美国及欧洲药典系统适应性要求
美国药典、欧洲药典、日本药典最新介绍

内容简介
共5卷,第1、2卷主要为药品各论,第3卷为膳食 补充剂各论,第4、5卷主要为通则(包括测试方 法及通用原则等)。共有各种测试方法178项和通 用信息168项(如离子色谱、拉曼等),其中测试 方法依次分为微生物方法6项(如无菌)、生物方 法25项(如活性和效价测定等)、化学方法69项 (一般鉴别试验1项、有机生物碱色谱鉴定以及限 度和含量测定等68项)、物理方法78项(如灰分、 pH、泄露率等);
内容简介
美国药典正文药品名录分别按法定药名字母顺序 排列,各药品条目大都列有药名、结构式、分子 式、CAS登记号、成分和含量说明、包装和贮藏 规格、鉴定方法、有关物质、含量测定等项目。
可根据书后所附的USP和NF的联合索引查阅本书。
欧洲药典
历史沿革
1977年出版第一版《欧洲药典》
从1980年到1996年期间,每年将增修订的项目与 新增品种出一本活页本,汇集为第二版《欧洲药 典》各分册,未经修订的仍按照第一版执行。
美国药典、欧洲药典、日本药 典最新介绍
一土三石 20200520
主要内容
➢药典简介 ➢美国药典(历史沿革、内容简介) ➢欧洲药典(历史沿革、内容简介) ➢日本药典(历史沿革、标准来保证人类和牲畜使用的药物的质量、 剂量和药物纯度和浓度标准的权威性出版物。它是在专业的,通常 是政府的权威人士的监督下进行编辑,并且是所有药物的制造、分 配和药物治疗所必须遵照的标准 。
年7月1日开始实施。
自1971年第8版起每五年更新一版,期
间会出版增补本。
内容简介
《日本药典》现行版为第17版,于2016年3月7日 颁布,目前共1卷和1增补本。
主要内容为凡例、生药通则、制剂通则、通用测 试处理及设备、各论、生药及成药、红外对照图 谱、紫外对照图谱、通用信息等,增补本主要为 增修订内容,类似中国药典。
美国药典标准

猪八戒照镜子歇后语
猪八戒照镜子歇后语
小编相信一提起猪八戒,没有人是不知道的,这是《西游记》贡献给我国文化长廊中的一个经典角色。
虽说那么的好吃懒做。
但也憨厚惹人爱。
民间就流传着很多关于猪八戒的歇后语,可见他的魅力哦:
猪八戒照镜子——里外不是人
【关于猪八戒的歇后语集锦】
猪八戒照镜子——里外不是人
猪八戒的后脊梁——无能之辈(悟能之背)
猪八戒戴花——自美
和尚打伞——无法无天
猪八戒读书——竟冲识字的
八戒保媒把把成功——猪连必合(珠联壁合)
猪八戒进女儿国——看花了眼
猪八戒娶媳妇——背着走
猪八戒背媳妇——舍得花力气
猪八戒不成仙——坏在嘴上
猪八戒吃黄连/猪八戒吃人参果——苦了大嘴的
猪八戒吃猪啼——自残骨肉
猪八戒充英雄——只是嘴皮子拱得欢
猪八戒戴耳环——自以为美
猪八戒戴花——越多越丑
猪八戒的武艺/猪八戒过火焰山/猪八戒耍把式——倒打一耙
猪八戒的嘴巴——自我欣赏
猪八戒掉进万花筒——丑态百出
猪八戒发眸气——又丑又恶
猪八戒拱帘子——嘴先进
猪八戒进了女儿国——看花了眼
猪八戒进屠场——自己贡献自己
猪八戒啃地梨——什么仙人吃什么果
猪八戒了天拜佛——掸心不稳
猪八戒买猪肝——难得心肠
猪八戒卖炒肝——这是哪道肺
猪八戒卖凉粉——样数不多,滋味不少
猪八戒三十六变——没有一副好嘴脸
猪八戒摔镜子——怕露丑
猪八戒西天取经——三心二意
猪八戒相亲——怕露嘴脸
猪八戒想娶媳妇——一厢情愿
猪八戒背媳妇——心甘情愿
猪八戒招亲——黑灯黑人
猪八戒照镜子/猪八戒照像——自找难堪(看)。
美国及欧洲药典系统适应性要求

电路、方法和样品组成一个整体 定的色谱条件下,n 表示洗脱物
系统,我们可以对这个系统进行 中相邻化合物的分离程度,可作
测试评估。
为衡量色谱系统柱效能的指标,
影响色谱系统的因素包括: 但是不如直接测试的结果可靠。
流动相的组成、离子 强度、温度和 pH 值
峰的尖锐程度部分反映柱效,这 个参数对检查微量物质至关重要。
系统适应性——美国药典
最常用,其它经过化学修饰
系统适应性是气相和液相色
的硅胶也有使用。
谱分析方法的重要组成部分,用 于证明色谱系统的分离度和重现 性能满足样品的分析要求。
分离度 Rs是理论塔板数 n 的 函数(也叫柱效),α是分离因子,k 是容量因子(所有符号的意义见前
测试基于这样的原理:仪器、 文“色谱定义和说明”部分)。在规
流动相的组成(HPLC):以下 围内改变。第三组分 5 的 30%是
调整限度适用于流动相中的小组 1.5%。加上组分一共同构成洗脱
分(比例小于等于 50%)。这些组 体系。因此三相体系的比例变化
分的量可在正负 30%范围内调整。 范 围 是 50:45:5 到 70:25:5 或 者
但是不能超过总流动相的 10%。 58.5:35:6.5 到 61.5:35:3.5。
流速(GC):流速可调范围在± 50%
流速(HPLC);柱子尺寸改变 时,流速可由下式计算:
F2=F1l2d22/l1d12
进样体积和分流体积(GC): 检测器和重现性较好时可做适当 调整。
柱温(HPLC):柱温波动范围 在±10º
进样口温度(GC):可调范围± 10º
程序升温(GC):温度可做适 当改变,需要保持一定温度或者 改变到另一温度值时,温度变化
中国、美国、欧洲药典比较

姓名:徐涛学号:14211020462 专业:中药生物技术学《中国药典》、《美国药典》、《欧洲药典》比较1、各国药典概况1.1 历史沿革《中国药典》英文名称Pharmacopoeia of The People’s Republic of China;简称Ch .P。
1950年4月,成立了第一届中国药典编纂委员会,药典委员会分设名词、化学药、制剂、植物药、生物制品、动物药、药理、剂量8个小组,第一版《中国药典》于1953年由卫生部编印发行。
1957年出版《中国药典》1953年增补本。
1953年药典共收载药品531中,其中化学药215种,植物药与油脂类65种,动物药13种,抗生素2种,生物制品25种,各类制剂211种。
1965年1月26日卫生部颁布《中国药典》1963年版(第二版)发行通知和实施办法。
本版药典收载药品1310种,分一、二部,各有凡例和有关的目录,一部收载中医常用的中药材446种和中药成方制剂197;二部收载化学药品667种。
此外,一部记载药品的“功能主治”,二部增加了药品的“作用与用途”。
1979年10月4日卫生部颁布《中国药典》1977年版(第三版),自1980年1月1日起执行。
本版药典共收载药品1925种,其中一部收载中草药材(包括少数民族药材)、中草药提取物、植物油脂以及单味药材制剂等882种,成方制剂(包括少数民族药成方)270种,共1152种;二部收载化学药品、生物制品等773种。
1985年9月出版《中国药典》1985年版(第四版),1986年4月1日起执行。
本版收载药品1489种,其中一部收载中药材、植物油脂及单味制剂506种,成方制剂207种,共713种,二部收载化学药品、生物制品等776种。
1990年12月3日卫生部颁布《中国药典》1990年版(第五版),自1991年7月1日起执行。
1990年版的第一、第二增补本先后于1992、1993年出版,英文版于1993年7月出版。
目前常用药典标准

目前常用的药典标准有英国药典(BP)、欧洲药典(EP)、美国药典(USP)、德国药典(DAP)、日本药典(JP)、中国药典(CP)British Pharmacopoeia(BP)英国药典英国药典是英国官方医学标准集,是英国药品委员会的正式出版物,是英国制药标准的重要出处。
该药典囊括了几千篇颇有价值医学专题论文,其中有几百篇是医学新论。
它不仅为读者提供了药用和成药配方标准以及公式配药标准,而且也向读者展示了所有明确分类并可参照的欧洲药典专著。
对于制药厂和化学工业、政府管理者、医学研究院及学习制药的学生都是一部必不可少的工具书。
英国The Stationery Office出版社在2001年五月出版发行的《英国药典2001》(British Pharmacopoeia 2001)是英国药典的最新版本。
Europe Pharmacopoeia(EP)欧洲药典欧洲药典委员会以协调欧洲药典的操作为目的,由二十六国组成:奥地利、比利时、波斯尼亚黑塞哥伟那、克罗地亚、塞普路斯、捷克、丹麦、芬兰、法国、德国、希腊、匈牙利、冰岛、爱尔兰、意大利、卢森堡、荷兰、挪威、葡萄牙、斯洛伐克、斯洛文利亚、西班牙、瑞典、瑞士、马其顿、土耳其、英国和欧共体。
欧洲药物质量理事会认证秘书处给予适应性证明书(COS),用以证明原料药生产商生产的产品的质量是以欧洲药典专论适当控制的。
要取得此证书,生产商需要提交一详细的档案,其中可能含有机密资料,此证书用户表明通过应用欧洲药典有关专论,能够检查药物原料和辅料是否适用于人用药品的生产,也就是说,从这一特殊方法(包括原材料)生产的药品的所有可能杂质和污染能够由有关专论的要求来完全控制。
United States Pharmacopoeia(USP)美国药典美国药典是一个非政府组织,是为完成促进公众健康的使命,通过建立起国家技术标准来保证人类和牲畜使用的药物的质量。
这些标准是从一个包含全体群众的独特的程序中发展起来的,并被全世界所接受。
EP USP CP质量标准

浅析EP(欧洲药典)、USP(美国药典)和CP(中国药典)的质量标准区别名词解读:EP: European Pharmacopeia欧洲药典,缩写为EP。
为欧洲药品质量检测的指导文献。
所有药品或原料的生产厂家在欧洲范围内销售和使用的过程中,必须遵循《欧洲药典》的质量标准。
USP:U.S. Pharmacopeia 美国药典,缩写为USP。
为美国药品质量检查的指导文献。
出版时为U.S. Pharmacopeia / National Formulary《美国药典/国家处方集》(简称USP/NF)。
CP:The People's Republic of China Pharmacopoeia中华人民共和国药典,缩写CP。
为中国制药行业中药、西药、生物制品等质量检查的指导文献,分一部:中药、二部:西药、三部:生物制品。
2010版将于10月1日起执行。
正文:由于其质量标准中药品的检测项目繁多,我就不一一来比较,只拿出相对较为复杂的“液相法含量测定”来进行简单的比较。
知识水平有限、时间也不是很多,因此他们之间的许多不同可能没有发掘出来,还请大家和我一道,继续发掘、一起分析!EP和USP在一定程度是相通的,也就是说他们对于同一个药品的质量标准是相同的,但也不能排除有不同的情况,比如在流动相比例、检测波长上就有的品种不同。
而他们对于CP来说,区别就很多了,我要描述的也主要是这个方面:色谱柱:CP一般的规定是填料,而对柱长、粒径等不做规定。
那么,对我们而言,色谱柱的选择就成了一个问题,我们需要花时间去找柱子。
而EP、USP,基本上就把柱子的长度、直径、粒径都规定好了。
洗脱方法:EP、USP多采用梯度洗脱,配制起来比较复杂,稍微的ph值不适当或比例问题就会影响实验结果,且在标准中大多规定了主峰或相关杂质峰的保留时间,因此难度较大。
而CP多采用等度洗脱,操作、试验都相对较容易。
无论是梯度还是等度,只要把相关的物质分离出来就可以了。
各国药典储存条件汇总

各国药典储存条件汇总————————————————————————————————作者: ————————————————————————————————日期:药典储存条件的比较2012-11-27美国药典34 欧洲药典7.0 中国药典2010二部冷冻储存-25℃~-10℃深冷低于-15℃冷处不超过8℃冷处2℃~10℃控制的冷处2℃~8℃,允许在0℃~15℃之间冰箱中储存2℃~8℃阴凉储存8℃~15℃阴凉储存8℃~15℃阴凉处不超过20℃凉暗处避光并不超过20℃室温工作区的一般温度控制下的室温储存20℃~25℃,允许在15℃~30℃之间室温储存15℃~25℃常温10℃~30℃温暖30~40℃过热40℃以上干燥储存控制室温下湿度不超过40%见:药品GMP指南《质量控制实验室与物料系统》P387-388药典加速、长期试验条件的比较中国药典二部附录试验名称试验条件备注1 备注2加速试验(一般情况)温度40℃±2℃、相对湿度75%±5%所用设备应能控制温度±2℃,相对湿度±5%,并能对真实温度和湿度进行检测溶液剂、混悬剂、乳剂、注射液等含有水性介质的制剂可不要求相对湿度加速试验(中间条件) 温度30℃±2℃、相对湿度 65%±5%在温度40℃±2℃、相对湿度75%±5%加速6个月内不符合标准则采用该条件加速试验(温度敏感) 温度25℃±2℃、相对湿度60%±10%预计只能在冰箱中保存(4-8℃加速试验30℃±2℃、相对湿度 65%±5%乳剂、混悬剂、软膏剂、乳膏剂、糊剂、凝胶剂、眼膏剂、栓剂、气雾剂、泡腾剂及泡腾颗粒宜直接采用加速试验40℃±2℃、相对湿度25%±5%包装在半透明容器中药物制剂,例如低密度聚乙烯制备的输液袋、塑料安瓿、眼用制剂容器等长期试验(一般情况)温度在25℃±2℃、相对湿度 60%±10%或温度在30℃±2℃、相对湿度 65%±5%南方、北方气候差异长期试验(温度敏感)6℃±2℃长期试验温度在25℃±2℃、相对湿度 40%±5%或温度在30℃±2℃、相对湿度35%±5%包装在半透明容器中药物制剂选择由研究者决定药典加速、长期试验条件的比较ICH试验名称试验条件备注1 备注2 加速试验(一般情况) 温度40℃±2℃、相对湿度 75%±5%加速试验(中间条件) 温度30℃±2℃、相对湿度 60%±5%在温度40℃±2℃、相对湿度75%±5%加速6个月内不符合标准则采用该条件长期试验(一般情况) 温度在25℃±2℃、相对湿度 60%±5%美国药典USP35试验名称试验条件备注1备注2 加速试验温度40℃±2℃、相对湿度 75%±5%允许解释数据和信息的短期峰值在储存条件下除了受控室温允许的偏离长期试验温度在25℃±2℃、相对湿度60%±5%欧洲药品局试验名称试验条件备注1 备注2 影响因素试验温度:以10℃的增长量(10℃、2可评估药品在一定PH范围内的溶液或(原料药) 0℃、……50℃、60℃)高于加速试验相对湿度:≥75%混悬液对水解的敏感性光稳定性研究可作为影响因素试验的必要部分(条件按ICHQ1B)储存条件一般情况长期试验温度在25℃±2℃、相对湿度 60%±5%或温度30℃±2℃、相对湿度 65%±5%选择由研究者决定中间条件温度30℃±2℃、相对湿度65%±5% 若长期试验条件为温度30℃±2℃、相对湿度 65%±5%,则无中间条件若长期试验条件为温度25℃±2℃、相对湿度 60%±5%,且加速6个月内发生显著变化则应增加中间条件试验以评估显著变化的标准,最初申请应包括12个月的中间条件研究中至少6个月的数据加速试验温度40℃±2℃、相对湿度 75%±5% 储存条件在冰箱保存长期试验温度5℃±3℃加速试验温度在25℃±2℃、相对湿度 60%±5%储存条件冷藏长期试验温度-20℃±5℃加速试验在评估温度下进行,5℃±3℃或25℃±2℃储存条件低于-20℃根据个别方案而定制剂储存条件一般情况长期试验温度在25℃±2℃、相对湿度60%±选择由研究者决定5%或温度30℃±2℃、相对湿度65%±5%中间条件温度30℃±2℃、相对湿度65%±5% 若长期试验条件为温度30℃±2℃、相对湿度65%±5%,则无中间条件若长期试验条件为温度25℃±2℃、相对湿度60%±5%,且加速6个月内发生显著变化则应增加中间条件试验以评估显著变化的标准,最初申请应包括12个月的中间条件研究中至少6个月的数据加速试验温度40℃±2℃、相对湿度75%±5%储存条件不透水的容器可在任何可控的或环境湿度条件下进行储存条件半透水的容器长期试验温度在25℃±2℃、相对湿度 40%±5%选择由研究者决定或温度30℃±2℃、相对湿度 35%±5%中间条件温度30℃±2℃、相对湿度 35%±5% 若长期试验条件为温度30℃±2℃、相对湿度 35%±5%,则无中间条件若长期试验条件为温度25℃±2℃、相对湿度40%±5%,且加速6个月内发生除水分流失外的显著变化则应增加中间条件试验以评估30℃时温度的影响若加速试验中发生的显著变化只是水分流失,则不需要增加中间条件试验,但是应有数据证明在提供的有效期内在温度25℃±2℃、相对湿度40%±5%的条件下不会发生显著的水分流失加速试验温度40℃±2℃、相对湿度不大于25%储存条件冰箱长期试验温度5℃±3℃加速试验温度在25℃±2℃、相对湿度60%±5%储存条件冷藏长期试验-20℃±5℃无加速条件试验但规定在冷藏条件下储存的,应取一批在评估温度(5℃±3℃或25℃±2℃)下进行适当时间的试验,以说明短期内偏离标示储存条件的影响储存条件低于-20℃根据个别方案而定。
欧洲药典系统适应性测试

yi 表示内部标准方法中的峰面积、 峰高或者 面积比的值 y表示平均值 n 表示值的个数
相对保留因子 r 相对保留因子的计算公式如下: r=
������ ������������ −������ ������ ������ ������ st +������ ������
ห้องสมุดไป่ตู้
但是不能增加填料粒径 色谱柱尺寸: 长度:可调范围±50% 内径:可调范围±25% 如果色谱柱尺寸改变, 流速也需按以下公式 做适当调整: F2 = F1 2
2 ������ ������ 2 2 ������ 1 ������ 1
F1:方法中规定的流速,单位 ml/min F2:改变后的流速,单位 ml/min l1:方法中规定的柱长,单位 mm l2:改变后的柱长,单位 mm d1:方法中规定的柱内径,单位 mm d2:改变后的柱内径,单位 mm 柱温:±10℃,除非另有说明,操作时的柱 温需明确规定 检测波长:不能改变 进样体积: 在满足检测限和重现性的情况下 可减少进样体积,但不能增加进样体积 液相色谱:梯度洗脱 梯度洗脱不像等度洗脱, 要改变洗脱条件的 话需要慎重考虑 流动相: 只有在满足下列条件时才可对流动 相组成做微小调整: 满足系统适应性要求 主峰的洗脱时间改变范围不超过方法 中规定的±15% 改变后的流动相洗脱能力不能比方法 中规定的差 如果改变条件不能满足系统适应性要求, 则 需考虑滞留体积和换色谱柱 滞留体积: 实验仪器的布局能显著改变分离 度、保留时间和相对保留时间等。如有此情 况,可能是滞留体积过大。在开始梯度洗脱 程序之前,会有一段等度洗脱过程。考虑到 规定的方法与实际使用方法中的滞留体积 有差异, 可在等度洗脱时调整开始梯度洗脱 的时间。 测试员有责任使等度洗脱时间适应 所用的分析仪器。 如果方法中规定了滞留体 积, 梯度洗脱的开始时间可根据下面公式换 成实际开始时间: tc = t������−D 0 ������
美国usp 800标准

美国usp 800标准美国usp 800标准是指美国药典委员会(USP)发布的一项关于药品在医疗保健环境中的安全使用和管理的标准。
该标准主要针对医疗保健机构内的药品管理和药品安全,特别是针对有毒药物的管理。
这些有毒药物可能对医护人员和患者造成危害,因此需要严格的管理和控制。
根据美国usp 800标准,医疗保健机构需要建立一套完善的有毒药物管理系统,包括采购、接收、存储、配制、分发、使用和废弃等环节。
这些环节需要严格的操作规程和管理措施,以确保有毒药物不会对人员和环境造成危害。
在采购环节,医疗保健机构需要选择可靠的供应商,确保采购的药品符合质量标准,并且具有相关的许可证和资质。
在接收环节,需要对药品进行验收,并及时处理异常情况。
在存储环节,需要建立专门的存储区域,对有毒药物进行单独存放,并严格控制温度、湿度和光线等环境因素。
在配制、分发和使用环节,需要严格按照操作规程进行操作,并使用个人防护装备,以降低职业暴露的风险。
在废弃环节,需要对废弃药品进行正确处理,以防止对环境造成污染。
美国usp 800标准的实施,对医疗保健机构提出了更高的要求,需要加强对有毒药物管理的重视,加强对医护人员的培训和教育,加强对药品管理的监督和检查。
只有通过严格的管理和控制,才能有效地保护医护人员和患者的安全,减少药品管理中的风险和事故发生。
总的来说,美国usp 800标准的实施,对于提高医疗保健机构的药品管理水平,保障医护人员和患者的安全,具有重要的意义。
医疗保健机构应当认真学习和贯彻这一标准,加强内部管理,提高工作人员的安全意识,建立健全的管理制度,确保有毒药物的安全使用和管理。
同时,相关部门应当加强监督和检查,及时发现和纠正存在的问题,推动美国usp 800标准的全面实施,为医疗保健环境的安全和健康作出贡献。
美国药典USP-FAQ-CompliancewiththeUSP-NF英中150828

美国药典USP-FAQ-CompliancewiththeUSP-NF英中150828Compliance with the USP-NF (药典符合性)1. What does compliance with USP–NF standards mean?An article of commerce that is recognized in the USP–NF complies with USP–NF standards when it meets all of the requirements stated in the article’s monograph, applicable General Chapters, and the General Notices (with monograph requirements superseding those of the General Chapters and General Notices, in any cases where requirements differ). Applicable standards apply at all times in the life of an article, from production to expiration. Thus, any official article is expected to meet the compendial standards if tested, and any official article actually tested as directed in the relevant monograph must meet such standards to demonstrate compliance. Frequency of testing and sampling are left to the preferences or direction of those performing compliance testing, and other users of USP–NF, including manufacturers, buyers, or regulatory authorities. (General Notices, section3.10) 问:什么是USP-NF标准的符合性?答:一个市售药品如果完全符合USP-NF中相应各论、适用通则,凡例的要求(若各论的要求与通则和凡例的要求不一致时,则以各论的要求为准),则该药品符合USP-NF标准。
美国及欧洲药典系统适应性要求

系统适应性——美国药典系统适应性是气相和液相色谱分析方法的重要组成部分,用于证明色谱系统的分离度和重现性能满足样品的分析要求。
测试基于这样的原理:仪器、电路、方法和样品组成一个整体系统,我们可以对这个系统进行测试评估。
影响色谱系统的因素包括:●流动相的组成、离子强度、温度和pH值●柱子大小、流速、柱温和压力●固定相特点,包括填料类型,载体形状、粒径、孔径、表面积等。
●常用固定相为反相硅胶,以十八碳烷基健合硅胶最常用,其它经过化学修饰的硅胶也有使用。
分离度R s是理论塔板数n的函数(也叫柱效),α是分离因子,k是容量因子(所有符号的意义见前文“色谱定义和说明”部分)。
在规定的色谱条件下,n表示洗脱物中相邻化合物的分离程度,可作为衡量色谱系统柱效能的指标,但是不如直接测试的结果可靠。
峰的尖锐程度部分反映柱效,这个参数对检查微量物质至关重要。
标准品或者标准溶液需要重复进样以确保精密度。
除非个论中有规定, 系统适用性五针的数据的相对标准偏差不超过2.0%, 如果超过2.0%的话, 需要进样六针。
在含量测定中,如果纯品含量100%,则相对标准偏差没有最大值限制,这个值可根据多次进样对照溶液来计算:%RSD=KB/t90%,n-1K为常数0.349,由公式k=(0.6/)×(t90%,5/)计算得来,表示B=1.0时六次进样的相对标准偏差。
B是个案中规定的上限。
n是对照溶液的进样次数(3≤n≤6),t90%,n-1是自由度为n-1、置信水平为90%,双侧检验时的t 值。
除非另有规定,RSD不能超过下表中的值。
此规定不适用于相关物质检测。
Relative Standard Deviation Requirents对称因子AS,用于衡量峰的对称性,完全对称时值为1。
拖尾越严重,AS的值越大(见图4)。
偶尔也会有值小于1的情况。
如果对称因子与1的差值越大,则积分的精密度越差。
信噪比(S/N)是系统适应性的一个重要参数,计算公式如下(图5):S/N = 2H/hH是峰高,即峰最高点到基线的距离;h 是噪音最大值和最小值之间的差值。
欧洲药典HPLC规定

±10%。
改变柱尺寸时,允许改变流速
柱参数
柱尺寸
固定相类型不得改变;粒径不得改变。
长度可以改变±70%;内径可以改变±25 %。
柱温
检测波长 进样量
±10℃。
不得改变 如果色谱峰的检测和重复性符合要求, 可以减少进样量,但不得增加。
±5℃
不得改变 如果色谱峰的检测和重复性合适,进样量 可以减少,不得增加 当仪器配置改变时,会影响分辨率和保留 时间,可能需要增加延迟体积,因此可以 改变梯度开始时等度运行的时间。
对仪器的一般要求和色谱条件
2.流动相 (1)流动相组成比例:±30%,最小组成的比例 可以调整30%,但是,任何组成的比例的改变不能 超过±10%. 例如:60:40乙腈/水,水的比例可以调整±12% (=40*30%),但是这超过了±10%的限制。因此, 这个条件下,水的比例只能在30%至50%之间进行调 整。 (2)流动相pH值:±0.2,pH7.6可以在7.4-7.8 之间调整
系统适用性试验
(一)关于系统适用性的具体规定
进行有关物质或含量测定时,除另有规定外,色谱图中 定量用的对照品溶液,其色谱峰对称因子(即《中国药典》 中拖尾因子)应为0.8-1.5。
进行有关物质检查时,定量限(以信噪比为10计算)应 小于或等于忽略限。
系统适用性试验
进行原料药的含量测定时,采用对照品溶液进样数次,按下式计算最 大允许相对标准偏差(Sr(%)max):
式中,K为常数0.349,从表达式
推导得出
表示B=1.0时,6次进样后获得的百分相对标准偏差。B为药典正文中各个品 种含量限度的上限与100%之差。n为对照品溶液的重复进样次数(3<=n<= 6)。T90%,n-1为当自由度为n-1,概率水平为90%,(双侧)时的t检验值。 除另有规定外,最大允许相对标准偏差不得超过下表相应的数值。
系统适用性

系统适用性系统适应性是在每天运行样品之前,需要做的一系列测试,保证系统运行正常,结果可靠。
就相当于对系统作一个mini版的认证,保证当天的分析结果准确有效,别人能认可。
这里的系统,包括了仪器软硬件,分析方法,样品制备,分析方法等等等等.各种法规都有相关的指导,但是也都没有给出特别具体的怎么做的步骤. 我们先来看看这些高屋建瓴地指导法规吧.1(USP(United States Pharmacopeia)是怎么说的呢?“System suitability tests are an in tegral part of gas and liquid chromatographic methods. They are used to verify that the resolution and reproducibility of the chromatographic system are adequate for the analysis to be done. The tests are based upon the concept that the equipment, electronics, analytical operations, and samples to be analyzed constitute an integral system that can be evaluated as such.”大概意思是说:系统适应性(System Suitability)是气相和液相色谱方法的重要组成部分,用来确认色谱系统的分离度和重复性能够满足当前分析的要求。
测试基于的原则是:整个系统是由仪器,电路,分析方法和样品所组成的,我们可以每个每个地去测试, 但我们更要将其作为一个整体去测试,从而验证整个系统的状态。
这也是为什么做了仪器的硬件,软件认证(Compliance),还要做系统适应性的原因:因为你需要将包含了仪器软硬件,方法,样品等这些方面的整个系统作为一个整体,再进行测试。
2020版药典标准

2020版药典标准
药典是指规范药品品质、药材质量及相关测试方法的权威性参考书。
每个国家或地区都有自己的药典标准,用于指导药品的研发、生产和监管。
2020年版的药典标准可能因国家和地区而异,因此具体内容需要根据特定的国家或地区来确定。
以下是一些常见的国际药典标准:
1. 欧洲药典(European Pharmacopoeia,简称EP):欧洲药典委员会出版的,适用于欧洲国家的药品和药材标准。
2. 美国药典(United States Pharmacopeia,简称USP):美国药典委员会出版的,适用于美国的药品和药材标准。
3. 英国药典(British Pharmacopoeia,简称BP):英国药典委员会出版的,适用于英国的药品和药材标准。
除了上述国际药典标准外,不同国家和地区还可能有自己独立的药典标准。
这些标准通常包括药品的命名、药品质量要求、药材的质量要求、制备方法、测试方法等内容。
药典标准的更新通常是根据科学研究进展和药物监管需要来进行的。
因此,不同年度的药典标准可能会有所更新和调整,以确保药品的质量和安全性能得到维护和提升。
2011.11.04 适用于美国药典的标准、实验、分析和其它规范说明

USP29-凡例凡例此颜色的为与USP28相比,新增的内容适用于美国药典的标准、实验、分析和其它规范说明。
凡例(后面提到的General Notices)和在通用章节中出现的general requirements以总则的形式提供美国药典中的标准、实验、分析和其它规范说明的解释与应用的基本指导,以消除整本书中与大量实例相关的那些要求的重复需要。
只要没有相反的特定说明,就应用凡例(General Notices)和通用章节(General Chapters)中的要求。
凡是不同于凡例(General Notices)和通用章节(General Chapters)时,将优先采用个论中的说法,并应特别指明其用法或内容。
为了强调这些例外的存在,在凡例(General Notices)和通用章节(General Chapters)的某些地方使用“除非另有规定”这样的措词。
在正文中,它(“除非另有规定”)应理解为标准、实验、分析或其它规范说明中与凡例(General Notices)和通用章节(General Chapters)中存在偏差的特殊规定,无论是否有例外的表述。
标题本出版物的标题,包括它的增补本,是美利坚合众国药典,第29版。
这个标题可被缩写为美国药典,第29版或USP 29。
美国药典,第29版取代了以前的所有版本。
凡是使用“USP”这个词的地方,没有更多的限制说明,在这本药典的法定期限内,仅指USP 29或相应的增补本。
相同的标题同样适用于包含这些内容的印刷版或电子版。
“OFFICIAL”AND“OFFICIAL ARTICLES”凡在药典或相应的参考中使用“official”,这个词,与“Pharmacopeial”,“USP”和“compendial”.是同义的。
若USP与法定名称相连,或在一物品标签上标上“USP”则表明USP收载了该个论并且该物品符合USP标准。
在标签上USP这个名称,既不可以也不能含生产商的标示的描写、签注或二者的合并,标示上有包含在USP正文中的信息材料,也不包含由USP保证这个物品符合USP标准。
美国药典系统适宜性试验及注射用水的检测
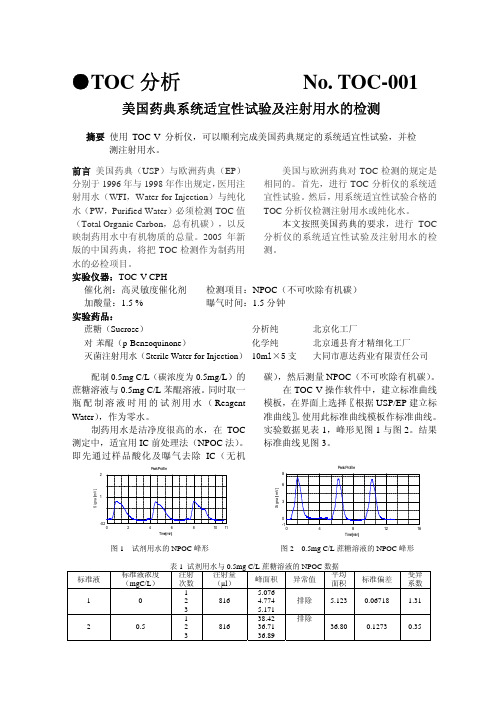
3
Signa l[mV ] Signa l[mV ]
-0.2
0
2
4
6
8
10 11
Time[min]
图 1 试剂用水的 NPOC 峰形
0
-1
0
4
8
12
16
Time[min]
图 2 0.5mg C/L 蔗糖溶液的 NPOC 峰形
标准液 1 2
标准液浓度 (mgC/L)
0
0.5
则此 TOC 分析仪通过系统适宜性测 试,可以用于检测制药用水。
取市售大同市惠达药业有限责任公司
观。 则此次试验回收率为: R = 34.33− 5.12 ×100% = 92.2% 36.80 - 5.12
生产的灭菌注射用水,批号 0403201,每支 10ml,取 5 支。在 TOC-V 操作软件中,将 其作为未知样品检测,选择〖根据 USP/EP 进行样品检测〗。实验数据见表 3。
模板,在界面上选择〖USP/EP 系统适宜性 试验〗。调用此监控样品模板,以 0.5mg C/L 苯醌溶液作监控样品,进行 USP 系统适宜性 试验。实验数据见表 2,峰形见图 4。
7.9 6
4
2
0
-1
0
3
Peak Profile
6
9
Time[min]
12
14
图 4 0.5mg C/L 苯醌溶液的 NPOC 峰形
机抽取的 5 支注射用水,尽管每支之间的
结论 使用 TOC-V 分析仪可以方便地进行 美国及欧洲药典规定的制药用水 TOC 检 测。TOC-V 操作软件上设计了对应美国及 欧洲药典制药用水检测的相关选项,使操作 非常简便。
EP USP CP质量标准
浅析EP(欧洲药典)、USP(美国药典)和CP(中国药典)的质量标准区别名词解读:EP: European Pharmacopeia欧洲药典,缩写为EP。
为欧洲药品质量检测的指导文献。
所有药品或原料的生产厂家在欧洲范围内销售和使用的过程中,必须遵循《欧洲药典》的质量标准。
USP:U.S. Pharmacopeia 美国药典,缩写为USP。
为美国药品质量检查的指导文献。
出版时为U.S. Pharmacopeia / National Formulary《美国药典/国家处方集》(简称USP/NF)。
CP:The People's Republic of China Pharmacopoeia中华人民共和国药典,缩写CP。
为中国制药行业中药、西药、生物制品等质量检查的指导文献,分一部:中药、二部:西药、三部:生物制品。
2010版将于10月1日起执行。
正文:由于其质量标准中药品的检测项目繁多,我就不一一来比较,只拿出相对较为复杂的“液相法含量测定”来进行简单的比较。
知识水平有限、时间也不是很多,因此他们之间的许多不同可能没有发掘出来,还请大家和我一道,继续发掘、一起分析!EP和USP在一定程度是相通的,也就是说他们对于同一个药品的质量标准是相同的,但也不能排除有不同的情况,比如在流动相比例、检测波长上就有的品种不同。
而他们对于CP来说,区别就很多了,我要描述的也主要是这个方面:色谱柱:CP一般的规定是填料,而对柱长、粒径等不做规定。
那么,对我们而言,色谱柱的选择就成了一个问题,我们需要花时间去找柱子。
而EP、USP,基本上就把柱子的长度、直径、粒径都规定好了。
洗脱方法:EP、USP多采用梯度洗脱,配制起来比较复杂,稍微的ph值不适当或比例问题就会影响实验结果,且在标准中大多规定了主峰或相关杂质峰的保留时间,因此难度较大。
而CP多采用等度洗脱,操作、试验都相对较容易。
无论是梯度还是等度,只要把相关的物质分离出来就可以了。
美国药典、欧洲药典、日本药典最新介绍
内容简介
美国药典正文药品名录分别按法定药名字母顺序 排列,各药品条目大都列有药名、结构式、分子 式、CAS登记号、成分和含量说明、包装和贮藏 规格、鉴定方法、有关物质、含量测定等项目。
可根据书后所附的USP和NF的联合索引查阅本书。
欧洲药典
历史沿革
1977年出版第一版《欧洲药典》
从1980年到1996年期间,每年将增修订的项目与 新增品种出一本活页本,汇集为第二版《欧洲药 典》各分册,未经修订的仍按照第一版执行。
内容简介
共5卷,第1、2卷主要为药品各论,第3卷为膳食 补充剂各论,第4、5卷主要为通则(包括测试方 法及通用原则等)。共有各种测试方法178项和通 用信息168项(如离子色谱、拉曼等),其中测试 方法依次分为微生物方法6项(如无菌)、生物方 法25项(如活性和效价测定等)、化学方法69项 (一般鉴别试验1项、有机生物碱色谱鉴定以及限 度和含量测定等68项)、物理方法78项(如灰分、 pH、泄露率等);
内容简介
通用测试方法89项,依次为化学方法15项、物理 方法34项、粉末特性测定5项、生物生化微生物测 定6项、生药测定2项、制剂测试13项、包材测试3 项、对照品、试剂、标准溶液、仪器等相关规定
11项。另有通用信息43项,依次为物化4项、固体 特性5项、生物制品12项、微生物限度11项、生药 测定7项、药物配方2项、包材要求2项、水的质控 2项、对照品1项、其他3项。
内容简介
《 欧洲药典》各论正文主要内容包括:品名、分 子结构式、CAS登录号、化学名称及含量限度、 性状、鉴别、检查、含量测定、贮藏、可能的杂 质结构等。
索引在第三卷后,用以检索所需内容。此外,每 一章(节)开始均有索引,用于检索。
日本药典
中国、美国、欧洲药典对HPLC方法的规定

中国、美国、欧洲药典对HPLC方法的规定所谓药典方法,顾名思义,药典专论所收载的方法,但是否每个人都知道,药典方法考虑到各个实验室的差异,有一定的可调范围。
下面我们具体的来探讨一下这个问题吧!一、中国药典2015规定0512高效液相色谱法规定如下:品种正文项下规定的条件除填充剂种类、流动相组分、检测器类型不得改变外,其余如色谱柱内径与长度、填充剂粒径、流动相流速、流动相组分比例、柱温、进样量、检测器灵敏度等,均可适当改变,以达到系统适用性试验的要求。
调整流动相组分比例时,当小比例组分的百分比例X 小于33%时,允许改变范围为0.7X 〜1.3X ; 当X大于33%时,允许改变范围为X —10%〜X+ 10%。
由上文可知,中国药典专论方法除填充剂种类、流动相组分(比例调整在规定范围内)、检测器类型不得改变外,其他条件改变了,还是认为是药典方法。
但中国药典没有规定方法确认之说,所以,即使是中国药典方法,也是应该按分析方法验证的相关规定进行全套的方法验证。
二、美国药典41规定USP41 <621> CHROMATOGRAPHY 规定如下:为符合系统适用性的要求而调整的操作条件是允许的,除非专论项下另有规定,下面为所列的最大的可调范围,这些调整需要额外的确认数据。
为确认新条件对方法的适用性,评估调整对相关分析性能特征存在的潜在影响。
多条件的调整,将对系统性能产生累积影响,实施前需仔细考虑。
1、pH of mobile phase(HPLC): 流动性缓冲液的pH:±0.2 units,适用梯度及等度。
2、Concentration of salts in buffer (HPLC): ±10%(pH允许情况下),适用梯度及等度。
3、Ratio of componentsin mobile phase (HPLC):流动相中组分(≤50%)可以在此比例上再调节±30%,且任一组分比例的调节不应超过±10%(相对于总的流动相),含三组分的流动相的调节可以分组分进行,含双组分的流动相的调节可在最小的组分进行,双组份和三组分的调节实例如下:双组分:例如流动相50:50,其中一组分50%的30%是15%,但是它超过了占总体比例±10%的规定,所以这个流动相比例的调节范围应以±10%为限,可以在不超过40:60~60:40的范围之间调节。
美国药典质量标准的起草技术指南

美国药典质量标准的起草技术指南下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!摘要:本文介绍了美国药典质量标准起草的技术指南,包括制定标准的基本原则、流程、要求和审核方法等内容,旨在为药品质量标准的制定提供指导和参考。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
系统适应性——美国药典系统适应性是气相和液相色谱分析方法的重要组成部分,用于证明色谱系统的分离度和重现性能满足样品的分析要求。
测试基于这样的原理:仪器、电路、方法和样品组成一个整体系统,我们可以对这个系统进行测试评估。
影响色谱系统的因素包括:●流动相的组成、离子强度、温度和pH值●柱子大小、流速、柱温和压力●固定相特点,包括填料类型,载体形状、粒径、孔径、表面积等。
●常用固定相为反相硅胶,以十八碳烷基健合硅胶最常用,其它经过化学修饰的硅胶也有使用。
分离度R s是理论塔板数n的函数(也叫柱效),α是分离因子,k是容量因子(所有符号的意义见前文“色谱定义和说明”部分)。
在规定的色谱条件下,n表示洗脱物中相邻化合物的分离程度,可作为衡量色谱系统柱效能的指标,但是不如直接测试的结果可靠。
峰的尖锐程度部分反映柱效,这个参数对检查微量物质至关重要。
标准品或者标准溶液需要重复进样以确保精密度。
除非个论中有规定, 系统适用性五针的数据的相对标准偏差不超过2.0%, 如果超过2.0%的话, 需要进样六针。
在含量测定中,如果纯品含量100%,则相对标准偏差没有最大值限制,这个值可根据多次进样对照溶液来计算:%RSD=KB/t90%,n-1K为常数0.349,由公式k=(0.6/)×(t90%,5/)计算得来,表示B=1.0时六次进样的相对标准偏差。
B是个案中规定的上限。
n是对照溶液的进样次数(3≤n≤6),t90%,n-1是自由度为n-1、置信水平为90%,双侧检验时的t值。
除非另有规定,RSD不能超过下表中的值。
此规定不适用于相关物质检测。
Relative Standard Deviation Requirents进样次数3456B%RSD上限2.00.410.590.730.852.50.520.740.92 1.063.00.620.89 1.10 1.27对称因子AS,用于衡量峰的对称性,完全对称时值为1。
拖尾越严重,AS的值越大(见图4)。
偶尔也会有值小于1的情况。
如果对称因子和1的差值越大,则积分的精密度越差。
信噪比(S/N)是系统适应性的一个重要参数,计算公式如下(图5):S/N = 2H/hH是峰高,即峰最高点到基线的距离;h是噪音最大值和最小值之间的差值。
系统适应性测试的数据通过重复进样标准品或者特定文件中规定的对照溶液而得到,此文件中对相关参数的定义同样适用于其它操作条件,以下情况可做相应调整:●标准品(包括参考物质)对适应性测试中的所有化合物均适用●在系统适应性测试中为改进色谱系统性能而作适当调整对色谱系统的调整不能弥补柱子和系统本身的缺陷。
为满足系统适应性要求而对分析方法调整时,除非另有规定,以下每个变量的最大值都应考虑;这些调整需要附加有效数据。
为验证新方法的系统适应性,需要对改变条件后的分析方法重新评价。
多处改动会对系统性能产生积累效果,在分析之前能仔细考虑。
在梯度洗脱中不推荐改变流动性组成,如果必须改变,则只对溶剂体积或滞后体积改变。
流动相pH(HPLC):在配备流动相时,水相缓冲液的pH波动范围在正负0.2个单位缓冲液盐浓度(HPLC):在满足pH值条件下,缓冲液中盐的浓度波动范围在±10%。
流动相的组成(HPLC):以下调整限度适用于流动相中的小组分(比例小于等于50%)。
这些组分的量可在正负30%范围内调整。
但是不能超过总流动相的10%。
下面是两相体系和三相体系组分的调整范围。
两相体系:溶剂比为50:50时,50的30%是15%,超过了10%的限度,因此流动相比例只能在40:60到60:40范围内调整。
容积比为2:98时,2的30%时0.6%,因此改变范围在1.4:98.6到2.6:97.4三相体系:溶剂比为60:35:5时,第二组分35的30%时10.5%,超过10%的限度,只能在25%到45%的范围内改变。
第三组分5的30%是1.5%。
加上组分一共同构成洗脱体系。
因此三相体系的比例变化范围是50:45:5到70:25:5或者58.5:35:6.5到61.5:35:3.5。
紫外-可见光检测器的波长:操作过程中不允许波长有误差,厂家或者其它机构对检测器波长的校正误差不超过3nm。
固定相:柱长:可变范围为原柱长的±70%柱内径(HPLC):如果线速度恒定可做适当调节,见流速项(HPLC)下.柱内径(GC):调整范围为原柱内径的±50%薄膜厚度(毛细管GC):调整范围为-50%到100%粒径(HPLC):填料颗粒尺寸可以减小50%,但是不能增大。
粒径(GC):在满足色谱系统适应性要求的情况下,粒径由大换小或者由小换大都可以,调料的粒径比率由最大颗粒的直径除以最小颗粒的直径而来。
流速(GC):流速可调范围在±50%流速(HPLC);柱子尺寸改变时,流速可由下式计算:F2=F1l2d22/l1d12F1是文件中规定的流速,单位是mL/min,F2是调整后的流速;l1是文件中规定的柱长,l2是使用柱长;d1为文件中规定的柱内径,d2为使用柱内径。
流速可变范围是±50%。
进样体积(HPLC):只要满足精密度和检测限,进样体积可以适当减少,但不能增加。
进样体积和分流体积(GC):检测器和重现性较好时可做适当调整。
柱温(HPLC):柱温波动范围在±10º进样口温度(GC):可调范围±10º程序升温(GC):温度可做适当改变,需要保持一定温度或者改变到另一温度值时,温度变化做大容许范围是±20º。
除非另有规定,系统适应性参数由待测物峰的数据计算、样品的Rr,RF,tR实测值应和对照品的实测值一致,文件中给的相对保留时间值是为了方便峰的指认,对于相对保留时间没有评判标准。
适应性测试时为了确保最终操作结果的有效性。
实验过程中为确保系统适应性,进样前应做适当的准备工作。
配置待测溶液时,溶液中包括一定量的待测物和一些其它物质(如药品辅料或者杂质)。
当色谱系统有显著变化时,或者要用特殊试剂,则要重新做系统适应性实验,否则不能进行样品分析。
欧洲药典2.2.46色谱分离技术色谱分离技术是一种多级分离方法,样品在固定相和流动相之间多次分配,达到动态平衡。
固定相一般是附在硅胶或者其它固体载体上的固体或液体,可填充在层析柱上或铺在薄层版/薄膜上,流动相可以是气体、液体或者超临界流体。
分离原理包括吸附色谱、分配色谱、离子交换色谱、分子排阻色谱等,也可根据样品分子的大小、空间体积、质量等物理化学性质来分离。
本章介绍了系统适应性测试涉及的参数定义及计算方法,分析方法和仪器介绍等已分别在以下章节做了介绍:●纸色谱2.2.26●薄层色谱2.2.27●气相色谱2.2.28●液相色谱2.2.29●排阻色谱2.2.30●超临界流体色谱2.2.45定义下面分别介绍系统适应性和验收标准的相关参数定义。
有些仪器的软件系统包含了一些特定参数的计算方法(例如性噪比、分离度)。
操作人员有责任确认该计算方法是否符合欧洲药典要求,如果有差别,应对该方法做适当调整以符合药典规定。
色谱图反应检测器的响应信号、洗脱剂浓度或体积和时间的关系等。
理想的色谱图是在平稳的基线上各色谱峰彼此分离,呈高斯分布。
色谱峰色谱柱中洗脱的单个组分或者未分离的多个组分在检测器上的响应信号,是色谱图的一部分。
一般用峰面积、峰高h和半高峰宽w h、峰高h和峰宽w i等参数来描述色谱峰。
高斯峰有如下关系(图2.2.46-1):w h = 1.18w i保留时间t R组分从进样到出现最大峰所需时间保留体积V R组分从进样到出现最大峰所需的流动相体积,可用保留时间乘以流速F。
V R = t R ×F死时间t M不被固定相滞留的组分,从进样到出现最大峰所需时间死体积V M不被固定相滞留的组分,从进样到出现最大峰所需流动相体积。
V M = t M ×F排阻色谱中用V o表示(见下面)。
保留因子k有时用容量因子k’或者分配比D m表示,计算公式如下:K = = k cK c表示分配常数,也叫平衡分配系数V s表示固定相体积V M表示流动相体积在色谱系统中某组分的保留因子也可按下式计算:K =流动相总时间t t,当组分粒径小于固定相胶体孔径的最小尺寸时,该组分从进样到出现最大峰所需时间(图2.2.46-2)流动相总体积V t,当组分粒径小于固定相胶体孔径的最小尺寸时,该组分从进样到出现最大峰所需流动相体积,可按下式计算V t = t t×F滞留时间t0:当组分粒径大于固定相胶体孔径的最大尺寸时,该组分从进样到出现最大峰所需时间滞留体积V0:当组分粒径大于固定相胶体孔径的最大尺寸时,该组分从进样到出现最大峰所需体积,可按下式计算V0 = t0×F分配常数k0:表示排阻色谱中某一组分的洗脱能力,也叫分配系数,可用下式计算:K0 =滞留因子R F:在平面色谱中也叫保留因子R f,表示组分在板上的点到原点之间的距离和溶剂前沿到原点的距离的比值(图2.2.46-3)。
R f=B是分析物的移动距离A是溶剂前沿的移动距离塔板数N:可反映柱效,又叫理论塔板数,计算公式如下:N = 5.54()2t R是组分的保留时间,w h是该物质的半高峰宽色谱柱类型、柱温、流动相以及化合物类型都会影响理论塔板数的值滞后体积D:也叫梯度延迟体积,指从流动相混合点至柱头的系统体积,可用以下方法计算:色谱柱:用适当的毛细管(如1 m×0.12 mm)代替流动相:A相是水,B相是0.1%(V/V)的丙酮水流速F:能达到足够的反压为准(如2ml/min)时间min A相体积百分比B相体积百分比0-20100→00→10020-300100检测器:检测波长265 nm记录吸光度到达最大值的50%时的时间t0.5(图2.2.45-4)D = t D×Ft D = t0.5-t G,t G预设为20 minF为流速,单位为ml/min对称因子A色谱峰的对称因子(图2.2.46-5)按下式计算A s =W0.05表示在二十分之一峰高处峰宽d表示二十分之一峰高处到峰顶点投射到水平线之间的距离A s值为1表示峰绝对对称,大于1则为拖尾峰,小于1则为前置峰分离度R s 两组分之间的分离度(2.2.46-1)计算公式如下R s =t R2 > t R1t R1和t R2是色谱峰的保留时间w h1和w h2是半高峰宽在定量平面色谱中,用移动距离代替保留时间来计算两组分之间的分离度,计算公式如下:R s =R F1和R F2是色谱峰的阻滞因子w h1和w h2是半高峰宽a是溶剂前沿到基线的距离峰谷比(p/v)做有关物质的系统适应性测试时,如果两峰之间的基线分离效果不好,可以用峰谷比作为评价依据(图2.2.46-6)。