HPLC-HPCE实验 毛细管 液相色谱
色谱5--HPCE

- - - - - - -
- - - -
石英表面 负电荷
++ +- +- +- +- +-
+ ++ + + + + - - - - - - - - - - - EOF
- - - - -
水合阳 离子在 表面积 聚
电场作用下 向负极运动
+
- -
电渗流的一个独特性质是其具有平面
流型,推动液体流动的力在毛细管内 均匀分布,平面流型的优点是对谱带 的扩散没有直接作用。
热称为焦耳热。受毛细管尺寸、溶液电导、 外加电压等影响。 不均匀的温度梯度和局部的黏度变化会引 起区带展宽。温度变化1℃→黏度变化 2%~3% →淌度变化2%~3% 。 进样塞长度——在进样过程中减少样品塞 长度非常重要。对进样长度的限制是低于 毛细管总长度的(1~2)%。例 70cm长 的毛细管,进样量应小于7mm。
溶质与管壁相互作用——可能导致峰拖
尾 或发生对溶质的完全吸附。对多肽和 蛋白质来说,这种吸附特别严重。可采 用多种方法来减少相互作用: 增加缓冲液浓度以降低有效表面电荷; 在极端pH值下进行分离,使石英表面 硅羟基以不带电的形式存在; 对毛细管壁进行涂层处理。 电分散作用——样品区带与操作缓冲液 的电导差异可产生峰型畸变。
表面活性剂
在毛细管电泳中常添加表面活性剂, 作为疏水性溶质的增溶剂、与溶质形 成离子对,或作为毛细管内壁的改性 剂等,以改善分离效率。常用表面活 性剂有 : 阴离子—十二烷基硫酸钠(SDS) 阳离子---十六烷基三甲基溴化铵 (CTAB) 两性离子---N,N’二甲基胺-3-丙烷-1磺酸
化合物 HCl NaCl 甘氨酸 柠檬酸 细胞色素C 人血红蛋白 烟草花叶病毒
扩散系数D 3.05 1.48 1.06 0.66 0.11 0.069 0.0046
毛细管电色谱应用在哪些方面

毛细管电色谱应用在哪些方面
毛细管电色谱(capillary electro chromatography,CEC)以内含色谱固定相的毛细管为分离柱,兼具毛细管电泳及高效液相色谱的双重分离机理,既可分离带电物质也可分离中性物质。
毛细管电色谱法是用电渗流或电渗流结合压力流来推动流动相的一种液相色谱法。
因此,毛细管电色谱法可以说是HPLC和HPCE 的有机结合,它不仅克服了HPLC 中压力流本身流速不均匀引起的峰扩展,而且柱内无压降,使峰扩展只与溶质扩散系数有关,从而获得了接近于HPCE 水平的高柱效,同时还具备了HPLC 的选择性。
HPLC是用压力驱动流动相。
流速是随填充微粒的大小和柱长而变化的。
流速在管中呈抛物线轮廓,因而造成了色谱峰谱带的展宽,降低了柱效。
而CEC是采用电场推动流动相。
其线速度是与柱的直径和填微粒的大小无关的,因而在毛细管中几乎没有流速梯度。
谱带展宽效应相应的就十分小。
这点是CEC与HPLC的本质差别,也是CEC效率高于HPLC的根本。
(上海通微)。
毛细管气相色谱和HPLC法检测头孢美唑中的残留溶剂

中,封 瓶 。其 中,标准溶 液I V的浓度 为 限量 浓度 。 供试 品溶液制备 :取头孢美唑约0 g . ,精密称 定, 2 置1m 顶空瓶 中,精密加入M Ⅳ 二 甲基 甲酰胺0 0L . . 3 水 1 m ,封瓶 ,超声振荡使溶解,即得供试 品溶液 。 . L 7
213 测 定 法 ..
分别 取标 准系 列溶液 ,供试 品溶液 ,回 收试验
溶 液按 照上 述条件 进样 ,记 录色 谱 图。按外 标法 以
峰 面积进 行 计算 。
Agln 8 0 i t 9 N气 相 色谱 仪 ,Agln 6 4 顶 空 e 6 i t 9E e 7 进 样 器 。 乙醇 ( 津 市 科 密 欧 化 学 试 剂 有 限 公 司) 天 、 二 氯 甲烷 ( 都 市科 龙 化 学试 剂 厂 ) 乙酸 乙酯 ( 成 、 成都 市 科 龙 化 学 试 剂 厂) 苯 甲醚( 都 市 科 龙化 学试 剂 、 成
wa r 5: 5 a bl h s n ed tcinwa ee ghwa 5 n Reut T el er ag f i s u l t ( 2) s e7 mo i p aea dt eet v l t s2 4 m. s l h n a n eo xr i a e h o n s i r s ed og ncs le t i emea oewee1 15 6 75 gmL r 09 9 ,, 5,1 98 9 89 gmL r 09 9 , = ) r a i ov ns n c f tz l r 2 . ̄ 0 .p / (= .9 7 2 ) 9 . ̄ 9 .p / (= .9 8 即 5, — 1 95 1 4 .g / (= . 9 , 5, 56 2 .g / ( O9 9 , ) 2 95 14 . ̄ / (= . 9 , 5 a d 9 . 2 71 gmL r O9 9 ) 1 . ~ 9  ̄1 52 gmL r .9 3 n 6, 4 . ̄ 2 77t mL r 09 3 = ) n = g 9
HPLC-ELSD测定五苓散中2种泽泻醇的含量

五1 散 4- 来源于中医 经典名著《 伤寒论》全方由 , 白术、 泽泻、 获等、 猪菩、 桂枝共5 味药组成It] 。 收载 于2005 年版《 国药典》 中 。五等散是以 泽泻为君药 的代表方。泽泻为 泽泻科植物泽泻[ A " orientail lism ( Sam. ) Juzep. 〕 的干燥块茎。所含的 泽泻醇A 乙 24一 酸醋、 泽泻醇 B23一 乙酸醋为其主要活性成分, 也是 其质 量控制的主 要指标hs: 目 泽泻定量分析只限于药材中有效成分的 前, 定量, 所使用 的方 法多为薄层 扫描法和 HPL CU V[21。复 方中 有关泽泻活性成分的定量分析未见 报道, 本实验采用 H PLC-EI SD测定五 等散中 所含泽 泻醇 B23一 乙酸醋、 泽泻醇 A 乙酸醋的含量, 24一 方法 简便、 可靠、 重现性好, 提高和完善了五等散的质量
过滤 , 置水浴 上蒸 干, 乙情 定容 至 2 ml 量瓶 中, 加
0. 45 lL m微孔滤膜过滤, 进样 2. 4 检测器 参数优化 根据流动相种类和比 在不同漂移管温度和 例, 不同载气流速条件下观察泽泻醇 A24一 乙酸醋、 泽泻 醇 B 乙酸醋对照品的信号 23一 强度, 最后确定漂移管 温度为82 ℃, 载气流速为2. 00 L - min 一 ’ 为最优参
HPLC-ELSD 测定五菩散中 2 种泽泻醇 的含量
李 潘馨」肖 或1, , 建平2, 瑜,陈建忠 ( 福 中 学 药 系中 学 建 等 校 点 验 ,州3 08:2 福 中 学 陈 , , 建 医 院 学 ,药 福 高 学 重 实 室福 501 - 建 医 院 I
附属第二人民医院药剂科, 350003) 福州 摘要 : 目的 采用 HPLC-ELSD 刚定五茶散 中泽泻醉 A24一 乙酸1'f , 泽 泻醉 B23一 乙酸醋的含 量 方法 色谱柱 :大连依 利特 Hy-
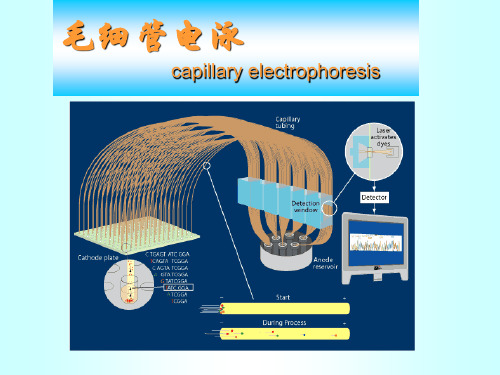
毛细管电泳

高效毛细管电泳(high performance capillary electrophoresis,HPCE)是近年来发展起来的一种分离、分析技术,它是凝胶电泳技术的发展,是高效液相色谱分析的补充。
该技术可分析的成分小至有机离子、大至生物大分子如蛋白质、核酸等。
可用于分析多种体液样本如血清或血浆、尿、脑脊液及唾液等,HPLC分析高效、快速、微量。
电泳迁移不同分子所带电荷性质、多少不同,形状、大小各异。
一定电解质及PH的缓冲液或其它溶液内,受电场作用,样本中各组分按一定速度迁移,从而形成电泳。
电泳迁移速度(v)可用下式表示:v=uE其中E为电场强度(E=V/L,V为电压,L为毛细管总长度)。
u 为电泳淌度。
电渗迁移:电渗迁移指在电场作用下溶液相对于带电管壁移动的现象。
特殊结构的熔合硅毛细管管壁通常在水溶液中带负电荷,在电压作用下溶液整体向负极移动,形成电渗流。
带电微粒在毛细管内实际移动的速度为电泳流和电渗流的矢量和。
分离分析类型根据其分离样本的原理设计不同主要分为以下几种类型:①毛细管区带电泳(capillary zone electrophoresis,CZE);②毛细管等速电泳(capillary chromatography,CITP);③毛细管胶速电动色谱(miceller electrokinetic capillary chromatography,MECC);④毛细管凝胶电泳(capillary gelelectrophoresis,CGE);⑤毛细管等电聚焦(capillary isoelectric focusing ,CIEF)。
毛细管区带电泳(CZE)为HPCE的基本操作模式,一般采用磷酸盐或硼酸盐缓冲液,实验条件包括缓冲液浓度、pH值、电压、温度、改性剂(乙腈、甲醇等),用于对带电物质(药物、蛋白质、肽类等)分离分析,对于中性物质无法实现分离。
毛细管胶束电动色谱(MECC)为一种基于胶束增溶和电动迁移的新型液体色谱,在缓冲液中加入离子型表面活性剂作为胶束剂,利用溶质分子在水相和胶束相分配的差异进行分离,拓宽了CZE的应用范围,适合于中性物质的分离,亦可区别手性化合物,可用于氨基酸、肽类、小分子物质、手性物质、药物样品及体液样品的分析。
HPLC-HPCE实验毛细管液相色谱

HPLC-HPCE实验毛细管液相色谱HPLC法测定水溶液中扑热息痛含量一、教学目标:1. 认识HPLC的结构,熟悉HPLC操作2. 了解HPLC进行分离、定性、定量的方法。
二、教学内容:1.高效液相色谱(HPLC)仪器简介色谱分离的基本原理:组分在固定相与流动相中的分配系数不同。
根据流动相不同,可以分为气相、液相色谱。
液相色谱最为常见。
有机合成实验中常使用常规尺寸的硅胶柱:分离量大、分离度低、分离速度慢、只能对部分物质做半定量。
提高分离效率、稳定性和定性、定量能力的办法:减小颗粒尺寸和粒径分布,增加管长度、均匀地填充固定相。
但这些措施往往增加柱传质阻力。
因此发展出使用微米级颗粒为固定相材料、泵驱动液体流动进行分离的方法即高效液相色谱法。
加上归一法标准曲线内标法原理见仪器分析书书上原理三、仪器组成:以我实验室分离二茂铁(Fc)标记的DNA溶液为例(上图右)。
HPLC系统一般应包括:流动相、泵、进样阀、分离柱、检测器直至废液瓶等部分。
预设提问,在实验中一起讨论,点名回答,计分。
1:从HPLC最基本的原理讲,分离过程中流动相组成逐渐变化能够改变什么?告诉我们为什么梯度洗脱往往比等度洗脱效率高?(提示:从色谱基本原理出发)2:HPLC中的泵应该具有什么样的性质才算是一台好的泵?3:流动相中进入颗粒状杂质会引起什么故障?怎样避免引入颗粒?4:微量进样器的使用还记得吗?如何驱除其中的气泡?5:什么是正向柱和反向柱?6:如果一个气泡进入色谱柱,对仪器是“致命”的损害吗?7:回忆HPLC检测器都可以是哪些?有什么特点?四、实验步骤1)分别移取0.1 0.15 0.2 0.25 mL扑热息痛stock溶液(1mg/mL)至50mL容量瓶,用二次水定容。
配制成4 6 8 10 ug/mL的扑热息痛标准溶液。
注意溶液应倒在干净的烧杯中然后用移液管量取,烧杯和移液管在使用前都应用储备液荡洗。
其他操作遵循“化学分析”实验要求。
毛细管反相液相色谱-串联质谱用于肽的鉴定和相对定量分析

832&,*)& :7 -AB%’1 3’# B%A .1A0B.3.$"B.’0 "01 #A("B.CA D5"0B.3.$"B.’0 ’3 2A2B.1AE FG $"2.(("#G #ACA#EA1+2%"EA (.D5.1 $%#’-"B’=#"2%G .’0+B#"2 B"01A- -"EE E2A$B#’-AB#G( 8H+9I+!J K !J )L"E AEB"F(.E%A1) 7B 3.#EB ,B%A 2A2B.1AE LA#A "5B’-"B.$"((G .1A0B.3.A1 FG $’##A("B.0= B%A B"01A- -"EE E2A$B#" L.B% B%A 2A2B.1A EAD5A0$AE 3#’- " 1"B"F"EA) 73BA# B%A D5"0B.B"B.CA .03’#-"B.’0 ’3 2A2+ B.1A .’0E LA#A A/B#"$BA1 3#’- B%A 35((+E$"0 !J "$$’#1.0= B’ B%A #AE5(BE ’3 1"B"F"EA EA"#$%.0= , B%A 2A"M .0BA0E.B.AE ’3 B%A .1A0B.3.A1 2A2B.1A .’0E L.B% 1.33A#A0B $%"#=A EB"BAE LA#A E5--A1 B’+ =AB%A# B’ 1A3.0A B%A B’B"( .0BA0E.BG ’3 B%A 2A2B.1A) I%A0 ,B%A 2A"M .0BA0E.B.AE ’3 B%A E"-A 2A2+ B.1A .0 B%A #A2(.$"BA "0"(GE.E ’3 B%A E"-A E"-2(A LA#A "CA#"=A1 "01 "EE.=0A1 "E B%A "F501"0$A ’3 B%A 2A2B.1A) N.0"((G ,B%A "F501"0$AE ’3 B%A $’--’0 2A2B.1A .0 B%A "0"(GE.E ’3 1.33A#A0B E"-+ 2(AE LA#A $’-2"#A1) I%.E "22#’"$% #A(.A1 ’0 B%A "0"(GB.$"( #A2#’15$.F.(.BG "01 (.0A"#.BG ’3 E.=+ 0"( CA#E5E -’(A$5("# $’0$A0B#"B.’0) 7E " -A"E5#A ’3 B%A "0"(GB.$"( #A2#’15$.F.(.BG 3’# B#G2B.$ 2A2B.1AE E"-2(.0= ,B%A -A1."0 ’3 B%A #A("B.CA EB"01"#1 1AC."B.’0 ’3 $- O L"E 1ABA#-.0A1 3’# -" $’--’0 2A2B.1AE 3#’- $ #A2(.$"BA "0"(GE.E ’3 !"" 3-’( F’C.0A EA#5- "(F5-.0( PJ7 ) 1.=AEB) I%A -AB%’1 L"E 35#B%A# .((5EB#"BA1 5E.0= 1.=AEBA1 -./B5#AE ’3 PJ7 "01 -G’=(’F.0 "E 3’((’LE) I%A PJ7 1.=AEB L"E =#"15"((G 1.(5BA1 L%.(A B%A -G’=(’F.0 1.=AEB L"E 2#AEA0B .0 B%A -./B5#AE "B $’0EB"0B (ACA() I%.E EB51G #ACA"(A1 B%"B B%A "F501"0$A ’3 B%A C"#."F(A PJ7 2A2B.1AE .0$#A"EA1 (.0A"#(G( B#A01 (.0A 4 ! . " ) ’* ) L.B% .0$#A"E.0= "-’50B 3#’- &" B’ & """ 3-’( , L%.(A B%A "F501"0$AE ’3 B%A $’0EB"0B 2A2B.1AE 3#’- -G’=(’F.0 #A-".0A1 "22#’/.-"BA(G B%A E"-A) 90 B%A 2#AEA0B -AB%’1 ,$%A-.$"( 1A#.C"B.4"B.’0 EBA2E "#A 0’B 0AA1A1 B’ $#A"BA "0 .0BA#0"( EB"01"#1 ,"E .0 .E’B’2A+$’1A1 "33.0.BG B"= ’# E.-.("# -AB%’1E) I%.E -AB%’1 2#’C.1AE "0 "(BA#0"B.CA "22#’"$% 3’# 1.33A#A0B."( "0"(GE.E ’3 2A2B.1AE .0 F.’(’=.$"( E"-2(AE) 9$4 :+,#2 : (.D5.1 $%#’-"B’=#"2%G+B"01A- -"EE E2A$B#’-AB#G ( 8H+!J K !J ) ; ("FA(+3#AA 2A2B.1A D5"0B.3.$"B.’0 ;2#’BA’-.$E
毛细管电泳的基本原理及应用

毛细管电泳的基本原理及应用摘要:毛细管电泳法是以弹性石英毛细管为分离通道,以高压直流电场为驱动力,依据样品中各组分之间淌度和分配行为上的差异而实现分离的电泳分离分析方法。
该技术可分析的成分小至有机离子、大至生物大分子如蛋白质、核酸等。
可用于分析多种体液样本如血清或血浆、尿、脑脊液及唾液等,比HPLC 分析高效、快速、微量。
关键词:毛细管电泳原理分离模式应用1概述毛细管电泳(Caillary Electrophoresis)简称CE,是一类以毛细管为分离通道,以高压直流场为驱动力的新型液相分离分析技术。
CE的历史可以追溯到1967年瑞典Hjerten最先提出在直径为3mm的毛细管中做自由溶液的区带电泳(Capillary Zone Electro-phoresis,CZE)。
但他没有完全克服传统电泳的弊端[1]。
现在所说的毛细管电泳(CE)是由Jorgenson和Lukacs在1981年首先提出,他们使用了75mm的毛细管柱,用荧光检测器对多种组分实现了分离。
1984年Terabe将胶束引入毛细管电泳,开创了毛细管电泳的重要分支: 胶束电动毛细管色谱(MEKC)。
1987年Hjerten等把传统的等电聚焦过程转移到毛细管内进行。
同年,Cohen 发表了毛细管凝胶电泳的工作。
近年来,将液相色谱的固定相引入毛细管电泳中,又发展了电色谱,扩大了电泳的应用范围。
毛细管电泳和高效液相色谱(HPLC)一样,同是液相分离技术,因此在很大程度上HPCE与HPLC可以互为补充,但是无论从效率、速度、样品用量和成本来说,毛细管电泳都显示了一定的优势毛细管电泳(C E)除了比其它色谱分离分析方法具有效率更高、速度更快、样品和试剂耗量更少、应用面同样广泛等优点外,其仪器结构也比高效液相色谱(HPLC)简单。
C E只需高压直流电源、进样装置、毛细管和检测器。
毛细管电泳具有分析速度快、分离效率高、试验成本低、消耗少、操作简便等特点,因此广泛应用于分子生物学、医学、药学、材料学以及与化学有关的化工、环保、食品、饮料等各个领域[2]。
毛细管电泳法测定阿司匹林中的水杨酸(精)

毛细管电泳法测定阿司匹林中的水杨酸一、实验目的1 进一步理解毛细管电泳的基本原理;2 熟悉毛细管电泳仪器的构成;3 了解影响毛细管电泳分离的主要操作参数。
二、实验原理:毛细管电泳又称高效毛细管电泳( High Performance Capillary Electrophoresis, HPCE) 是一种仪器分析方法。
通过施加10-40kV 的高电压于充有缓冲液的极细毛细管,对液体中离子或荷电粒子进行高效、快速的分离。
现在,HPCE 已广泛应用于氨基酸、蛋白质、多肽、低聚核苷酸、DNA 等生物分子分离分析,药物分析,临床分析,无机离子分析,有机分子分析,糖和低聚糖分析及高聚物和粒子的分离分析。
人类基因组工程中DNA 的分离是用毛细管电泳仪进行的。
1.仪器结构毛细管电泳较高效液相色谱有较多的优点。
其中之一是仪器结构简单(见图1)。
它包括一个高电压源,一根毛细管,紫外检测器及计算机处理数据装置。
另有两个供毛细管两端插入而又可和电源相连的缓冲液池。
high-v oltagepower supply BufferV ialBuffer V ial Detector Recording dev icecapillaryElectrode Electrode2.分离原理毛细管中的带电粒子在电场的作用下,一方面发生定向移动的电泳迁移,另一方面,由于电泳过程伴随电渗现象,粒子的运动速度还明显受到溶液电渗流速度的影响。
粒子的实际流速 V 是泳流速度 Vep 和渗流速度 Veo 的矢量和。
即:V = V ep + Veo (1)电渗是一种液体相对于带电的管壁移动的现象。
溶液的这一运动是由硅/水表面的Zeta 势引起的。
CE 通常采用的石英毛细管柱表面一般情况下(pH>3)带负电。
当它和溶液接触时,双电层中产生了过剩的阳离子。
高电压下这些水合阳离子向阴极迁移形成一个扁平的塞子流,如图2。
毛细管管壁的带电状态可以进行修饰,管壁吸附阴离子表面活性剂增加电渗流,管壁吸附阳离子表面活性剂减少电渗流甚至改变电渗流的方向。
一种亲水性毛细管液相色谱整体柱的制备及应用

1973年,Ishii等【3J用湿法填装聚四氟乙烯柱管的微填充柱,在自制的微柱液相系统 上成功地分离了多环芳烃化合物。1976年,Scott和Kuerca等【4】用改装的紫外检测器(检 测池为2.4肛L),在10
in×1.0 mm
I.D的色谱柱上分离了一系列烷基苯。同年,日本
JASCO公司推出了第一台商品微柱液相色谱系统,这些标志着微柱液相色谱进入了一 个新的发展时期。在接下来的近十年的时间里,以Ishii,Novotony,Yang和Scott为代 表的科研小组在微柱液相色谱领域的研究十分活跃。在1984到1985年间,出版了三本 专著【5.7l并且发表了大量综述性文章【8-101。1990年9月在日本神户召开了第十二届国际毛 细管色谱会议,详细讨论了常规液相转换为微柱液相所涉及的技术问题。 90年代以来,随着分离科学与技术的发展,特别是电子工业和微制造业的进步,为 分析仪器微型化注入了新的生命力。微型分析仪器能减少仪器的重量、体积、物耗与能 耗,提高稳定性和响应速度,降低了制造成本。微制造技术于70年代术渗透入微型气 相色谱仪器研究领域,于80年代中期在气相色谱微型化方面取得成功,这种理念和技 术又迅速扩展到色谱仪器的其他方面和整个分析仪器领域。近年来,微型液相色谱的研 究也得到迅速发展【11。。毛细管色谱柱是微分离分析的核心,也是毛细管分析技术的瓶颈。 作为分离的场所和工具,色谱柱性能的优劣,从根本上决定了分离效果的好坏。因此, 有人将色谱柱誉为色谱仪的心脏,应是当之无愧的。发生在色谱柱中的分离过程,受热 力学因素和动力学因素的控制。为了获得满意的分离效果,首先必须考虑固定相的性质
1.2.2.1干法 在制备常规液相色谱柱(4.6 mm I.D.)时j通常认为,当固定相填料的粒度如<20
高效毛细管电泳法测定复方阿昔洛韦滴眼液的含量

高效毛细管电泳法测定复方阿昔洛韦滴眼液的含量
梁改玲;连莹
【期刊名称】《华西药学杂志》
【年(卷),期】2006(21)3
【摘要】目的建立测定复方阿昔洛韦滴眼液含量的方法。
方法采用高效毛细管电
泳(HPCE)法。
用非涂层渍石英毛细管,运行缓冲溶液为30mmol·L^-1硼砂缓冲液(pH9.6);运行电压25kV;柱温25℃;检测波长240nm;压力进样5s。
结果阿昔洛韦和地塞米松磷酸钠分别在0.05~0.6mg·ml^-1和0.0084—0.1008mg·ml^-1范围内呈线性关系;r分别为0.9996、0.9992;平均回收率分别为99.6%(RSD=1.36%)、99.2%(RSD=1.54%。
结论方法简便、灵敏、准确、经济,专属性强,可用于复方阿昔洛韦滴眼液质量控制。
【总页数】2页(P286-287)
【关键词】阿昔洛韦;地塞米松磷酸钠;含量测定;高效毛细管电泳;滴眼液
【作者】梁改玲;连莹
【作者单位】河南省药品检验所
【正文语种】中文
【中图分类】R917
【相关文献】
1.高效毛细管电泳法测定复方氯霉素洗剂中醋酸曲安奈德和氯霉素的含量 [J], 王
学艳;林英
2.高效液相色谱法测定复方阿昔洛韦滴眼液中阿昔洛韦含量 [J], 李宏斌;刘海宏;毕雪艳
3.HPLC法测定复方阿昔洛韦滴眼液中阿昔洛韦的含量 [J], 吴伟;何梅凤;唐细兰
4.高效毛细管电泳法测定复方茵黄解毒汤中咖啡酸的含量 [J], 蔡茁;杨芳;俞发;楼陆军
因版权原因,仅展示原文概要,查看原文内容请购买。
维生素的检测方法

维生素的检测方法
维生素有不同的检测方法,根据不同的维生素种类和检测目的,选择不同的方法。
下面介绍几种常见的维生素检测方法:
1.高效液相色谱法(HPLC)
HPLC是目前最常用的维生素分析方法之一,可以高效、准确地定量维生素,特别适用于复杂样品中的多种维生素的同时测定。
2.气相色谱法(GC)
GC法适用于对脂溶性维生素的分析,如维生素A、D、E、K等,它们在气相色谱柱上的保留时间较长,使其分离效果良好。
3.荧光光谱法
荧光光谱法是一种快速、准确、灵敏的方法,对于生物样品中的维生素测定特别适用。
该方法通过受激态分子辐射和自发荧光光谱衰减分析样品中的维生素含量。
4.高效毛细管电泳法(HPCE)
HPCE法能够有效地分离、测定样品中的多种水溶性维生素,如维生素B族成员,其检测灵敏度高,分析速度快,适用于复杂的生物样品中。
5.生物检测法
生物检测法利用生物感受器(如酵母、细菌、动物组织等)来检测维生素,具有灵敏度高、选择性好、可再现性好等优点,可用于药物残留检测和补充剂中的维生素分析。
毛细管电泳原理

毛细管电泳原理作者:admin 发表时间:2008-8-28 14:55:41 阅读:次毛细管电泳原理毛细管电泳基本原理分离的原因:电泳迁移,电渗迁移电泳迁移:在高压电场下,带电离子向相反的方向移动。
电渗迁移:当毛细管内充满缓冲溶液时,毛细管壁上的硅羟基发生解离,生成氢离子溶解在溶液中,这样就使毛细管壁带上负电荷与溶液形成双电层,在毛细管的两端加上直流电场后,带正电的溶液就会整体的向负极端移动,这就形成了电渗流。
cE在操作缓冲溶液中,带电粒子的运动速度等于电泳速度和电渗速度的矢量和,电渗速度一般大于电泳速度,因此即使是阴离子也会从阳极端流向阴极端。
加大缓冲溶液的酸度、在缓冲溶液中加入有机试剂都会减少硅羟基的解离,减小电渗流分离模式毛细管电泳的分离模式有以下几种。
(1)毛细管区带电泳将待分析溶液引入毛细管进样一端,施加直流电压后,各组分按各自的电泳流和电渗流的矢量和流向毛细管出口端,按阳离子、中性粒子和阴离子及其电荷大小的顺序通过检测器。
中性组分彼此不能分离。
出峰时间称为迁移时间,相当于高效液相色谱和气相色谱中的保留时间。
(2)毛细管凝胶电泳在毛细管中装入单体和引发剂引发聚合反应生成凝胶,这种方法主要用于分析蛋白质、DNA等生物大分子。
另外还可以利用聚合物溶液,如葡聚糖等的筛分作用进行分析,称为毛细管无胶筛分。
有时将它们统称为毛细管筛分电泳,下分为凝胶电泳和无胶筛分两类。
(3)毛细管等速电泳采用前导电解质和尾随电解质,在毛细管中充入前导电解质后,进样,电极槽中换用尾随电解质进行电泳分析,带不同电荷的组分迁移至各个狭窄的区带,然后依次通过检测器。
(4)毛细管等电聚焦电泳将毛细管内壁涂覆聚合物减小电渗流,再将样品和两性电解质混合进样,两个电极槽中分别加入酸液和碱液,施加电压后毛细管中的操作电解质溶液逐渐形成pH梯度,各溶质在毛细管中迁移至各自等电点时变为中性形成聚焦的区带,而后用压力或改变检测器末端电极槽储液的pH值的办法使溶质通过检测器。
毛细管电泳分析方法的工作原理介绍

毛细管电泳分析方法的工作原理介绍毛细管电泳(capillary electrophoresis, CE)又叫高效毛细管电泳(HPCE), 是近年来发展最快的分析方法之一。
1981年Jorgenson和Lukacs首先提出在75μm内径毛细管柱内用高电压进行分离, 创立了现代毛细管电泳。
1984年Terabe等建立了胶束毛细管电动力学色谱。
1987年Hjerten 建立了毛细管等电聚焦, Cohen和Karger提出了毛细管凝胶电泳。
1988~1989年出现了第一批毛细管电泳商品仪器。
短短几年内, 由于CE符合了以生物工程为代表的生命科学各领域中对多肽、蛋白质(包括酶,抗体)、核苷酸乃至脱氧核糖核酸(DNA)的分离分析要求, 得到了迅速的发展。
CE是经典电泳技术和现代微柱分离相结合的产物。
CE和高效液相色谱法(HPLC)相比, 其相同处在于都是高效分离技术, 仪器操作均可自动化, 且二者均有多种不同分离模式。
二者之间的差异在于:CE用迁移时间取代HPLC中的保留时间, CE的分析时间通常不超过30min, 比HPLC速度快;对CE而言, 从理论上推得其理论塔板高度和溶质的扩散系数成正比, 对扩散系数小的生物大分子而言, 其柱效就要比HPLC高得多;CE所需样品为nl级, 最低可达270fl, 流动相用量也只需几毫升, 而HPLC所需样品为μl 级, 流动相则需几百毫升乃至更多;但CE仅能实现微量制备, 而HPLC可作常量制备。
CE和普通电泳相比, 由于其采用高电场, 因此分离速度要快得多;检测器则除了未能和原子吸收及红外光谱连接以外, 其它类型检测器均已和CE实现了连接检测;一般电泳定量精度差,而CE和HPLC相近;CE操作自动化程度比普通电泳要高得多。
总之, CE的优点可概括为三高二少:高灵敏度, 常用紫外检测器的检测限可达10-13~10-15mol,激光诱导荧光检测器则达10-19~10-21mol;高分辨率, 其每米理论塔板数为几十万;高者可达几百万乃至千万, 而HPLC一般为几千到几万;高速度, 最快可在60s内完成, 在250s内分离10种蛋白质, 1.7min分离19种阳离子, 3min内分离30种阴离子;样品少, 只需nl (10-9 L)级的进样量;成本低, 只需少量(几毫升)流动相和价格低廉的毛细管。
高效毛细管电泳测定α-乳白蛋白、β-乳球蛋白A及β-乳球蛋白B的纯度

高效毛细管电泳测定α-乳白蛋白、β-乳球蛋白A及β-乳球蛋白B的纯度宋宝花;赵广莹;杨媛媛;李芸;丁晓静;王志【摘要】采用毛细管电泳分析手段,用校正峰面积归一化法同时测定了本实验室购得的α-Lac、β-LgA及β-LgB三个蛋白参考物的纯度,其值分别为75.4%、84.5% 和 76.9%,相对标准偏差分别为1.6%、0.7% 及1.4%.所得结果与应用十二烷基硫酸钠-聚丙烯酰胺平板凝胶电泳法的测定结果进行了比较,并讨论了造成二者差异的原因.结果表明毛细管电泳法是分析此类蛋白的有效潜在手段.【期刊名称】《河北农业大学学报》【年(卷),期】2010(033)004【总页数】4页(P124-127)【关键词】高效毛细管电泳;纯度;α-乳白蛋白;β-乳球蛋白A;β-乳球蛋白B【作者】宋宝花;赵广莹;杨媛媛;李芸;丁晓静;王志【作者单位】河北农业大学,理学院,河北,保定,071001;北京市疾病预防控制中心,北京,100013;河北农业大学,理学院,河北,保定,071001;北京市疾病预防控制中心,北京,100013;河北农业大学,理学院,河北,保定,071001;北京市疾病预防控制中心,北京,100013;北京市疾病预防控制中心,北京,100013;首都医科大学,公共卫生与家庭医学学院,北京,100069;北京市疾病预防控制中心,北京,100013;首都医科大学,公共卫生与家庭医学学院,北京,100069;河北农业大学,理学院,河北,保定,071001【正文语种】中文【中图分类】O652.1标准物质是影响分析测试结果准确性的主要因素之一,色谱分析的新方法开发中,由于有证标准物质或参考物质(CRM)的缺乏,分析工作者有时从国际知名和大的试剂供应商处购得的已知纯度的、性质较稳定的实验室试剂、工业化学品试剂等作为参考物质来使用,并按供应商所提供的试剂纯度进行折算后对未知样进行定量分析,而对于一些性质不稳定、易降解的参考物质如维生素A和维生素E等,则需要进行浓度测定校正后再进行定量[1]。
毛细管色谱法3讲解
3. 电解质溶液性质的影响
(1)溶液pH的影响
对于石英毛细管,溶液pH增高时,表面电离多,电荷密 度增加,管壁zeta电势增大,电渗流增大,pH=7,达到最大; pH<3,完全被氢离子中和,表面电中性,电渗流为零。分析 时,采用缓冲溶液来保持pH稳定。
(2)阴离子的影响
在其他条件相同,浓度相同而阴离子不同时,毛细管中 的电流有较大差别,产生的焦耳热不同。
2.进样的影响
当进样塞长度太大时,引起的峰展宽大于纵向扩散。分 离效率明显下降;理想情况下,进样塞长度:
Winj= (24D t )1/2 实际操作时进样塞长度小于或等于毛细管总长度的1%~2%。
3.焦耳热与温度梯度的影响
电泳过程产生的焦耳热可由下式计算:
Q
VI π r2L
Λm cb
E
2
m—电解质溶液的摩尔电导;I—工作电流:cm—电解质浓度;
在HPCE中,控制电渗流非常重要。
三、HPCE中影响电渗流的因素
factors influenced electroosmosis 1.电场强度的影响
电渗流速度和电场强度成正比,当毛细管长度一定时, 电渗流速度正比于工作电压。
2.毛细管材料的影响
不同材料毛细管的表面电 荷特性不同,产生的电渗流大 小不同;
3.操作方便、消耗少
进样量极少,水介质中进行;
4.应用范围极广
有机物、无机物、生物、中性分子;生物大分子等; 分子生物学、医学、药学、化学、环境保护、材料等;
2高效毛细管电泳理论基础
一、高效毛细管电泳(HPCE)基本原理
basic principles of PHCE
电泳是指带电离子在电场中的定向移动,不同离子具有 不同的迁移速度,迁移速度与哪些因素有关?
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
新仪器:点击“分析”/“分析”,之后点击“报告”/“面积百分比报告”,查找并记录样品对应的峰位置和峰面积。
老仪器:点击“预览”,查找并记录样品对应的峰位置和峰面积。
2:使用毛细管进行电泳时,为什么要加高电压?高电压下进行实验两个电泳电极表面接触水的部分会不会爆炸?进行高电压分离的实验是不是很危险?
3:显然,使用常规尺寸石英管在pH>4时也可以形成双电层。为什么一定要在小的管径下才出现电渗流?小结形成电渗流的必要条件?
4:根据电渗流产生的原因和条件,说说如何控制电渗流方向?我们实验里要检测Cl-,如果改变上图的电渗流方向,离子表观运动速度如何变化?实验里将用什么物质进行表面改性?
5)新建方法:设定1.冲洗:CTAB缓冲液冲洗;冲洗方式:压力,压力值:20psi,时间2min;
(
2.进样:选择某一样品位置处进样,进样方式:压力进样,0.5psi,5s
3. auto zero自动基线调零
3.电泳分离:时间设为0,电压8kV,时间5min,入口BID6,出口BOD1,选择“反向”加压。
2.电渗流大小和方向:
电渗流速度比电泳快5-7倍,因此正负离子和中性分子都向电渗流方向运动,并且产生速度差异,实现对各组分的分离。
Vap=VefVeo(efeo)E=apE
3.电渗流的流形与压力驱动流形:
4.电渗流方向的控制:如果测定阴离子,通过表面修饰CTAB(十六烷基溴化铵)进行改性,可使离子电泳方向与电渗流方向一致,从而加快出峰速度。与由于离子流方向变化,我们在分离时还要改变电压施加方向,变为左负右正。否则电渗流不经过检测器。
6)重复4、5至标准溶液和样品全部测定完毕。
7)用乙腈清洗移液器数次。
8)利用校准曲线法对未知样中的扑热息痛进行定量分析。需要将数据在坐标纸上绘出。
用高效毛细管电泳法测定样品中Cl-含量(教案)
一、教学目标:
1.掌握电泳与电渗流概念,了解其在毛细管电泳分离过程中起到的作用
2.掌握利用校准曲线法进行定量检测的过程
2)将配好的5瓶溶液、扑热息痛未知样品带回408实验室。同时带两个烧杯装洗微量注射器的废液。
3)检查流动相溶液是否充足,若溶液量少于200mL应在教师指导下更换流动相。设置HPLC参数,使流速为1mL/min,检测器工作波长为254nm,柱温为30oC,将基线校正归零。打开紫外-可见检测器的光源。(老仪器所有参数设置均在仪器面板上,光源已经打开。新仪器所有参数设置均在软件界面内的“仪器状态”里。新仪器同时需要设置乙腈与水比例为20%:80%)。
加上归一法标准曲线内标法原理见仪器分析书
书上原理
三、仪器组成:
以我实验室分离二茂铁(Fc)标记的DNA溶液为例(上图右)。
HPLC系统一般应包括:流动相、泵、进样阀、分离柱、检测器直至废液瓶等部分。
预设提问,在实验中一起讨论,点名回答,计分。
1:从HPLC最基本的原理讲,分离过程中流动相组成逐渐变化能够改变什么?告诉我们为什么梯度洗脱往往比等度洗脱效率高?(提示:从色谱基本原理出发)
3)CTAB缓冲共需过滤3瓶,其中两瓶作为电泳分离时电极和毛细管插入的溶液。为避免虹吸现象,应保持两瓶缓冲液面高度大致相同。领取3个空瓶盖好盖,一个作为废液瓶。另外两个备用。
4)将盛放上述11个小瓶的塑料盒带回408。点击“load”,待样品托盘处于装样品位置后打开仪器盖。在仪器BI(bottle inlet)托盘上,将标准溶液从小浓度到大浓度依次放在C1、C2、C3、C4、C5位置上。C6放置待测Cl-。B6放置CTAB缓冲液,用于冲洗、平衡毛细管。BI D6,BO D1 (BO代表outlet方向的小瓶托盘)分别放置两瓶CTAB缓冲液,用于电泳分离样品。BI B1放置废液小瓶。在记录纸上记录所有位置的样品浓度,防止搞错。装样时注意转动小瓶,使小瓶上橡皮塞上的分隔为前后方向,不要沿左右方向。防止电极和毛细管叉在瓶盖上。(此要求不严格,实际上我们看到仪器自身震动就会改变瓶塞方向。也没有看到电极卡在瓶盖上的情况)
4)检查所有设置的参数是否正确,开始进样分离过程。新老仪器开始进样分离的步骤略有不同:
新仪器:点击“控制”中“单次运行”。在“方法”中检查应为“…method/WK01…”。设定保存路径为“E:/仪分/wk/当天日期(YY-mm-dd)/group1”(当天第二组使用仪器的改为group2)。样品名为<001><ID>,样品ID可根据实际样品命名,如:“1 ug injection 1st”,(当天第二组使用仪器的改为group2)之后点击“开始”。此时软件会显示“正在平衡方法”,可利用此时间在盛放“HPLC专用乙腈”的小玻璃瓶中清洗微量注射器数次,尽量除去其中的气泡。之后在待注射样品溶液中多次清洗,除去可能残留的乙腈。吸取~30uL样品,尽量避免吸入气泡。确认进样阀位置在“load”位置,待“正在平衡方法”变为“等待触发”后,将进样针轻轻插入进样孔,直至能感觉到到底后的阻力。将~25uL样品推入进样器,由于采样环为20uL,多余的溶液会从一个出口流出,不必担心进样量前后不一致。如果“等待触发”已经出现而洗针尚未结束,不用理会。仪器会一直等待。每次进样前微量注射器都应该利用样品清洗几次,以保证浓度一致。乙腈清洗仅在第一次使用前和实验结束后才需要。每个浓度进两个样,误差应在5%以内。
电泳速度与淌度的关系:v=E物理化学中曾经有电泳实验。
电泳分离:不同带电粒子泳动速度不同,从而得到分离。
常规尺寸电泳的弊端:电泳速度慢、分离效率低、消耗样品量大、扩散严重。
当把电泳通道逐渐减小至几十微米的毛细管,出现一种新的效应:电渗流
电渗流:容器壁(这里为石英毛细管)在pH>4,表面硅羟基≡Si-OH离解生成阴离子≡Si-O-。从而吸引溶液中正电荷形成双电层。正电荷在外电场作用下向阴极移动,由于体积小,管中溶液被带动一起向阴极移动,形成电渗流。
2:HPLC中的泵应该具有什么样的性质才算是一台好的泵?
3:流动相中进入颗粒状杂质会引起什么故障?怎样避免引入颗粒?
4:微量进样器的使用还记得吗?如何驱除其中的气泡?
5:什么是正向柱和反向柱?
6:如果一个气泡进入色谱柱,对仪器是“致命”的损害吗?
7:回忆Hห้องสมุดไป่ตู้LC检测器都可以是哪些?有什么特点?
四、实验步骤
3.掌握间接紫外法的基本原理,利用间接紫外法测定Cl-离子含量
4.了解毛细管表面改性与电渗流的关系
二、教学内容
1.电泳现象:带电粒子在外电场作用下向与其电性相反的电极发生定向移动的现象叫电泳。
普通尺寸的管道中只能观察到电泳微管道中还能观察到电渗流
离子电泳速率:正比于粒子的电荷-半径比(q/r),电场强度E,而与溶液粘度成反比。
7.由于毛细管尺寸小易堵塞、内壁性质受到杂质颗粒的干扰大,所以所有用水都是二次水,并且样品要用滤头过滤。
8.定量方法:校准曲线法。
预设问题:
1:假设电泳溶液为NaCl,在电泳过程中Na+向阴极移动,Cl-向阳极移动。一段时间后是不是说阴极附近只有Na+一种离子?而阳极周围由于Na+离去形成了阳离子的“真空”?
1)分别移取0.1 0.15 0.2 0.25mL扑热息痛stock溶液(1mg/mL)至50mL容量瓶,用二次水定容。配制成4 6 8 10ug/mL的扑热息痛标准溶液。注意溶液应倒在干净的烧杯中然后用移液管量取,烧杯和移液管在使用前都应用储备液荡洗。其他操作遵循“化学分析”实验要求。(在414室)
三、实验步骤
1)分别移取0.5 1.0 1.5 2.0 2.5mL 10mM Cl-储备液,用二次水定容至刻度,配得离子浓度为0.2 0.4 0.6 0.8mmol/L的Cl-标准溶液。所有溶液均用二次水。
加上书上第二步
2)将5份标准溶液和CTAB缓冲液(实验员已配好,溶液中已含有CTAB、NaOH与NaCrO4)分别倒入相应标签的小烧杯中(之前用该溶液荡洗烧杯),用具有相应标签的针筒抽取3-4mL溶液。取孔径为0.22 um的滤头装在针筒头上。装紧,手掌握住针筒,拇指按压针筒,过滤溶液。前数滴过滤出的溶液滴入HPCE的样品小瓶,荡洗小瓶。之后将过滤后溶液滴入小瓶至溶液约充满小瓶2/3以上。(由于电泳电极和毛细管都会插入小瓶中,内充溶液过满可能导致溶液溢出。)盖上红色橡胶瓶盖。将小瓶放在CE专用的白色塑料盒中。
老仪器:在“参数设置”栏内填入样品名称,如:“1 ug injection 1st”,在“保存路径”内设定“E:/仪分/10仪分试验/王康/当天日期(YY-mm-dd)/group1”(当天第二组使用仪器的改为group2)。在盛放“HPLC专用乙腈”的小玻璃瓶中清洗微量注射器数次,尽量除去其中的气泡。之后在待注射样品溶液中多次清洗,除去可能残留的乙腈。吸取~30uL样品,尽量避免吸入气泡。确认进样阀位置在“load”位置,将进样针轻轻插入进样孔,直至能感觉到到底后的阻力。将~25uL样品推入进样器,然后立即点击“采集数据”(可以两人配合)。由于采样环为20uL,多注入的溶液会从一个出口流出,不必担心进样量前后不一致。老仪器每次进样前微量注射器都应该利用样品清洗几次,以保证浓度一致。乙腈清洗仅在第一次使用前和实验结束后才需要。每个浓度进两个样,误差应在5%以内。
HPLC法测定水溶液中扑热息痛含量
一、教学目标:
1.认识HPLC的结构,熟悉HPLC操作
2.了解HPLC进行分离、定性、定量的方法。
二、教学内容:
1.高效液相色谱(HPLC)仪器简介
色谱分离的基本原理:组分在固定相与流动相中的分配系数不同。根据流动相不同,可以分为气相、液相色谱。液相色谱最为常见。
有机合成实验中常使用常规尺寸的硅胶柱:分离量大、分离度低、分离速度慢、只能对部分物质做半定量。提高分离效率、稳定性和定性、定量能力的办法:减小颗粒尺寸和粒径分布,增加管长度、均匀地填充固定相。但这些措施往往增加柱传质阻力。因此发展出使用微米级颗粒为固定相材料、泵驱动液体流动进行分离的方法即高效液相色谱法。