构建分子进化树
分子进化树构建方法

MP法建树流程
Sequence1 Sequence2 Sequence3
Sequence4
Position 1
Position 1 2 3 T G C T A C A G G A A G
If 1 and 2 are grouped a total of four changes are needed.
5
genetic change
系统发生树术语
Rooted tree vs. Unrooted tree
无 A 有 根 根 树 B 树 two major ways to root trees:
A
10 3 2 5
C D
By midpoint or distance
d (A,D) = 10 + 3 + 5 = 18 Midpoint = 18 / 2 = 9
Distance Uses only pairwise distances Minimizes distance between nearest neighbors Very fast Easily trapped in local optima Good for generating tentative tree, or choosing among multiple trees Maximum parsimony Uses only shared derived characters Minimizes total distance Maximum likelihood Uses all data Maximizes tree likelihood given specific parameter values Very slow Highly dependent on assumed evolution model Good for very small data sets and for testing trees built using other methods
分子进化树构建方法
C B
2
D
outgroup
外群、外围支
Rooted tree vs. Unrooted tree
plant animal
plant
plant animal
Unrooted tree
fungus
animal
bacterium
plant plant plant
animal
Rooted tree
Monophyletic group
Cat Dog Rat Cow 3 4 6 5 7 6 Dog Dog Rat Cat
1
2 2 1 4
计算序 列的距 离,建 立距离 矩阵
Rat
通过距 离矩阵 建进化 树
Cow
Step1. 计算序列的距离,建立距离矩阵
对位排列, 去除空格 (选择替代模型)
Uncorrected “p” distance (=observed percent sequence difference) Kimura 2-parameter distance (estimate of the true number of substitutions between taxa)
A
节点 Node
祖先节点/树 根
Root
内部节点/分歧点
该分支可能的祖先 HTU
系统发生树术语
A clade(进化支) is a group of organisms that includes an ancestor and all descendents of that ancestor. 分支树
Step2. 通过矩阵建树 由进化距离构建进化树的方法有很多,常见有:
1. Unweighted Pair Group Method with Arithmetic mean (UPGMA)
分子系统发育树构建的简易方法

分子系统发育树构建的简易方法
分子系统发育树的构建是根据分子序列的差异来推断不同物种之间的进化关系。
下面是一个简易的分子系统发育树构建方法:
1. 选择目标基因序列:选择与所研究物种相关的基因序列(如核糖体RNA或蛋白质编码基因)作为目标序列。
2. 数据收集:收集各个相关物种的目标基因序列数据。
可以通过公共数据库(如NCBI)或研究文献中的已有数据进行获取。
3. 序列比对:使用序列比对软件将收集到的序列进行比对,找出相同和不同的碱基或氨基酸位置。
常用的比对软件有CLUSTALW和MAFFT。
4. 构建进化树:根据序列比对结果,使用进化树构建软件(如MEGA)进行系统发育树的构建。
常用的进化树构建方法包括最大简约法(UPGMA)和最大似然法(ML)。
5. 进化树评估:对构建的系统发育树进行评估,可以使用Bootstrap方法进行支持值分析,提高树的可靠性。
6. 结果解读:根据构建的系统发育树,可以解读不同物种之间的进化关系和群体间的分化程度。
需要注意的是,分子系统发育树是基于目标基因序列的进化关系推断,仅仅代表目标基因的进化历史,并不一定能完全反映
整个物种的进化历史。
因此,在研究中还需要综合考虑其他重要因素,如形态特征和生态行为等。
分子进化树算法

分子进化树算法分子进化树算法是一种用于研究生物进化关系的计算方法。
通过分析DNA、RNA或蛋白质序列的差异和相似性,可以构建出生物物种的进化树。
本文将介绍分子进化树算法的原理、应用和局限性。
一、原理分子进化树算法的原理基于遗传变异和进化。
生物个体的遗传信息通过DNA、RNA或蛋白质序列传递给后代,而在这个过程中会出现突变和重组等变异事件。
这些变异事件积累起来,形成了不同物种之间的差异。
分子进化树算法通过比较不同物种之间的序列差异和相似性,来推断它们之间的进化关系。
具体而言,分子进化树算法首先收集不同物种的DNA、RNA或蛋白质序列数据,然后利用计算方法计算它们之间的差异和相似性。
常用的计算方法包括序列比对、距离计算和进化模型推断。
通过这些计算,可得到一个差异矩阵或距离矩阵,它描述了不同物种之间的关系。
接下来,算法会利用这个矩阵来构建进化树,常见的构建方法有最小进化树、最大似然法和贝叶斯推断等。
二、应用分子进化树算法在生物学研究中有着广泛的应用。
首先,它可以帮助研究者揭示不同物种之间的进化关系。
通过构建进化树,可以了解物种的亲缘关系、起源时间和地理分布等信息。
这对于研究物种的进化历史和生态演化具有重要意义。
分子进化树算法可以用于物种鉴定和系统学研究。
在分类学中,鉴定物种是一个基础性任务。
通过分析物种的分子序列,可以判断它们是否属于同一物种,进而指导分类学的研究和实践。
分子进化树算法还可以用于研究基因功能和基因家族的进化。
通过比较不同物种中的基因序列,可以推断基因的功能和进化过程。
这对于深入理解基因的演化和功能具有重要意义。
三、局限性尽管分子进化树算法在生物学研究中有广泛应用,但也存在一些局限性。
首先,算法的结果受到数据质量和选择的进化模型的影响。
如果数据质量不高或选择的进化模型不合适,可能会导致结果的不准确性。
分子进化树算法无法解决样本不完整或有限的情况。
如果物种样本有限或者存在缺失数据,算法可能无法准确地构建进化树。
分子进化学中的进化树构建方法

分子进化学中的进化树构建方法随着科技的进步和生物技术的广泛应用,分子生物学的研究逐渐深入,成为生物学、生物技术和医药学等领域的重要研究方向。
而分子进化学作为分子生物学中的一个重要分支,研究物种间的分子差异和进化关系。
其中,构建进化树是分子进化学研究中的重要工作,下面我们来了解一下进化树构建的方法。
一、进化树的基本概念进化树是描述不同物种、不同基因或不同蛋白质之间进化关系的图形化表示。
在进化树中,每一个分支代表了一个物种、一个基因或一个蛋白质序列,分支的长度表示了物种、基因或序列的进化距离,而进化距离则是衡量不同物种或不同序列之间关系的基本参数。
而构建进化树的过程则是根据分子序列数据的重构得到物种或基因的进化树。
二、进化树的构建方法构建进化树有多种方法,主要有距离矩阵法、系统发育学法、最大似然法和贝叶斯法等。
下面我们逐一介绍这些方法的基本原理。
1.距离矩阵法距离矩阵法是最早采用的一种构建进化树的方法,它基于序列之间的距离矩阵计算和聚类方法来得到进化树。
该方法首先计算所有分子序列之间的距离(距离可由序列相似性计算得出),然后根据聚类方法构建进化树。
聚类方法包括单链接聚类、均链接聚类和最大链接聚类等。
距离矩阵法的优点是构建速度快、适用性广,但是对于高变异的序列来说,该方法可能会产生误导性的结果。
2.系统发育学法系统发育学法是基于系统学原理,采用系统发生学的理论和方法来构建进化树。
该方法主要是通过分子序列的相似性构建系统发育分析矩阵,然后利用不同的计算方法(如UPGMA、NJ和ML等)推断进化树。
系统发育学法的优点是能够更准确地反映分子序列的演化,并且可以通过不同的方法比较结果,但是该方法需要大量的计算资源和长时间的计算。
3.最大似然法最大似然法是一种统计学上的方法,通过最大化序列数据与观测数据的相似度,来推断出最可能的进化树。
该方法需要整合进化模型和数据,然后计算不同进化模型下数据的似然函数,最终选择似然度最大的进化树。
phylophlan构建进化树的原理

一、phylophlan的介绍phylophlan是一种用于建立进化树的工具,它利用基因组学数据来推断生物进化关系的工具。
通过比较不同物种的基因组序列,phylophlan可以帮助研究者理解生物物种之间的遗传差异和亲缘关系。
二、phylophlan的原理1. 建立物种基因组数据库phylophlan需要建立一个包含各种物种基因组数据的数据库。
这些基因组数据可以来自公共数据库或者用户自己的实验数据。
这些基因组数据包含了各种物种的DNA序列信息。
2. 提取共同的基因片段phylophlan会从不同物种的基因组数据中提取共同的基因片段。
这些共同的基因片段通常被称为核心基因组。
这些核心基因组在不同物种中存在,并且具有一定的保守性,可以在物种之间进行比较和分析。
3. 构建物种间的进化模型接下来,phylophlan利用这些核心基因组数据来构建不同物种之间的进化模型。
进化模型可以反映不同物种之间的亲缘关系和演化历史。
phylophlan利用这些进化模型来推断物种之间的共同祖先和演化路径。
4. 构建进化树phylophlan根据构建的进化模型,利用一定的算法来建立进化树。
进化树可以显示物种之间的亲缘关系和演化路径。
通过进化树,研究者可以更好地理解不同物种之间的遗传差异和演化关系。
三、应用举例1. 物种分类和演化研究phylophlan可以帮助生物学家更好地理解不同物种之间的遗传差异和亲缘关系,为物种分类和演化研究提供重要的工具和数据支持。
2. 药物研发和生物技术应用在药物研发和生物技术应用领域,phylophlan可以帮助科研人员对不同物种的基因组进行比较分析,找到相关的基因和信号通路,为药物研发和生物技术的应用提供重要的参考和支持。
四、结论phylophlan作为一种用于建立进化树的工具,可以帮助生物学家更好地理解不同物种之间的遗传差异和亲缘关系,为物种分类和演化研究提供重要的工具和数据支持。
phylophlan在药物研发和生物技术应用领域也具有重要的应用前景。
怎样使用MEGA建立进化树

怎样使用MEGA建立进化树在进行生物信息学研究中,建立进化树是一项非常重要的任务。
MEGA (分子进化遗传学分析)是一款常用的软件,专门用于进行进化树和多序列分析。
下面将详细介绍如何使用MEGA建立进化树。
安装完成后,打开MEGA软件。
在MEGA的主界面上,有几个常用的功能选项,包括「File」、「Edit」、「View」、「Tools」、「Align」、「Phylogeny」和「Help」。
我们主要关注「Phylogeny」(进化树)选项。
在新窗口中,我们需要选择构建进化树的方法。
MEGA支持多种构建进化树的方法,包括Neighbor Joining、Maximum Parsimony、Maximum Likelihood和Bayesian等。
在这里,我们以Neighbor Joining方法为例进行演示。
在Neighbor Joining方法中,我们需要先选择计算进化距离的方法。
MEGA支持许多计算进化距离的方法,如P-distance、Kimura 2-parameter、Tamura 3-parameter等。
在这里,我们选择P-distance方法。
在选择了计算进化距离的方法后,我们还需要选择树的标准。
MEGA支持Bootstrap(Bootstrap方法是统计学中一种用于评估统计性信号和树的可靠性的方法)和Nearest-Neighbor Interchange等标准。
在这里,我们选择Bootstrap标准。
在选择了进化距离的方法和树的标准后,我们需要选择输入序列数据的文件格式。
MEGA支持多种格式的序列文件,如FASTA、PHYLIP和MEGA 等。
选择相应的格式后,我们需要导入序列数据。
可以通过从文件中导入或从剪贴板中粘贴来导入序列数据。
MEGA是一款非常强大的进化树分析软件,但对于初学者来说,可能需要一些时间去了解其中的各种选项和功能。
因此,建议在使用MEGA之前,先阅读相关文档和教程,以便更好地使用MEGA进行进化树的构建和分析。
建立进化树的方法

建立进化树的一般步骤:MEGA 的全称是Molecular Evolutionary Genetics Analysis 分子进化遗传分析。
MEGA 可用于序列比对、进化树的推断、估计分子进化速度、验证进化假说等。
MEGA 还可以通过网络(NCBI)进行序列的比对和数据的搜索。
打开软件选择Alignment ---- Alignment Explorer/CLUSTAL,出现一个对话框:根据提示内容,进行选择,在此我选择第一个“Create a new alignment”,出现:根据自己的序列是核酸还是氨基酸序列进行选择,在此我选择“Yes”,出现:Date --- Open --- Retrieve Sequences from File ,选择已在Clustal X中已对齐的格式文件[CLUSTAL文件(.aln)],如下图:选择之后,得到:双击文件名可以进行修改(某些Clustal X版本无法识别原FASTA文件名的,在这里就可以修改了,就像我用汉化版的Clustal X 1.81不可以识别某些序列文件名) ,修改后如下:右键菜单点击删除Clustal X中附带的“※”号行,修改文件名后可以保存“当前比对结果”,以便下次再用。
然后再补充一下,此软件整合了Clustal X程序,菜单Alignment中选择“Align by ClustalX”即可。
选择所要比对的序列,单击后出现下面这个对话框:选择默认设置,点击OK就进行比对了。
此后会出现一个过渡对话框,显示的是两两比对和多序列比对的过程:等待其运行完成后,可以保存,也可以直接删除,出现对话框:选择Yes,出现:输入一个名称,如SIV-N2,接下来几步类似,保存后点YES出现:当这个序列数据界面出来后,注意软件的主界面发生了一定的变化,多出了几个功能菜单:选择主界面中的Phylogeny菜单,Bootstrap Test of Phylogeny --- Neighbor-joining…Bootstrap选择1000次重复,模型选择核酸---p-distance。
分子进化树构建的简要步骤(以蛋白为例)

分⼦进化树构建的简要步骤(以蛋⽩为例)PhyML利⽤氨基酸序列建树步骤(核酸建树也可以作为参考)前⾔:本⽂阅读对象适合建树新⼿,⽣物信息学⾼⼿请勿嘲笑,其中有什么错误还恳请指点。
为什么要建树及其你要解决什么问题这⾥不做讨论,只是⼀个纯粹的建树过程,前期的序列收集过程⾃⼰费⼼,根据⾃⼰的需要来做。
这⾥主要是最⼤似然法来建树,NJ法像mega这些软件中都有集成,最新的mega7也集成ML法,不过模型及各种参数不⼀定适合你,所以学习多种多种⽅法也是有⽤的,PhyML速度较慢,如果数列数量较多、步长检验次数多,等待时间会很长,有可能达到⼏⼗⼩时,也与电脑配置有关,⼀般时间都是以⼩时计数,所以要有⼼理准备,如果数据量⼤,推荐⽤RaxML或其他⽅法建树,它处理速度要⽐PhyML 快,不过RaxML是纯命令操作,对不熟悉命令及参数意义的⼈有⼀定难度,我只在linux 下操作过,在win下没有使⽤过。
本⽂是⽤氨基酸建树过程,如果你是⽤核酸序列建树,也可以参考这个过程,核酸替代模型请⽤jmodeltest或其他同功软件计算。
由于PhyML计算过程⽐较长,做⼀遍⽐较耗时,推荐你⽤其他软件⽤NJ法先⾏试验建树,看看你选择的序列是否有效及符合你的预期结果,调整好序列后再⽤PhyML跑⼀遍看结果是否符合⾃⼰的要求。
PhyML有线上版本,只需要提交序列⽐对结果,设置模型参数,留下邮箱等待就会给你返回结果,不过时间不可控,根据⾃⾝情况选择线上还是本地⾃⼰建树。
⽔平有限,如有错误遗漏恳请各位指点。
如果在⽂库不能下载,可以去⽹盘下载,见⽂末。
●建树过程:序列准备-模型选择-建树及树的验证。
●环境准备:电脑^-^Windows或者Linux都可以(没试过mac,如果是mac环境,请参考具体的操作⼿册)、ProtTest、PhyMl及序列⽐对的软件,线上或本地都可以。
1.序列准备:在⾃⼰熟悉的数据库中(我⾃⼰⽐较熟悉Ncbi)上做blast,选取跟要建树蛋⽩同源的各物种序列,下载到本地,整合到⼀个fasta⽂件中,注意修改物种名称,字数最好不要太长,序列⽐对后.phy格式⽂件对⽂件名长度有限制(这个可能跟软件有关系,只要⾃⼰知道是什么物种,不⾄于混淆就⾏),注意规范性,fasta⽂件中最好除了>头标,字母及下划线不要有其他不相关的字符,因为如果后⾯你要⽤软件分析.phy⽂件的时候这些软件对.phy的格式要求⽐较变态,有其他多余字符它都会报错的(你如果在dos 下⽤命令合并⽂件请注意⽂件中最后⼀⾏的字符,请删除)。
分子进化:系统树的构建_图文(精)

计:ˆ1 + v ˆ 2 = K 12 v ˆ1 + v ˆ3 = K 13 v ˆ2 + v ˆ3 = K 23 v 估值为 1 ( K 12 + K 13 − K 23 2 1 ˆ2 = ( K 12 + K 23 − K 13 v 2 1 ˆ3 = (K 13 + K 23 − K 12 v 2 实际序列并非具有相等的碱基频率,因而 Jukes-Cantor 距离不会使似然值最大,但它们的确为迭代法提供了很好的初始值。
Newton-Raphson 迭代法为找 -vi 到最大似然值的数值解提供了直接的方法,且从寻求 pi=1-e 的估值来看,这一方法在描述上是最为简单的。
表 5.7 给出了图 5.4 中人类(1、大猩猩(2、长臂猿(3线粒体序列收敛过程的例子。
三个序列间的平均碱基频率用作模型中的概率项πi。
ˆ1 = v 表 5.7 图 5.4 中人类、大猩猩和长臂猿线粒体序列非约束型最大似然树分枝长度的连续迭代 v2 v3 迭代 v1 初始值 0.0423 0.0174 0.2215 1 0.0420 0.0196 0.2230 2 0.0420 0.01990.2299 3 0.0420 0.0199 0.2299 标准差 0.0297 0.0218 0.0600 用几个序列作为树端来构建系统树时,可采用以上所述的一般方法。
先指定一种系统树,然后对来自该系统树似然函数的方程进行 Newton-Raphson 迭代来估计分枝长度。
在理论上,应研究所有可能的系统树来寻找具有最大似然值的系统树。
Fukami 和 Tateno(1989证实至多存在一组对于 L 给出平稳值的分枝长度,且这组分枝长度提供了所需的最大似然估计。
将这一方法应用于图 5.4 所列的 5 种线粒体序列,获得了图 5.16 所示的无根树状图。
117人类 0.015 0.030 1 0.000 黑猩猩大猩猩 0.000 0.051 0.045 2 3 0.138 猩猩长臂猿图 5.16 利用 Felsenstein 的 PHYLIP 软件构建的图 5.4 线粒体序列资料的最大似然树四.对系统树 Bootstrap 抽样在任一特定的树状拓扑结构内,已知最大似然值提供了分枝长度的一致估计值,这意味着随着资料量的增加,估计值逐渐接近真值。
几款软件构建生物进化树的方法比较

生物进化树的构建目录前言 (2)一、 NCBI (6)二、 Mega (9)三、 DNAMAN (15)四、DNAStar (18)五、 Bio edit (21)前言1.背景资料进化树(evolutionary tree)又名系统树(phylogenetie tree)进化树,用来表示物种间亲缘关系远近的树状结构图。
在进化树中,各个分类单元(物种)依据进化关系的远近,被安放在树状图表上的不同位置。
所以,进化树简单地表示生物的进化历程和亲缘关系。
已发展成为多学科(包括生命科学中的进化论、遗传学、分类学、分子生物学、生物化学、生物物理学和生态学,又包括数学中的概率统计、图论、计算机科学和群论)交叉形成的一个边缘领域。
归纳总结生物进化的总趋势有以下几类:①结构上:由简单到复杂②生活环境上:由水生到陆生③进化水平上:由低等到高等一般来说,进化树是一个二叉树。
它由很多的分支和节点构成。
根据位置的不同,进化树的节点分为外部节点和内部节点,外部节点就是我们要进行分类的分类单元(物种)。
而物种之间的进化关系则用节点之间的连线表示。
内部节点表示进化事件发生的地方,或表示分类单元进化的祖先。
在同一个进化树中,分类单元的选择应当标准一致。
进化树上不同节点之间的连线称为分支,其中有一端与叶子节点相连的分支称为外枝,不与叶子节点相连的分支称为内枝。
进化树一般有两种:有根树和无根树。
有根树有一个鲜明的特征,那就是它有一个唯一的根节点。
这个根节点可以理解为所有其他节点的共同祖先。
所以,有根树能可以准确地反映各个物种的进化顺序,从根节点进化到任何其他节点只有能有一条惟一的路径。
无根树则不能直接给出根节点,无根树只反映各个不同节点之间的进化关系的远近,没有物种如何进化的过程。
但是,我们可以在无根树种指派根节点,从而找出各个物种的进化路径。
无根树有根树放射树分子进化树(以分子数据为依据构建的进化树)不仅精确地反映物种间或群体间在进化过程中发生的极微细的遗传变异(小至一个氨基酸或一个核昔酸差异),而且借助化石提供的大分子类群的分化年代能定量地估计出物种间或群体间的分化年代,这对进化论的研究而言无疑是一场革命。
利用mega构建树原理

利用mega构建树原理
Mega构建树的原理主要基于系统发育树(又称分子进化树)的概念。
这是一种描述一群有机体发生或进化顺序的拓扑结构,用于在生物信息学中描述不同生物之间的相关关系。
拓扑结构将讨论范围内的事物之间的相互关系表示出来,将这些事物之间的关系通过图表示出来。
Mega软件可用于序列比对、进化树的推断、估计分子进化速度、验证进化假说等。
在构建系统发育树时,它采用了一系列的算法和模型,如邻接法(NJ)、最大似然法(ML)、最大简约法(MP)和贝叶斯法(Bayes)等。
这些方法和模型的选择取决于具体的数据和研究目标。
构建系统发育树的一般过程包括以下几个步骤:
1. 数据准备:收集需要研究的物种的基因或蛋白序列,并进行比对,以确保它们的同源性。
比对的结果可以保存为特定的格式,如FASTA。
2. 模型选择:根据数据的特性,选择一个合适的进化模型。
例如,对于DNA序列,可以选择GTR、TN93、HKY等模型;对于蛋白序列,可以选择JTT、WAG、LG等模型。
3. 树的构建:使用选择的模型和方法,构建系统发育树。
这个过程可能包括搜索最优的树结构、计算分支长度等。
4. 树的评估和优化:通过一些统计方法,如自展值(Bootstrap)等,对构建的树进行评估和优化,以提高其可靠性。
需要注意的是,构建系统发育树是一个复杂的过程,需要一定的专业知识和经验。
同时,由于生物进化的复杂性,构建的树可能并不完全准确,需要结合其他证据进行解释和验证。
单倍型的分子系统树

单倍型的分子系统树一、概述单倍型的分子系统树是一种基于DNA序列数据构建的进化树,它能够反映物种间的亲缘关系和演化历史。
在单倍型分子系统树中,只考虑一个个体所拥有的一套染色体中某一个位点上的等位基因,这就是所谓的单倍型。
本文将从单倍型分子系统树的构建方法、应用领域、优缺点等方面进行详细介绍。
二、构建方法1. 样本收集:首先需要收集不同物种或个体之间相同位点上的DNA 序列数据。
2. 序列比对:将收集到的DNA序列进行比对,以确定它们之间的异同。
3. 构建进化模型:通过比对结果来确定不同物种或个体之间遗传差异程度,并选择合适的进化模型。
4. 构建进化树:利用选择出来的进化模型构建进化树。
三、应用领域1. 生物分类学研究:单倍型分子系统树可以被用来探究不同物种或亚种之间的亲缘关系和演化历史,为生物分类学研究提供了有力支持。
2. 种群遗传学研究:单倍型分子系统树可以被用来研究种群间的遗传结构和遗传多样性,为种群遗传学研究提供了有力工具。
3. 进化生物学研究:单倍型分子系统树可以被用来探讨不同物种或个体之间的进化历史和演化模式,为进化生物学研究提供了有力支持。
四、优缺点1. 优点:(1)能够准确反映物种间的亲缘关系和演化历史;(2)数据收集方便,成本较低;(3)能够对不同物种或个体之间的遗传差异进行量化比较。
2. 缺点:(1)只考虑一个个体所拥有的一套染色体中某一个位点上的等位基因,无法全面反映整个基因组的信息;(2)在构建进化树时需要选择合适的进化模型,选择不当可能会导致结果产生误差。
五、结论总之,单倍型分子系统树是一种重要的分子生物学工具,在生物分类学、种群遗传学和进化生物学等领域都有广泛应用。
虽然它也存在一些缺点,但是其优点仍然使它成为研究生物进化和演化历史的重要工具之一。
分子进化树构建及数据分析的简介

分子进化树构建及数据分析的简介开始动笔写这篇短文之前,我问自己,为什么要写这样的文章?写这样的文章有实际的意义吗?我希望能够解决什么样的问题?带着这样的疑惑,我随手在丁香园(DXY)上以关键字“进化分析求助”进行了搜索,居然有289篇相关的帖子(2006年9月12日)。
而以关键字“进化分析”和“进化”为关键字搜索,分别找到2,733和7,724篇相关的帖子。
考虑到有些帖子的内容与分子进化无关,这里我保守的估计,大约有3,000~4,000篇帖子的内容,是关于分子进化的。
粗略地归纳一下,我大致将提出的问题分为下述的几类:1.涉及基本概念。
例如,“分子进化与生物进化是不是一个概念”,“关于微卫星进化模型有没有什么新的进展”以及“关于Kruglyak的模型有没有改进的出现”,等等。
2.关于构建进化树的方法的选择。
例如,“用boostrap NJ得到XX图,请问该怎样理解?能否应用于文章?用boostrap test中的ME法得到的是XXX树,请问与上个树比,哪个更好”,等等。
3.关于软件的选择。
例如,“想做一个进化树,不知道什么软件能更好的使用且可以说明问题,并且有没有说明如何做”,“拿到了16sr RNA数据,打算做一个系统进化树分析,可是原来没有做过这方面的工作啊,都要什么软件”,“请问各位高手用clustalx做出来的进化树与phylip做的有什么区别”,“请问有做过进化树分析的朋友,能不能提供一下,做树的时候参数的设置,以及代表的意思。
还有各个分支等数值的意思,说明的问题等”,等等。
4.蛋白家族的分类问题。
例如,“搜集所有的关于一个特定domain的序列,共141条,做的进化树不知具体怎么分析”,等等。
5.新基因功能的推断。
例如,“根据一个新基因A氨基酸序列构建的系统发生树,这个进化树能否说明这个新基因A和B同源,属于同一基因家族”,等等。
6.计算基因分化的年代。
例如,“想在基因组水平比较两个或三个比较接近物种之间的进化年代的远近,具体推算出他们之间的分歧时间”,“如何估计病毒进化中变异所需时间”,等等。
利用MEGA4构建分子系统进化树
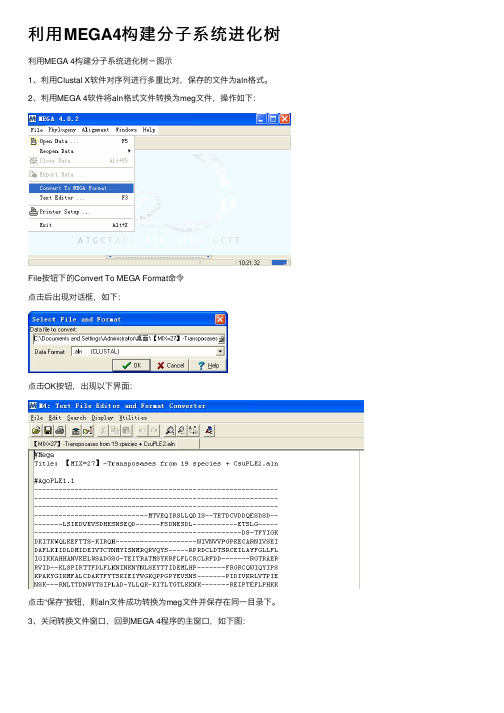
利⽤MEGA4构建分⼦系统进化树利⽤MEGA 4构建分⼦系统进化树-图⽰
1、利⽤Clustal X软件对序列进⾏多重⽐对,保存的⽂件为aln格式。
2、利⽤MEGA 4软件将aln格式⽂件转换为meg⽂件,操作如下:
File按钮下的Convert To MEGA Format命令
点击后出现对话框,如下:
点击OK按钮,出现以下界⾯:
点击“保存”按钮,则aln⽂件成功转换为meg⽂件并保存在同⼀⽬录下。
3、关闭转换⽂件窗⼝,回到MEGA 4程序的主窗⼝,如下图:
点击“Click me to activate a data file”按钮,选择之前转换好的meg⽂件并打开,如下图:
选择所输⼊的数据类型(核酸or蛋⽩),之后点击OK即可。
此时,在MEGA4主程序窗⼝的底部出现了我们所输⼊的⽂件名(如下图),之后就可以构建分⼦系统进化树了。
4、通常选择邻接法(neighbor-joining,NJ)构建分⼦系统进化树。
分子进化树

(2)-(3)+(1)
d=4,e=6
dD eE
=> C最接近DE!
分成三组:C, DE, 以及AB
c+g+(e+d)/2=19 (1) c+f+(a+b)/2=40 (2) (e+d)/2+(a+b)/2+f+g=41 (2) (1)+(2)-(3) => c=9 => g=5
由:(a+b)/2+f+g+(d+e)/2=41 得:f=20 由:a+f+c=39 得:a=10,则b=12
最大简约法(maximumparsimony,MP)
根据信息位点提供的各序列间的 替换情况,在所有可能的树中筛 选含最小替换数的树的方法。
最大简约法(MP)
该方法的理论基础是奥卡姆剃刀原理,即如无必要,勿 增实体(解释一个过程的最好的理论是所需假设数目最 少的那一个)。
主要思想:构造一个反映分类单元之间最小变化的系统 发育树,即选择核苷酸序列全部位点最小核苷酸替代数 之和最小的树作为最优树。
– 可用于任何相关序列集合
• 计算速度:
– 距离法 >最大简约法 >最大似然法
2.1 距离法
• 又称距离矩阵法,首先通过各个物种之间 的比较,根据一定的假设(进化距离模型 )推导得出分类群之间的进化距离,构建 一个进化距离矩阵。再依据进化距离,分 别依次将序列合并聚类,构建进化树。
简单的距离矩阵
• Tree 1最为简约 • MP tree的最优结果为tree 1
最大简约法
如:用最大简约法构建下面4组序列的系统树 AAG AAA GGA AGA
分子进化:系统树的构建

~ =0.7237 所得 Jukes-Cantor Kimura 距离为 0.3513。这与只根据相同碱基比例 q
距离 0.3446 没有本质上的差异。
图 5.3 兔和鸡的β-球蛋白序列。每两条序列上下两行星号表示由转换 (I 型变化)或颠换(Ⅱ型变化)造成的碱基差异。 DNA 序列距离 K 又可称为 DNA 序列间的分歧度(sequence divergence),即 序列间相异性的一个指标。蛋白质序列的分歧度分为两序列同义变化的分歧度 (KS)和非同义变化的分歧度(KA),根据 Jukes-Cautor 单参数模型和 Kimura 两参 数模型等遗传模型,可以分别计算得到两序列的分歧度(或称为蛋白质序列间的 距离)。
98
ห้องสมุดไป่ตู้
祖先序列
A C T G A A C G T A A C G C
A C T G A→C→T A C→G G T→A A A→C→* T C G C
A C→A T G A A C→A G T→A A A * →T C G C→ +T→C
单一置换 (single substitution) 多重置换 (multiple substitutions) 同义置换 (coincidental substitutions) 平行置换 (parallel substitutions) 趋同置换 (convergent substitution) 反转置换 (back substitution)
OUT 数
1 2 3 … t
t d1t d2t d3t … -
用这些距离对 OUT 进行表型意义的分类可借助于聚类分析(clustering), 聚 类过程可以看作是鉴别具有相近 OUT 类群的过程。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
monkey
dog hamster bovine
PQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
LQVRDVELAGAPGEGGLQPLALEGALQKRGIVEQCCTSICSLYQLENYCN PQVAQLELGGGPGADDLQTLALEVAQQKRGIVDQCCTSICSLYQLENYCN PQVGALELAGGPGAGG-----LEGPPQKRGIVEQCCASVCSLYQLENYCN
生物信息学
第五章
多序列对位排列和进化分析 ( I)
多序列对位排列
Multiple Sequence Alignment (MSA)
chicken
xenopቤተ መጻሕፍቲ ባይዱs human
PLVSS---PLRGEAGVLPFQQEEYEKVKRGIVEQCCHNTCSLYQLENYCN
ALVSG---PQDNELDGMQLQPQEYQKMKRGIVEQCCHSTCSLFQLESYCN LQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
Using ClustalX for multiple sequence alignment
by Jarno Tuimala
两种工作模式:
Multiple Alignment
Profile Alignment
第一步:输入序列
File
Load sequences
1、序列为多重fasta格式(可进行编 辑,保存为txt文件)
Clustal使用方法
Clustal:目前应用最广泛的 MSA 方法
可在线分析
可在本地计算机运行 序列输入、输出格式
Input FASTA
NBRF/PIR EMBL/SWISSPROT ALN GCG/MSF GCG9/RSF GDE
/
>sequence 1 ATTGCAGTTCG CA …… >sequence 2 ATAGCACATCG CA…… >sequence 3 ATGCCACTCCG CC……
guinea pig PQVEQTELGMGLGAGGLQPLALEMALQKRGIVDQCCTGTCTRHQLQSYCN
Bring the greatest number of similar characters into the same column of the alignment
为什么要做MSA?
Gene tree
a b
A B
Species tree
c
C
We often assume that gene trees give us species trees
为什么要做MSA?
Contig assembly
怎么做MSA?
动态规划算法(dynamic programming):MSA 改进算法(heuristic algorithm):
3、为便于识别 每条序列,可在 >后输入物种名 称,并用空格和 其它描述内容分 开,如: 2、序列文件所在路径不能有空格和 中文字符(如放在系统桌面),否则 ClustalX无法载入
>Human gi|301129180|ref|NP_001180303.1| resistin [Homo sapiens]
用于描述一组序列之间的相似性关系,以便了解一个基因家族 的基本特征,寻找motif,保守区域等。用于预测新序列的二 级和三级结构,进而推测其生物学功能。
Find out which parts “do the same thing”
为什么要做MSA?
用于描述同源序列之间的亲缘关系的远近,应用到分子进化 分析中。是构建分子进化树的基础。
/Tools/msa/clustalw2/
粘贴或上载序列
调整参数 多序列对位排列结果 Alignments
Result Summary
/Tools/msa/clustalw/help/
Clustal离线分析方法(ClustalX) 下载安装 自带Help文件
可进一步对排列好的序列进行修饰(2)
ESPript 多种修饰 功能,突出相同或相似位点 http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi 在EBI ClustalW结果网页下载“Alignments”(CLUSTALW format)
Output ALN
NBRF/PIR GCG/MSF PHYLIP NEXUS GDE/FASTA
Clustal W/X算法基础
两两比对 构建距离矩阵
构建指导树 (guide tree)
将距离最近的两条 序列用动态规划的 算法进行比对; “渐进”的加上其 他的序列
Clustal在线分析方法(ClustalW) EBI的ClustalW分析网页
第二步:设定比对参数
第三步:进行序列比对,得到结果
第四步:评价比对质量
打开比对结果: 1、可在ClustalX中直接输出打印 2、可用写字板打开aln文件
3、可将aln文件以图形展示,更直观
更改参数、手动编辑,使之具有生物学意义
可进一步对排列好的序列进行修饰(1)
Boxshade 突出相同或相似位点 (/software/BOX_form.html)
1. 渐进法(progressive methods):Clustal, T-Coffee, MUSCLE 2. 迭代法(iterative methods):PRRP, DIALIGN 3. 其它算法:Partial Order Algorithm、profile HMM、 meta-methods (MAFFT)… /Tools/msa/ Current Opinion in Structural Biology 2006, 16:368–373
在EBI ClustalW结果网页复制序列比对结果
在“Boxshade”网页粘贴序列,在“Input sequence format”栏目选择“ALN”,在“Output format”栏目 选择“RTF_new”
在结果网页点击“here is your output number 1”
修饰过的排列结果