构建系统发育树需要注意的几个问题
系统发育进化树构建

系统发育进化树构建【实用版】目录一、什么是系统发育进化树二、系统发育进化树的构建方法三、系统发育进化树的应用四、总结正文一、什么是系统发育进化树系统发育进化树是一种用来表示物种或基因间亲缘关系的树状图,它可以利用树状分支图形来展示生物之间的进化关系。
系统发育进化树主要用于研究物种或序列的进化和系统分类,其研究对象通常包括碱基序列或氨基酸序列。
二、系统发育进化树的构建方法系统发育进化树的构建过程称为分支系统发育分析,它通过数理统计算法来计算生物间的进化距离,并以此为基础构建进化树。
以下是构建系统发育进化树的主要步骤:1.选择研究对象:首先需要选择合适的研究对象,例如碱基序列或氨基酸序列。
2.获取数据:搜集研究对象的相关数据,这通常需要通过实验或数据库获取。
3.计算进化距离:利用数理统计算法(如距离法、最大似然法等)计算不同生物间的进化距离。
4.构建进化树:根据进化距离构建树状分支图,通常使用聚类方法或最小生成树算法。
5.检验树状图:对构建好的进化树进行检验,以确保其符合生物学实际情况。
三、系统发育进化树的应用系统发育进化树在生物学研究中有广泛的应用,主要包括:1.物种分类和演化关系研究:通过构建进化树,可以了解不同物种之间的亲缘关系和演化历史。
2.基因功能预测:根据基因在进化树上的位置,可以推测基因的功能和作用。
3.基因调控关系分析:进化树可以帮助研究者了解基因之间的调控关系,从而揭示生物过程的调控机制。
4.病原体演化研究:对于病原体,进化树可以揭示其演化历程,有助于疫苗设计和疾病防治。
四、总结系统发育进化树是一种重要的生物学研究方法,它可以帮助研究者揭示物种或基因间的亲缘关系和演化历史。
系统发育进化树构建

系统发育进化树构建1. 什么是系统发育进化树?系统发育进化树(Phylogenetic Tree),也称为系统树或进化树,是生物学中常用的一种图形表示方法,用于展示不同物种之间的亲缘关系以及它们的进化历史。
系统发育进化树可以帮助我们理解生物多样性的起源、演化以及物种之间的关系。
2. 构建系统发育进化树的方法2.1 形态学特征比较法形态学特征比较法是构建系统发育进化树最早也是最常用的方法之一。
通过比较不同物种的形态特征,如体型、颜色、器官结构等,来推断它们之间的亲缘关系。
这种方法适用于无法进行分子遗传学研究的古生物学领域。
2.2 分子遗传学方法分子遗传学方法是目前构建系统发育进化树的主要手段之一。
它利用DNA、RNA、蛋白质等分子的序列信息来推断不同物种之间的亲缘关系。
常用的方法包括序列比对、构建进化模型、计算进化距离等。
2.3 组织化石记录法组织化石记录法是通过研究化石中的细胞结构、细胞组织等信息,来推断不同物种之间的亲缘关系。
这种方法适用于无法获取分子遗传学信息的古生物学领域。
3. 构建系统发育进化树的步骤3.1 收集相关数据构建系统发育进化树的第一步是收集相关的数据,包括形态学特征数据、分子序列数据或化石记录数据。
数据的准确性和全面性对于构建准确的进化树非常重要。
3.2 数据处理与分析在收集到数据后,需要对数据进行处理和分析。
对于形态学特征数据,可以通过比较不同物种的特征值来计算相似性矩阵;对于分子序列数据,可以进行序列比对和计算进化距离等操作。
3.3 构建进化模型在数据处理与分析的基础上,需要选择合适的进化模型来描述不同物种之间的进化关系。
常用的进化模型包括NJ(Neighbor-Joining)方法、ML(Maximum Likelihood)方法和Bayesian方法等。
3.4 构建进化树在选择了合适的进化模型后,可以利用计算机软件或在线工具来构建进化树。
常用的软件包括MEGA、PAUP*和MrBayes等。
植物基因家族进化树的构建

植物基因家族进化树的构建一、数据收集在构建植物基因家族进化树之前,需要收集相关的基因序列数据。
这些数据可以通过各种数据库,如NCBI、Ensembl等获取。
在收集数据时,需要注意以下几点:1. 选择具有代表性的物种,覆盖尽可能多的系统发育分支;2. 确保所收集的基因序列数据质量可靠,无测序错误和拼接错误;3. 对于每个基因家族,应尽可能收集多个成员的序列,以便进行多序列比对和树的构建。
二、序列比对在获得基因序列数据后,需要进行多序列比对。
比对的目的是为了找到不同物种间基因序列的相似性和差异性,从而确定它们之间的系统发育关系。
常用的多序列比对软件有MUSCLE、CLUSTAL W等。
在进行多序列比对时,需要注意以下几点:1. 选择合适的比对参数,以保证比对结果的准确性和可靠性;2. 在比对过程中,需要注意保持基因序列的原始阅读框,避免引入不必要的拼接错误;3. 对于较长的基因序列,可以分段进行比对,以提高计算效率和准确性。
三、距离矩阵计算在多序列比对的基础上,需要计算不同物种间基因序列之间的距离。
距离矩阵的计算是树构建的重要步骤之一。
常用的距离矩阵计算方法有:1. 欧氏距离法:直接计算不同物种间基因序列的差异数目,得到距离矩阵;2. Kimura距离法:基于Kimura模型计算不同物种间基因序列的差异概率,得到距离矩阵;3. Jukes-Cantor距离法:考虑基因序列的突变率和进化速率,计算不同物种间基因序列的差异概率,得到距离矩阵。
在选择距离矩阵计算方法时,需要根据具体情况选择适合的方法。
如果数据量较大或序列较短时,可以考虑使用欧氏距离法;如果数据量较小或序列较长时,可以考虑使用Kimura或Jukes-Cantor距离法。
四、树构建方法选择在获得距离矩阵后,需要选择合适的树构建方法来构建进化树。
常用的树构建方法有:1. UPGMA(Unweighted Pair Group Method with Arithmetic Mean):将距离矩阵中的行或列进行聚类分析,根据聚类结果构建树;2. Neighbor Joining:基于距离矩阵中的最近邻关系构建树;3. Maximum Parsimony:基于树的构建准则函数(如最小改变数、最小代价等)构建树。
构建系统发育树的步骤

构建系统发育树的步骤
1. 收集种群样本:首先要选择一组相关的生物种群作为研究对象,并从不同地理区域或时间点采集具有代表性的样本。
2. 提取DNA:从样本中提取DNA,通常使用PCR技术扩增并纯化DNA。
3. 测序:对DNA进行测序,可以通过Sanger测序或高通量测序方法。
4. 序列比对:将不同样本的DNA序列进行比对,查找共有的相同或不同点。
5. 构建系统发育树:根据不同生物之间DNA序列的相似度,对各种生物类群进行分类和排列,以此建立系统发育树。
6. 验证树的假设:进行统计推断和深入分析,验证系统发育树的假设,确保树的枝条和叶节点都具有生物学意义。
7. 解释结果:解释树的拓扑结构和演化关系,关注不同物种或层级之间的相似性和差异性,并从中探究生物的进化历史和机制。
MEGA软件——系统发育树构建方法
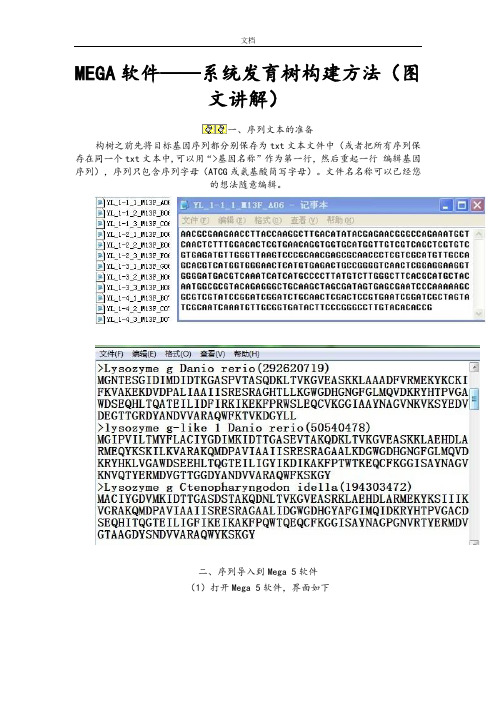
MEGA软件——系统发育树构建方法(图
文讲解)
一、序列文本的准备
构树之前先将目标基因序列都分别保存为txt文本文件中(或者把所有序列保存在同一个txt文本中,可以用“>基因名称”作为第一行,然后重起一行编辑基因序列),序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您
的想法随意编辑。
二、序列导入到Mega 5软件
(1)打开Mega 5软件,界面如下
(2)导入需要构建系统发育树的目的序列
OK
选择分析序列类型(如果是DNA序列,点击DNA,如果是蛋白序列,点击Prot
ein)
出现新的对话框,创建新的数据文件
选择序列类型
导入序列
导入序列成功。
(3)序列比对分析
点击工具栏中“W”工具,进行比对分析,比对结束后删除两端不能够完全对齐
碱基
(4)系统发育分析
关闭窗口,选择保存文件路径,自定义文件名称
三、系统发育树构建
根据不同分析目的,选择相应的分析算法,本例子以N—J算法为例
Bootstrap 选择1000,点击Compute,开始计算
计算完毕后,生成系统发育树。
文档
根据不同目的,导出分析结果,进行简单的修饰,保存。
系统发育树的序列的选择标准

系统发育树的序列的选择标准系统发育树(Phylogenetic tree)是一种用于描述物种之间进化关系的图形表示方法。
它通过比较不同物种的遗传信息(如DNA序列、蛋白质序列等),来确定它们之间的亲缘关系和进化历史。
在构建系统发育树时,选择合适的序列是非常重要的,因为它直接影响到树的准确性和可靠性。
以下是选择系统发育树序列的一些标准:1. 高质量序列:选择高质量的序列可以提高系统发育树的准确性。
高质量的序列通常具有较低的错误率、较高的覆盖率和较少的重复区域。
此外,高质量的序列还应该来自可靠的数据源,如公开数据库或经过同行评审的文献。
2. 物种代表性:为了构建一个全面的系统发育树,需要选择尽可能多的物种进行比较。
在选择序列时,应尽量选择各个分支上的物种,以便更好地反映物种之间的进化关系。
同时,还应注意选择具有代表性的物种,避免选择过于相似或过于不同的物种。
3. 基因特异性:系统发育树的构建通常基于特定基因的序列比较。
因此,在选择序列时,应考虑基因的特异性。
例如,如果研究的是某种特定的生物学过程,可以选择与该过程相关的基因进行比较;如果研究的是某种特定的生态位,可以选择与该生态位相关的基因进行比较。
4. 序列长度:序列长度对系统发育树的准确性有很大影响。
一般来说,较长的序列具有较高的分辨率,可以更好地区分物种之间的差异。
然而,过长的序列可能会导致计算复杂度增加,从而影响系统发育树的构建速度。
因此,在选择序列时,应根据实际需求和计算能力来选择合适的序列长度。
5. 数据可用性:在选择序列时,应考虑数据的可用性。
首先,应尽量选择已经公开的数据进行比较,以减少不必要的重复工作。
其次,应尽量选择易于获取的数据进行比较,以节省时间和成本。
最后,应尽量选择最新的数据进行比较,以提高系统发育树的准确性。
6. 数据一致性:在选择序列时,应尽量选择一致性较高的数据进行比较。
一致性较高的数据通常具有较低的错误率和较高的可信度。
系统发育树的构建与分析方法

系统发育树的构建与分析方法概述系统发育树是生物学中重要的研究工具,通过构建系统发育树可以探究生物之间的关系,研究进化过程和生物多样性。
本文将介绍系统发育树的构建和分析方法。
系统发育树的构建方法系统发育树的构建方法可以分为以下几种:1. 相似性分析法相似性分析法是最简单和常用的构建系统发育树的方法之一。
该方法通过比较不同物种的形态、行为、生理等特征的相似性,判断它们之间的亲缘关系。
这种方法的局限性在于很多特征可能出现多次独立进化,而不是从共同祖先继承的。
2. 分子序列分析法由于DNA或蛋白质序列的进化是按照分子钟模型进行的,因此分子序列分析成为当前构建系统发育树的最常用和最准确的方法之一。
该方法通过比较生物体DNA或蛋白质序列的差异,建立相似度矩阵,并在此基础上运用数学模型进行树的构建。
3. 基因组分析法基因组分析法通过直接比较不同生物体的基因组,从而确定它们之间的进化关系。
这种方法包括全基因组比较和重构古基因组。
4. 形态-分子组合分析法形态-分子组合分析法是将形态特征和分子特征结合起来分析生物之间的进化关系。
在这种方法中,形态特征通常用于解决分子序列存在误差的问题。
系统发育树的分析方法系统发育树的分析方法包括静态分析和动态分析两种。
静态分析静态分析是指对系统发育树形态和拓扑关系的分析,这种方法主要依靠人工分析和软件分析两种方式。
1. 人工分析法人工分析法主要是通过比较不同树之间的拓扑结构和相应的节点值,判断它们之间的相关性。
人工分析法需要手动绘制树,并用统计方法比较不同树之间的相似性。
2. 软件分析法软件分析法主要是应用多种专业软件进行计算和模拟,比如molecular evolution software suite (MEGA)、PAUP和PhyML等。
这种方法可以减少人力工作,提高分析准确性。
动态分析动态分析是指以时间序列为基础,考察系统发育树演化的过程和趋势。
这种方法主要依靠统计分析方法,如马尔科夫链蒙特卡罗(MCMC)、Bayesian标记链蒙特卡罗(MCMC)等。
初二生物系统发生树构建方法

初二生物系统发生树构建方法生物系统发生树是一种用来描述生物进化关系的图形工具,它可以帮助我们理解不同物种之间的亲缘关系和进化历史。
构建生物系统发生树需要收集大量的生物学数据,并运用一系列的分析方法。
本文将介绍初二生物系统发生树构建的方法。
一、确定研究对象要构建生物系统发生树,首先需要确定研究对象。
可以选择一组具有亲缘关系的物种作为研究对象,比如不同种类的昆虫、鱼类或者植物。
在选择研究对象时,需要考虑它们的进化关系已经被广泛研究并且有可靠的分类信息可供参考。
二、收集分类信息为了构建生物系统发生树,我们需要收集各个物种的分类信息。
这包括它们的科、属、种等分类级别。
分类信息可以通过查阅专业的生物学书籍和数据库获得。
三、收集形态和分子数据除了分类信息,形态和分子数据是构建生物系统发生树的重要依据。
通过观察物种的外部形态特征,比如体型、花朵形状、翅膀结构等,可以推测它们的亲缘关系。
此外,通过分析物种的DNA序列,尤其是核酸或蛋白质序列,可以揭示它们的遗传关系。
因此,我们需要收集和记录物种的形态和分子数据。
四、进行系统发生树分析系统发生学是构建生物系统发生树的主要工具。
它利用形态和分子数据进行计算,通过比较不同物种之间的相似性和差异性,推断它们之间的进化关系。
常用的系统发生学方法包括距离法、最大简约法和最大似然法等。
在进行系统发生树分析前,需要选择合适的方法,并准备好相关的计算软件。
五、树构建在进行系统发生树分析后,我们可以通过计算机软件生成生物系统发生树。
树的构建过程会考虑到物种之间的进化距离和分支长度等因素。
树的构建可以使用专业的系统发生学软件,如MEGA和PHYLIP 等。
生成的树结构可以通过图像输出和基因组数据展示。
六、树解读和分析生成生物系统发生树后,我们需要对树进行解读和分析。
树的分枝结构和分枝长度等特征可以帮助我们理解物种之间的进化关系。
同时,树还可以用来预测物种的共同祖先和演化路径等信息。
通过对树的分析,我们可以深入理解生物的进化历史和多样性。
构建系统发育树的方法

构建系统发育树的方法
系统发育学是生物学中的一个重要分支,它研究的是生物种类之间的亲缘关系。
构建系统发育树是系统发育学的核心内容之一,它可以帮助我们更好地了解生物种类之间的演化历史和亲缘关系。
下面我们来介绍一下构建系统发育树的方法。
1. 形态学比较法
形态学比较法是最早被使用的构建系统发育树的方法之一。
它通过比较不同生物种类的形态特征,来推断它们之间的亲缘关系。
这种方法的优点是简单易行,但是它的缺点也很明显,即不同生物种类的形态特征可能会受到环境因素的影响,从而导致推断结果的不准确性。
2. 分子生物学方法
分子生物学方法是目前最常用的构建系统发育树的方法之一。
它通过比较不同生物种类的DNA序列或蛋白质序列,来推断它们之间的亲缘关系。
这种方法的优点是准确性高,但是它的缺点也很明显,即需要大量的实验数据和计算资源。
3. 综合方法
综合方法是将形态学比较法和分子生物学方法结合起来使用的一种方法。
它通过同时考虑不同生物种类的形态特征和分子序列,来推
断它们之间的亲缘关系。
这种方法的优点是可以克服单一方法的缺点,但是它的缺点也很明显,即需要更多的实验数据和计算资源。
构建系统发育树是一个复杂的过程,需要综合考虑多种因素。
不同的方法各有优缺点,我们需要根据具体情况选择合适的方法来进行研究。
随着科技的不断进步,我们相信未来会有更多更准确的方法被开发出来,来帮助我们更好地了解生物种类之间的亲缘关系。
构建系统发育树的方法

构建系统发育树的方法
构建系统发育树的方法
一、定义
系统发育树(Phylogenetic Tree)又称为系统种群学树,是一
种描述物种演化的树型结构,从根节点开始描述物种主要进化分支结构,树上的每条边则表示两个物种在进化的历史中距离彼此更近或来自同一进化祖先的关系。
二、建立系统发育树的方法
1.收集数据:系统发育树的建立首先要收集数据,作为建立树的基础,这些数据一般是利用各种实验技术来收集,比如遗传学实验和物种形态的实验。
2.选取特征:从收集的大量数据中,应选取尽可能多的可靠特征,作为建立树的材料,这些特征要有规律性,有可靠性,可以容易发现物种之间的内在关系,有利于在研究中可靠地比较各物种之间的相似程度。
3.分类比较:将所有待比较的物种或实体按照类似的特征进行分类,根据同一物种种的特征之间的差异,可以比较出物种之间的相似度,确定出有利于建立树的特征。
4.描绘树枝:根据比较的结果,可以依次将物种分类编码,从根节点开始,逐级分细枝条,最后得出系统发育树的图形结构。
5.校正树枝:检查系统发育树的构建结果,如果发现有一些物种不太符合物种演化过程的规律,可以根据其他数据和结果来校正树枝,
从而得出最终的发育树结构。
系统发育树构建的三种方法

系统发育树构建的三种方法
系统发育树(Systems 发育 Tree,简称Stree)是一种用于描述生物系统进化的图形化工具,通常用于模拟生物系统行为的演化过程。
以下是三种构建系统发育树的方法:
1. 基于规则的方法:这种方法使用预定义的规则和偏好来构建
系统发育树。
例如,可以使用遗传算法或人工神经网络等机器学习方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法需要大量
的人工工作,但可以生成较为准确的演化树。
2. 基于统计方法的方法:这种方法使用统计学方法来推断物种
之间的演化关系。
例如,可以使用最大似然估计或贝叶斯推断等方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法不需要人工工作,但需要更多的计算资源和时间,才能得到比较准确的演化树。
3. 基于模型的方法:这种方法使用已经建立的模型和数据来构
建系统发育树。
例如,可以使用层次结构模型(如生物进化树、社会网络模型等)来预测一个物种的遗传特征或行为演化轨迹。
这种方法可
以快速构建系统发育树,但需要更多的人工工作来验证模型的准确性。
系统发育进化树构建

系统发育进化树构建系统发育进化树(Phylogenetic tree)是一种用于描述物种或群体之间进化关系的图形表示。
通过构建系统发育进化树,我们可以了解不同物种之间的亲缘关系,以及它们的共同祖先。
本文将介绍系统发育进化树的构建方法和其在生物学领域中的应用。
一、系统发育进化树的构建方法1. 选择合适的基因或序列:构建系统发育进化树需要选择适当的基因或序列进行分析。
常用的基因包括核糖体RNA(rRNA)和线粒体DNA(mtDNA)等。
2. 收集物种样本:从不同物种中收集样本,并提取相应的基因或序列。
3. 序列比对:将收集到的序列进行比对,找出它们之间的相同和差异。
4. 构建进化模型:根据序列比对的结果,选择适当的进化模型,如最大似然法或贝叶斯推断等。
5. 构建进化树:利用选定的进化模型,根据序列的相似性和差异性,构建系统发育进化树。
二、系统发育进化树的应用1. 物种分类:系统发育进化树可用于物种分类,帮助我们理解不同物种之间的亲缘关系。
通过比较进化树上的分支长度和节点位置,我们可以判断物种之间的相似性和差异性。
2. 进化研究:系统发育进化树可用于研究物种的进化历史和进化速率。
通过比较不同物种之间的进化树,我们可以了解它们的共同祖先以及它们之间的演化路径。
3. 分子演化研究:系统发育进化树在分子演化研究中起着重要的作用。
通过比较不同物种的基因或序列,我们可以推断它们的演化历史和演化速率。
4. 物种保护:系统发育进化树可用于指导物种保护工作。
通过研究物种的进化关系,我们可以了解哪些物种是濒危物种或有特殊保护需求的物种。
5. 药物开发:系统发育进化树可用于药物开发。
通过比较不同物种的基因或序列,我们可以了解它们之间的差异,并找到可能具有药用潜力的物种。
总结:系统发育进化树是一种重要的工具,用于描述物种或群体之间的进化关系。
通过构建系统发育进化树,我们可以了解不同物种之间的亲缘关系,以及它们的共同祖先。
系统发育进化树在物种分类、进化研究、分子演化研究、物种保护和药物开发等领域都有着广泛的应用。
系统发育树的构建与分析方法

系统发育树的构建与分析方法系统发育学是一门研究物种进化关系的学科,通过对不同物种的形态特征、生理生态特性、分子遗传信息等数据进行分析,可以得出它们在进化树中的位置。
而系统发育树则是用来表示各物种间进化历程和亲缘关系的图形表示。
它不仅可以揭示物种之间的起源和进化演化,而且可以对生物多样性的保护和利用产生重要影响。
本文将介绍系统发育树的构建和分析方法。
一、系统发育树的构建方法1. 形态学方法形态学方法是最早用于构建系统发育树的方法。
它是根据物种的形态特征进行比较研究,如昆虫的翅膀、花的形态、动物的身体部位等。
这种方法的优点是简单易行,但缺点是由于不同物种形态特征的相似性,并不能完全反映它们在进化树上的亲缘程度,有一定局限性。
2. 分子系统发育学方法随着分子生物学的发展,分子数据也开始被应用于系统发育树的构建。
这种方法通过对物种不同基因的序列进行修建的,如rRNA、DNA、蛋白质序列等来了解它们进化的历程和亲缘关系。
因为DNA和蛋白质在进化过程中往往较为保守,且具有一定统一性,因此这种方法比形态学方法更为精确,同时也能够构建更多样化的进化模型。
3. 固定标记法固定标记法是结合形态学和分子方法的一种新技术。
它利用生物体细胞核DNA中的高度变异的区域,如微卫星(Simple Sequence Repeats, SSR)等,通过引物特异性地扩增这些标记区域,将扩增产物的大小与数量(即大小因素和数量因素)组成一个确定的数字序列,用于构建系统发育树。
这种方法因能够同时反映形态和分子信息,且具有标记简单、修建渐进、多样性较高等优点,逐渐成为研究系统发育的新方法。
二、系统发育树的分析方法一旦构建了系统发育树,便需要进行分析以研究物种间的亲缘关系、进化历程等信息。
1. 树形态分析树形态分析包括节点分支的长度、角度、直线距离等进化遗传参数的分析。
通过对树形态的分析,可以更全面地了解不同物种亲缘关系的演化特点。
2. 分子位点分析分子位点分析是指对系统发育树上指定基因位点的序列进行分析,如进化速率、突变次数、转换和替换等信息。
叶绿体基因组系统发育树的构建

叶绿体基因组系统发育树的构建
叶绿体基因组是一种常用的分子标记,可用于构建植物的系统发育树。
构建叶绿体基因组系统发育树的步骤如下:
1. 叶绿体基因组测序:首先进行叶绿体基因组的测序,获取叶绿体基因组的完整序列信息。
2. 叶绿体基因组序列比对:将测得的叶绿体基因组序列与已知的参考序列进行比对,找出相似的序列区域。
3. 序列选择:根据不同研究目的和物种特点,选择一些适当的叶绿体基因组序列作为研究对象。
一般选择高度保守的基因,如rbcl、matK等。
4. 序列比对:将选定的叶绿体基因组序列进行多序列比对,找出序列间的相似性和差异性。
5. 构建系统发育树:利用多序列比对结果,采用系统发育学分析的方法,如最大似然法、最小进化法等,构建叶绿体基因组的系统发育树。
6. 树的评估和解读:对构建的系统发育树进行评估,包括支持值(bootstrap值)和相似度等。
根据系统发育树的拓扑结构和分支长度,解读物种间的亲缘关系和进化历史。
需要注意的是,叶绿体基因组系统发育树的构建是一种较为复杂和精细的分析过程,需要较高的数据分析能力和专业知识。
同时,样本选择、序列比对和系统发育分析方法的选择也会对结果产生一定影响,因此需要进行合理的设计和操作。
三种方法构建系统发育树学习笔记

三种方法构建系统发育树学习笔记所用数据为一个属内不同种不同群体的叶绿体基因组序列,数量为80条。
发现用全长序列建树的时候,不适合选用太多外类群,否则ML法中会导致属内分枝的枝长特别短。
原因应该是基因间隔区和内含子区域序列位点的差异较大。
枝长含义NJ:表示遗传距离;MP:性状状态变换的替换数;ML/BI:该分枝上的相对进化数量(遗传变异量);每个位点上的替换数(一般以每位点多少次核苷酸替换或氨基酸取代来表示)。
遗传距离大多数情况以序列来说遗传距离就是两个OTU(个体、群体、物种或基因家族)之间序列的差异值。
序列比对多序列比对用mafft得到的结果较为准确,muscle比对的速度较快。
多序列比对的绝大多数算法都是基于渐进比对的概念。
简单来说就是先从两个序列的比对开始,逐渐添加新序列,直到所有的序列都加入为止。
但是不同的添加顺序会产生不同的比对结果。
所以由最相似的两个序列开始比对,由近到远逐步完成最为可靠。
mafft --thread 15 --auto 80-AcoeOut.fasta > 80-AcoeOut_aln.fasta##比对时如果不清楚什么参数合适,加个参数--auto,软件可以自动帮你处理挑选保守位点进行下一步建树序列比对完后,用于建树的序列位点必须保证具有良好的同源性。
所以需要删除序列分歧很大的区域和gap区域。
我用的软件为Gblocks,主要目的是把有gap的位点全部去除,参数为-b5=n,其余的选项有-b5=h,h表示half 指去除在大于50%的序列中出现gap的位点。
Gblocks 80-AcoeOut_aln.fasta -t=d -b5=n最大简约法(软件PAUP)最大简约法的树长指所有性状在一棵树上的进化改变总数。
计算得到的结果可能会有许多树长相等的简约树,此时需要计算它们的一致树。
分为strict consensus和semistrict consensus等,strict表示100%,在所有简约树中都出现的分枝,才会出现在一致树中,否则为梳子。
系统发育树的构建方法

系统发育树的构建方法
嘿,朋友们!今天咱来聊聊系统发育树的构建方法,这可有意思啦!
你想想啊,系统发育树就像是一棵大树,上面挂满了各种生物,它们之间有着千丝万缕的联系。
那怎么把这棵大树给建起来呢?
首先得有数据呀!就像盖房子得有砖头一样。
这些数据可以是各种生物的特征啦、基因序列啦等等。
这可不能马虎,得仔细收集。
然后呢,就是选择合适的方法啦。
这就好比做菜,得选对调料和烹饪方法才能做出美味的菜肴。
不同的方法有不同的特点,得根据实际情况来选。
接下来,就是分析数据啦!这就像是侦探在破案,要从一堆线索中找出真相。
得仔细琢磨每个数据的意义和关系。
在这个过程中,可不能瞎搞哦!得有耐心,就像绣花一样,一针一线都要精细。
要是马马虎虎,那建出来的树可就歪七扭八啦。
还有哦,要不断地调整和优化。
就像雕刻一件艺术品,得不断地打磨才能让它更完美。
建系统发育树可不是一件容易的事儿,但当你看到那棵清晰地展示出生物之间关系的大树时,那种成就感,哇,简直没法形容!
你说,这是不是很神奇?通过这样的方法,我们就能更好地了解生物的演化历程,就像穿越时空看到了它们的过去一样。
这不就像是我们在探索一个神秘的世界吗?每一个数据都是一个线索,每一次分析都是一次冒险。
所以啊,朋友们,别小看了系统发育树的构建方法,它可是打开生物奥秘大门的一把钥匙呢!让我们一起努力,去构建出更漂亮、更准确的系统发育树吧!
原创不易,请尊重原创,谢谢!。
系统发育进化树构建

系统发育进化树构建
系统发育进化树是一种用来表示生物种类和它们之间进化关系的图表。
它基于共有衍征和演化关系的分析,可以帮助我们理解物种的起源和演化过程。
下面是一个示例的系统发育进化树构建过程:
1. 收集数据:我们需要收集关于不同物种的特征和遗传信息的数据。
这些数据可以包括形态特征、分子序列等。
2. 数据处理:接下来,将收集到的数据进行处理,例如进行序列比对、计算相似性指数等。
这些处理会将数据转化为可以进行系统发育树分析的形式。
3. 构建系统发育树:通过使用系统发育树构建软件,如MEGA、PHYLIP等,我们可以利用处理后的数据构建系统发育树。
这些软件通常使用一些统计模型和算法来计算物种之间的相似性和进化关系。
4. 评估树的可靠性:构建系统发育树后,还需要对树的可靠性进行评估。
这可以通过计算支持值或进行自举分析等方法来实现。
支持值表示构建树的数据集中的信息支持树的某个分支。
5. 进行树的修正:如果评估发现树的某些分支的可靠性较低,我们可以根据需要进行进一步的分析,例如添加更多的数据或调整分析的参数。
6. 结果解读:在构建了系统发育树之后,可以通过对树的结构和分支进行解读,了解物种的起源和演化过程。
树的结构可以显示物种之间的近缘关系和进化路径。
注意:以上只是一个概括的系统发育进化树构建过程,具体的步骤和方法可能会因不同的研究目的和数据类型而有所不同。
在实际研究中,还需要根据具体情况选择适合的分析方法和工具。
构建系统发育树需要注意的几个问题

构建系统发育树需要注意的几个问题1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。
2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。
3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。
4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。
5 枝长可以用来表示类间的真实进化距离。
6 重要的是理解系统发育分析中的计算能力的限制。
任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。
7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。
然而,有些方法可以检测系统发育树检测的可靠性。
第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。
分子进化研究的基本方法对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。
表型的(phenetic)和遗传的(cladistic)数据有着明显差异。
Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。
这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。
表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。
进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。
文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。
系统进化树分有根(rooted)和无根(unrooted)树。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
构建系统发育树需要注意的几个问题1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。
2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。
3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。
4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。
5 枝长可以用来表示类间的真实进化距离。
6 重要的是理解系统发育分析中的计算能力的限制。
任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。
7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。
然而,有些方法可以检测系统发育树检测的可靠性。
第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。
分子进化研究的基本方法对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。
表型的(phenetic)和遗传的(cladistic)数据有着明显差异。
Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。
这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。
表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。
进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。
文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。
系统进化树分有根(rooted)和无根(unrooted)树。
有根树反映了树上物种或基因的时间顺序,而无根树只反映分类单元之间的距离而不涉及谁是谁的祖先问题。
用于构建系统进化树的数据有二种类型:一种是特征数据(character data),它提供了基因、个体、群体或物种的信息;二是距离数据(distance data)或相似性数据(similarity data),它涉及的则是成对基因、个体、群体或物种的信息。
距离数据可由特征数据计算获得,但反过来则不行。
这些数据可以矩阵的形式表达。
距离矩阵(distance matrix)是在计算得到的距离数据基础上获得的,距离的计算总体上是要依据一定的遗传模型,并能够表示出两个分类单位间的变化量。
系统进化树的构建质量依赖于距离估算的准确性。
一.clustal X建树1) 打开clustal X,载入上述序列,“load sequences”→“output format options”:“CLASTAL FORMAT”; CLASTAL SEQUENCES NUMBERS:ON;ALIGNMENT PARAMETERS:“RESET NEW GAPS BEFOR ALIGNMENT”“MULTIPLE ALIGNMENT PARAMETERS”→设置相关参数2)“DO COMPLETE ALIGNMENT”→FILE→SA VE AS,掐头去尾。
3) 打开MEGA 4,FILE→CONVERT TO MEGA FORMATE→SA VE→FILE→OPEN DATA→CONTAINING PROTAIN SEQUENCES NO →PHYLOGENY→BOOTSTRAP TEST OF PHYLOGENY→N J →设置相关参数。
最后看到系统发育树二.这里要介绍的是Bioedit-Mega建树法,简单实用,极易上手。
1 将所测得的序列在NCBI上进行比对,这个就不多讲了。
2 选取序列保存为text格式。
3 运行Bioedit,使用其中的CLUSTAL W进行比对。
4 运用MEGA 4 建树,首先将前面的文件转化格式为mega格式,然后进行激活,最后进行N-J建树。
此法简单实用,树形美观。
构建系统进化树的详细步骤1.建树前的准备工作1.1相似序列的获得——BLASTBLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。
国际著名生物信息中心都提供基于Web的BLAST服务器。
BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为内核向两端延伸,以找出尽可能长的相似序列片段。
首先登录到提供BLAST服务的常用网站,比如国内的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。
这些网站提供的BLAST服务在界面上差不多,但所用的程序有所差异。
它们都有一个大的文本框,用于粘贴需要搜索的序列。
把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。
如果是DNA序列,一般选择BLASTN搜索DNA数据库。
这里以NCBI为例。
登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。
BLASTN结果如何分析(参数意义):>gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequenceScore = 2020 bits (1019), Expect = 0.0Identities = 1382/1497 (92%), Gaps = 8/1497 (0%)Strand = Plus / PlusQuery: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60 Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58 Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120 Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118Score :指的是提交的序列和搜索出的序列之间的分值,越高说明越相似;Expect:比对的期望值。
比对越好,expect越小,一般在核酸层次的比对,expect 小于1e-10,就比对很好了,多数情况下为0;Identities:提交的序列和参比序列的相似性,如上所指为1497个核苷酸中二者有1382个相同;Gaps:一般翻译成空位,指的是对不上的碱基数目;Strand:链的方向,Plus / Minus意味着提交的序列和参比序列是反向互补的,如果是Plus / Plus则二者皆为正向。
1.2 序列格式:FASTA格式由于EMBL和GenBank数据格式较为复杂,所以为了分析方便也出现了十分简单的FASTA数据格式。
FASTA格式又称为Pearson格式,该种序列格式要求序列的标题行以大于号“>”开头,下一行起为具体的序列。
一般建议每行的字符数不超过60或80个,以方便程序处理。
多条核酸和蛋白质序列格式即将该格式连续列出即可,如下所示:>E.coli 1 aaattgaaga gtttgatcat ggctcagatt gaacgctggc ggcaggccta acacatgcaa 61 gtcgaacggt aacaggaaga agcttgcttc tttgctgacg agtggcggac …… >AY631071 Jiangella gansuensis YIM 002 1 gacgaacgct ggcggcgtgc ttaacacatg caagtcgagc ggaaaggccc tttcgggggt 61 actcgagcgg cgaacgggtg agtaacacgt gggtaacctg ccttcagctc tgggataagc ……其中的…>‟为Clustal X默认的序列输入格式,必不可少。
其后可以是种属名称,也可以是序列在Genbank中的登录号(Accession No.),自编号也可以,不过需要注意名字不能太长,一般由英文字母和数字组成,开首几个字母最好不要相同,因为有时Clustal X程序只默认前几位为该序列名称。
回车换行后是序列。
将检测序列和搜索到的同源序列以FASTA格式编辑成为一个文本文件(例:C:\temp\jc.txt),即可导入Clustal X等程序进行比对建树。
2. 构建系统树的相关软件和操作步骤构建进化树的主要步骤是比对,建立取代模型,建立进化树以及进化树评估。
鉴于以上对于构建系统树的评价,结合本实验室实际情况,以下主要介绍N-J Tree 构建的相关软件和操作步骤。
2.1 用Clustal X构建N-J系统树的过程(1) 打开Clustal X程序,载入源文件. File-Load sequences- C:\temp\jc.txt.(2) 序列比对Alignment - Output format options - √ Clustal format;CLUSTALW sequence numbers: ON Alignment -Do complete alignment (Output Guide Tree file,C:\temp\jc.dnd;Output Alignment file, C:\temp\jc.aln;) Align → waiting……等待时间与序列长度、数量以及计算机配置有关。
(3) 掐头去尾File-Save Sequence as…Format: ⊙CLUSTALGDE output case: LowerCLUSTALW sequence numbers: ONSave from residue: 39 to 1504 (以前后最短序列为准)Save sequence as: C:\temp\jc-a.alnOK将开始和末尾处长短不同的序列剪切整齐。