美国FDA药物审批PPT
fda培训资料ppt课件

行政审批
FDA会对通过技术评审的申请进行行政审 批,审批周期一般需要几个月左右。
技术评审
FDA会对申请资料进行技术评审,评审周 期根据产品类别和风险等级而有所不同, 一般需要几个月到一年不等。
医疗器械不良事件监测与报告
报告义务
医疗器械生产企业有义务向FDA 报告医疗器械不良事件,包括产
品缺陷、故障、质量问题等。
企业应建立专门的法规合规部门,负 责跟踪和研究FDA的法规更新,并及 时向相关部门传达。
提升员工素质
企业应定期对员工进行FDA法规培训 ,提升员工的法规意识和操作技能。
合理规划生产和质量控制
企业应根据FDA的监管要求,合理规 划生产和质量控制环节,确保产品符 合要求。
与FDA保持良好沟通
企业应与FDA保持良好沟通,及时反 馈产品在市场上的表现,以及遇到的 问题和挑战。
FDA建立了一套完善的监测 机制,以收集和整理生物制 品使用过程中出现的不良事 件。这些信息来源于多个渠 道,包括医生报告、患者投 诉、生产商报告等。
根据法规要求,生产商必须 在获知不良事件后的一段时 间内向FDA提交报告。这些 报告必须包含详细的事件描 述、可能的原因分析以及采 取的措施等内容。同时, FDA也会对不良事件进行调 查和分析,以评估事件的真 实性和因果关系。
食品不良反应监测与报告
不良反应监测
FDA对进口和在美销售的食品进行不良反应监测,包括食品中毒、过敏反应、化学物质超标等。
报告义务
FDA要求企业、消费者和医疗专业人士报告疑似食品不良反应事件,以便及时展开调查和采取措施。
06
FDA对生物制品的监管
生物制品注册与审批流程
申请与受理
申请人需提交完整的申请材料,包括生物制品的名称、活性成分、剂型、适应症等信息, 并缴纳相应的费用。FDA对申请材料进行审核,并对申请进行分类,将符合要求的申请转 至评审部门。
美国FDA批准上市的药物(2019年)
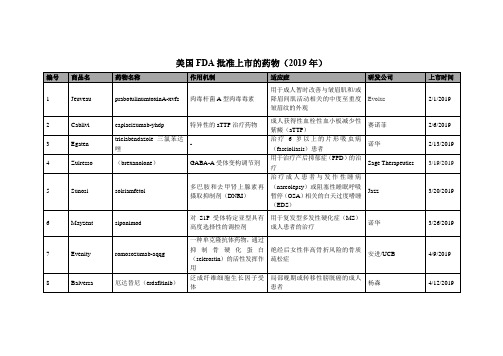
用于眼科手术
Dutch Ophthalmic, USA
12/20/2019
45
Caplyta
lumateperone tosylate
多巴胺受体磷酸蛋白调节剂(DPPM),在D2受体上充当突触前部分激动剂和突触后拮抗剂。
精神分裂症
Kah-PLY’-tah
Intra-Cellular Therapies, Inc.
-
用于治疗成人由于人工型或遗传性转甲状腺素卵白介导的淀粉样变性引起的心肌病(ATTR-CM)
辉瑞
5/3/2019
11
Piqray
Alpelisib
PIK3抑制剂
治疗绝经后女性及男性特定晚期或转移性乳腺癌患者
诺华
5/24/2019
12
Polivy
polatuzumab vedotin-piiq
CD79b
全球结核病药物开发联盟(TB Alliance)
8/14/2019
20
Wakix
pitolisant
用于治疗成年发作性睡病(narcolepsy)患者的白日过度嗜睡(EDS)
Bioprojet
8/14/2019
21
Rozlytrek
entrectini恩曲替尼
选择性酪氨酸激酶抑制剂
用于ROS1阳性转移非小细胞肺癌和所有NTRK融合实体瘤(12岁以上)
12/20/2019
46
Dayvigo
lemborexant
食欲素受体拮抗剂
失眠症
卫材公司
12/20/2019
47
Enhertu
fam-trastuzumab deruxtecan-nxki
HER2靶点的抗体偶联药物
FDA新药审批流程简述

FDA新药审批流程简述FDA(美国食品药品监督管理局)负责监管并审批新药的上市。
下面将对FDA新药审批流程进行简述。
1.阶段Ⅰ临床试验:新药首先在健康志愿者身上进行,评估药物的安全性和耐受性,并确定药物的剂量范围。
2.阶段Ⅱ临床试验:新药在患者身上进行,评估药物的疗效和副作用。
试验时间较长,研究人员需要收集更多的数据,以确定新药的安全性和效力。
3.阶段Ⅲ临床试验:新药在大规模患者群体身上进行,以证明其疗效和安全性是否持久有效,并与现有的治疗方法进行比较。
4.新药申请:药企将试验结果提交给FDA,并申请新药上市批准。
包括药物的数据和试验结果,使用方法等。
FDA会评估申请材料。
5.NDA审批:NDA(新药申请)包括对药物研究的细节、试验结果等的描述。
FDA对NDA进行评估,确定药物是否符合上市标准。
此过程可能需要数月或数年。
6.审查:FDA将药物进行详细审查,并与药企进行沟通,以充分了解药物的性质和潜在的风险。
7.审查会议:FDA可能会召开药物审查会议,邀请专家、学者和公众就药物的疗效和风险发表意见。
8.确认上市:如果FDA认为新药符合上市标准,将发出批准通知,允许药企将新药投入市场销售。
以上是FDA新药审批流程的简要概述。
在流程中,FDA扮演着保障公众健康和安全的角色,确保新上市的药物是安全有效的。
FDA New Drug Approval Process OverviewThe FDA (U.S. Food and Drug Administration) is responsible for regulating and approving new drugs for market. The following is an overview of the FDA's new drug approval process.1. Phase I Clinical Trials: The new drug is first tested on healthy volunteers to assess its safety and tolerability and determine the dosage range.5. NDA Approval: The NDA (New Drug Application) includes a detailed description of the drug's research, trial results, etc. The FDA evaluates the NDA to determine if the drug meets the approval standards. This process can take months or years.7. Review Meetings: The FDA may hold drug review meetings where experts, scholars, and the public are invited to provide opinions on the drug's efficacy and risks.9. Post-Market Surveillance: The FDA continues to monitor the drug's safety and effectiveness, collects feedback from the market, and takes necessary actions, including further research and updating warning labels.The above is a brief overview of the FDA's new drug approval process. Throughout the process, the FDA plays a crucial role insafeguarding public health and safety, ensuring that newly marketed drugs are safe and effective.。
FDA新药审批流程简述

FDA新药审批流程简述FDA(美国食品药品监督管理局)是负责监督和管理美国食品和药品安全的联邦机构。
它负责确保市场上的药品是安全有效的,并且符合严格的审批标准。
新药审批是FDA的主要职责之一,它是一个复杂且漫长的过程,通常包括以下几个步骤:1.提交新药申请(NDA)首先,药物制造商需要通过NDA向FDA提交有关新药的完整资料。
这些资料通常包括药物的化学成分、制造工艺、药理学研究、临床试验数据以及用于治疗的适应症等信息。
此外,还需要提供药物的质量、安全和有效性的证据。
2.评估申请文件一旦FDA收到NDA,会对申请文件进行评估。
这个过程通常包括对文件的完整性和合规性的审查,例如核实所有必需的资料是否齐全。
如果缺少必要的信息,FDA可能会要求制造商提供补充材料。
3.优先审批对于一些药物,FDA可能会给予优先审批待遇。
例如,对于治疗一些严重疾病的新药,FDA可能会加快审批速度,以符合患者的迫切需求。
4.临床试验阶段一旦FDA确认NDA文件完整无误,药物制造商可以开展临床试验。
临床试验是评估药物疗效和安全性的关键步骤,通常分为三个阶段(I、II、III)。
这些试验需要遵守严格的方案和伦理规定,以确保患者的安全和药物的有效性。
5.申请审核委员会的审查在临床试验结束后,药物制造商将向FDA提交试验结果,并要求FDA审核审查委员会对其进行审查。
审查委员会是由FDA专家组成的独立机构,他们会仔细评估试验结果以及相关数据和文献,并发表意见。
这些意见对于FDA的最终决策具有重要影响。
6.申请批准在经过临床试验和审查后,FDA将根据收集到的数据和顾问委员会的意见,决定是否批准新药上市。
如果FDA认为药物的风险和益处之间的平衡是积极的,它将批准新药,并颁发批准证书。
7.监督上市后安全性一旦新药获得批准,FDA对其进行监督,以确保其安全性和真实性。
制造商需要持续向FDA提供有关药物的安全性和有效性的信息。
此外,FDA还通过药物安全盛会进行监测,并与制造商和医疗专业人员合作,收集和分析有关药物的副作用和其他安全问题的信息。
中英文-美国FDA 药品监管

Conduct Inspections, monitor relevant events 执行检查,监控相关事件
16
Approval of a Drug Application Cont. 药品申请的批准 续
GMP inspection GMP检查 Domestic firms: district inspectors 国内企业:区域检查官 Foreign firms: international inspector cadres 国外企业:国际检查官骨干
Increase collaboration and coordination of product safety with relevant US Govt agencies 与相关美国政府机关一起加强产品安全性方面的协调与合作
5
Jurisdiction 管辖权力
What products do we regulate? 哪些产品属于我们的监管范围?
Chapter I: Short Title 21 USC 301 简称
Chapter II: Definitions 21 USC 321 定义
Chapter III: Prohibited Acts and Penalties 21 USC 331-337 禁止的行为和处罚
Chapter IV: Food 21 USC 341-350 食品
外来动物(进口啮齿类动物、龟等) Combination products 组合产品
美国FDA批准上市的药物
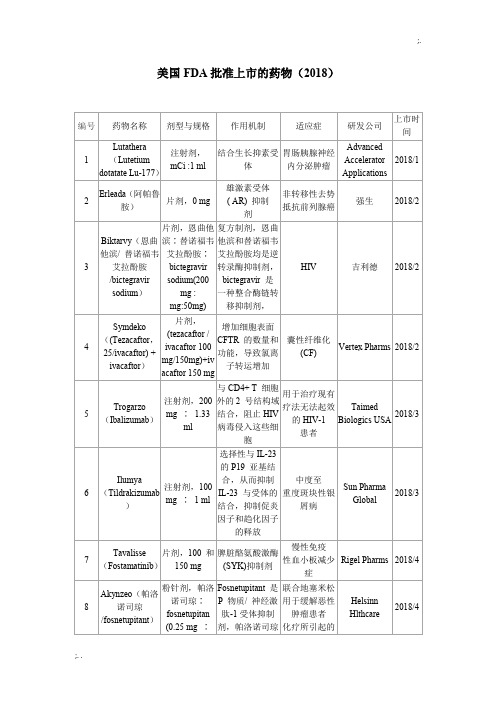
Siga Technologies
2018/7
23
Krintafel(Tafenoquine)
片剂,150 mg
-
疟疾
葛兰素史克
2018/7
24
Orilissa(Elagolix sodium)
片剂,150和200 mg
促性腺激素释放激素(GnRH)受体拮抗剂
治疗子宫内膜异位引起的中度至重度疼痛
基因泰克
2018/10
47
Tegsedi(Inotersen)
皮下注射,284 mg∶1.5 ml
是一种反义寡核苷酸,通过与TTRmRNA结合导致突变体和野生型TTR mRNA的降
解,从而使组织血清TTR蛋白和TTR蛋白沉积物减少
成人遗传性转甲状腺素介导
的淀粉样变性多发性神经病
Akcea Theraps公
口服悬浮液,每包5 g和每包10 g
本品是一种不被吸收的锆硅酸盐,可以捕获钾离子并进行氢、钠的交换,通过结合胃肠道中游离的钾离子,促进钾离子的排放
成人高钾血症
阿斯利康
2018/5
12
Doptelet(Avatrombopag maleate)
片剂,20 mg
血小板生成素(TPO)受体激动剂
计划接受手术的慢性肝病患者的血小板减少症
艾伯维
2018/7
25
Omegaven(Fish oil triglycerides)
注射剂(乳液),5 g∶50 ml和
10 g∶100 ml
脂肪酸补充剂
治疗患有肠外营养相关胆汁淤积(PNAC)的儿童患者
Fresenius Kabi USA
2018/7
26
Mulpleta(鲁索曲波帕)
美国FDA药物分析程序及方法验证指导原则(中文版)

药品及生物制品的分析方法和方法验证指导原则目录1.介绍...................... (1)2.背景..................... .. (2)3.分析方法开发. ..................... . (3)4.分析程序内容.............................................. ......... ..................................... .. 3A.原则/范围 (4)B.仪器/设备............................................. . (4)C.操作参数.............................................. .. (4)D.试剂/标准............................................. . (4)E.样品制备.............................................. .. (4)F.标准对照品溶液的制备............................................ .. (5)G.步骤......... ....................................... (5)H.系统适应性..... (5)I.计算 (5)J.数据报告 (5)5.参考标准和教材............................................ (6)6分析方法验证用于新药,仿制药,生物制品和DMF (6)A.非药典分析方法............................................. (6)B.验证特征 (7)C.药典分析方法............................................. .. (8)7.统计分析和模型 (8)A.统计 (8)B.模型 (8)8.生命周期管理分析程序 (9)A.重新验证 (9)B.分析方法的可比性研究............................................ . (10)1.另一种分析方法............................................... .. (10)2.分析方法转移的研究 (11)C.报告上市后变更已批准的新药,仿制药,或生物制品 (11)9.美国FDA方法验证............................................... . (12)10.参考文献前言本指导原则草案,定稿后,将代表美国食品和药物管理局(FDA)目前关于这个话题目前的想法。
DMF文件简介.ppt

2
背景
根据美国的“食品、药品和化妆品法”,药品(制剂)在上市之前,必需要向FDA 提出新药注册申请,提供拟上市新药及其成分在“安全性、有效性和质量”三方 面的全部信息,FDA做出全面评价认为满足要求后,才能给与批准上市。
3
分类
在FDA发布的《DMF指南》中,DMF分为以下五个类型: (官方网址:/Drugs/GuidanceComplianceRegulator yInformation/Guidances/ucm122886.htm) I型,生产地点、厂房设施、操作步骤和人员;(2000年7月12日已取消) II型,原料药、原料药中间体、生产前述物质使用的原材料,或药品; III型,包装物料; IV型,赋形剂、着色剂、香料、香精,或生产这些物质的原材料; V型,FDA接受的其它信息
(原料药生产企业向FБайду номын сангаасA申报的DMF文件属于II型。)
4
分类
其它:
● 欧 盟: 仅有一种DMF,即 EDMF
● 加拿大: 有四种类型的DMF 第一类:药品物质 第二类:包装材料 第三类:赋形剂 第四类:剂型
5
各类DMF的差别
在美国,整个DMF文件均被视为机密保存. 该产品的部分DMF摘要资料 也许会提供给客户,此部分的DMF资料称为“技术资料档”. 在欧盟的EDMF及加拿大的DMF分为公开及保密管制两个部分.
6
DMF注册流程
7
注释:
CDER:Center for drug evaluation and research 美国FDA药物评价及研究中心
GCP培训PPT课件

某基因治疗临床试验项目。该案例重点介绍了GCP在基因治疗临床试验中的实践,涉及试验设计、供体筛选、 细胞制备、给药方式、安全性监测等方面的规范操作,以确保受试者安全和试验质量。
GCP在药物研发中的应用案例
GCP在药物研发中应用案例一
某新药临床试验项目。该案例详细介绍了GCP在药物研发过程中临床试验阶段的 实施要点,包括试验设计、伦理审查、受试者招募、药物发放、数据采集与分析 等环节的规范要求,以确保药物研发的科学性和安全性。
结果总结
对收集到的数据进行深入分析,挖掘数据背 后的意义和规律,为研究提供有力支持。
报告审核
对撰写的报告进行审核和修改,确保报告的 质量和准确性。
04
GCP案例分析
GCP在生物医学研究中的应用案例
GCP在生物医学研究中应用案例一
某疫苗临床试验项目。该案例介绍了GCP在疫苗临床试验中的实施过程,包括试验设计、伦理审查、受试者招 募、数据采集与分析等方面的规范要求,确保试验结果的科学性和可靠性。
临床研究
除了药品和医疗器械,GCP还应用于生物制 品、体外诊断试剂等的临床研究。
02
GCP培训内容
GCP基本原则与要求
GCP基本原则
科学、公正、透明、尊 重受试者权利和尊严。
GCP要求
确保临床试验数据的可 靠性、完整性和安全性 ,保障受试者权益和安
全。
GCP实施主体
药物研发机构、临床试 验机构、申办方等。
感谢您的观看
THANKS
数据整理
对收集到的数据进行整理、分类和筛选,为 后续的数据分析和报告撰写提供便利。
数据备份
定期备份数据,防止数据丢失或损坏。
GCP实验报告撰写
FDA新药审批流程简述(中英文)

FDA新药审批流程美国的新药审批可以说是世界上最严格和规范的,作为一个公司通常需要花费5亿美元资金,用 12到15年的时间才能将一个新药从试验室走入市场。
在5000个临床前化合物中大约只有5个化合物可以进入临床试验(Clinical Trials),而这5个化合物中只有一个才能被批准用于临床治疗病人,成为真正的药物。
从一个实验室发现的新化合物发展成为一个治疗疾病的药物,需要经过如下开发阶段:一、临床前试验将一个新发现的化合物经过实验室和动物试验,证明该化合物针对特定目标疾病具有生物活性,并且要评估该化合物的安全性。
二、新药临床研究申请当一个化合物通过了临床前试验后,需要向FDA提交新药临床研究申请,以便可以将该化合物应用于人体试验。
如果在提交申请后30天内FDA没有驳回申请,那么该新药临床研究申请即被视为有效,可以进行人体试验。
新药临床研究申请需要提供先前试验的材料;以及计划将在什么地方,由谁以及如何进行临床试验的说明;新化合物的结构;投药方式;动物试验中发现的所有毒性情况;该化合物的制造生产情况。
所有临床方案必须经过机构审评委员会(Institutional Revuew Board,IRB)的审查和通过。
每年必须向FDA 和IRB 汇报一次临床试验的进程和结果。
三、一期临床试验这一阶段的临床试验一般需要征集20-100名正常和健康的志愿者进行试验研究。
试验的主要目的是提供该药物的安全性资料,包括该药物的安全剂量范围。
同时也要通过这一阶段的临床试验获得其吸收、分布、代谢和排泄以及药效持续时间的数据和资料。
四、二期临床试验这一期的临床试验通常需要征集100-500名相关病人进行试验。
其主要目的是获得药物治疗有效性资料。
五、三期临床试验这一期的临床试验通常需1000-5000名临床和住院病人,多在多个医学中心进行,在医生的严格监控下,进一步获得该药物的有效性资料和鉴定副作用,以及与其他药物的相互作用关系。
药事管理组织机构PPT课件

五、SFDA药品审评中心
CDE(Center for Drug Evaluation)
六、SFDA药品认证管理中心
CCD(certification Committee for Drugs)
GLP药物非临床研究质量管量规范 GCP药品临床实验管理规范 GMP药品生产质量管理规范 GAP药材生产管理规范 GSP药品经营质量管理规范 GUP药品使用管理规范
二、我国医药行业管理机构的职责
1、我国医药行业管理 机构的建立 SFDA 国家中医药管理局 国家经贸委医药司 2、我国医药行业管理 机构的职责(P15)
第三节
药学教育、科研组织和社会团体
随着改革的深入发展,我国药学教育和药物 科研的机构和体制,发生了较大变化。药物科研 机构处于从事业性组织向企业化过渡阶段。政府 机构改革以来,部分原有职能委托药学社团机构 办理,药学社团的行业管理职能有所加强。
一、药学教育组织
有药学类专业的高校大多为综合性大学,仅 有独立药科大学2所,另有中医药大学和中医学院 23所。96所设置有药学类专业的高校。 药学继续教育主要由设有药学类专业的高校、 中等学校和药学会承担。
二、药学科研组织
我国的药学科研组织有独立的药物研究院、所 以及附设在高等药学院校、大型制药企业、大型医 院中的药物研究所、室两种类型。
县级FDA(派出机构)
三、药品检验机构
国家级、省级、市级、县级 中国药品生物制品检定所 省、自治区、直辖市药品检验所
四、国家药典委员会
(The Pharmacopoeia commission of the
People’s Requblic of China) 性质: 组成: 全体委员会(主任委员、副主任委员等) 专业委员会 常设机构
美国 FDA批准靶向药物

美国食品和药物管理局(FDA)批准上市的靶向抗肿瘤药物---表1 FDA批准的单抗药物名称靶点获批适应症/获批时间Rituximab(Mabthera)利妥昔单抗美罗华CD20非霍奇金淋巴瘤/1997年Trastuzumab(Herceptin)曲妥珠单抗(赫赛汀)HER2(ERBB2/neu)乳腺癌/1998年胃癌/2010年Bevacizumab(Avastin)贝伐珠单抗(安维汀)VEGF结直肠癌/2004年非小细胞肺癌/2006年肾癌/2009年脑癌/2009年Cetuximab(Erbitux)西妥昔单抗(爱必妥)EGFR(HER1/ERBB1)头颈部鳞状细胞癌/2006年KRAS野生型结直肠癌/2009年Panitumumab(Vectibix)帕尼单抗(维克替比)EGFR(HER1/ERBB1)KRAS野生型结直肠癌/2006年Ipilimumab(Yervoy)CTLA-4黑素瘤/2011年Obinutuzumab(Gazyva)CD20慢性淋巴细胞白血病/2013年Ado-trastuzumabemtansine(Kadcyla)/T-DM1HER2 (ERBB2/neu)HER2阳性的晚期(转移性)乳腺癌/2013年Ramucirumab(Cyramza)VEGF晚期胃癌或胃食管连接部腺癌收起表2 FDA批准的小分子靶向抗肿瘤药物名称靶点获批适应症/获批时间Imatinib(Gleevec)伊马替尼(格列卫)KIT,PDGFR,ABL多种恶性血液病/2001年胃肠道间质肿瘤/2002年Gefitinib(Iressa)吉非替尼(易瑞沙)EGFR非小细胞肺癌/2003年Erlotinib (Tarceva)厄洛替尼(特罗凯)EGFR (HER1/ERBB1)非小细胞肺癌/2004年胰腺癌/2005年Crizotinib克唑替尼(赛可瑞)ALK, METALK阳性的非小细胞肺癌/2011年Bosutinib(Bosulif)博舒替尼ABL慢性髓细胞白血病/2012年Cabozantinib (Cometriq)卡博替尼FLT3, KIT, MET, RET, VEGFR2 甲状腺髓样癌/2012年Axitinib(Inlyta)阿昔替尼KIT, PDGFRβ, VEGFR1/2/3肾癌/2012年Dasatinib(Sprycel)达沙替尼(施达赛)ABL慢性髓细胞性白血病/2006年急性淋巴细胞白血病/2006年Sorafenib(Nexavar)索拉非尼(多吉美)VEGFR, PDGFR, KIT, RAF肾癌/ 2005年肝癌/2007年甲状腺癌/2013年Sunitinib(Sutent)舒尼替尼(索坦)VEGFR, PDGFR, KIT, RET胃肠道间质肿瘤/2006年肾癌/2006年胰腺神经内分泌肿瘤/2011年Lapatinib(Tykerb)(泰立沙)HER2 (ERBB2/neu), EGFR (HER1/ERBB1)HER2阳性乳腺癌/2007年Nilotinib(Tasigna)尼洛替尼(达希纳)ABL慢性髓细胞性白血病/2007年Temsirolimus (Torisel)替西罗莫司mTOR肾癌/2007年Everolimus (Afinitor)依维莫司(飞尼妥)mTOR肾癌/2009年肾移植后预防器官排斥/2010年室管膜下巨细胞星形细胞瘤与结节性硬化症/2010年胰腺神经内分泌肿瘤/2011年与依西美坦联用治疗乳腺癌/2012年肝脏移植手术后预防器官排斥/2013年Pazopanib(Votrient)帕唑帕尼VEGFR, PDGFR, KIT肾癌/2009年Ponatinib(Iclusig)ABL, FGFR1-3, FLT3, VEGFR2慢性髓细胞性白血病/2012年急性淋巴细胞白血病/2012年Regorafenib (Stivarga)瑞戈非尼KIT, PDGFRβ, RAF, RET, VEGFR1/2/3结直肠癌/2012年胃肠道间质瘤/2013年Ruxolitinib(Jakafi)JAK1/2骨髓纤维化/2011年Tofacitinib托法替尼JAK3风湿性关节炎/2012年Vandetanib (Caprelsa)凡德他尼EGFR (HER1/ERBB1), RET, VEGFR2 甲状腺髓样癌/2011年Vemurafenib (Zelboraf)维罗非尼BRAFBRAF V600突变的黑素瘤/2011年Dabrafenib(Tafinlar)达拉非尼BRAFBRAF V600突变的黑素瘤/2013年Trametinib(Mekinist)曲美替尼MEK1,MEK2BRAF V600突变的黑素瘤/2013年Afatinib(Gilotrif)阿法替尼EGFR,HER2非小细胞肺癌/2013年Ibrutinib(Imbruvica)依鲁替尼BTK套细胞淋巴瘤/2013年慢性淋巴细胞白血病/2014年。
中美药品注册审批制度比较研究

中美药品注册审批制度比较研究随着全球医药产业的快速发展,药品注册审批制度在各国药品监管体系中的地位日益重要。
中美两国作为全球最大的药品市场之一,其药品注册审批制度对药品研发、生产和上市具有重大影响。
本文旨在比较中美药品注册审批制度的异同,探讨其影响,为完善我国药品注册审批制度提供借鉴和参考。
国内外学者针对中美药品注册审批制度进行了一系列研究。
这些研究主要集中在制度框架、审批流程、监管机构等方面。
然而,大多数研究仅某一方面的比较,缺乏对整体框架和最新改革的系统分析。
中美药品注册审批制度在申请要求方面存在一定差异。
美国食品药品监督管理局(FDA)对药品申请人的资格没有特殊要求,但申请人需提交完备的技术资料、临床试验报告等相关文件。
而中国药品审评中心(CDE)则对申请人资格有严格的规定,同时要求申请人提交相关证明文件和临床试验报告等资料。
美国FDA的药品审批流程包括申请、审评、审批、上市后监测等多个环节。
在审评环节,FDA采用科学、公开、透明的审评标准,对药品的安全性、有效性和质量进行全面评估。
而中国CDE的药品审批流程相对简单,主要包括申请、审评、审批三个环节,但在审批环节中缺乏明确的审评标准和公开透明的机制。
美国药品注册审批制度由FDA负责,其独立于政府其他部门,具有较高的权威性和专业性。
中国药品注册审批制度则由CDE负责,隶属于国家药品监督管理局,独立性相对较低。
中美药品注册审批制度的差异对两国药品产业的发展产生了一定影响。
美国药品注册审批制度在促进新药创新、保障药品质量和安全方面具有显著优势,而中国药品注册审批制度在简化程序、提高审评效率方面具有一定优势。
然而,两国制度也存在一些问题,例如审批流程繁琐、监管机构独立性不足等。
为完善我国药品注册审批制度,可采取以下对策:完善相关法律法规,加大对药品产业的扶持力度,鼓励药品创新。
本文比较了中美药品注册审批制度的异同,发现两国在申请要求、审批流程和监管机构方面存在一定差异。
新药注册的申请与审批课件

二、新药监测期的管理
(一)新药监测的目的 根据保护公众健康的需要,国家药品监督管理部门可以对
批准生产的新药品种设立监测期,对该新药的安全性继续进 行监测。
(二)监测期时间设置 监测期自新药批准生产之日起计算,最长不得超过5年。对
原料新药不设监测期。不同种类新药具体时间设置见下表。
监测 中药、天然药物
五、新药的申报与审批
(三)新药审批有关规定
3.联合研制的新药申报 多个单位联合研制的新药,可以由其中的一个单
位申请注册,其他的单位不得重复申请。需要联合 申请注册的,应当共同署名作为该新药的申请人。 新药申请批准后每个品种只能由一个单位生产,同 一品种的不同规格不得分由不同单位生产。
五、新药的申报与审批
生物制品。
1.实行特殊 审批
治疗艾滋病、恶性肿瘤、 罕见病等疾病且具有明 显临床治疗优势的新药。
治疗尚无有效治疗手 段的疾病的新药。
(三)新药审批有关规定
2.对报送材料的要求 申请新药注册所报送的资料应当完整、规范
,数据必须真实、可靠;引用文献资料应当注 明著作名称、刊物名称及卷、期、页等;未公 开发表的文献资料应当提供资料所有者许可使 用的证明文件。外文资料应当按照要求提供中 文译本。
(11)药品标准及起草说明,并提供 粘膜、肌肉等)刺激性等特殊安全性
标准品或者对照品。
试验资料和文献资料。
(12)样品的检验报告书。
(22)复方制剂中多种成份药效、毒性、
(13)原料药、辅料的来源及质量标 药代动力学相互影响的试验资料及文
准、检验报告书。
献资料。
(14)药物稳定性研究的试验资料及 (23)致突变试验资料及文献资料。
【知识链接】美国的临床研究申请(IND)
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
•
病例报告
– 包含第二期、第三期临床试验的每一个病人的病例报告 – 每一个已死亡和因不良反应退出临床试验对象的病例报告
Squire, Sanders & Dempsey L.L.P.
新药申请
•
其他
– Right of Reference or Use:引述或使用已经提交FDA的信息的 权利,如果该信息不是新药申请人所提交的,必须获得书面授 权
Squire, Sanders & Dempsey L.L.P.
FDA药物申请的基本组织结构
• IRB
– 功能:确保临床试验的安全性与临床试验对象的权 益 • 利益必须基于临床试验对象本身的直接利益
– 不能基于对社会的利益
– 不能是远期利益 – 组成:至少5人,必须有一位非科学背景的成员(一 般是律师或商业人员、必须有一位具科学训练的成 员) – 职责:审批并持续审查每一项临床试验(功能与职责 相当于FDA,但立场必须独立与专业) – 利益冲突 • 临床试验人员不能成为IRB成员
Squire, Sanders & Dempsey L.L.P.
修改临床试验蓝本
临床试验申请
•
–
FDA终止令
试验对象受伤害的风险太高,IND误导性或错误, 或者不完备,试验方法、设备及药物制造设备等 不能实验对象的安全,试验偏离IND,没有及时报 告负面试验经历/结果,药物被商业性买卖,IND 在非活动状态达五年以上,无令人信服的证据证 明药物安全…… 申请方必须在30天内就上述缺失提出说明、解释 如果FDA不接受该说明,申请方可以在10天内提 出听证要求
– 如果是抗感染类药物,总述相关微生物信息
– 临床试验数据及分析 – 儿童使用试验
Squire, Sander药申请
•
样品及标签
– 四份下列各样品:药物,药品,试验空白样品 – 药品包装 – 存档用副本必须包括
• 三份在药物的制造化学、制造方法、及分析控制手段部分 的分析方法和分析
Squire, Sanders & Dempsey L.L.P.
新药申请
• 新药申请(New Drug Application) (NDA)
– 总述(Summary):NDA各种数据(有效性、安全性、药理、 毒理、代谢数据等等)及信息
• 标签,包括用药指南 • 药理类别,包括药理、用途和药物的医疗作用
•
专利信息
– 所有包涵药物、药物形态、药品组成、以及药物使用的专利信 息 – 专利报告要求(FDA Forms 3542和3542a): • 新药申请号、申请方、商品名、活性成分、药性强度、剂 型、美国专利号、批准日和过期日、专利拥有人的联系信 息等等 • 在新药申请批准后30天内必须提出专利报告 • 新的相关专利必须在批准后30天之内报告FDA
Squire, Sanders & Dempsey L.L.P.
新药申请
• 专利证言(Patent Certification)
– 药物专利、药品专利、药物使用专利 • 专利信息未提交FDA (Paragraph I Certification) • 专利已过期 (Paragraph II Certification)
临床试验申请 – 临床试验蓝本(Protocols) -- 每项临床试验必须有一个蓝本, 包含 • • 该临床试验的目的和目标 每一个试验人员及其资质(含简历),试验场所及设施, IRB每一个成员的姓名地址
•
• • •
试验病人的选择标准及大略人数
临床试验研究设计,包括控制组设计以及对源于试验对 象、试验人员、以及试验分析人员的偏差的控制 决定剂量的方法、最大剂量以及试验对象的用药期 描述临床试验程序、实验测试、以及其他该药药效及控 制试验危险的方式
Squire, Sanders & Dempsey L.L.P.
FDA药物申请的基本组织结构
(1) FDA (2) 内部审查委员会(Institutional Review Board) (IRB) (3) 药物申请方(Sponsor) (4) 药物临床试验研究人员(Investigator) (5) 药物临床试验对象(subject) (1) 需要保护的对象
Squire, Sanders & Dempsey L.L.P.
新药申请
• 技术部分 – 该药物的制造化学、制造方法、及分析控制手段 • 药物的化学、物理、生物特性,制造商及制造过程,分析质控 及生效性控制方法, • 药物的化学、物理、生物特性,制造商及制造过程,分析质控 及生效性控制方法 – 包含用来试验生效性和生体等同性的batch record及总制 造记录 • 环境影响分析 – 非临床试验药理及毒理试验结果 – 该药物的人体药理动力学和生效性(bioavailability)
Squire, Sanders & Dempsey L.L.P.
FDA药物申请的基本种类
• • •
– – – –
研究性药物申请 (Investigational Drug Application) 新药申请(New Drug Application) 生物药物许可申请
DNA类药物(virus plasmid, etc) 血清制品 毒素(toxin)和毒素抗体(antibody) 饱含或少于40个氨基酸的合成多肽类药物
美国FDA药物审批
主讲人:李兆阳(Zhaoyang Li) 化学博士、律师 美国翰宇国际律师事务所 Squire, Sanders & Dempsey LLP zli@ 001-415-393-9885
FDA药物审批的基本法律
• •
食品、药物及美容物品法(Food, Drug, and Cosmetic Act)第5章第505分章 联邦政府法规(Code of Federal Register)第21章第 312、第314和第514分章
Squire, Sanders & Dempsey L.L.P.
临床试验
• 控制试验的方法
– 安慰剂对照试验(placebo concurrent control) – 剂量对照试验(dose-comparison concurrent control) – 非治疗对照试验(no-treatment concurrent control) – 积极治疗对照实验(active-treatment concurrent control) – 病史对照试验(historical control)
Squire, Sanders & Dempsey L.L.P.
新药申请
• 市场独占要求
– 对新化合物(NCE, new chemical entity)新药,自新药申请 批准后3年内,FDA不接受就该药物提出的generic新药申 请(505(b)(2)申请或者简捷新药申请(abbreviated new drug application -- ANDA),如果一项ANDA新药申请含
• 国外上市的历史
• 该药物的制造化学、制造方法、及分析控制手段(称 CMC) • 该药物的非临床试验药理及毒理试验
• 该药物的人体药理动力学和生效性(bioavailability)
• 如果是抗感染类药物,总述相关微生物信息 • 临床试验数据及分析 • 结论,包含benefit/risk,及提出附加试验和上市后监 测手段
• 专利将在某日过期 (Paragraph III Certification)
• 该专利无效、不可执行、或者不会被本新药申请的新药 制造、销售和使用行为侵权(Paragraph IV Certification) – 无相关专利 – 未按时提交的专利信息对后叙generic新药申请的影响 • 如果专利信息在专利颁布后30天后提交FDA,之前提交 的generic新药申请可以不修正它自己的专利信息,之 后提交的generic新药申请必须按上述规定提交有关该 专利的专利信息
– –
•
–
IND非活动状态
试验活动暂停一年或一年以上
–
两年没有试验对象进行临床试验
Squire, Sanders & Dempsey L.L.P.
临床试验申请
•
–
FDA/申请方的第二期临床试验结束会议
讨论第三期临床实验
•
– – – –
药物上市前期会议(Pre-NDA Meeting)
讨论临床试验未解决的问题 确定有关药物有效性的研究 确定还需要进行的研究以确定该药物对孩童的有效性 讨论药物上市申请
•
–
关键是临床试验(Clinical Trials)
试验对象的风险大
Squire, Sanders & Dempsey L.L.P.
临床试验
• 第一期临床试验(Phase 1 clinical trial)
– 确定人体代谢机制和药学行为(20-80个对象),了解 增加药剂量的副作用,以及(如果可能)有效性 • 为后面的临床试验作准备
Squire, Sanders & Dempsey L.L.P.
FDA药物申请的基本组织结构
• 临床试验对象
– 临床试验对象必须同意(Informed consent) • 解释药效 • 解释风险 – 紧急情况下可以免除同意 • 有立即死亡风险 • 同意无法获得 – 比如病人昏迷等
Squire, Sanders & Dempsey L.L.P.
• 有关安慰剂组成、制造以及控制的简单描述
• 标签 • 环境影响评估或者声明环境影响甚微
Squire, Sanders & Dempsey L.L.P.
临床试验申请
– 药理和毒理信息 – 已有的经历 – 其他信息 • 药物依赖性及可能的滥用 • 孩童使用的有效性与安全性 • 其他