单拷贝直系同源基因构建进化树
单拷贝直系同源基因

单拷贝直系同源基因引言单拷贝直系同源基因是指在一个生物体的基因组中只有一个拷贝的同源基因。
同源基因是指具有相似DNA序列和功能的基因。
单拷贝直系同源基因在生物体的进化和功能发育中扮演着重要的角色。
本文将介绍单拷贝直系同源基因的定义、特点、重要性以及相关研究。
定义单拷贝直系同源基因是指在一个生物体的基因组中只存在一个拷贝的同源基因。
同源基因是指具有相似DNA序列和功能的基因。
单拷贝直系同源基因与多拷贝同源基因相对应,多拷贝同源基因是指在一个生物体的基因组中存在多个拷贝的同源基因。
特点单拷贝直系同源基因具有以下特点:1.稳定性:由于只有一个拷贝,单拷贝直系同源基因在基因组中的存在相对稳定,不容易发生拷贝数变异。
2.功能保守性:单拷贝直系同源基因在物种进化过程中通常具有保守的功能,其在不同物种中的序列和功能相似。
3.重要性:单拷贝直系同源基因在生物体的功能发育中扮演着重要的角色,对于生物体的正常发育和生理功能具有重要影响。
重要性单拷贝直系同源基因在生物体的进化和功能发育中具有重要的作用:1.进化:单拷贝直系同源基因的保守性使得其在物种进化过程中起到重要的指示作用。
通过研究单拷贝直系同源基因的序列和功能变化,可以推断不同物种之间的进化关系和进化速率。
2.功能发育:单拷贝直系同源基因在生物体的功能发育中起到关键的调控作用。
它们参与调控生物体的正常发育过程,如胚胎发育、器官形成和细胞分化等。
3.疾病研究:单拷贝直系同源基因的突变和变异与一些遗传性疾病的发生和发展密切相关。
通过研究单拷贝直系同源基因的突变和功能异常,可以深入了解疾病的发病机制,并为疾病的诊断和治疗提供依据。
相关研究对单拷贝直系同源基因的研究主要包括以下几个方面:1.序列分析:通过比对不同物种中的单拷贝直系同源基因的序列,可以推断它们的进化关系和进化速率。
此外,还可以通过比对同一物种中的不同个体的单拷贝直系同源基因序列,研究个体间的遗传差异和种群遗传结构。
3个基因构建进化树的方法

3个基因构建进化树的方法基因是生物体内部的遗传物质,它们携带着生物体的遗传信息,并且决定了生物体的性状和特征。
在生物学研究中,通过研究基因的变化和演化关系,可以揭示生物种群之间的进化历程和亲缘关系。
构建进化树是研究基因演化的重要方法之一,它可以帮助我们了解不同物种之间的演化关系以及共同祖先的存在。
构建进化树的方法有很多种,其中比较常用的方法之一是基于DNA 或RNA序列的系统发育分析。
DNA和RNA是生物体内的核酸分子,它们携带着基因信息,并且在生物进化过程中会发生变异和演化。
通过比较不同物种之间的DNA或RNA序列差异,可以推断它们之间的亲缘关系和进化历程。
在构建进化树的方法中,一种常用的方法是基于单个基因的系统发育分析。
通过选择一个具有高变异性的基因,如线粒体DNA或核基因的特定区域,可以对不同物种之间的进化关系进行推断。
这种方法的优点是操作简单,成本低廉,但由于只考虑了单个基因的信息,可能会导致结果的不准确性。
为了提高进化树的准确性,还可以使用多个基因进行系统发育分析。
多个基因可以提供更多的信息,从而增加了结果的可靠性。
同时,使用多个基因还可以减少单个基因由于突变等原因引起的误差。
然而,选择哪些基因进行分析是一个关键问题,需要考虑基因的稳定性、变异速率以及在不同物种之间的保守性。
另一种构建进化树的方法是基于基因组数据的系统发育分析。
随着基因组测序技术的发展,我们可以获取到更多物种的基因组序列。
通过比较不同物种的基因组序列,可以揭示它们之间的进化关系。
基因组数据具有更高的分辨率和更全面的信息,可以提供更准确的进化树。
除了基于DNA或RNA序列的系统发育分析,还有其他一些方法可以用于构建进化树。
例如,可以利用蛋白质序列的相似性进行系统发育分析。
蛋白质是基因的产物,它们在不同物种之间可能存在相似性。
通过比较不同物种的蛋白质序列,可以推断它们之间的亲缘关系。
还可以利用形态学特征进行系统发育分析。
形态学特征是生物体外部的形状、结构和功能等方面的特征。
单倍型的分子系统树

单倍型的分子系统树一、概述单倍型的分子系统树是一种基于DNA序列数据构建的进化树,它能够反映物种间的亲缘关系和演化历史。
在单倍型分子系统树中,只考虑一个个体所拥有的一套染色体中某一个位点上的等位基因,这就是所谓的单倍型。
本文将从单倍型分子系统树的构建方法、应用领域、优缺点等方面进行详细介绍。
二、构建方法1. 样本收集:首先需要收集不同物种或个体之间相同位点上的DNA 序列数据。
2. 序列比对:将收集到的DNA序列进行比对,以确定它们之间的异同。
3. 构建进化模型:通过比对结果来确定不同物种或个体之间遗传差异程度,并选择合适的进化模型。
4. 构建进化树:利用选择出来的进化模型构建进化树。
三、应用领域1. 生物分类学研究:单倍型分子系统树可以被用来探究不同物种或亚种之间的亲缘关系和演化历史,为生物分类学研究提供了有力支持。
2. 种群遗传学研究:单倍型分子系统树可以被用来研究种群间的遗传结构和遗传多样性,为种群遗传学研究提供了有力工具。
3. 进化生物学研究:单倍型分子系统树可以被用来探讨不同物种或个体之间的进化历史和演化模式,为进化生物学研究提供了有力支持。
四、优缺点1. 优点:(1)能够准确反映物种间的亲缘关系和演化历史;(2)数据收集方便,成本较低;(3)能够对不同物种或个体之间的遗传差异进行量化比较。
2. 缺点:(1)只考虑一个个体所拥有的一套染色体中某一个位点上的等位基因,无法全面反映整个基因组的信息;(2)在构建进化树时需要选择合适的进化模型,选择不当可能会导致结果产生误差。
五、结论总之,单倍型分子系统树是一种重要的分子生物学工具,在生物分类学、种群遗传学和进化生物学等领域都有广泛应用。
虽然它也存在一些缺点,但是其优点仍然使它成为研究生物进化和演化历史的重要工具之一。
单拷贝直系同源基因

单拷贝直系同源基因什么是单拷贝直系同源基因?单拷贝直系同源基因是指在一个物种的基因组中,存在只有一份副本且高度相似的基因。
这些基因通常具有相似的序列和功能,并且在进化过程中保持了较高的保守性。
单拷贝直系同源基因的重要性单拷贝直系同源基因在生物学研究中具有重要的意义。
首先,它们可以作为物种鉴定和系统发育研究的理想标记。
由于这些基因在物种内部高度保守,但在不同物种之间存在差异,通过比较不同物种之间这些基因的序列差异,可以揭示它们之间的亲缘关系和进化历史。
其次,单拷贝直系同源基因还可以用于研究人类遗传变异与疾病之间的关联。
许多遗传性疾病都与特定基因突变相关联,通过对这些相关基因进行深入研究,可以揭示其与疾病发生发展的机制。
此外,单拷贝直系同源基因还可以作为分子演化研究的重要工具。
通过比较不同物种之间这些基因的序列差异,可以了解物种的进化速度和模式,揭示生物进化的规律。
单拷贝直系同源基因的鉴定方法基于序列相似性的方法BLAST(Basic Local Alignment Search Tool)BLAST是一种常用的基于序列相似性进行基因鉴定的方法。
它通过将待鉴定基因序列与已知数据库中的序列进行比对,计算二者之间的相似性得分,并根据得分进行排序和筛选。
HMM(Hidden Markov Model)HMM是一种基于统计模型的方法,常用于蛋白质家族成员鉴定。
它通过构建蛋白质家族的隐马尔可夫模型,将待鉴定蛋白质序列与模型进行比对,并计算其与模型之间的匹配得分。
基于表达谱数据的方法RNA-seqRNA-seq是一种高通量测序技术,可以用来研究转录组中基因表达水平的变化。
通过比较不同组织或条件下基因表达谱中单拷贝直系同源基因的差异,可以鉴定出与特定组织或条件相关的基因。
基于基因组结构的方法基因家族分析基因家族分析是一种常用的鉴定单拷贝直系同源基因的方法。
它通过比对物种基因组中相似的基因序列,并进行进化分析,确定哪些基因是单拷贝直系同源基因。
单拷贝直系同源基因构建进化树

单拷贝直系同源基因构建进化树摘要:I.引言- 介绍单拷贝直系同源基因- 说明其在进化树构建中的重要性II.直系同源基因的定义和作用- 定义直系同源基因- 阐述其在进化过程中的作用III.单拷贝直系同源基因的构建方法- 介绍构建进化树的基本流程- 重点讲解单拷贝直系同源基因的构建方法IV.单拷贝直系同源基因在进化树构建中的应用- 实例分析单拷贝直系同源基因在进化树构建中的应用- 阐述其对进化树准确性的影响V.总结- 总结单拷贝直系同源基因在进化树构建中的作用和意义- 展望未来研究方向正文:I.引言在生物进化研究中,进化树是一种重要的分析工具,能够帮助我们理解物种间的进化关系。
而单拷贝直系同源基因(single copy orthologousgenes)作为进化树构建的重要依据,对于进化树的准确性和可靠性起着至关重要的作用。
本文将围绕单拷贝直系同源基因的构建方法以及在进化树构建中的应用进行详细阐述。
II.直系同源基因的定义和作用直系同源基因(orthologous genes)是指在进化过程中,起源于同一祖先基因,分别位于不同物种基因组中的基因。
它们具有相同的基因序列和功能,是生物进化的基本遗传单位。
在构建进化树时,通过比较不同物种间的直系同源基因序列,可以推测物种间的进化关系。
单拷贝直系同源基因是指在基因组中仅存在一个拷贝的直系同源基因。
与其他多拷贝直系同源基因相比,单拷贝直系同源基因在进化过程中受到的选择压力较小,因此更可能在不同物种间保持稳定的序列相似性和功能。
III.单拷贝直系同源基因的构建方法构建单拷贝直系同源基因的方法有很多,其中较为常用的方法是基于基因序列比对和进化模型。
首先,通过基因序列比对软件(如BLAST)比较不同物种的基因序列,找到具有较高相似性的基因对。
然后,利用进化模型(如Maximum Likelihood,Bayesian Inference 等)对这些基因对进行进化树重建,推测它们的进化关系。
生物大数据分析中的进化遗传树构建方法与技巧

生物大数据分析中的进化遗传树构建方法与技巧进化遗传树(Phylogenetic Tree)是生物学研究中用于分析物种关系和演化历程的重要工具。
通过构建进化树,我们可以了解不同物种之间的进化关系,揭示物种的演化历史以及预测它们之间的共同祖先。
在生物大数据分析中,构建进化遗传树有着重要的意义,因为它可以帮助我们理解生物的遗传多样性、物种起源以及群体分化等重要生物学问题。
在构建进化遗传树的过程中,我们需要根据生物学数据来推断物种间的关系。
这些生物学数据可以是DNA或RNA序列、蛋白质序列、形态特征等。
为了准确地构建进化遗传树,我们需要选择合适的方法和技巧。
下面将介绍一些常用的进化遗传树构建方法和技巧。
1. 距离法(Distance-based methods):距离法是通过计算物种间的相似度或差异度来构建进化遗传树的方法。
常用的距离法包括最邻近法(Neighbor Joining)、最小进化法(Minimum Evolution)和最大简约法(Maximum Parsimony)等。
这些方法根据不同的算法和模型,通过计算物种间的距离矩阵来构建进化关系。
2. 贝叶斯方法(Bayesian methods):贝叶斯方法是一种基于统计模型和概率推断的进化遗传树构建方法。
它通过采用贝叶斯推断和蒙特卡洛马尔科夫链蒙特卡洛算法(MCMC)来估计进化树的拓扑结构和参数。
贝叶斯方法具有高度灵活性和更准确的模型,适用于复杂的进化树推断问题。
3. 最大似然方法(Maximum likelihood methods):最大似然方法是一种常用的基于概率统计的进化遗传树构建方法。
它通过最大化观测到的数据出现的概率,推断出可能的进化树。
最大似然方法考虑了模型中的参数估计问题,并用参数化的模型来描述进化过程,从而提高了推断结果的准确性。
在进行进化遗传树构建时,还有一些技巧需要注意,以保证结果的准确性和可靠性:1. 数据质量的控制:数据质量是构建进化遗传树的关键因素之一。
小麦族植物单拷贝核基因DMC1的进化式样

摘要基因复制是多倍体植物基因组结构的重要特征。
全基因组复制即多倍化,是促进生物进化改变的重要驱动力,也是植物适应自然环境的主要方式。
在植物多倍化过程中,衍生了大量的复制基因,为植物进化提供了以复制基因为基础的潜在资源库。
随着植物多倍化过程,复制基因可能在群体中消失或被保留并在群体中扩散,其经历不同的进化方向为多倍体植物产生更丰富的遗传多样性和更大的生存竞争力。
DMC1基因编码的减数分裂重组蛋白,是在真核生物中发现的大肠杆菌(E.coli)RecA蛋白的同源蛋白,在减数分裂同源重组期间的同源性搜索和DNA链交换反应中起到了至关重要作用。
DMC1基因的遗传变异式样分析将有助于了解该基因在植物物种形成及分化过程中的分子进化动力及成因。
小麦族是禾本科植物中一个十分重要的类群,约30个属465个物种,主要分布于北半球的温带,个别属种分布至寒带、热带高山,可生长于山坡、沼泽、荒漠等环境中。
小麦族植物物种繁多、起源复杂,在生物学机制和遗传系统上存在着较大变异,基因组供体来源较为清晰,因此该族对于物种形成、分类学、系统发育和分子进化及遗传多样性等方面的研究具有重要意义。
本研究通过小麦族25个属159个种4个变种26个亚种的382条单拷贝核基因DMC1序列,对多倍体与二倍体间的同源基因位点进行多态性比较、选择压力、基因表达等分析,探讨DMC1基因在小麦族中进化动力,获得主要结果如下:1.小麦族直系起源的属种之间,DMC1基因的核苷酸多态性存在显著差异,天然杂交使得多倍体中基因座多态性通常较其二倍体供体更高,表明DMC1基因在多倍体中进化更快。
2.小麦属植物B基因组多倍化后核苷酸多态性降低,进化速率减慢,遗传基础变得狭窄,是人类长期驯化导致遗传瓶颈的结果;StSt基因组组成的四倍体物种形成后降低的核苷酸多态性可能是群体历史扩张的结果。
3.小麦族中A、B、Ns、Xm、St、H和P基因组的选择压分析dN/dS均小于1,表明DMC1基因在进化过程中经历了纯化选择的影响,且各基因组所受选择压力存在显著差异,随着多倍化过程发生改变,驱使DMC1基因进化。
基因BLAST及系统进化树构建操作步骤
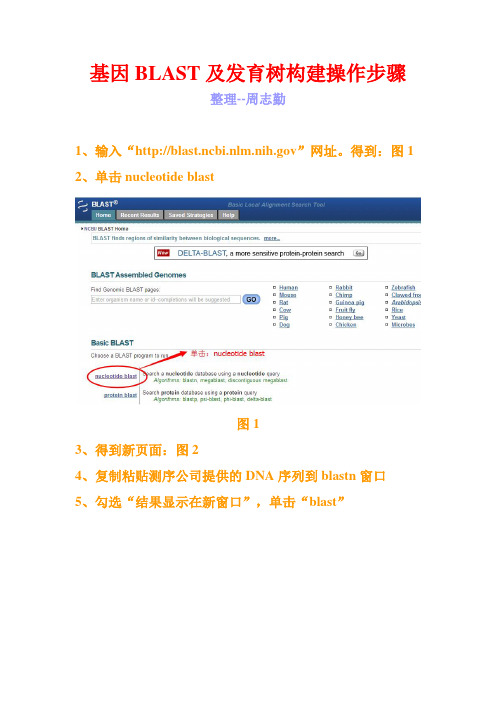
基因BLAST及发育树构建操作步骤整理--周志勤1、输入“”网址。
得到:图12、单击nucleotide blast图13、得到新页面:图24、复制粘贴测序公司提供的DNA序列到blastn窗口5、勾选“结果显示在新窗口”,单击“blast”图2 6、得到新窗口:图3图37、下拉页面,如:图48、选择Accession下的基因编码。
入选第一个“HQ711983.1”图49、得到新窗口如:图510、单击FASTA图511、得到如:图612、复制菌株种名跟16SrDNA序列,并粘贴到“.txt”文件中图613、新建“.txt”文件,如:图714、把测序菌株16SrDNA序列和选取blast所得的匹配度高的菌株16SrDNA序列粘贴在“.txt”文件下,在菌株种名前加“>”,每个序列后空一行图715、复制“.txt”文件,改“.txt”文件为“.fasta”文件,如:图8图816、用MEGA 4.0.2软件打开“.fasta”文件,如:图9图917、单击Alignment里的Alinment Explorel/CLUSAC,如:图10图1018、单击Alignment里的Align by clustalW,如:图11图1119、在弹出窗口中单击OK,如:图12图1219、在弹出窗口中单击OK,如:图13图1320、等待数据运算,如:图14图1421、运算结束,关闭当前窗口。
如:图1522、在弹出的第一个窗口单击NO图1523、在弹出的第一个窗口单击YES,如:图16图1624、将文件以“.meg”格式保存,如:图17图1725、关闭其余窗口,打开刚刚保存的“.meg”文件,如:图18图1826、选择phyloeny下的Bootsteap Test of phylogeng下的Neighbor-Joining...,如:图19图1927、在弹出窗口单击Compulte,如:图20图2028、单击树状图,得到系统发育树,建议截图保存,如:图21图21。
单拷贝直系同源基因构建进化树

单拷贝直系同源基因构建进化树单拷贝直系同源基因(Single-copy orthologous genes)是指在多个物种间具有高度保守性和功能相似性的基因。
它们在进化研究中具有重要意义,可以帮助我们揭示物种间的亲缘关系和演化历程。
本文将介绍如何利用单拷贝直系同源基因构建进化树,以及这一方法的优势和应用实例。
一、单拷贝直系同源基因的定义和作用单拷贝直系同源基因是指在多个物种间具有唯一对应关系的基因,它们在进化过程中保持相对稳定。
这是因为这些基因通常对生物体的生存具有重要作用,因此在自然选择的作用下,它们不容易发生大规模的变异。
单拷贝直系同源基因在构建进化树中起到关键作用,因为它们可以在不同物种间进行对比,揭示物种间的亲缘关系。
二、构建进化树的方法和步骤1.收集数据:首先需要收集多个物种的基因组数据,找出单拷贝直系同源基因。
这些基因通常具有较高的保守性,可以在多个物种间找到。
2.比对基因组:将不同物种的基因组序列进行比对,找出保守的单拷贝直系同源基因。
比对工具如BLAST(Basic Local Alignment Search T ool)和Clustal Omega等可用于这一步骤。
3.构建进化树:利用生物信息学方法,如最大似然法(Maximum Likelihood)或贝叶斯法(Bayesian Inference)构建进化树。
进化树中的每个节点代表一个物种,每个分支代表两个物种之间的亲缘关系。
4.评估结果:分析进化树的可信度,如评估树拓扑结构的稳定性、基因序列相似性等。
三、单拷贝直系同源基因在进化树构建中的优势1.准确性:单拷贝直系同源基因在多个物种间具有较高的保守性,使得进化树构建具有较高的准确性。
2.稳定性:这些基因在进化过程中不易发生变异,有助于揭示更古老的演化关系。
3.分辨率:单拷贝直系同源基因可揭示低分辨率的进化关系,有助于研究物种间的亲缘关系。
四、应用实例及意义1.物种亲缘关系研究:通过单拷贝直系同源基因构建进化树,可以揭示物种间的亲缘关系,为生物分类学和系统发育提供依据。
3个基因构建进化树的方法

3个基因构建进化树的方法进化是生物学中一个重要的概念,它描述了生物种群随时间的演化过程。
进化树是一种用来表示不同物种之间演化关系的图表,它可以帮助我们理解生物的演化历史和亲缘关系。
构建进化树的方法有很多种,其中一种常用的方法是基于基因序列的比较。
本文将介绍基于3个基因的构建进化树的方法。
基因是生物体内用来传递遗传信息的分子,它们以DNA的形式存在于细胞中。
每个物种的基因组中都有很多基因,其中一些基因在不同物种之间保持高度保守,也就是说它们的序列变化很小。
这些保守的基因可以用来构建进化树。
在构建进化树的过程中,我们需要选择适合的基因进行比较。
一般来说,选择的基因应该满足以下几个条件:首先,基因在不同物种中的序列变化应该相对较小,这样才能准确地反映物种之间的演化关系;其次,基因在不同物种中应该有足够的变异,这样才能提供足够的信息来推断进化关系;最后,基因的比较应该能够得到可靠的结果,这就要求我们选择那些已经被广泛研究和验证的基因。
在基因选择完毕后,我们需要获取各个物种的基因序列。
这可以通过DNA测序技术来实现,现代的测序技术已经非常高效和准确,可以快速得到大量的基因序列数据。
在获取到基因序列后,我们需要对这些序列进行比对和分析,以便得到物种之间的差异。
比对可以使用一些开源的软件来完成,比如BLAST和ClustalW等。
通过比对,我们可以得到物种之间基因序列的异同点,这些差异点可以用来推断进化关系。
基于比对结果,我们可以使用一些计算模型来构建进化树。
常用的计算模型有距离法、最大简约法和最大似然法等。
这些方法都是基于不同的原理来进行计算的,它们可以根据基因序列的差异程度来计算物种之间的进化距离,并将这些距离用树状图的形式展示出来。
进化树的构建过程是一个迭代的过程,通过不断调整模型参数,我们可以得到更准确的进化树。
基于3个基因的构建进化树的方法可以提高进化树的准确性。
因为多个基因的比较能够提供更多的信息,可以避免单个基因的局限性。
标准分析内容及文章思路

一、(物种)比较基因组学及分子进化研究1、基因组进化地位研究通过和近缘物种基因组直接比较,构建物种的进化树,确定其进化地位。
上图是构建的海马跟斑马鱼等其他鱼类构建的海马的进化分析(Qiang Lin,Shaohua Fan,Yanhong Zhang,Meng Xu,et al.The seahorse genome and the evolution of its specialized morphology.Nature.14 December 2016)2、基于比较基因组挖掘功能基因通过与xxxx等基因组进行比较,挖掘(物种名)特有基因,然后进行功能注释,最后结合(物种名)自身表型特征,挖掘功能基因。
特有基因或基因组家族分析示意图如下:例如通过比较基因组学分析之后,发现非洲爪蟾与人、鸡等其他物种相比,mix家族存在显著的扩张(Kang Y J, Kim S K, Kim M Y, et al. Genome sequence of mungbean and insights into evolution within Vigna species.[J]. Nature Communications, 2014, 5(5543):5443-5443.)通过与xxx等基因组进行比较,挖掘(物种名)相对于其它物种存在收缩与扩张的基因家族,并结合基因家族注释与性状表型挖掘功能基因。
基因组家族收缩扩张示意图如下:图五步蛇家族收缩扩张分析例如上图五步蛇基因组文章中对四个蛇和蜥蜴的基因组进行比较分析。
(Cai J, Liu X, Vanneste K, et al. The genome sequence of the orchid Phalaenopsis equestris.[J]. NatureGenetics, 2014, 47(1):186-186.)通过与xxxx等基因组进行比较,选取直系同源基因,并针对每个直系同源基因进行选择压力分析,挖掘(物种名)相对于其它物种存在正选择或负选择的基因,并结合基因注释与性状表型挖掘功能基因。
鼩鼱科三物种转录组初步分析

兽类学报,2021, 41 (2): 173-181Acta Theriologica Sinica D O I:10. 16829/j. slx b. 150403齣鼠青科三物种转录组初步分析周婧晶1丘银彬1万韬2何水旺3蒋学龙3潘星华h何锴(1南方医科大学基础医学院,广东省单细胞技术与应用重点实验室,广州510515)(2陕西师范大学生命科学学院,西安710119) (3中国科学院昆明动物研究所,昆明650223)摘要:中国是駒鼷科物种十分丰富的国家,已知有12属61个物种在中国分布,并演化出多种生态适应型。
本研究采用高通量测序技术对小纹背駒鼯、云南长尾齣鼯和蹼足駒3个物种的心和肺组织样本进行转录组测序。
其中小纹背和云南长尾齣营地表生活,蹼足駒适应半水生生活。
3个物种的转录组测序一共获得20.5 G b的Clean Data,拼接后分别获得131 045条、153 621条和135 838条Unigenes,BUSCO分析显示其中完整的保守基 因最高为82. 2%,共有同源基因家族超过8 000个。
此外,我们发现通过优化分析方案,可以将单拷贝基因的比 例提高18% ~25%,同时将多拷贝基因比例从28.4% ~31. 8%降为0.7% ~1.1%。
同物种不同组织间的基因差 异表达分析找到各组织的标志性基因。
转录组差异表达分析发现,蹼足駒心脏和肺均高表达W/f M基因,该基 因是缺氧诱导因子信号通路中的关键基因,调节下游与缺氧应激相关基因的表达,可能与水生适应有关。
关键词:劳亚食虫类•,駒_科;转录组从头组装;低氧适应中图分类号:Q75 文献标识码:A文章编号:1000 -1050 (2021) 02 - 0173 -09Transcriptome analyses for three Soricid shrewsZHO U Jin g jin g1,QIU Y in b in1 ,W AN Tao2,H E Shuiw ang3,JIA N G X uelong3,PAN X in g h u a1* ,H E K ai1'3*(1 Southern Medical Uniiv:rsity, School of Basic Medical Science,Guangdong Provincial Key Laboratory of Single Cell Technology and Application,Guangzhou 510515, China)(2 College of Life Sciences, Shaanxi Normal University, X i' an 710119, China)(3 Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223 , China)A b s t r a c t:C h in a h a rb o rs h ig h s p e c ie s d iv e rsity o f S o ric id sh rew s. S ix ty-o n e s p e c ie s r e p re s e n tin g12 g e n e ra a re d is tr ib u te din C h in a, in h a b itin g v a rio u s h a b ita ts. In th is s tu d y, w e s e q u e n c e d th e tra n sc rip to m e o f h e a rt a n d lu n g tis s u e from th re es p e c ie s,n a m e ly,Episoriculus u m b rin u s,Nectogale ele g a n s,a n d Sorex b ed fordiae,a n d o b ta in e d20. 5 G h c le a n d ata. A fter de novo a s s e m b ly, w e o b ta in e d131 045, 153 621 , a n d135 838u n ig e n e s for E. u m brinus, N. ele g a n s, a n d S. bedfordiae,re sp e c tiv e ly. B U S C O a n a ly s e s su g g e ste d w e re c o v e re d a s m an y a s82.2%fu ll-le n g th tra n s c r ip ts ofg en es. O rth o F in d e r re c o g n iz e d m o re th a n8 000h o m o lo g o u s g e n e fa m ilie s a s s h a re d b y so ric id a n d ta lp id sp e c ie s. W e c o mp a re d tw o p o st-T rin ity p ip e lin e s to o p tim iz e th e q u a lity o f u n ig e n e s. W h e n c o m b in in g T rin ity s c r ip ts, C D-H it a n d T ra n-s R a te, th e p ro p o rtio n s o f sin g le-c o p y g e n e s in c re a se d18%-25%c o m p a re d w ith u sin g C D-H it a lo n e, a n d th e p ro p o rtio n sof m u lti-c o p y g e n e s d e c r e a s e d from28. 4%-31.8%to 0. 7%- 1. 1%. A n a ly sis o f d iffe re n tia lly e x p re s s e d g e n e s ( D E G s)id e n tifie d tis s u e e n r ic h e d g e n e s, v a lid a tin g o u r a n a ly s is p ip e lin e. O u r a n a ly s e s H/F1A h ig h ly e x p re ss e d in b o th h e a rt a n dlu n g o f N. elegans. H IF1A is a k e y g e n e in th e h y p o x ia-in d u c ib le fa c to r sig n a lin g p ath w ay a n d re g u la te s th e e x p re ss io n ofd o w n stre a m g e n e s r e la te d to h y p o x ia, p ro b a b ly a s so c ia te d w ith a q u a tic a d a p ta tio n. T h is is th e first re p o rt of sh rew tr a n s c r ipto m e, p ro v id in g im p o rta n t d a ta fo r u n d e rs ta n d in g th e ev o lu tio n a n d p h y sio lo g y in S o ric id a e. O u r a n a ly s is p ip e lin e c o u ld a ls oin sp ire tra n sc rip to m ic stu d y in o th e r n o n-m o d e l o rg an ism s.Key words:E u lip o ty p h la;S o r ic id a e;T ra n s c rip to m e d e novo a s s e m b ly;A d a p ta tio n to h y p o x ia基金项目:国家自然科学基金面上基金(31970389);中国博士后科学基金面上项目(2009M652952);广东省珠江人才计划本土创新科研团队(2017B T01S131)作者简介:周婧晶(1995 -),女,硕士研究生,主要从事进化生物学研究.收稿日期:2019-12-30;接受日期:2020-10 -14* 通讯作者,Corresponding authors, E-ma i l:*****************.cn;****************174兽类学报齣_是劳亚食虫目(Eulipotyphla)駒黯科 (Soricidae)动物的总称。
基因进化树构建

基因进化树构建
基因进化树构建是一种重要的生物学研究方法。
它可以通过比较不同
物种之间的基因序列或蛋白质结构来推断它们的演化关系。
这种方法
可以帮助科学家们了解不同物种之间的亲缘关系,研究物种的起源和
演化,以及预测未来的进化方向。
基因进化树构建的基本原理是通过比较不同物种之间的基因或蛋白质
序列的异同,来推断它们的演化关系。
这种方法可以用来研究不同物
种之间的亲缘关系,比如说研究人类和大猩猩之间的关系,以及研究
鸟类之间的演化历程。
基因进化树构建的方法主要有两种:分子钟方法和序列比较方法。
分
子钟方法是一种基于时间的方法,它通过比较不同物种之间的基因或
蛋白质序列变异程度,来推断它们的演化时间和速率。
而序列比较方
法则是一种基于比较的方法,它主要通过比较不同物种之间的基因或
蛋白质序列相似程度,来推断它们的演化关系。
基因进化树构建的过程中,需要先收集不同物种的基因或蛋白质序列,并将它们进行比较分析。
通常会使用一些专门的生物信息学软件,比
如Clustal,Mega等来进行序列比对和进化分析。
在分析过程中,还需要对数据进行校准和修正,以确保分析结果的准确性和可靠性。
最
终,得到的结果可以用树状图的形式来表示不同物种之间的亲缘关系和演化历程。
总的来说,基因进化树构建是一种重要的生物学研究方法,它可以帮助我们了解物种之间的演化关系和进化历程。
随着生物信息学技术的不断发展和改进,基因进化树构建的分析方法也在不断演化和改进,这将有助于我们更深入地研究生命的起源和演化。
单拷贝直系同源基因构建进化树

单拷贝直系同源基因构建进化树【原创实用版】目录1.直系同源基因的概念和重要性2.单拷贝直系同源基因的构建方法3.进化树的构建和分析4.直系同源基因在生物学研究中的应用正文1.直系同源基因的概念和重要性直系同源基因是指在不同物种间具有相同功能和相似序列的基因。
它们在生物体内扮演着相似的角色,并在进化过程中通过基因复制和突变产生。
研究直系同源基因有助于我们了解物种间的进化关系和基因功能。
2.单拷贝直系同源基因的构建方法构建单拷贝直系同源基因的方法主要包括以下两个步骤:(1)寻找直系同源基因:通过比对不同物种的基因组序列,利用生物信息学方法找到具有相似序列和功能的基因。
(2)构建进化树:利用生物统计学方法,如最大似然法、贝叶斯法等,根据直系同源基因的序列差异构建进化树,以揭示物种间的进化关系。
3.进化树的构建和分析进化树是一种图形化的表示方法,用于展示物种间的进化关系。
它通常由树根、树枝和树叶组成,其中树根表示共同祖先,树枝表示进化分支,树叶表示现存物种。
通过分析进化树,我们可以了解不同物种之间的亲缘关系、进化距离和演化历史。
此外,进化树还可以为物种分类和基因功能研究提供重要依据。
4.直系同源基因在生物学研究中的应用直系同源基因在生物学研究中具有广泛的应用价值,例如:(1)基因功能研究:通过比较直系同源基因在不同物种中的表达模式和功能,可以推测基因在生物体内的作用。
(2)物种分类:利用进化树中的物种分枝关系,可以辅助对物种进行分类和演化地位的判断。
(3)基因家族分析:直系同源基因可以作为基因家族研究的基础,通过分析基因家族的扩张和收缩,可以揭示基因家族在进化过程中的变化规律。
比较基因组学中-利用单拷贝基因构建进化树

⽐较基因组学中-利⽤单拷贝基因构建进化树(1)同源基因的查找 OrthoMCL or Orthofinder;(2)多序列⽐对 Muscle / MAFFT / ClustalW / T-coffee, Muscle 效果好点(3)调取保守区域,并收尾连接,形成supergene Gblocks (4)进化树构建 RaxML MEGA 等,很多⽂献⽤RaxML,PhyML或Mrbayes,因为ML树和贝叶斯进化树对核苷酸 / 氨基酸替代模型的选择⾮常敏感,故在进⾏进化树或分化时间构建之前,需对核苷酸 / 氨基酸替代模型进⾏选择。
(jModelTest 对cDNA进⾏替代模型选择,ProtTest 对蛋⽩进⾏替代模型选择)(5)分化时间分析 divergence time mcmctree. PAML中的⼀个程序, BEAST2(6)基因扩张收缩分析 CAFE (7)基因是否收到正选择 codeML PAML中⼀个程序⼀、为什么需要选择核苷酸替换模型构建进化树可以通过同源 DNA序列或蛋⽩质分⼦的氨基酸序列来实现,其具体的步骤基本上是先选取⽣物数据(同源 DNA 序列或蛋⽩质分⼦的氨基酸序列数据)与进化距离模型,然后对不同物种DNA 或蛋⽩质的序列进⾏⽐对,再应⽤距离模型和⽐对结果计算进化距离,最后通过进化距离构建进化树。
因此,选择进化距离模型是构建进化树的基础,DNA分⼦中基因的进化距离是通过对核苷酸替代数进⾏估计获得的(当遗传信息从⽗代复制到⼦代时,往往会发⽣⼀些改变,这些改变称为突变。
突变是DNA进化的动⼒。
常见的突变模式有:替代,即⼀个核苷酸被另⼀个核苷酸所替代;插⼊,即插⼊⼀个或多个核苷酸;删除,即删除⼀个或多个核苷酸。
但是在分析进化时,⼀般只考虑替代。
),要估计核苷酸替代数,就必须应⽤核苷酸替代的数学模型。
由于核苷酸替换模型的选择直接影响进化距离的计算,进⽽对所构建的系统树是否合理起决定作⽤。
单拷贝直系同源基因系统发育树的构建

标题:单拷贝直系同源基因系统发育树的构建摘要:随着基因测序技术的不断发展,越来越多的基因序列得到了公开发布,为研究者提供了丰富的遗传信息。
在众多研究中,通过构建系统发育树来揭示不同物种的亲缘关系和进化历史是一项重要的工作。
而单拷贝直系同源基因系统发育树的构建,对于了解不同物种之间的关系和进行进化分析具有重要意义。
本文将介绍单拷贝直系同源基因系统发育树的构建方法及相关应用。
正文:1. 单拷贝直系同源基因系统概述单拷贝直系同源基因即同一基因家族中的每个成员都只有一个拷贝,且这些拷贝是由同一个祖先基因直接演化而来,因此它们在不同物种之间具有较高的同源性和拓展性。
而单拷贝直系同源基因系统则是指由这些单拷贝直系同源基因所构成的系统。
这些基因在不同物种之间的保守程度较高,因此常被用于物种之间亲缘关系的研究和系统发育树的构建。
2. 单拷贝直系同源基因系统发育树构建的重要性单拷贝直系同源基因系统发育树的构建对于揭示不同物种之间的亲缘关系具有重要意义。
通过比较单拷贝直系同源基因在不同物种中的序列差异和演化速率,可以推断这些物种之间的亲缘关系和进化历史。
单拷贝直系同源基因系统发育树的构建还可以为物种的分类和系统发育关系提供重要参考。
3. 单拷贝直系同源基因系统发育树构建的方法a. 基因家族的筛选与挑选需要从目标物种的基因组序列中筛选出单拷贝直系同源基因家族。
可以利用基因同源性分析工具如BLAST、HMMER等进行筛选和挑选,确保所选择的基因家族符合单拷贝直系同源基因的特征。
b. 序列比对与进化树构建选定合适的单拷贝直系同源基因后,需要对这些基因序列进行比对。
可以利用一些专业的序列比对软件如ClustalW、MAFFT等进行多序列比对,得到基因序列的保守区域和变异区域。
利用分子进化树构建软件如PHYLIP、MEGA等构建系统发育树,并进行进化分析。
4. 单拷贝直系同源基因系统发育树构建的应用单拷贝直系同源基因系统发育树的构建在生物学领域有着广泛的应用。
单拷贝同源基因

单拷贝同源基因单拷贝同源基因是指在染色体上存在两个或多个相同的基因序列。
这些同源基因在物种进化过程中的复制事件中产生。
单拷贝同源基因在遗传学和进化生物学研究中具有重要的意义。
单拷贝同源基因在物种进化中起着重要的作用。
通过基因复制事件,同源基因在染色体上形成多个拷贝,这些拷贝可以发生功能分化和结构变异。
一些拷贝可能会发生突变,导致功能差异。
这种差异可以促进物种的适应和进化。
例如,单拷贝同源基因的复制事件在人类基因组中非常常见,这些基因的复制和功能分化可能与人类智力和疾病的发生有关。
单拷贝同源基因在基因组结构和功能的调控中起着重要的作用。
同源基因的存在可以增加基因组的稳定性,并提供基因组结构的可塑性。
同源基因的拷贝可以作为起始点,驱动基因的重组和重排。
这种基因的结构变异可以导致基因在不同条件下的表达模式发生改变。
例如,在植物中,一些同源基因的拷贝可能在不同组织或发育阶段中表达,从而调节植物的生长和发育。
单拷贝同源基因还可以用于物种的亲缘关系分析和进化树构建。
通过比较不同物种间的同源基因序列差异,可以评估物种的进化距离和亲缘关系。
同源基因的拷贝数目和差异程度可以用于构建进化树,并推断不同物种的演化历史和亲缘关系。
这种方法被广泛应用于分子进化学和系统发育学领域。
单拷贝同源基因的研究对于遗传疾病的诊断和治疗也具有重要意义。
一些遗传疾病与特定基因的突变或缺失有关,而同源基因的存在可以提供替代的功能。
通过研究同源基因的结构和功能,可以开发针对遗传疾病的治疗策略。
例如,通过引入同源基因来修复突变基因的功能缺陷,可以治疗一些遗传性疾病。
单拷贝同源基因在生物学研究中具有重要的地位和作用。
通过研究同源基因的复制、功能分化和结构变异,可以深入理解物种的进化机制和基因组的调控网络。
同时,同源基因的研究也为遗传疾病的诊断和治疗提供了新的思路和方法。
随着基因组学和生物信息学的快速发展,相信单拷贝同源基因的研究将会在未来取得更加重要的突破和进展。
单基因建树流程

单基因建树流程一、啥是单基因建树呢。
简单来说哈,单基因建树就像是给一个家族画族谱,不过这个家族是由基因组成的呢。
咱们想知道不同生物或者同一生物不同个体之间,某个特定基因的关系有多近或者多远,就可以通过单基因建树来实现。
这就好比你想知道你和你远房亲戚到底有多亲,那就得去查一查家族的族谱是一个道理啦。
二、准备工作。
1. 数据收集。
咱们得先找到想要研究的那个单基因的序列数据。
这些数据可以从好多地方来呢,比如说公共数据库,像NCBI(美国国立生物技术信息中心),这里面就像是一个超级大的基因宝藏库,啥基因数据都有。
不过在从这里找数据的时候呀,可得小心点儿,要找那种质量比较高的数据哦,就像挑水果一样,得挑那些新鲜又没坏的。
还有就是如果是自己实验室做的测序,那就更好啦,自己的数据就像是自己种的水果,知根知底的。
2. 序列比对。
拿到数据之后呢,就像把一堆拼图碎片整理一下,咱们要进行序列比对。
这时候就需要一些好用的工具啦,像ClustalW或者MAFFT这些软件就特别棒。
它们能把相似的序列片段对齐,就像把拼图按照形状和图案对齐一样。
这个过程有时候可能会遇到一些小麻烦,比如说有些序列不太完整呀,或者有一些特殊的变异,不过没关系,多调整调整参数,就像调收音机找台一样,总能找到合适的设置让比对结果看起来比较完美。
三、建树方法。
1. 距离法。
距离法就像是用尺子量距离一样来确定基因之间的关系。
比如说有个软件叫MEGA (分子进化遗传学分析软件),它就可以用距离法来建树。
咱们把比对好的序列放进去,它就会根据序列之间的差异程度算出距离,然后根据这个距离来构建树的形状。
这个树的形状就像一棵真正的树一样,有树干有树枝,树枝的长短就代表了基因之间的距离远近。
不过呢,这个方法也有它的小缺点,有时候它可能会把一些关系判断错,就像你有时候看走眼了,把长得像的两个人当成双胞胎了一样。
2. 最大简约法。
这个方法就比较有趣啦,它是基于一个原则,就是在进化过程中,发生的变化越少越好。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
单拷贝直系同源基因构建进化树
摘要:
I.引言
- 介绍单拷贝直系同源基因
- 说明其在进化树构建中的应用
II.单拷贝直系同源基因的定义和性质
- 定义单拷贝直系同源基因
- 描述其性质
III.单拷贝直系同源基因在进化树构建中的作用
- 介绍进化树构建的基本原理
- 说明单拷贝直系同源基因在进化树构建中的作用
IV.单拷贝直系同源基因构建进化树的算法
- 介绍单拷贝直系同源基因构建进化树的算法
- 描述算法的步骤和流程
V.应用实例
- 举例说明单拷贝直系同源基因构建进化树在实际研究中的应用VI.结论
- 总结单拷贝直系同源基因构建进化树的意义和价值
- 展望其在未来的应用和发展
正文:
I.引言
在生物进化研究中,构建进化树是一种重要的方法,可以揭示物种间的进化关系。
单拷贝直系同源基因,作为一种特殊的基因类型,在进化树构建中具有重要的应用价值。
本文将介绍单拷贝直系同源基因的概念、性质以及在进化树构建中的应用。
II.单拷贝直系同源基因的定义和性质
单拷贝直系同源基因是指在基因组中仅存在一个拷贝的基因,它来自于物种的祖先,并在后代物种中保持不变。
单拷贝直系同源基因具有以下性质:
1.仅存在一个拷贝:在基因组中,单拷贝直系同源基因只存在一个拷贝,这使得它在进化分析中具有较高的可靠性。
2.祖先起源:单拷贝直系同源基因来源于物种的祖先,因此可以揭示物种间的进化关系。
3.保守性:单拷贝直系同源基因在进化过程中保持相对保守,这使得它成为研究生物进化的关键线索。
III.单拷贝直系同源基因在进化树构建中的作用
进化树构建的基本原理是通过比较不同物种的基因序列,找出它们之间的相似性和差异性,从而确定它们在进化过程中的关系。
单拷贝直系同源基因在进化树构建中的作用主要体现在以下几个方面:
1.确定进化关系:单拷贝直系同源基因可以作为进化分析的标志,确定物种间的进化关系。
2.提高分析可靠性:由于单拷贝直系同源基因在基因组中仅存在一个拷贝,因此在进化分析中具有较高的可靠性。
3.揭示进化过程:通过比较单拷贝直系同源基因在不同物种中的序列差
异,可以揭示物种间的进化过程。
IV.单拷贝直系同源基因构建进化树的算法
单拷贝直系同源基因构建进化树的算法主要包括以下几个步骤:
1.寻找单拷贝直系同源基因:在基因组中寻找具有单一拷贝的基因,并将其作为进化分析的候选基因。
2.提取候选基因的序列:从基因组中提取候选基因的序列信息,以便进行后续的进化分析。
3.构建进化树:根据候选基因的序列信息,利用系统发育分析方法构建进化树。
4.评估进化树:通过比较进化树与已知物种间的关系,评估进化树的准确性和可靠性。
V.应用实例
单拷贝直系同源基因构建进化树的方法在实际研究中具有广泛的应用。
例如,研究人员利用这种方法对果蝇的进化关系进行了研究,揭示了果蝇间的进化历程。
此外,这种方法还被应用于植物、动物等其他生物类群的研究,为揭示物种间的进化关系提供了重要的依据。
VI.结论
单拷贝直系同源基因构建进化树是一种重要的生物进化分析方法,具有较高的可靠性和准确性。
通过揭示物种间的进化关系,这种方法为生物进化研究提供了重要的线索。