突变源自其母(图2b);例3(图3)亦为ATM基因复合杂合突变,包括无义突变c.8911C>T(p.Gln2971X)和缺失突变c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18),其中,c.8911C>T(p.Gln2971X)突变源自其父(图4a),c.7141_7151delAATGGAAAAAT (p.Asn2381GlufsX18)突变源自其母(图4b),此2种突变均未见诸报道,经Mutation Taster软件[5]预测其致病率均为1,且未在200例正常对照者中检出该突变;例4(图5)为ATM基因纯合突变c.1402_1403delAA(p.Lys468GlufsX18,图6a),其父母均为该突变携带者(图6b). ATM基因定位于11q22~q23,覆盖长度160 kb的基因组DNA,包含66个外显子,该基因编码丝氨酸/苏氨酸蛋白激酶(包含3056个氨基酸,相对分子质量为315X103),属磷酸肌醇3⁃激酶相关蛋白激酶(PIKK)家族成员,参与DNA损伤应答、细胞周期调控等信号转导通路,在儿童和成人各组织器官中均有表达[6].ATM基因突变具有极高的异质性,突变形式和种类具有多样性.突变形式包括错义突变、无义突变、剪切位点突变、插入或缺失突变等,其中70%为无义突变[7].突变位点遍布ATM基因全长,无明显热点突变[8].共济失调毛细血管扩张症系ATM基因纯合突变或复合杂合突变所致,其中85%系错义突变或剪切位点突变导致蛋白截短或蛋白功能缺失[8⁃9].本研究例1、例2和例3均存在ATM基因无义突变,导致编码蛋白提前终止;例4存在ATM基因纯合移码突变,改变氨基酸序列,既往文献报道该突变具有致病性[10].本研究4例患者均以步态性共济失调(2岁前)为首发症状,伴进行性加重的小脑共济失调、构音障碍,至儿童晚期(约12岁)需轮椅辅助行走,符合典型共济失调毛细血管扩张症表现[11].Moin等[12]对104例共济失调毛细血管扩张症患者进行研究,发现所有患者均伴小脑共济失调,87例(83.65%)可见球结膜毛细血管扩张、73例(70.19%)可见皮肤毛细血管扩张,78例(75.00%)伴感染,尤以肺炎、中耳炎和鼻窦炎常见.共济失调毛细血管扩张症临床表现具有异质性,部分患者神经系统症状进展缓慢或仅轻度受累,统称为变异型共济失调毛细血管扩张症,此类患者神经系统症状较轻,头部MRI通常正常,神经系统以外受累少见,无毛细血管扩张,内分泌功能和呼吸功能正常,免疫球蛋白水平正常,生存期较长,肿瘤发生较晚[13].临床表型与ATM蛋白活性相关,ATM蛋白活性越高、临床症状越轻[13].变异型共济失调毛细血管扩张症多系ATM基因错义突变所致,与残留部分功能性ATM蛋白有关[1,13].随着ATM基因的克隆,诸多研究显示,并非所有患者均伴小脑共济失调、球结膜和皮肤毛细血管扩张,部分患者甚至无神经系统受累表现[14⁃15].一项对成年共济失调毛细血管扩张症患者运动障碍的研究共纳入14例患者,结果显示,10/14例存在小脑共济失调,2/14例存在构音障碍和肌张力障碍,所有患者均存在运动障碍.运动障碍以肌张力障碍和皮质下肌阵挛最为常见(86%),其次为震颤(6/14)[14].Teive等[11]总结50例变异型共济失调毛细血管扩张症患者的临床表现,结果显示,86%(43/50)患者存在运动障碍,78%(39/50)存在小脑共济失调,仅16%(8/50)存在球结膜和皮肤毛细血管扩张.Verhagen等[1]总结13例变异型共济失调毛细血管扩张症患者的临床表现,发现常见神经系统症状包括小脑共济失调(12例)、肌张力障碍(10例)、舞蹈手足徐动症(9例)、静止性震颤(9例)和脊髓前角细胞性周围神经病(3例).变异型共济失调毛细血管扩张症通常于儿童期发病,毛细血管扩张可能并非其主要临床症状,共济失调进展缓慢,至成年期仅表现为轻中度共济失调,运动障碍更为常见,主要表现为肌张力障碍、肌阵挛、舞蹈样动作,帕金森样症状和震颤等. 共济失调毛细血管扩张症的早期诊断对遗传咨询具有重要意义,但以共济失调发病的患者早期诊断有一定难度,应注意与其他疾病引起的共济失调相鉴别.共济失调毛细血管扩张症是5岁前发病的最常见的常染色体隐性遗传性共济失调,Friedreich 共济失调(FRDA)通常于儿童晚期发病,且伴深反射消失,Romberg征阳性和病理征阳性,可资鉴别.小脑萎缩通常在婴幼儿期不明显,至儿童晚期(约10岁)可见明显小脑萎缩.实验室检查具有一定的鉴别诊断价值,约95%共济失调毛细血管扩张症患者血清甲胎蛋白水平升高[7],核型分析可见第7号染色体与第14号染色体易位,T淋巴细胞数目减少、B淋巴细胞数目正常或仅轻度增加,IgE(80%)、IgG2(80%)和IgA(60%)明显缺陷且随疾病进展相对稳定,而IgM在病程中波动较大[7].在临床诊断基础上,结合ATM基因检测发现复合杂合突变或纯合突变有助于明确诊断.在本研究中,例1和例2存在2种已报道的复合杂合突变,其中,c.8287C>T为无义突变,位于第57外显子,由Cavalieri等[16]于2008年率先报道,该突变可以导致1个未成熟的截短蛋白(p.Arg2763X);c.9139C>T亦为无义突变,导致蛋白羧基末端(C末端)10 个氨基酸缺失(p.Arg3047X)[17⁃18],上述2种突变位于磷酸肌醇3⁃激酶相关蛋白激酶结构域内或附近,影响磷脂酰肌醇3⁃激酶(PI3K)活性.尽管例1和例2存在相同致病性突变,但二者临床表型不尽一致,如足内翻、斜视、扭转性痉挛、肌阵挛、原发性闭经和口角流涎等,表现出疾病的遗传异质性.例3存在2种未报道的突变,包括无义突变c.8911C>T和缺失突变c.7141_7151delAATGGAAAAAT,均导致截短蛋白(p.Gln2971X,p.Asn2381GlufsX18),但这2种突变在200例正常对照者中未检出,且突变氨基酸在不同物种间高度保守,经Mutation Taster软件预测其严重影响蛋白功能,提示这2种突变均为例3的致病性突变,明确诊断尚待进一步分子功能学检测.研究显示,共济失调毛细血管扩张症患者父母均为突变基因携带者,虽无共济失调毛细血管扩张症症状与体征,但与正常对照者相比,其肿瘤易感性(乳腺癌、白血病)明显增加[19⁃20].本研究4例共济失调毛细血管扩张症患者父母目前均无肿瘤表现,尚待进一步随访研究.本研究经基因检测明确诊断4例共济失调毛细血管扩张症患者,均为儿童期发病的进行性进展的小脑共济失调、球结膜和皮肤毛细血管扩张、免疫缺陷导致反复感染;血清甲胎蛋白水平升高;头部MRI显示小脑萎缩;同时报道2种ATM基因新突变和3种已知致病性突变,扩大ATM基因突变谱,为进一步研究奠定基础.【相关文献】[1]VerhagenMM,AbdoWF,WillemsenMA,HogervorstFB,Smeets DF,Hiel JA,Brunt ER,van Rijn MA,Majoor Krakauer D,Oldenburg RA,Broeks A,Last JI,van't Veer LJ,Tijssen MA,Dubois AM,Kremer HP,Weemaes CM,Taylor AM,van Deuren M.Clinicalspectrumofataxia⁃telangiectasia in adulthood.Neurology,2009,73:430⁃437.[2]Gatti RA,Berkel I,Boder E,Braedt G,Charmley P,Concannon P,Ersoy F,Foroud T,Jaspers NG,Lange K,Lathrop GM,Leppert M,Nakamura Y,O'Connell P,Paterson M,Salser W,Sanal O,Silver J,Sparkes RS,Susi E,Weeks DE,Wei S,White R,Yoder F.Localization of anataxia⁃telangiectasia gene to chromosome 11q22-23.Nature,1988,336:577⁃580.[3]Becker⁃Catania SG,Chen G,Hwang MJ,Wang Z,Sun X,SanalO,Bernatowska⁃Matuszkiewicz E,Chessa L,Lee EY,GattiRA.Ataxia⁃telangiectasia:phenotype/genotype studies ofATM proteinexpression,mutations,and radiosensitivity.Mol Genet Metab,2000,70:122⁃133.[4]Harding AE.The hereditary ataxiasand related disorders.Edinburgh:Churchill Livingstone,1984:325⁃326.[5]Schwarz JM, Cooper DN, Schuelke M, Seelow D.MutationTaster2:mutation prediction for the deep⁃sequencing age.Nat Methods,2014,11:361⁃362.[6]Chaudhary MW,Al⁃Baradie RS.Ataxia⁃telangiectasia:future prospects.Appl Clin Genet,2014,7:159⁃167.[7]Perlman S,Becker⁃Catania S,Gatti RA.Ataxia⁃telangiectasia:diagnosis and treatment.Semin Pediatr Neurol,2003,10:173⁃182.[8]Chun HH,GattiRA.Ataxia⁃telangiectasia,an evolving phenotype.DNARepair,2004,3:1187⁃1196.[9]Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia.J ClinPathol,2005,58:1009⁃1015.[10]Broeks A,de Klein A,Floore AN,Muijtjens M,Kleijer WJ,Jaspers NG,van'tVeerLJ.ATM germline mutationsin classical ataxia⁃telangiectasia patients in the Dutch population.Hum Mutat,1998,12:330⁃337.[11]Teive HA,Moro A,Moscovich M,Arruda WO,Munhoz RP,Raskin S,AshizawaT.Ataxia⁃telangiectasia⁃A historical review and a proposal for a new designation:ATM syndrome.J Neurol Sci,2015,355:3⁃6.[12]Moin M,Aghamohammadi A,Kouhi A,Tavassoli S,Rezaei N,Ghaffari SR,Gharagozlou M,Movahedi M,Purpak Z,Mirsaeid Ghazi B,Mahmoudi M,Farhoudi A.Ataxia⁃telangiectasia in Iran:clinical and laboratory features of 104 patients.Pediatr Neurol,2007,37:21⁃28. [13]Verhagen MM,Last JI,Hogervorst FB,Smeets DF,Roeleveld N,VerheijenF,Catsman⁃Berrevoets CE,Wulffraat NM,Cobben JM,Hiel J,Brunt ER,Peeters EA,Gómez Garcia EB,van der Knaap MS,Lincke CR,Laan LA,Tijssen MA,van Rijn MA,Majoor⁃Krakauer D,Visser M,van't Veer LJ,Kleijer WJ,van de Warrenburg BP,Warris A,de Groot IJ,de Groot R,Broeks A,Preijers F,Kremer BH,Weemaes CM,Taylor MA,vanDeurenM,WillemsenMA.PresenceofATM proteinand residual kinase activity correlates with the phenotype in ataxia⁃telangiectasia:a genotype⁃phenotype study.HumMutat,2012,33:561⁃571.[14]Méneret A,Ahmar⁃Beaugendre Y,Rieunier G,Mahlaoui N,Gaymard B,ApartisE,Tranchant C,Rivaud⁃Péchoux S,Degos B,Benyahia B,Suarez F,Maisonobe T,Koenig M,Durr A,Stern MH,Dubois d'Enghien C,Fischer A,Vidailhet M,Stoppa⁃Lyonnet D,Grabli D,Anheim M.The pleiotropic movement disorders phenotype of adultataxia⁃telangiectasia.Neurology,2014,83:1087⁃1095.[15]Trimis GG,Athanassaki CK,Kanariou MM,Giannoulia⁃Karantana AA.Unusual absence of neurologic symptoms in a six⁃year old girl with ataxia⁃telangiectasia.J PostgradMed,2004,50:270⁃271.[16]Cavalieri S,Funaro A,Pappi P,Migone N,Gatti RA,Brusco rge genomic mutations within the ATM gene detected by MLPA,including a duplication of 41 kb from exon 4 to 20.Ann Hum Genet,2008,72:10⁃18.[17]Toyoshima M,Hara T,Zhang H,Yamamoto T,Akaboshi S,Nanba E,Ohno K,Hori N,Sato K,Takeshita K.Ataxia⁃telangiectasia without immunodeficiency:novel point mutations within and adjacent to the phosphatidylinositol 3⁃kinase⁃like domain.Am J Med Genet,1998,75:141⁃144.[18]McConville CM,Stankovic T,Byrd PJ,McGuire GM,Yao QY,LennoxGG,TaylorMR.Mutationsassociated with variant phenotypes in ataxia⁃telangiectasia.Am J Hum Genet,1996,59:320⁃330.[19]GattiRA,Tward A,Concannon P.Cancerrisk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations.Mol Genet Metab,1999,68:419⁃423.[20]Ahmed M,Rahman N.ATM and breast cancersusceptibility.Oncogene,2006,25:5906⁃5911.。
案例分享基因检测助力罕见病诊断——白化病

案例分享基因检测助⼒罕见病诊断——⽩化病⽩化病(Albinism)是⼀组以⾊素缺失和视⼒差为主要临床表现的遗传性疾病。
与外胚层来源的组织(最明显的是⽪肤,⽑发和眼睛)中的⿊⾊素减少或缺失相关,产⽣特征性的苍⽩。
眼部缺陷是⽩化病的共有特征,也是功能上影响最严重的器官,主要包括视⽹膜中央凹发育不良、视觉通路的视神经叉投射异常、虹膜透明、畏光以及眼球震颤等。
⽩化病总体发病率约为1/17000-20000。
⼤约每70个⼈中就有⼀个携带OCA突变的等位基因,其中OCA2突变是世界上最常见的。
⽩化病依据临床表型特征主要分为:⾮综合征型的眼⽪肤⽩化病(Oculocutaneous albinism,OCA)、仅累及眼部的眼⽩化病(Ocular albinism,OA)以及综合征型⽩化病(如Hermanskyr Pudlak综合征、Chediak-Higashi综合症)三类。
值得⼀提的是由于在OCA 2型中相同基因的缺失⽽导致其⽩化样特征的⼀对综合征:Angelman(AS)和Prader-Willi(PWS)综合征。
(⼤约1%的AS和PWS患者具有连续的基因缺失,导致类似OCA2的呈现。
)2018年5⽉11⽇,国家卫⽣健康委员会等5部门联合制定了《第⼀批罕见病⽬录》,⽩化病被收录其中。
临床表型⽩化病临床和遗传异质性很⼤,着⾊相关表型可以从类似OCA1A到接近正常,如从⽩⾊到⾦黄直⾄深褐⾊。
OCA1A由于完全缺乏⿊⾊素,出现⽩头发,⽩睫⽑,⽩⾊⽪肤和粉红⾊的眼睛等⼀般⼈群认为⽩化病的典型特征。
最极端的例⼦就是OA1,其⽪肤和⽑发的表现⼏乎与野⽣型⼀致,如果不做细致的眼部检查,⼏乎很难作出诊断。
⽩化病最常见表型注:OCA=眼⽪肤⽩化病,OA=眼⽩化病,HPS=Hermansky-Pudlak综合症,CHS=Chediak-Higashi综合症,AS=Angelman综合征,PWS=Prader-Willi综合征。
犕犈犆犘2重复综合征的产前临床表型及遗传学分析

《中国产前诊断杂志(电子版)》 2024年第16卷第1期·论著· MECP2重复综合征的产前临床表型及遗传学分析何凌 韦庆芳 卢庆(南宁市第二人民医院产科,广西南宁 530031)【摘要】 目的 探讨1例新发MECP2重复综合征产前病例和遗传咨询。
方法 应用常规G显带染色体核型分析和拷贝数变异测序(copynumbervariationsequencing,CNV Seq)对胎儿羊水样本检测,对胎儿进行系统超声检查,夫妇行外周血CNV seq明确胎儿基因组变异来源,并对相关文献资料进行复习。
结果 系统超声检查提示胎儿右侧肾盂分离(12mm)。
胎儿染色体核型分析未见异常。
羊水CNV seq结果:seq[hg19]dup(X)(q28q28)chrX:g.153140000_153400000dup,Xq28区段存在约260kb的拷贝数重复,夫妇外周血CNV seq检查未见异常,提示胎儿为新发MECP2重复综合征。
结论 MECP2重复综合征在胎儿期缺乏特异的超声表现,CNV seq可有效检出MECP2重复综合征,为其产前诊断和遗传咨询提供依据。
【关键词】 MECP2重复综合征;产前诊断;CNV seq;胎儿【中图分类号】 R715.5 【文献标识码】 A犇犗犐:10.13470/j.cnki.cjpd.2024.01.006犘狉犲狀犪狋犪犾犮犾犻狀犻犮犪犾狆犺犲狀狅狋狔狆犲犪狀犪犾狔狊犻狊犪狀犱犾犻狋犲狉犪狋狌狉犲狉犲狏犻犲狑狅犳犕犈犆犘2犱狌狆犾犻犮犪狋犻狅狀狊狔狀犱狉狅犿犲犎犲犔犻狀犵,犠犲犻犙犻狀犵犳犪狀犵,犔狌犙犻狀犵(犇犲狆犪狉狋犿犲狀狋狅犳犗犫狊狋犲狋狉犻犮狊,犖犪狀狀犻狀犵犛犲犮狅狀犱犘犲狅狆犾犲’狊犎狅狊狆犻狋犪犾,犖犪狀狀犻狀犵犌狌犪狀犵狓犻530031,犆犺犻狀犪)【犃犫狊狋狉犪犮狋】 犗犫犼犲犮狋犻狏犲 ToexploretheprenataldiagnosisandgeneticcounselingforacaseofdenovoMECP2duplicationsyndrome.犕犲狋犺狅犱狊 RoutineG bandingkaryotypingandCNV seqwereusedtodetectfetalamnioticfluidsamples,andsystematicultrasoundexaminationwasperformedonthefetus.CNV seqofperipheralbloodofthecouplewasperformedtodeterminethesourceoffetalgenomicvariation,andtherelevantliteraturewasreviewed.犚犲狊狌犾狋狊 Systematicultrasoundexaminationsuggestedrightrenalpelvisseparationinthefetus.Fetalchromosomekaryotypeanalysisshowednoabnormality.TheCNV Seqresultsofthefetuswereseq[hg19]dup(X)(q28q28)chrX:g.153140000_153400000dup,revealinga260KbduplicationatXq28.CNV seqofthecouple'speripheralbloodshowednoabnormalities,suggestingthatthefetuswasadenovoMECP2duplicationsyndrome.犆狅狀犮犾狌狊犻狅狀 MECP2duplicationsyndromelacksspecificultrasoundmanifestationsinfetus.CNV seqcaneffectivelydetectMECP2duplicationsyndrome,whichprovidesabasisforprenataldiagnosisandgeneticcounseling.【犓犲狔狑狅狉犱狊】 MECP2duplicationsyndrome;prenataldiagnosis;CNV seq 甲基化CpG结合蛋白 2基因(methyl CpGbindingprotein2gene,犕犈犆犘2)重复综合征是一种罕见的X连锁隐性遗传性染色体疾病,主要累及男性患者,临床特征为反复呼吸道感染和严重神经发育障碍,包括严重智力障碍、全面发育迟缓、癫痫、肌张力低下等,最早于1999年由Lubs等报道[1],故也叫LubsX连锁智力障碍综合征(LubsX linkedmentalretaradationsyndrome),2005年被证实为MECP2基因重复所致[2],2010年被命名为MECP2重复综合征(犕犈犆犘2duplicationsyndrome,MDS,92·论著·《中国产前诊断杂志(电子版)》 2024年第16卷第1期OMIM:300260)[3]。
OMIM使用简要说明【论坛精选优秀帖】
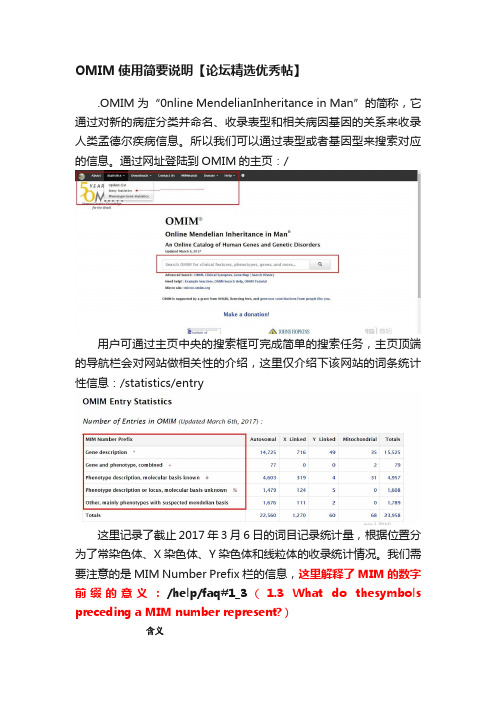
OMIM使用简要说明【论坛精选优秀帖】.OMIM 为“0nline MendelianInheritance in Man”的简称,它通过对新的病症分类并命名、收录表型和相关病因基因的关系来收录人类孟德尔疾病信息。
所以我们可以通过表型或者基因型来搜索对应的信息。
通过网址登陆到OMIM的主页:/用户可通过主页中央的搜索框可完成简单的搜索任务,主页顶端的导航栏会对网站做相关性的介绍,这里仅介绍下该网站的词条统计性信息:/statistics/entry这里记录了截止2017年3月6日的词目记录统计量,根据位置分为了常染色体、X染色体、Y染色体和线粒体的收录统计情况。
我们需要注意的是MIM Number Prefix栏的信息,这里解释了MIM的数字前缀的意义:/help/faq#1_3(1.3 What do thesymbols preceding a MIM number represent?)含义符号星号(*)该记录是一个基因井号(#)这是一个描述性的记录,常常是一个表型,而不是一个唯一的基因座,加号(+)该记录包含了已知序列的基因和表型的描述百分号(%)该记录描述了一个经过证实的孟德尔表型或表型的基因座,但是潜在的分子基础未知无符号前缀该符号表明这个记录是一个表型的描述,但是它的孟德尔疾病基础还未被清楚地证实,或者该表型与其他搜索记录的区别尚不清晰脱字符(^)该记录已经不复存在,可能已经从数据库中删除或换成了其他词目另外针对OMIM的收录条目的数字使用也有区别:(/help/faq#1_11 1.2 What numbering system is used in the OMIM database?)每一个收录条目都使用了一个唯一的6位数字表示:数字说明1-----(1000000);2-----(200000)常染色体基因位点或表型(创建于1994年5月15日前)3-----(300000)X染色体关联的基因位点或表型4-----(400000)Y染色体关联的基因位点或表型5-----(500000)线粒体基因座或表型6-----(600000)常染色体基因位点或表型(创建于1994年5月15日后)另外OMIM数据库的内容主要包括如下信息:表型示例单个基因的孟德尔疾病、失调和表型囊性纤维化,镰状细胞性贫血,软骨发育不全,表型性特征例如头发眼睛颜色不同,药物反应例如恶性高体热和华法林敏感性,可变的感染反应例如单纯疱疹(性)脑炎和发展为艾滋病的HIV感染,癌症的胚系易感基因例如BRCA1和乳腺癌卵巢癌,等等具有显著性的单基因致病的复杂疾病补体因子H和年龄相关的黄斑变性复发性缺失和重复综合症Potocki-Shaffer综合症和染色体10q26缺失综合症对于一个查询网站,我觉得最重要的是搜索功能,通过主页的导航栏的Help链接我们可以查看到OMIM的搜索规则:/help/search这里介绍了很多搜索帮助,简述如下:搜索方式说明示例基本搜索简单输入词目duchenne muscular dystrophy加号运算符做前缀(+)确保结果包含输入的搜索条目+duchenne减号运算符做前缀(-)搜索结果不包含搜索条目-duchenne短语搜索只返回包含该短语的搜索结果"duchenne muscular dystrophy"通配符搜索(*?)作为搜索条目的通配符,*表示搜索条dystroph*目接多个字符,?表示搜索条目接单个字符dystrophi?dystro??i? 搜索域搜索(title :) 指定搜索该搜索域的内容title: Duchenne title:duchenne title:muscular 布尔运算符支持布尔运算符的功能 AND NOT OR 等同于加减号前缀和或者 染色体位置搜索 指定包含的起始区域1p36-p32 基因组坐标搜索 指定基因组的起始位置1:12,000,000-48,000,000 2.通过前面的简单介绍说明我们尝试搜索下,例如:Parkinson 打开基本搜索结果界面(/search/?index=entry&sort=score+desc%2C+prefix_sort+des c&start=1&limit=10&search=parkinson ):图示是基本搜索返回界面的部分截图,在图中1号框位置我们可以对该搜索条目进行进一步的Gene Map Table 和Clinical Synopsis 搜索,这里分别会反馈搜索条目(Parkinson)相关染色体基因位置和临床简介信息,下面2号框(Hightlights)我们可以选择在结果中高亮搜索条目;在3号框位置的第一个返回结果为前缀为#号的描述性结果(#168600,PARKINSO DISEASE,LATE-ONSET; PD),下面描述了该疾病相关的染色体位置:1q22,4q23等等。
权威知识库-OMIM

权威知识库-化学信息尖源
CHEMINFO:Chemical Information Resources (Wiggins at Indiana) /~cheminfo/ 印第安娜大学化学信息资源有:Chemical Acronyms Database(CAD)–化学物质缩写词库,共11000多条款目链 至缩写词的全称;Chemical Reference Sources Database (CRSD)–化学参考材料库,其中包括印本参考 资源、商业数据库和软件;Faculty Publications of the Indiana University Department of Chemistry (FACPUB);Indiana University Molecular Structure Center (IUMSC)–可溶性晶体结构数据库及使其可视的 Java程序。 WHO - Guide to Good Prescribing http://www.med.rug.nl/pharma/ggp.htm 《WHO良方实践手册》,有图解和纯文本两种版本。另有 《WHO基本药物标准目录》2005年14版,《WHO模式处方 集》2004年版。
权威知识库-PhysioBank
(二)PhysioBank PhysioBank是一个大型的逐渐扩增的生理学信号和相关数据 的数字化记录文档,目前包括多参数的心肺、神经和其他生 物医学信号,信号来自健康受试者和各种疾病的患者,涉及 到的患者疾病包括心脏猝死、充血性心衰、癫痫、步态不稳、 睡眠呼吸暂停和衰老。 这些数据库有三类,一类是完全参考数据库(completed reference databases,Class 1),二类是支持已发表研究 的原始数据的文档拷贝,由作者和期刊投交(archival copies of raw data that support published research, contributed by authors or journals,Class 2),第三 类是其它来源数据集,包括进行中的工作(other contributed collections of data, including works in progress,Class 3)。作为对研究团体的服务,第二类和 第三类数据可通过PhysioNet获得。投交的数据一经接受, 都放在第二类和第三类,经同行评议和公众评论期后可能转 入第一类。 练习:按数据特征例举介绍这些数据库。
OMIM使用简要说明

OMIM使用简要说明OMIM(Online Mendelian Inheritance in Man,在线人类遗传疾病目录)是一种数据库,致力于收集和整理关于人类遗传疾病的信息。
该数据库提供了有关遗传疾病的描述、遗传模式、疾病的分子基础以及相关的文献引用。
OMIM为研究人类遗传疾病的科学家、医生和患者提供了宝贵的资源和参考资料。
OMIM的主要内容包括以下几个方面:遗传疾病的描述:OMIM提供了详细的遗传疾病描述,包括疾病的症状、表型特征和患病率等。
这些信息帮助科学家和医生了解疾病的临床表现和特点。
遗传模式:OMIM描述了疾病的遗传模式,包括常染色体显性遗传、常染色体隐性遗传、性连锁遗传和线粒体遗传等。
这些遗传模式的了解有助于我们理解疾病的遗传规律以及在家族中的传播方式。
分子基础:OMIM提供了疾病的分子基础信息,包括与该疾病相关的基因、基因突变和基因功能等。
这些信息有助于我们理解疾病的发生机制和病理过程,也为疾病的诊断和治疗提供了重要的依据。
文献引用:OMIM提供了与每种疾病相关的文献引用列表。
这些文献引用包括了关于该疾病的研究论文、临床案例和科学文章等。
科学家和医生可以通过查阅这些文献,获取最新的关于疾病的研究进展和治疗方法等信息。
除了以上内容,OMIM还提供了一系列与遗传疾病相关的资源和工具,包括基因序列、突变数据库、药物信息等。
这些资源和工具为科学家和医生提供了方便和支持,有助于他们进行遗传疾病的研究和诊疗工作。
OMIM的使用非常简便,用户可以通过浏览器访问OMIM的官方网站,进行和浏览相关信息。
用户可以通过输入疾病的名称、基因的名称或其他相关关键词,来与之相关的信息。
结果将呈现出相关的疾病描述、遗传模式、分子基础和文献引用等内容。
用户可以通过点击链接进一步查看详细信息。
总的来说,OMIM是一个非常有用的工具,为研究人员、医生和患者提供了丰富的关于遗传疾病的信息。
通过使用OMIM,我们可以更好地了解并研究人类遗传疾病,为其诊断、治疗和预防提供科学依据。
基于语义的疾病表型相似性

基于语义的疾病表型相似性王志刚;谢丽芳;陈鑫;杨啸林;彭屹;张正国【摘要】If a drug can treat a specific disease,this drug can probably treat the diseases with similar phenotype. Therefore,large - scale computing similarity of the disease phenotype can help to find the new treatment. We downloaded 3742 diseases phenotype information from OMIM database,and 13721 Mesh words related to anatomy and disease symptoms from Mesh vocabulary thesaurus. Each Mesh word was searched in all of 3742 diseases phenotype annotation text. Finally,we got a Mesh vocabulary list for every disease. Then disease phenotype pairwise similarity matrix was systematically calculated using semantic analysis approach. We.found that most of the diseases associated biological pathways include tumor biological pathways,insulin signaling,hypertrophic cardiomyopathy pathways and cell adhesion pathway. The probability of involving same KEGG biological pathway increases with the disease phenotype similarity. This also shows our method is reliable. Disease phenotype similarity can be used as the supplement of disease genomic space similarity,and may have potential application value in drug discovery.%已知一种药物可用于治疗某疾病,则该药物可能对与该疾病具有相似表型的其他疾病有疗效.因此,大规模地计算疾病表型相似性可辅助发现的疾病新的治疗方法.我们从OMIM下载了3742种疾病的表型信息,从Mesh词库下载13721个关联解剖学和疾病症状的注释词.我们将以上的Mesh词逐一在3742种疾病的表型信息文本中搜索,得到每种疾病涉及的Mesh词汇列表,进而基于语义分析的方法系统地计算了疾病表型的两两相似性矩阵.我们发现疾病关联生物通路最多的有肿瘤生物通路,胰岛素信号通路,肥大心肌病通路和细胞粘附通路等.随疾病对表型相似度的增加,其更涉及相同KEGG生物通路的概率亦增加,证明了本文方法的可靠性.疾病表型相似性可作为疾病在基因水平相似性的补充,有望为药物发现研究提供一条新途径.【期刊名称】《生物信息学》【年(卷),期】2012(010)003【总页数】4页(P154-157)【关键词】疾病表型;语义;相似性;生物通路【作者】王志刚;谢丽芳;陈鑫;杨啸林;彭屹;张正国【作者单位】中国医学科学院基础医学研究所,北京协和医学院基础学院,北京100005;中国医学科学院信息中心,北京100009;中国医学科学院基础医学研究所,北京协和医学院基础学院,北京100005;中国医学科学院基础医学研究所,北京协和医学院基础学院,北京100005;中国医学科学院基础医学研究所,北京协和医学院基础学院,北京100005;中国医学科学院基础医学研究所,北京协和医学院基础学院,北京100005【正文语种】中文【中图分类】Q518.2已知一种药物可用于治疗某疾病,则该药物可能对与该疾病具有相似表型的其他疾病有疗效[1]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
omim对表型系列的分类
Omim是一种用于分类表型系列的方法。
表型系列是指一组具有类似临床特征的疾病。
Omim通过将表型系列分为不同的分类,帮助医生和研究人员更好地理解和诊断疾病。
下面将介绍一些常见的Omim分类及其特点。
1. 先天性心脏病
先天性心脏病是一种在出生时就存在的心脏结构异常。
这些异常可以影响心脏的结构、功能或血液流动。
Omim将先天性心脏病分为不同的亚型,如房间隔缺损、室间隔缺损、动脉导管未闭等。
每个亚型都有特定的临床特征和遗传机制。
2. 遗传性肌肉疾病
遗传性肌肉疾病是一组由基因突变引起的肌肉功能异常。
这些疾病可以影响肌肉的力量、协调性和耐力。
Omim将遗传性肌肉疾病分为不同的亚型,如肌营养不良、肌无力症、肌强直等。
每个亚型都有不同的临床表现和遗传模式。
3. 遗传性耳聋
遗传性耳聋是一种由基因突变引起的听觉功能障碍。
这些突变可以影响内耳、听神经或中枢神经系统。
Omim将遗传性耳聋分为不同的亚型,如先天性耳聋、幼年性耳聋、成年性耳聋等。
每个亚型都有不同的临床特征和遗传机制。
4. 先天性盲
先天性盲是指在出生时就存在的视觉功能障碍。
这些障碍可以是由眼球、视神经或大脑异常引起的。
Omim将先天性盲分为不同的亚型,如先天性白内障、视神经发育异常、视网膜疾病等。
每个亚型都有特定的临床特征和遗传机制。
5. 先天性发育迟缓
先天性发育迟缓是指在出生时就存在的发育延迟。
这些延迟可以涉及多个方面,如智力、语言、运动等。
Omim将先天性发育迟缓分为不同的亚型,如智力迟钝、语言障碍、运动发育迟缓等。
每个亚型都有不同的临床表现和遗传模式。
6. 先天性代谢病
先天性代谢病是一组由基因突变引起的代谢异常。
这些异常可以影响机体对营养物质的代谢和利用。
Omim将先天性代谢病分为不同的亚型,如酮症、氨基酸代谢异常、糖代谢异常等。
每个亚型都有特定的临床特征和遗传机制。
7. 先天性免疫缺陷
先天性免疫缺陷是一组由基因突变引起的免疫功能异常。
这些异常可以导致机体易感染、免疫反应异常或自身免疫疾病。
Omim将先天性免疫缺陷分为不同的亚型,如免疫缺陷病、免疫反应异常病、自身免疫病等。
每个亚型都有不同的临床表现和遗传机制。
8. 先天性骨骼畸形
先天性骨骼畸形是一组由基因突变引起的骨骼结构异常。
这些异常可以影响骨骼的形态、生长或骨骼的发育。
Omim将先天性骨骼畸形分为不同的亚型,如骨发育不良、骨骼畸形、骨骺发育异常等。
每个亚型都有特定的临床特征和遗传机制。
以上是一些常见的Omim分类及其特点。
通过Omim的分类,医生和研究人员可以更好地了解和诊断不同的表型系列,促进疾病的研究和治疗。
在未来的研究中,我们可以期待更多的表型系列被发现和分类,为人类健康带来更多的进展。