Thomson Reuters Pharma:全球综合医药信息数据库——如何有效的使用
Thomson-pharma药物报告Telaprevir
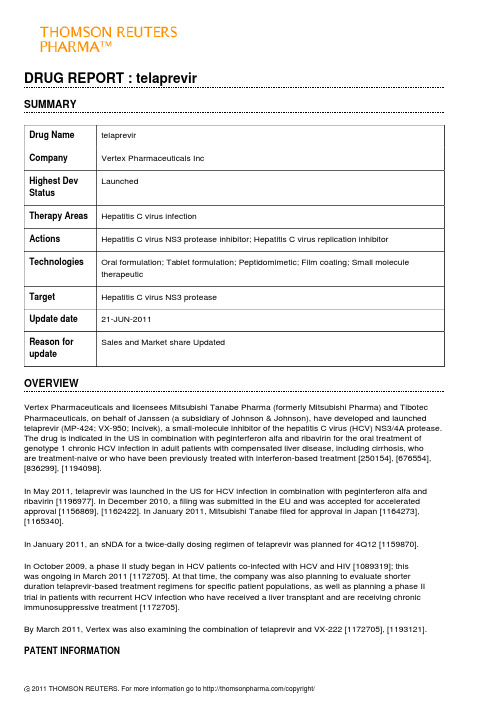
DRUG REPORT : telaprevirSUMMARYDrug Name telaprevirCompany Vertex Pharmaceuticals IncLaunchedHighest DevStatusTherapy Areas Hepatitis C virus infectionActions Hepatitis C virus NS3 protease inhibitor; Hepatitis C virus replication inhibitorTechnologies Oral formulation; Tablet formulation; Peptidomimetic; Film coating; Small moleculetherapeuticTarget Hepatitis C virus NS3 proteaseUpdate date21-JUN-2011Reason forSales and Market share UpdatedupdateOVERVIEWVertex Pharmaceuticals and licensees Mitsubishi Tanabe Pharma (formerly Mitsubishi Pharma) and Tibotec Pharmaceuticals, on behalf of Janssen (a subsidiary of Johnson & Johnson), have developed and launched telaprevir (MP-424; VX-950; Incivek), a small-molecule inhibitor of the hepatitis C virus (HCV) NS3/4A protease. The drug is indicated in the US in combination with peginterferon alfa and ribavirin for the oral treatment of genotype 1 chronic HCV infection in adult patients with compensated liver disease, including cirrhosis, whoare treatment-naive or who have been previously treated with interferon-based treatment [250154], [676554], [836299], [1194098].In May 2011, telaprevir was launched in the US for HCV infection in combination with peginterferon alfa and ribavirin [1196977]. In December 2010, a filing was submitted in the EU and was accepted for accelerated approval [1156869], [1162422]. In January 2011, Mitsubishi Tanabe filed for approval in Japan [1164273], [1165340].In January 2011, an sNDA for a twice-daily dosing regimen of telaprevir was planned for 4Q12 [1159870].In October 2009, a phase II study began in HCV patients co-infected with HCV and HIV [1089319]; thiswas ongoing in March 2011 [1172705]. At that time, the company was also planning to evaluate shorter duration telaprevir-based treatment regimens for specific patient populations, as well as planning a phase II trial in patients with recurrent HCV infection who have received a liver transplant and are receiving chronic immunosuppressive treatment [1172705].By March 2011, Vertex was also examining the combination of telaprevir and VX-222 [1172705], [1193121].PATENT INFORMATIONThe US patent covering the composition-of-matter for telaprevir was granted in 2010 with a term that expiresin 2025. In the EU, Vertex expected to obtain extensions to the term of the patent covering the composition-of-matter of telaprevir, thus extending expiry from 2021 to 2026; the company is required to apply separately for the extensions in the EU on a country-by-country basis [1172705].REGULATORY INFORMATIONThe USIn December 2005, the FDA granted telaprevir Fast Track status for HCV treatment [639367].By July 2010, a rolling NDA submission had been initiated; at that time, the CMC section of the NDA had been filed [1119626]. In November 2010, the filing was completed, which included data from both treatment-naive and -experienced HCV patients. The company also requested priority review [1149931], [1159870]. In January 2011, the NDA was accepted for priority review by the FDA, with a PDUFA date of May 23, 2011 [1162422].In April 2011, the FDA's Antiviral Drugs Advisory Committee recommended approval for genotype 1 chronic HCV infection [1187825]. In May 2011, telaprevir was approved for the treatment of chronic HCV infection in combination with peginterferon alfa and ribavirin in adult patients who had either not received interferon-based treatment or who had not responded adequately to prior therapy [1193767]. The drug was launched in the US later that month [1196977].In January 2011, an sNDA for a twice-daily dosing regimen of telaprevir was planned for 4Q12 [1159870]. EuropeIn December 2010, a filing was submitted in the EU for chronic HCV infection, which was accepted by the EMA for accelerated approval [1156869], [1162422]. In May 2011, a decision was anticipated in the second half of 2011 [1188291].JapanIn July 2010, filing was slated for early 2011 [1121439]. In January 2011, Mitsubishi Tanabe filed for approval in Japan [1164273].Rest of the worldIn January 2011, Vertex completed submission of its NDS for telaprevir in Canada; the drug was granted priority review in Canada [1162422].PREMARKETING STUDIESTreatment-naivePhase IIIIn March 2011, Vertex was planning a study to evaluate a short-duration, 12-week telaprevir-based regimen in treatment-naive patients with HCV with the CC IL28B genotype [1180816].In March 2011, a phase III HCV and HIV co-infection study of telaprevir was planned for the end of that year [1172677].In October 2010, the phase IIIb trial (OPTIMIZE) to compare twice-daily telaprevir (1125 mg) with three times-daily telaprevir (750 mg) was initiated in 700 treatment-naive patients with genotype-1 HCV in the US and the EU [1141995]. The randomized, open-label, study would have a primary endpoint of sustained viral response (SVR) 24 weeks after the end of all treatment. The primary objective would be to demonstrate non-inferiorityof twice-daily telaprevir versus telaprevir dosed every 8 h as measured by SVR. SVR data were expected in 2012. Patient screening for enrollment was expected to begin in November 2010 [1141996]. In January 2011, enrollment was ongoing [1159870].In October 2008, a randomized, open-label, active-control, parallel-group, safety and efficacy phase III trial (NCT00780416; G060-A6) was initiated in treatment-naive patients (expected n = 165) with genotype-1 HCV infection in Japan. Patients were to receive telaprevir in combination with peginterferon-alfa-2b (PEG-IFN-a-2b) plus ribavirin, or PEG-IFN-a-2b plus ribavirin alone. The primary endpoint was a sustained viral response(SVR) , defined as an undetectable HCV RNA level 24 weeks after treatment. The trial was expected to be completed in July 2011 [956738]. Development was ongoing in July 2009 [1047222].In September 2008, a randomized, open-label, active control, parallel group supplemental phase III trial(NCT00758043; VX08-950-111; ILLUMINATE) was initiated in treatment-naive patients (expected n = 500) with HCV at sites in the EU and US. Patients who achieved a rapid viral response to a telaprevir-based regimen were to be evaluated after extending total treatment duration from 24 to 48 weeks. The primary endpoint was SVR rate [956721], [1172705]. By January 2009, enrollment was complete [975836]. By August 2009, the telaprevir dosing section of the study had been completed and all patients were beyond week 24 [1031951]. In August 2010, data were reported, which demonstrated that there was no benefit from extending the telaprevir-based treatment to 48 weeks. The SVR rates were 92% (n = 149/162) and 88% (n = 140/160) for the 24 and 48-week treatment groups respectively. Overall, in the intent-to-treat (ITT) analysis, 72% of patients (n = 388/540) in the trial achieved a viral cure, and the safety profile was similar to the phase III ADVANCE study. The most common adverse events reported included fatigue, pruritus, nausea, anemia, rash and headache, and majority were considered mild or moderate [1123095]. In October 2010, data from the trial were presented at the 61st AASLD meeting in Boston, MA. It was demonstrated that 60% of African-Americans and 63% of all patientswith advanced liver fibrosis or cirrhosis achieved SVR with telaprevir-based therapy. Of African-Americans whose virus was undetectable at weeks 4 and 12, 88% achieved SVR in the 24- and 48-week randomized treatment arms. There was no control arm of PEG-IFN and ribavirin alone in the study. A total of 65% of people met the criteria for 24-week total treatment; there was no benefit in extending therapy to 48 weeks. Adverse events resulted in 17% of patients discontinuing the study, mainly due to a severe rash or anemia [1143419], [1146345].In March 2008, patient screening began in a randomized, double-blind, placebo-controlled, parallel-group, three-arm safety and efficacy phase III trial (NCT00627926; ADVANCE; VX07-950-108), which was expected to enroll 1050 treatment-naive patients with genotype-1 chronic HCV. The trial was to be conducted at 114 sites worldwide, including sites in the EU and US. The patients were to receive one of two regimens of telaprevir(8 weeks or 12 weeks) administered in combination with PEG-IFN-a-2a and ribavirin, or placebo. The primary endpoint was an SVR, defined as undetectable HCV RNA, 24 weeks after treatment completion [885649], [885933]. By October 2008, enrollment was completed [956858]. In February 2009, dosing of telaprevir or placebo, 8 or 12 weeks, had been completed [996403]. Data were reported in May 2010, showing that telaprevir was superior to control; SVR was achieved in 69 and 75% of telaprevir-treated patients after 8 and 12 weeks, respectively. After 48 weeks, 44% of patients in the control arm achieved SVR. Rapid viral responses were achieved by 68, 66 and 9% of patients in the 12-week telaprevir arm, the 8-week telaprevir arm and the control arm, respectively. Viral relapse was observed in 9, 10 and 28% of patients in the 12-week telaprevir arm, the8-week telaprevir arm and the control arm, respectively [1103157]. In October 2010, further data from the trial were presented at the 61st AASLD meeting in Boston, MA. It was demonstrated that 62% of African-Americans achieved SVR with telaprevir, compared to 25% of African-Americans treated with PEG-IFN and ribavirin alone.A total of 62% of people with advanced liver fibrosis or cirrhosis achieved SVR with telaprevir, compared to 33% treated with PEG-IFN and ribavirin alone. In the study, 58% of patients met the criteria for 24-week total treatment [1143419]. Further data presented at the meeting concluded that the 12-week regimen of teleprevir (150 mg/8 h) plus PEG-IFN (18 microg/week) and ribavirin (1000 to 1200 mg/day) provided the best benefitto risk profile. The most frequently observed adverse events were nausea, anemia, diarrhea, fatigue and rash [1148314].Phase IIIn May 2011, a US phase II trial to assess the efficacy of telaprevir in combination with PEG-IFN and ribavirin in patients with recurrent HCV following a liver transplant was planned for the fourth quarter of 2011 [1188291].In October 2009, a phase II multicenter, randomized, double-blind, placebo-controlled, two part (part A andpart B) parallel-group, safety and efficacy study (NCT00983853; VX08-950-110) began in the US in patients(n = 68) co-infected with HCV and HIV, who were treatment-naive for HCV. The study was to determine the efficacy of treatment with telaprevir plus peginterferon alfa-2a and ribavirin, with primary endpoints of safety and plasma HCV RNA level. The study was due to complete in June 2012 [1089319]. By October 2010, enrollment of 60 patients had been completed [1141995]. Patients in Part A and Part B of the study were randomized to receive either 12 weeks of telaprevir or placebo in combination with peginterferon alfa-2a and ribavirin followedby 36 weeks of peginterferon alfa-2a and ribavirin alone. Part A (n = 13) of the study enrolled patients who were not receiving antiretroviral therapy for HIV. Part B (n = 47) enrolled patients who were being treated for HIVwith either Atripla (n = 24) or a Reyataz-based regimen (n = 23). In March 2011, interim data (from 4 weeksof treatment) were presented at the 18th Conference on Retroviruses and Opportunistic Infections (CROI) in Boston, MA. The results demonstrated that 70% of patients in the study (Parts A and B) had undetectable HCV after treatment with telaprevir-based combination therapy compared to 5% in those on pegylated-interferonand ribavirin alone at week 4. In addition, HIV viral load and CD4 counts were stable in patients on a telaprevir-based regimen. Final data for sustained viral response (SVR) were expected in 2012 [1172677].In August 2009, a combination study of telaprevir and VX-222 was planned [1031951]. In October 2009, the trial was still expected to start that year, and data were expected by mid-2010 [1052205]. In January 2010, the trial was to begin pending completion of discussions with regulatory authorities [1067607]. In February 2010, the trial was planned to start in the first quarter of 2010 and interim data were expected in the third quarter of 2010. The trial would evaluate the rate of undetectable HCV RNA 24 weeks after the end of treatment with multiple VX-222 and telaprevir dosing regimens [1073316]. In March 2010, the phase II multicenter, randomized, parallel-group, dose-ranging trial (; VX09-222-103) began. The trial was designed to evaluate the safety and efficacy of telaprevir plus VX-222 in treatment-naive patients with genotype I HCV infection (expected n = 150), using a response-guided trial design. The primary endpoint of the trial was safety and tolerability, with a secondary endpoint of proportion of patients achieving SVR; the company stated that it may elect to enroll upto two additional treatment arms. At that time, initial data were expected in 2H10 [1078227], [1089256]. In April 2010, enrollment in the study was expected to be completed in the second quarter of 2010 and interim data were expected in the second half of 2010 [1091125]. In October 2010, Vertex discontinued arm A of the trial due to patients meeting a pre-defined stopping rule related to viral breakthrough in the first 4 weeks of dosing. At that time, an additional three cohorts were continuing and no viral breakthrough had been observed. Data were expected in the first half of 2011 with SVR data in the second half of 2011 [1141995]. In November 2010, the company planned to begin an additional treatment arm evaluating the triple combination regimen of telaprevir, VX-222 and ribavirin (po, bid) for 12-week using a response-guided regimen. Enrollment was expected to begin in the first quarter of 2011. The phase II study also included treatment arms evaluating 12-week, response-guided regimens of four-drug (telaprevir/VX-222 with or without Pegasys and ribavirin) combination therapy. Enrollment in the four drug treatment arm was completed in October 2010. Vertex may add an additional arm based on further results from the ongoing treatment arms [1146979]. In December 2010, the second two-drug treatment arm of telaprevir plus VX-222 (1125mg/400mg) alone was discontinued as a result of a pre-defined stopping rule related to viral breakthrough. The study would continue as planned with three treatment arms of the four-drug combinations, two of which were fully enrolled. At that time, enrollment in the three-drug treatment arm and data from the four-drug treatment arms were expected in the first quarter of 2011 [1157421]. In March 2011, interim data from the 106-patient ZENITH study were presented at the 46th annual meeting of the European Association for the Study of the Liver in Berlin, Germany. At week 12, 90 and 83% of patients treated with 400 and 100 mg VX-222, respectively, in combination with telaprevir, pegylated-interferon and ribavirin, showed undetectable hepatitis C virus. At that time, 50 and 38% of patients in the two groups, respectively, were able to stop all treatment. The remaining patients received a further 12 weeks of pegylated-interferon and ribavirin. At that time, enrollment was underway in an additional arm assessing VX-222, telaprevir and ribavirin [1180638].By February 2008, a phase II viral kinetics trial for HCV genotype-4 had begun [875818]. By July 2008, enrollment was complete [930471].In January 2008, an open-label, single-group, safety and efficacy phase II trial (NCT00621296) was initiatedin subjects (expected n = 16) with chronic HCV in Japan. The subjects were to receive telaprevir 750 mgthree times daily, for 24 weeks. The primary endpoint was to establish HCV RNA kinetics and other viral characteristics. The study was expected to be completed in September 2009 [907396], [904699]. Patient recruitment was ongoing in August 2008 [907396].In October 2007, a randomized, open-label, parallel-group, safety and efficacy phase II trial (NCT00528528;VX-950-TiDP24-C208; CR013516) was initiated in treatment-naive patients (expected n = 160) with genotype-1 chronic HCV in Europe. The patients were randomized to 1 of 4 treatment groups and received telaprevir (750 mg tid or 1125 mg bid) for 12 weeks in combination with PEG-IFN-a-2a plus ribavirin or PEG-IFN-a-2b plusribavirin for 24 weeks [956710]. In July 2008, data were released from the study. No substantial differences were seen in the safety profiles of the twice- and three-times-daily regimens, and both resulted in > 80% of patients with undetectable HCV RNA at weeks 4 and 12. A complete analysis was to be carried out when the study finished in 2009 [930471]. In October 2008, similar data were presented at the 59th Annual Meeting of the American Association for the Study of Liver Diseases in San Francisco, CA [958470]. In April 2009, all dosingin the study had been completed and in August 2009, patients were in the follow-up period. The number of patients with an SVR24 in the bid and tid dose groups would be compared [1031951]. In October 2009, further data were presented . An intent-to-treat analysis of 161 patients showed that SVR rates were 82 and 83% for patients receiving peg-IFN-alfa-2b bid and peg-IFN-alfa-2a bid, respectively, and 81 and 85% for those on peg-IFN-alfa-2b tid and peg-IFN-alfa-2a tid, respectively. RVR rates for the four groups were 67, 83, 69 and 80%, respectively. The most common adverse events were pruritis, rash, nausea, flu-like illness, anemia, headache and fatigue. These were similar in the bid and tid regimens. Of 161 patients, 5% discontinued treatment because of serious adverse events, namely rash and anemia. The data were to be presented at the 60th Annual Meeting of the American Association for the Study of Liver Diseases in Boston, MA [1053696], [1056258]. By January 2010, data from the trial had been submitted to the FDA and discussions were underway with bothUS and EU regulatory authorities regarding development of the drug as part of an every 12 h dosing regimen [1067607].In May 2006, Vertex was planning the phase II study PROVE 2 study in Europe. This would incorporate four,80-patient treatment arms: subjects were to receive telaprevir plus PEG-IFN for 12 weeks; telaprevir with PEG-IFN and ribavirin for 12 weeks; 12 weeks treatment with the triple combination followed by 12 weeks withPEG-IFN and ribavirin alone; or PEG-IFN and ribavirin for 48 weeks. The primary objectives were the sameas for PROVE 1 [668896]. By July 2006, PROVE 2 had started and by January 2007, recruitment had been completed [680324], [755789]. At that time, the company expected to expand clinical development of telaprevir into important HCV sub-populations in 2007, including patients with genotype-2 and genotype-3 HCV infection. The company also anticipated initiating clinical trials exploring twice-daily dosing of telaprevir some time in 2007 [755789]. In June 2007, preliminary data from the PROVE 2 study were reported to be consistent with PROVE 1 study results. Patients treated with telaprevir, PEG-IFN and ribavirin had undetectable HCV RNA levels at 4 and 12 weeks. At 12 weeks, treatment with telaprevir only was associated with antiviral activity that was lower than that in patients that received telaprevir with ribavirin and PEG-IFN, but was higher than that observed in the control arm. Rash, gastrointestinal events and anemia were the most common adverse events that led to discontinuation in the telaprevir arms. Fewer discontinuations were observed in patients that received telaprevir and PEG-IFN [803933]. In November 2007, similar data were presented at the 58th Annual Meeting of the American Association of the Study of Liver Diseases in Boston, MA. The SVR rates were 29, 59 and 65% in the 12-week (without ribavirin), 12-week (plus ribavirin) and 24-week treatment arms, respectively [847055]. In April 2008, similar data from the trial were presented at the 43rd Annual Meeting of the European Association for the Study of the Liver in Milan, Italy [899125]. Similar data were also reported in May 2008 at the DDW meetingin San Diego, CA [906212]. In October 2008, similar data were presented at the 59th Annual Meeting of the American Association for the Study of Liver Diseases in San Francisco, CA. The SVR rates were 30, 60 and69 and 46% in the 12-week (without ribavirin), 12-week (plus ribavirin), 24-week, and 48-week treatment arms, respectively [958470]. In April 2009, similar data were published [1004842], [1004581].In October 2005, Vertex was planning one- and three-month phase II studies of telaprevir in combination with PEG-IFN [626769]. By November 2005, Vertex had submitted an IND to the FDA [634055]. In December 2005, Vertex began a 28-day, US phase II study to evaluate the safety, tolerability and pharmacodynamics of 750 mg of telaprevir given every 8 h in combination with PEG-IFN and ribavirin in 12 treatment-naive subjects [638638]. In January 2006, enrollment was complete [644067]. In February 2006, Vertex reported preliminary results showing that at the end of week 1, plasma HCV RNA was below the limit of quantitation in half of the subjects and undetectable in 2 of the patients. After 4 weeks of dosing, HCV RNA was undetectable in plasma from all 12 patients. There were no treatment discontinuations and no serious adverse events reported [648823]. In May 2006, similar data were presented at the Discovery & Selection of Successful Drug Candidates meeting in Cambridge, MA [667947]. Also in May 2006, similar data were presented at the DDW meeting in Los Angeles, CA. All patients reached undetectable plasma HCV RNA levels within 28 days of dosing and levels remained undetectable after 12 weeks follow-up [667643], [668532].Treatment-experiencedPhase IIIIn October 2008, an open-label, uncontrolled, single-group, safety and efficacy phase III trial (NCT00781274;G060-A9) was initiated in patients (expected n = 30) with genotype-1 HCV who did not respond to previous therapy in Japan. Patients were to receive telaprevir for 12 weeks and peginterferon alfa-2b (PEG-IFN-a-2b) plus ribavirin for 24 weeks. The primary endpoint was an SVR, defined as an undetectable HCV RNA level 24 weeks after treatment. The trial was expected to be completed in January 2011 [956789].In October 2008, an open-label, uncontrolled, single-group, safety and efficacy phase III trial (NCT00780910;G060-A8) was initiated in patients (expected n = 100) with genotype-1 HCV who relapsed following previous therapy in Japan. Patients were to receive telaprevir for 12 weeks and PEG-IFN-a-2b plus ribavirin for 24 weeks. The primary endpoint was a SVR, defined as an undetectable HCV RNA level 24 weeks after treatment. The trial was expected to be completed in January 2011 [956781].In August 2008, Vertex reached agreement with US and EU regulatory authorities to proceed with its planned REALIZE trial (NCT00703118; CR014842) [936309], [956718]. In the 48-week, 650-patient, three-armed multicenter study patients with genotype-1 HCV who had failed to achieve a SVR with prior therapy of PEG-IFN plus ribavirin, would receive 750 mg of telaprevir q8 h plus PEG-IFN-a-2a and ribavirin for 12 weeks, followed by 36 weeks of PEG-IFN-a-2a plus ribavirin alone. Patients in the control arm would receive PEG-IFN-a-2a plus ribavirin alone for 48 weeks. The primary endpoint was undetectable HCV RNA levels 24 weeks after the endof treatment [936309]. By October 2008, patient dosing had begun; the trial was to include null responders, partial responders and relapsers to prior therapy [952629], [956585], and in November 2008, enrollment in Europe began [965253]. In February 2009, enrollment in REALIZE was complete [982641]. By August 2009,the telaprevir dosing section of the study had been completed and all patients were beyond week 24 [1031951]. In September 2010, top-line data were reported, which demonstrated that 65% of HCV patients (total n = 622) treated with telaprevir achieved a SVR compared to 17% in the control group at 24 weeks. In a subgroup analysis, data showed that 86% of relapsed patients treated with telaprevir achieved a SVR compared to 24% for the control. In addition, the SVR rates were 57% and 31% for partial and null responders, respectively, compared to 15% and 5% for the corresponding control arms [1143647]. In March 2011, further data were presented at the International Liver Congress 2011/46th annual meeting of the European Association for the Study of the Liver (EASL) in Berlin, Germany. No significant improvement in SVR rates and no significant reduction in virologic failure and relapse rates were observed in patients treated with a 4-week lead-in of PEG-IFN-a-2a and ribavirin alone compared with a simultaneous start of telaprevir, PEG-IFN-a-2a and ribavirin [1180788], [1184383].In February 2007, phase III trials were planned, and development of the drug was to be expanded to include HCV genotype-2 and -3 and twice-daily dosing schedules [762718], [762718]. In April 2007, Vertex and Tibotec planned to meet with regulatory authorities to discuss the design of phase III trials [784124], and by October 2007, Vertex and Tibotec were in discussions with the US and EU regulatory authorities to review clinical data and to ascertain how best to evaluate the drug in future trials [845344]. By January 2008, Vertex had submitted a phase III protocol to the FDA. The design included a 48-week control arm and 8 and 12 weeks of telaprevir treatment as part of 24-week combination treatment regimens. Vertex planned to determine which patients should cease treatment at week 24 using rapid viral response (RVR). A meeting with the FDA was scheduled for January 2008 to discuss the protocol [865541].In May 2006, the company began PROVE 1, a randomized, US phase II study, which was expected to enroll 280 patients into 4 treatment arms: 20 patients would receive 750 mg of telaprevir three times a day with PEG-IFN and ribavirin for 12 weeks; two 80-patient groups would take the same combination for 12 weeks followed by 12 or 36 weeks of treatment with PEG-IFN and ribavirin alone; the fourth group of 80 patients would receive PEG-IFN and ribavirin alone for 48 weeks. The primary objective was to determine the number of patients who achieved SVR, defined as undetectable HCV RNA levels 24 weeks after completing treatment. Patients in the 12- and 24-week treatment arms who had undetectable viral levels by the end of week 4 and who maintained this until week 10 or 20, respectively, would stop treatment at 12 or 24 weeks, respectively. Patients who had detectable HCV RNA at the initial time point would continue on PEG-IFN and ribavirin for 48 weeks. Enrollment began in June 2006 [668896]; by September 2006, enrollment in the trial had been completed [735053]. In December 2006, Vertex reported interim data from the study. An independent board compared pooled data from the three telaprevir arms with the control arm. At 12 weeks, 88% telaprevir-treated patients had undetectable HCV RNA levels, compared with 52% patients in the control group. In the telaprevir arms, the discontinuationrate was 9%, compared with 3% in the control arm. Serious adverse events occurred in 3% of telaprevir-treated patients and 1% of patients in the control group [751574], [751209]. In April 2007, further data were presented at the 42nd annual meeting of the European Association for the Study of the Liver in Barcelona, Spain, showing 88 and 79% of patients that received telaprevir achieved plasma HCV RNA levels of < 30 IU/ml and < 10 IU/ ml, respectively, at 4 weeks. Furthermore, 6 of 9 patients in one treatment arm continued to have undetectable HCV RNA 20 weeks after stopping treatment [784124]. In September 2007, data were presented at the 47th ICAAC meeting in Chicago, IL. Interim, 24-week PROVE 1 data demonstrated that telaprevir patients (n = 175) achieved undetectable HCV RNA at weeks 4 (79%) and 12 (70%); in the control arm (n = 75) 11% achieved undetectable HCV RNA at weeks 4 and 39% at week 12. Discontinuations due to adverse events were more frequent in the telaprevir arm at 11%, versus 35% in the control arm [820890]. In November 2007, similar data were presented at the 58th Annual Meeting of the American Association of the Study of Liver Diseases in Boston, MA [847055]. In April 2008, final data from the trial were presented at the 43rd Annual Meeting of the European Association for the Study of the Liver in Milan, Italy. SVRs were seen in 61% of telaprevir recipients, compared with 41% of control patients [899125]. Similar data were reported in May 2008 at the DDW meeting in San Diego, CA [907429]. In April 2009, similar data were published [1004828], [1004581].Phase IIIn September 2007, a non-randomized, open label, uncontrolled, single-group, phase II study (NCT00535847; study 107; VX06-950-107) was initiated in the US, Canada, Germany, France, the Netherlands and Puerto Rico. In the trial, 170 patients with genotype-1 hepatitis C who did not achieve an SVR with previous interferon-based treatment in the control arms of the PROVE 1, PROVE 2 or PROVE 3 trials were to receive telaprevir plus PEG-IFN-a-2a plus ribavirin for 12 weeks, followed by the same regimen without telaprevir for 12 weeks. The trial was expected to be completed in May 2009 [898809], [898686]. Interim data were reported in April 2008 at the 43rd Annual Meeting of the European Association for the Study of the Liver in Milan, Italy. Of the 60 patients treated with the telaprevir combination, 49 achieved HCV RNA levels < 25 IU/ml. This response was maintained, and no viral breakthrough was observed in 36 and 16 patients who had completed 4 and 12 weeks of treatment, respectively. Two patients discontinued treatment due to adverse events, which were similar to those commonly observed with PEG-IFN-a-2a and ribavirin. They included fatigue, nausea, rash and headache [899404]. In October 2008, further data were presented at the 59th Annual Meeting of the American Association for the Study of Liver Diseases in San Francisco, CA. At 4, 12 and 24 weeks, 65.4, 65.6 and 56.9% patients had HCV RNA <10 IU/ml [958470]. In October 2009, further interim data from 94 patients were reported. An SVR was achieved after 24 or 48 weeks of telaprevir-based treatment, by 90% of prior treatment responders and 55% of prior treatment partial responders. After 48 weeks of telaprevir-based treatment, an SVR was achieved by 57% of prior treatment null responders. At that time, adverse events had caused 7% patientsto discontinue treatment [1052655]. In April 2010, data were presented at the 45th Annual Meeting of the European Association for the Study of the Liver in Vienna, Austria. An overall SVR rate of 59% and an overall relapse rate of 16% were observed. In addition, it was reported that 10 patients had discontinued all therapy due to adverse events, with rash being the most common adverse event [1091068]. Further data were presented in May 2010 at the DDW meeting in New Orleans, LA. The viral breakthrough and relapse rates were 25 and 23% in prior null responders, 10 and 22% in prior partial responders, 13 and 0% in prior viral breakthroughs, and 0 and 4% in prior relapsers, respectively [1091233].By January 2007, Tibotec had begun a European phase II viral kinetics trial. The study would evaluate telaprevir in patients with HCV genotypes 2 and 3. At that time, patients were undergoing screening [865541]. By July 2008, enrollment was complete [930471].By October 2006, Vertex was planning PROVE 3, a 400-patient trial in patients with genotype-1 HCV who had failed prior treatment [735053]. In February 2007, Vertex began the phase IIb PROVE 3 trial. The randomized, multicenter, 440-patient, North American and European study assessed 750 mg of telaprevir every 8 h in patients with interferon-refractory, genotype-1 HCV. Patients received one of four treatment regimes: 12 weeks of telaprevir, PEG-IFN alfa-2a and ribavirin, followed by 12 weeks of PEG-IFN-a-2a and ribavirin; 24 weeksof telaprevir and PEG-INF-a-2a; 24 weeks of telaprevir, PEG-IFN-a-2a and ribavirin, followed by 24 weeks of interferon PEG-IFN-a-2a and ribavirin; or 48 weeks of PEG-IFN-a-2a and ribavirin. Patients in the final arm had the option to receive telaprevir if they did not respond to PEG-IFN-a-2a plus ribavirin [762718]. Enrollment of 440 patients was completed in June 2007 [803933]. In January 2008, Vertex was planning to meet with the FDA to discuss the telaprevir development program, once interim results from PROVE 3 were available [865541]. In June 2008, interim data were reported. By that time, patient dosing had been completed. In the first treatment。
Thomson_Pharma介绍

Thomson PharmaThomson Pharma是面向制药和生物技术公司的动态信息化解决方案的平台。
它含有Thomsom公司提供的生物制药、公司、专利以及金融方面的最前沿的信息。
这些大量而优质的信息以强大的检索工具为载体并被整合为一个完整的解决方案,令医药企业不同领域的专业人员都可获取他们所需要的信息。
Thomson Pharma通过门户网站的方式提供用户浏览、检索和分析多种数据的功能。
通过Thomson Pharma用户可以方便地整合所有可获得的药物相关的资源信息,并建立以下七大核心领域的动态报告:药物知识产权文献和新闻公司(财务金融,新闻,股市,药物注册信息等)靶标化学基因序列使用Thomson Pharma,用户可以:评估潜在的市场开展世界级的研究工作进军新的药物研发领域取得市场领先地位更快更好地决策有关汤姆森(Thomson)公司汤姆森公司是全球专业信息服务和出版领域最大、最领先的跨国企业,为全球130 多个国家二千多万用户提供服务,协助他们更好、更快地决策和发展。
汤姆森公司由汤姆森法律与条例信息集团、汤姆森金融信息集团、汤姆森学习出版集团、以及汤姆森科技与医疗卫生信息集团组成,在全球拥有38,000 名雇员,在46 个国家和地区设有分支机构与办事处,2004 年全年营业收入81 亿美元,位居福布斯全球500 强企业榜。
相关报道Thomson Pharma是Thomson在医药领域的一个旗舰级产品。
自2005年1月上市以来已有100 多家机构选择Thomson Pharma 作为他们在医药领域的信息化解决方案。
最初购买Thomson Pharma 的公司之一是总部设在新泽西州上萨德尔河的Davos化学公司——Davos是制药业客户化API 和cGMP 媒介的供应商。
“我们的客户是新兴和跨国的制药公司,客户的要求差异很大,从生产供潜伏期研究使用的克数量活性配料(API)到成吨的后期投放市场的产品而各不相同,” Davos Chemical 公司副总裁Brian Robins称。
国内外药品说明书查询步骤

国内外药品说明书查询步骤
对于个人而言主要是查询药品的适应症、用法用量,对于药品有个大概的了解,对于想要了解国内外药品说明书的立项人员来说,查询药品说明书知识了解剂型、规格、适应症等,同时对比多个国家的上市药品说明书(主要是中、美、日、欧)对比国家所批准的规格和适应症有什么不同,那么药品的说明书在哪里查询比较好呢?
推荐查询了解方式就是在药融云中查询了解国外药品的说明书,在上市药品数据库中查询,国内外的上市药品说明书,对比各个国家药品说明书了解批准的规格和适应症有什么不同,全球上市数据库包含40多个主流国家的上市药品说明书,对关键数据进行了多语言标准汉化,通过搜索即可快速了解目标产品在各国上市的详细内容(包含药品说明书),为药物研究和使用提供参考比对。
可以通过药品名称、公司名称、申请号、批准日期、上市日期等进行关键词的搜索,还能根据国家/地区/类型、ATC编码进行条件筛选,筛选出各个国家上市药品数据,包含药品说明书。
还可以通过高级检索功能“是(and)或(or)非(not)”三种检索方式,快速查询了解国内外上市药品说明书。
药品说明书主要是药品的重要来源之一,是医生、护士、病人在适用药物时的科学依据,是药物供应部门向医药卫生人员和人民群众,宣传和介绍药物特性、指导安全用药和普及药物知识的媒介,对于药企是了解规格、剂型,对比各个国家所批准的规格和适应症的不同,理清适应症对某种药物研发有个大概的了解,查询药品说明书是非常重要的。
Thomson Pharma数据库使用详解及实例解析

Thomson Pharma数据库使用详解及实例解析
李明辉
【期刊名称】《医学信息学杂志》
【年(卷),期】2010(031)007
【摘要】简述Thomson pharma数据库在药物研发各阶段的重要作用、资源体系结构以及主要功能,通过实例分析重点介绍其强大的检索和分析功能,由此引导用户更便捷、更有针对性地使用该数据库.
【总页数】4页(P31-34)
【作者】李明辉
【作者单位】中科院上海药物研究所图情室,上海,201203
【正文语种】中文
【相关文献】
1.Thomson Reuters Integrity数据库在药物研发中的应用 [J], 佟岩;汪新久;吴明智
2.新一代制药和生物技术企业动态信息服务系统——Thomson Pharma [J], 马建玲
3.创新药物研发信息资源:Thomson Reuters Integrity数据库 [J], 盛彧欣;李兰燕;毛雪石
4.新药研发数据库Thomson Pharma分析 [J], 马淑芹;张玢
5.扬州电视台SQL数据库维护实例解析——使用作业功能实现数据库同步 [J], 朱斌
因版权原因,仅展示原文概要,查看原文内容请购买。
医药信息数据库

医药信息数据库Dialog中与医药相关的数据库有192个,与化学相关的数据库有65个,与知识产权相关的数据库有25个。
Dialog数据库对于医药研发的全部过程提供完整的信息支持。
药物的生命循环通常,从药物研发到普通药品上中须经过以下几个过程:①研发筛选(R&D Screening),包括市场凋查(Market Survey)与专利调查(Patent Survey);②临床前研究(Preclincal Studies);③临床阶段(Clinical Phases);④新药批准上市(New Drug Approval);整个研究是一个循环往复的过程,缺一环而不可。
在药物研究过程中,更多的是依赖精心加上处理过的专业信息。
我们应该选择针对性强、质量高、覆盖面大、有权威性的检索工具。
另外,信息源的可靠性、获取数据的方便性、检索的效率都是是我们要考虑的首要因素。
DIALOG系统具有600多个数据库,其中和制药相关的数据达200个,这些数据库在为制药企业提供各个环节数据和信息的同时,还利用其功能庞大的指令检索系统为企业提供了优秀的信息和情报的解决方案。
常见的医药数据库介绍如下:ADIS R&D Insight (ADIS 药物研发数据库)ADIS药物研发数据库是Adis International公司的产品。
该数据库的信息来源InPharma、Reactions、PharmacoEconomics & Outcomes News、Clinical Trial Insights等2300种以上的药物、生物专业期刊,国际会议,公司年报和新闻报道等公开资料和非公开资料。
数据库内容包括每种药品的普通名,同义名,商品名,开发公司,国家及开发阶段,所有权信息,峰期销售额,专利失效期,不良事件,药理学,药动力学,药效学,副反应,治疗实验,开发历史,注册信息和参考文献等等。
IMS R&D Focus (IMS 药物研发数据库)IMS药物研发数据库是IMS HEALTH公司的产品。
五大医学文献数据库:只知道PubMed你就out了

五大医学文献数据库:只知道PubMed你就out了循证医学时代,写文章无论是背景、讨论、引用研究,一定要参考文献。
作为医学文献老大的Pubmed,基本无人没用过,而下面介绍的这些文献数据库,你可不能错过了。
1. 中国生物医学文献数据库(CBM)一个集检索、免费获取、个性化定题服务、全文传递服务于一体的生物医学中外文整合文献服务系统。
由中国医学科学院医学信息研究所开发研制。
收录1978 年以来1600 多种中国生物医学期刊,以及汇编、会议论文的文献题录,年增长量约40 余万篇,数据总量达350 余万篇。
学科范围涉及基础、临床、预防学等生物医学的各个领域。
CBM 注重数据的规范化处理和知识管理,全部题录均根据美国国立医学图书馆最新版《医学主题词表》、中国中医研究院中医药信息研究所《中国中医药学主题词表》等进行主题标引和分类标引。
此外,检索系统与PUBMED 具有良好兼容性:CBM 检索系统(CBMWEB) 具有检索入口多,检索方式灵活,以及主题、分类、期刊、作者等多种词表辅助查询功能,可满足简单检索和复杂检索的需求,与 PUBMED 具有良好兼容性。
可获得良好的查全率和查准率。
2. Embase主要特点:近 2000 种 Medline 以外的特有期刊,11 00 万条记录,覆盖更多疾病和药物的信息,无可匹敌的欧洲、亚洲文献汇总,使用 EMTREE 主题词表(包含了所有 MeSH 词)。
检索内容除了文章外,还支持药物和疾病检索。
embase 数据库(Excerpt Medica Database)是由荷兰 Elsevier Science 出版公司建立的 EM 的书目型数据库,收录 70 多个国家出版的 4550 种左右期刊的医药文献,每年约 50 多万条文献记录,累积约994 万条,80% 的文献带有文摘。
EMBASE 医学外文文献数据库中收录药物方面的文献量大,占 40% 左右,并设置了与药物有关的副主题词(连接词)17 个,与疾病有关的副主题词(连接词)14 个,2000 年又新增与给药途径的副主题词(连接词)47 个,设了许多与药物有关的字段,如药物主题词字段(DR),药物分类名称字段(EL),药物商品名字段(TN)等。
Thomson Reuters Pharma:全球综合医药信息数据库——如何有效的使用

单击此处编辑母版标题样式
单击此处编辑母版副标题样式
创建个性化主页
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
创建个性化主页
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
CORTELLIS for CI主页
https:///
实验室信息管理系统性
• Watson LIMS • SampleManager LIMS
5
内容介绍-Thomson Reuters Pharma
药物
交易
知识产 权
• 各种期刊文献
• 专利文献
单击此处编辑母版标题样式 临床实
验报告
Thomson
文献和 新闻
• 新闻 • 科技会议论文
Reuters
• 公司站点
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
战略品种销售数据和预测数据
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Deals和Patents的分析图表及详情
单击此处编辑母版标题样式
单击此处编辑母版副标题样式 点击右下角View list即 可调阅详细的结果列表
查看报告更新历史
单击此处编辑母版标题样式
比如选择横轴是Originator, 纵 轴是Technology
单击此处编辑母版副标题样式
据此了解不同的公司的技术分布
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
3、Star Tree
药物列表和专利列表页面都增加了Star Tree 的分析功能。这是针对Gefitinib药物检索的 专利的结果;点击Star Tree
单击此处编辑母版标题样式 药物检索新增Active和 Inactive的区分,可以 直接区分目前处于这两 种状态的药物。 新增按照专利到期 单击此处编辑母版副标题日样期式进行查询
Pharmaprojects-介绍

7
2.1
Tree Search 目录树检索
在目录树检索中包括11 种路径: (1) Main Details (2) Company/ Status (3) Activity Data (4) Pharmacokinetics (5) Chemical Data (6) Patent Data (7) Country Data (8) Ratings (9) Major Events (10) Alert Service (11) Latest change
10
(2) Company/ Status Data (共11个单项)
World Status Company Company R&D pipeline Company research focus Originator Originator Country Originator Status Licensee Licensee Country Licensee Status Licensed-Outing Drug 通过公司及药品市场状况检索某公司的情况或者某一特定的市场 被批准的所有药物。
4
2 Pharmaprojects 光盘数据库检索方法 打开光盘阅览室主界面, 选中“Pharmaprojects”,双击鼠标左键,出现界面。
5
6
2.1
左栏检索
左栏里列出了8 个检索途径,分别为: Tree Search 目录树检索 Browse Search 索引检索 Form Search 分子式检索 Structure Search 结构检索 Trend Analysis 趋势分析 Company Profile 公司信息 Therapy Profile 治疗信息
汤森路透Thomson+Reuters+Pharma数据库功能与价值

Thomson Reuters Pharma整合性信息平台功能与价值
Thomson Reuters Pharma 是世界领先的综合医药信息平台,客户遍及全球Top100药企及国内各大新药研发企业及单位。
Thomson Reuters Pharma 提供10个面向的信息,包括:药物;专利;公司;化学;临床试验方案;临床试验报告;靶标;
基因序列;交易;文献和新闻。
Thomson Reuters Pharma 直接以报告的方式提供以上10个方面的信息,其中每一份报告都由专业人员围绕主题将所有相关信息进行了整合和综合,并附有业界专家撰写的综述和评论,并且十大报告之间可以很好的相互链接。
通过Thomson Reuters Pharma 可以帮助山东绿叶制药集团:✓制定公司的战略决策;
✓进行新药的项目调研;
✓识别新的药物靶标、药物和基因序列;
✓研究并评估相关的临床试验;
✓寻找有利的注册时机;
✓获得竞争对手产品及交易活动的详细信息;
✓研究当前的药物销售和市场份额数据及对未来的预测数据;
✓查明药物专利何时在某些国家失效;。
国外医药文摘数据库的检索方法和技巧

其他数据库(L) – Derwent Biotechnology Abstracts
BIOSIS Previews
简介:
– BIOSIS Previews包含Biological Abstracts, Biological Abstracts/RRM (Reports, Reviews, and Meetings)的内容。 它收录了90多个国家的 5,000 多种生物学和生命科学的期刊, 及相关的国际会议、综述、书籍和专利信息。BIOSIS Previews 包括1969年以来的大约1300 万条记录。数据库每 周更新,每年增加560,000 多条新记录。BIOSIS Previews覆 盖所有生命科学的领域,包括:生物学,生物化学,生物工 程学,植物学,临床和实验医学,药理学,动物学,农学和 兽医学。
检索的注意事项
接连输入两个检索词,系统默认为词组进行检索。如: Burn* treatment
在进行Topic检索时,系统将自动在以下的字段进行检索:
Title(标题)、Abstract(文摘)、Organisms(生物体)、Major
Concepts(学科分类)、Super Taxa(上位生物分类)、Taxa Note(上位分
医学方面,如果在MEDLINE中不能检到的,可在LSC一试。
国际药学文摘数据库
IPA
IPA简介
IPA是国际药学文摘(International Pharmaceutical Abstracts简称IPA)英 文缩写,于1964年发行印刷版,1970年后实现了计算机化服务。国际 药学文摘(IPA)数据库主要用于检索药学文献,范围包括药物临床和技术 信息、药学实践、药学教育、药学和药物的相关法律。国际药学文摘 (IPA)数据库数据由美国医药卫生系统药剂师协会(American Society of Health-System Pharmacists 简称ASHP)提供 。
药物综合数据库介绍及使用指南(侧重市场数据查询)

药物综合数据库介绍及使用指南(侧重市场数据查询)数据库介绍中国医药数字图书馆在国家经济贸易委员会、财政部支持下(列入国经贸技术[2002]845号文件,2002年度国家技术创新项目),依托上海医药工业研究院信息中心强大的人力优势和资源优势,全新推出药物综合数据库(Pharmaceutical DataBase,简称PDB),力图为企业提供正确的、符合中国实际情况的医药数据平台。
该数据库从药品和企业入手,汇集药品基础信息、药品生产情况、药品销售情况、国家管理药品目录、治疗类别、药品生产企业信息等领域的详细数据,并具有强大的动态分析功能,能够方便、灵活地对数据进行分析、管理,为企业产品研发、市场策划、并购等决策提供第一手数据。
一、药品板块基础信息创新是民族的灵魂,产品乃企业的生命。
制药企业的发展更是离不开产品的创新。
药物综合数据库收载了30000个药品的详细内容,是企业了解国内外药品开发动态最理想的资源之一。
不同阶段的药品信息:数据库收录了临床前、I、II、III、IV期临床、上市、撤市、终止开发等各个阶段的药物基础数据,包含基本情况、合成工艺、制剂配方、临床开发、知识产权、市场动态、药品标准、药品说明书、行保事项、药典涵盖情况、基本药物与医保、新药批准情况等板块生产数据“知己知彼,百战不殆”,“全球化采购”,是企业成功的最重要因素之一。
两者的精髓都是掌握准确的信息。
药物综合数据库汇集了中间体、原料药、制剂的国内外供应商名录,是企业创出品牌的最有效途径之一。
290000个中间体、原料药、制剂的国内外供应商名录主要品种的产能、产量、出口量等销售情况中国市场是21世纪全球最具潜力的市场,我国经济持续高速发展带动了人们对健康的投资与消费。
因此了解药品的销售情况对于企业调整产品结构、营销策略、研发新产品至关重要。
全国重要城市医院用药销售数据国内药物批发数据全球历年畅销药品情况国内主要产品销售额美国畅销OTC/HBC美国畅销非专利药数据美国处方量TOP数据管理情况基本药物、国家医保、行政保护、非处方药等,药品的所属关系决定了其生产、营销、市场等的侧重点与地位的不同,了解基本面的情况是掌握制高点的基础。
药智网数据查询

药智网数据查询药智网数据查询临床前研究药智网数据查询批文上市药智网数据查询全球药物药智网数据查询药品标准药智网数据查询专利药智网数据查询app药智网数据查询须知无论是药智网数据查询临床前研究、批文上市、全球药物、药品标准、专利还是其它数据查询,都需要登录药智网数据网页版或app进行查询。
没有账号的可以去注册一个会员账号,药智网数据会员分为:普通会员、初级会员、中级会员、高级会员、特级会员。
每个会员等级对应查询权限也相对不一,有些等级会员能查询一个子数据库的部分数据,有些则没有权限惊醒查询,开通药智网数据企业版(最高级)能查询全部数据内容。
药智网数据查询技巧注册登陆:标准全文查询需要先注册登陆(免费)。
药品名称查询:本查询系统支持模糊查询:如果你需要搜索“维生素C片”,只需要在”药品名称”搜索框内输入其中的关键字,如“维生素”或“C片”都可查询。
正文搜索:本系统部分标准提供全文查询,如在“正文搜索”搜索框内输入关键字“高效液相”,就可以查出所有正文中含有“高效液相”的标准。
标准来源选择:查询时可以对标准类型选择,如输入在药品名称“分散片”,同时在“标准来源”的第一个下拉框中选择“中国药典”标准类型,第二个下拉框中选择“中国药典2010”输入“2010”,则可以查询出2010药典所有的分散片标准。
PDF阅读说明:部分标准原件采用是PDF格式,如果你没有安装相关软件,请下载并安装PDF阅读软件。
药智网数据竞品分析判断一个数据库查询数据是否可靠、精准的最好方法是对比其它数据的查询结果,从数据结果的丰富度、精准度、来源追溯等维度去对比分析判断。
选择一个功能和数据全面的药融云医药数据库作为对比源目标。
可以从药品研发、全球上市、药品销售、市场信息、一致性评价、原料药、生产检验、合理用药、医疗器械等版块数据分别对比分析数据。
药融云个人版,目前是开放程度最高的中文界面医药数据库,包含了全球药品研发管线、审评审批进度、全球临床试验、中国临床试验、药品招投标、集采、一致性评价等大量整合信息。
汤森路透Cortellis 药物研发的综合情报平台介绍

wlccCortellis™ for Competitive Intelligence药物研发的综合情报平台是Thomson Reuters Pharma的升级平台,底层数据与Pharma一致。
为您提供药物研发管线、交易、专利、公司内容及行业最新新闻。
具有多来源信息收集、自带强大分析工具、每天更新。
以报告的方式提供信息一般情况下,我们进行信息调研,最终总会将收集到的所有有用的信息整理为一份调查报告。
信息收集的过程是艰巨而繁琐的,而将信息整理为报告的过程也是十分具有挑战性的工作。
而Cortellis™ for Competitive Intelligence便是直接以报告的方式提供信息。
它将平台下数十个数据库提供的丰富信息整理为八种类型的报告:药物报告,公司报告,专利报告,文献报告、新闻报告、会议报告、交易报告和临床试验报告。
其中每一份报告都由工作人员围绕主题将所有相关信息进行了整理和综合,并附有业界专家撰写的综述和评论。
深入的药物报道通过Cortellis™ for Competitive Intelligence,科研工作者可以了解在研药物和已批准药物从开发、临床试验到上市和销售的各个阶段重要的科研、专利、商业和金融信息。
基于这些信息,研发人员可以对药物的研发过程和现状进行全面的了解,也可以对研发工作的最新进展进行紧密的跟踪,还可以对某个治疗领域的所有药物进行比较和分析。
通过药物报告可以获取以下信息:w w.i n ki n fo.o m.nccw w.i n ki n fo.o m.nwlc c其中“Development Profile”和“SWOT Analysis”是由业界专家根据收集的所有信息撰写的总结和竞争分析。
“Literature Review”是由业界专家根据收集的药物相关文献撰写的文献评论。
“Change History”可对药物的信息更新进行跟踪,详细了解药物不同时段的研发进展情况。
汤姆森数据库功能介绍

汤姆森三大数据库功能简介1Thomson Reuters Pharma®是一个整合了汤森路透提供的所有科学、医疗卫生和金融信息数据库的工作流工具。
Thomson Reuters Pharma允许您自由地浏览重要的市场情报,这些经过深加工的信息整合在由我们的行业专家团队所撰写的独一无二的摘要、总结、评论及分析报告中。
通过Thomson Reuters Pharma®,您能够:及时掌握最新的药物、试验、新闻报道及专利信息获取重大的公司新闻,并链接至全文报告和新闻发布稿查看会议报告在各种内容中交互链接汇集众多来源的数据定制检索,过滤内容∙Thomson Reuters Pharma提供∙40,162个药物专论(每月增加300个以上)∙7,600多个详尽的公司报告(共涵盖75,867 个机构)∙7,636个会议报告(每年增加约500个会议报告)∙11,000个医学期刊和100个有机化学期刊∙3,889,193个核心专利报告(覆盖87个专利授权机构)∙3,505,665种化合物(每月增加约5,000种)∙26,578个交易报告∙70,861个临床方案报告(每月增加800个以上新的临床方案)∙28,204个临床结果报告(每月增加200个以上新的临床结果报告)∙24,000个药物靶标为何选择Thomson Reuters Pharma®?∙权威性:Thomson Reuters Pharma®涵盖药物发现和开发流程全过程–最新药物、化合物、基因序列和靶标、临床试验、专利、期刊、会议、学术文章等∙∙相关性:直观的引导式检索可提供您所需要的情报个性化:个性化地定制您的检索条件,满足您不同的实际需求市场意识:有深度的竞争情报,包含涉及超过7,500家制药和生物科技公司的详细研发流程、财务及营销概况以用户为中心:易于使用的个人信息中心,与您的日常工作流需求整合在一起协作:直观的工作流和导出工具帮助您与团队进行数据共享,推动创新。
PharmaProjects 数据库简介及检索方法

PharmaProjects数据库简介及检索方法一、数据库简介Pharmaprojects是世界药物研制开发处于领先地位的智能型数据库。
它监控着国际上处于开发阶段的每一个重要新药,跟踪着国际上处于研发活跃阶段的侯选药物,提供给客户产品开发的全面资料。
Pharmaprojects数据库包括自1980年以来超过30000个处于开发阶段的药物,并且每月都有1000多个药物的更新信息。
Pharmaprojects中的每个药物都包含以下信息:1、主要信息:包括药物名称、开发阶段、各国上市情况。
2、该药物开发公司的情况:包括原始开发公司、国家、开发状况、上市国家。
3、药理依据:包括药效分类及代号、药物用于该适应证的开发状况、药理作用描述、适应证描述、给药途径等。
4、学依据:包括化合物代号、CA注册号、分子量、分子式、化学名、结构式。
5、利情况:包括专利国家、专利号码、专利优先号、优先日期等。
6、各国上市情况:包括上市国家、上市情况、上市时间、批准情况等。
7、主要事件:记录了该药物开发过程中的重大事件。
8、开发进度:记录了药物开发的进度、市场评测。
9、细节信息:详细记录了该药物的市场和临床前以及临床情况。
10、除了药物信息之外,Pharmaprojects还提供了世界上1300余家主要制药和生物技术企业的相关文件,通过组合查询,可以轻松地获得所需的开发信息。
总之,Pharmaprojects是跟踪国际上的新药开发动态、寻找新药报批机会及市场合作开发伙伴和分析市场收益的最佳工具!二、检索方法打开主界面选中【Pharmaprojects】,双击鼠标左键,出现如下界面:点击【accept】进入检索主界面,如下图,左栏是主要的检索途径;中间栏为【Tree Search】(目录树检索);右栏是检索框。
1、目录树检索在目录树检索中包括10种路径:(1)Main Details (4)Chemical Data (7)Ratings(2)Company/Status (5)Patent Data (8)Major Events(3)Activity Data (6)Country Data (9)Alert Service(10)Latest Change点击各路径前(+)可以看见每个路径又分为许多分支路径,如点击“Main Details ”可见其包括:Drug NameDrug Name IncludesActive, Ceased, Fully LaunchedAccession NumberDetailed Information如下图每一个子标题都可以作为一个检索途径。
医药信息数据库

医药信息数据库Dialog中与医药相关的数据库有192个,与化学相关的数据库有65个,与知识产权相关的数据库有25个。
Dialog数据库对于医药研发的全部过程提供完整的信息支持。
药物的生命循环通常,从药物研发到普通药品上中须经过以下几个过程:①研发筛选(R&D Screening),包括市场凋查(Market Survey)与专利调查(Patent Survey);②临床前研究(Preclincal Studies);③临床阶段(Clinical Phases);④新药批准上市(New Drug Approval);整个研究是一个循环往复的过程,缺一环而不可。
在药物研究过程中,更多的是依赖精心加上处理过的专业信息。
我们应该选择针对性强、质量高、覆盖面大、有权威性的检索工具。
另外,信息源的可靠性、获取数据的方便性、检索的效率都是是我们要考虑的首要因素。
DIALOG系统具有600多个数据库,其中和制药相关的数据达200个,这些数据库在为制药企业提供各个环节数据和信息的同时,还利用其功能庞大的指令检索系统为企业提供了优秀的信息和情报的解决方案。
常见的医药数据库介绍如下:ADIS R&D Insight (ADIS 药物研发数据库)ADIS药物研发数据库是Adis International公司的产品。
该数据库的信息来源InPharma、Reactions、PharmacoEconomics & Outcomes News、Clinical Trial Insights等2300种以上的药物、生物专业期刊,国际会议,公司年报和新闻报道等公开资料和非公开资料。
数据库内容包括每种药品的普通名,同义名,商品名,开发公司,国家及开发阶段,所有权信息,峰期销售额,专利失效期,不良事件,药理学,药动力学,药效学,副反应,治疗实验,开发历史,注册信息和参考文献等等。
IMS R&D Focus (IMS 药物研发数据库)IMS药物研发数据库是IMS HEALTH公司的产品。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
单击此处编辑母版副标题样式
针对该Gefitinib的专利从治疗领域进行分析,发现可以治疗 神经系统、呼吸系统、内分泌系统等疾病。选择自己感兴趣 的领域,比如神经系统疾病,用鼠标拖拽呼吸系统疾病的结 点,展开在该大的分类下,涉及到哪些具体的病症。树状结 构图很容易让我们观察这种层级结构。
单击此处编辑母版标题样式 药物检索新增Active和 Inactive的区分,可以 直接区分目前处于这两 种状态的药物。 新增按照专利到期 单击此处编辑母版副标题日样期式进行查询
培训提纲
一、内容介绍 二、主页介绍
单击此处编辑母版标题样式
三、检索 四、单分击析此处编辑母版副标题样式 五、生成报告 六、跟踪与管理
1、快速检索(Quick Search)
可以快速查找某一特定靶标、药物、公司、专利等; Quick Search
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
例1、快速检索药物及其药物报告
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
检索结果--药物报告
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Top10的交易和靶标
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
强大的集成分析功能
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
集成Spotfire Web Player分析工具
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
药物报告
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
战略品种销售数据和预测数据
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Deals和Patents的分析图表及详情
单击此处编辑母版标题样式
单击此处编辑母版副标题样式 点击右下角View list即 可调阅详细的结果列表
查看报告更新历史
单击此处编辑母版标题样式
实验室信息管理系统性
• Watson LIMS • SampleManager LIMS
5
内容介绍-Thomson Reuters Pharma
药物
交易
知识产 权
• 各种期刊文献
• 专利文献
单击此处编辑母版标题样式 临床实
验报告
Thomson
文献和 新闻
• 新闻 • 科技会议论文
Reuters
• 公司站点
比如选择横轴是Originator, 纵 轴是Technology
单击此处编辑母版副标题样式
据此了解不同的公司的技术分布
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
3、Star Tree
药物列表和专利列表页面都增加了Star Tree 的分析功能。这是针对Gefitinib药物检索的 专利的结果;点击Star Tree
2、Rank by
单击此处编辑母版标题样式
打开下拉菜单,可以对药物的研发阶段 分布、作用机制、技术类型、研发机构 分布、靶点等进行二次分析
单击此处编辑母版副标题样式
对药物的最高研发状态进行二次分析
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式 选择矩阵图(Matrix Chart) 中纵 轴、横轴的分析对象。
……
培训提纲
一、内容介绍 二、主页介绍
单击此处编辑母版标题样式
三、检索 四、单分击析此处编辑母版副标题样式 五、生成报告 六、跟踪与管理
Thomson Reuters Pharma的登录页面
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Thomson Reuters Pharma登录后的主页
单击此处编辑母版副标题样式
培训提纲
一、内容介绍 二、主页介绍
单击此处编辑母版标题样式
三、检索 四、单分击析此处编辑母版副标题样式 五、生成报告 六、跟踪与管理
五、生成报告
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
药物报告内容涵盖与该药物相关的:研发全过程;涉及的技术授权交易;药物作用 机制;化学/生物相关试验信息;处方信息;专利清单;销售数据;市场份额;临床 试验;SWOT分析;专利等等
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
例6:浏览公司信息
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Cortellis for Competitive Intelligence
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
CORTELLIS for CI高级检索
例5:专利检索—检索药物的化合物和衍生物的专利
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
例2、快速检索靶点及其对应的药物
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
通配符与逻辑运算符
通配符
符号
含义
举例
%
代替0到任意多个字符
药物名称:%tinib;
单击此处编逻辑辑运算母符版标题样式
获取专利原文
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
向导式检索
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
3、浏览(Browse)
可设定条件进行高级检索与结构式检索。
单击此处编辑母版标题样式
汤森路透集团简介
• 汤森路透集团是全球最大的专业与智能信息提供商; • 其产品包括:Pharma, Integrity, Newport Premium,
IDRA单C, I击nno此vat处ion,编Ge辑neG母o等版; 标题样式
单击此处编辑母版副标题样式
创腾科技是汤森路透集团在中国的战略合作伙 伴,是其数据库产品在中国的销售代理商。
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
培训提纲
一、内容介绍 二、主页介绍
单击此处编辑母版标题样式
三、检索 四、单分击析此处编辑母版副标题样式 五、生成报告 六、跟踪与管理
1、主页设置Alert
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
选择关心的领域
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
创建个性化主页
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
创建个性化主页
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
CORTELLIS for CI主页
https:///
单击此处编辑母版标题样式 财务/股市、合作伙伴、涉及到的交易、临床试验、期刊文献等)
• 3,400,000 个以上专利 • 13,000 个以上药物靶标
• 60,000条单以击上的此药物处临编床实辑验母方案版;2副0,0标00条题以样上的式药物临床实
验结果; • 5,000,000 个以上药物相关基因、蛋白序列 • 20,000 多个交易信息
3
服务于药物研发周期的不同阶段
专利保护期
专利保护过期
单击此处编辑母版标题样式 基础研究 药物发现
临床前研究
I-III期临床
报批 发布上市 仿制药
Thomson Reuters Integrity(早期研发)
GeneGo MetaCo单re/Dr击ug(此系统处生物编学)辑母版副标题样式
IDRAC (政策法规)
• Thomson Reuters Pharma
单击此处编辑母版标题样式 • Newport Premium
计算机模拟软件
• Material Studio • Discovery Studio
单击此信处息管编理、辑分析母与决版策 副标题样式
• Pipline Pilot • Spotfire • E-Notebook
65个心血管疾病领域的天然产物药物
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
排序不同药物的各项内容
按住鼠标左键的同 时左右拖动表格可
以查看更多项目
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
按照2010年实际销售额降序排序
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
图示分析公司、机制、适应症等信息
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
CORTELLIS for CI 主页
某个时间段内各研发状 态的药物汇总
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
培训提纲
一、内容介绍 二、主页介绍
单击此处编辑母版标题样式
三、检索 四、单分击析此处编辑母版副标题样式 五、生成报告 六、跟踪与管理
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式