贴壁细胞慢病毒转染步骤及方法
慢病毒转染

慢病毒转染
1、实验前一天,以每毫升3-5×10 4个目的细胞接种96孔培养板中的12孔,体积为90ul。
(不使用边上的孔,保持细胞状态良好)
2、感染预实验共分为四组,每组三个不同的MOI:1×108TU/ml、1×107TU/ml、1×106TU/ml
3、实验开始,配备1×108TU/ml、1×107TU/ml、1×106TU/ml,如病毒滴度为1×108TU/ml,取5ul病毒液稀释到45ul的ENi.S.中,在进行一次10被稀释即可。
4、取2ul10mg/mlpolybrene稀释到400ul,备用。
5、将10ul三个不同梯度的病毒加到各组的相应孔中。
6、再设定需要添加polybrene的孔中加入10ulpolybrene稀释液。
7、混匀后继续培养,8-12小时候观察细胞生长状态,并更换为新鲜培养基。
8、感染3-4天后,观察荧光表达情况。
9、通过细胞感染效果,确认目的细胞的感染条件和感染参数。
选取效果最优的一组实验条件按相同的实验方法进行正式转染实验。
整理)慢病毒稳转细胞株步骤

稳转慢病毒一、所需试剂1、慢病毒载体(详细信息见附录及《质粒得扩增提取》)(大肠杆菌-80℃保存2-3年,质粒-20℃保存2—3年,病毒液—80℃保存1年)(1)载体质粒:两端得LTR、剪切位点、包装信号Ψ以及抗性或荧光基因、gag基因5′端350bp 得序列及位于env序列中得RRE,含宿主RNA聚合酶识别部分(2)包装质粒(psPAX2):包含了pol、gag包装成分(3)包膜质粒(pMD2、G):用其她病毒得包膜蛋白代替了env基因、三种质粒共同转染产生不具有自我复制能力得病毒载体。
2、包装细胞:293T细胞3、菌株:大肠杆菌,用于提取质粒4、转染试剂:XTREME—GENE(-20℃保存,不可分装),一种脂质与其她组份构成得混合物5、浓缩试剂(配好后4℃保存,原材料室温保存):5XPEG8000/NaCl溶液(聚乙二醇):NaCl 8、766 g; PEG800050g溶解在200mlMilli-Q纯水中,高压蒸汽灭菌**也可直接从公司买来病毒液(—80℃封口膜封口冻存管保存,4℃保存3天):滴度一般为108TU/ml6、10mg/ml polybrene(-20℃分装保存):溴化己二甲铵。
就是带正电得小分子,与细胞表面得阴离子结合,提高慢病毒对细胞得感染效率,通常加入polybrene能提高感染效率2~10倍。
有一定细胞毒性,需要摸索浓度(1~10μg/ml)7、无血清培养基:optimen8、贴壁细胞(复苏后3代以上得细胞)9、puromycin:嘌呤霉素,用于筛选稳转细胞二、具体步骤<一〉病毒包装与收集(中皿,转染步骤类似于瞬转)第一天1、种板,10×105个293T细胞,加入全培养基双抗DMEM4—5ml,过夜2、配制5XPEG8000/NaCl溶液称取NaCl8、766 g; PEG8000 50g溶解在200ml Milli-Q纯水中;121摄氏度30min湿热灭绝 30min;保存在4℃第二天1、配管2、加入2ml全培养基DMEM3、将1加入2,孵育10h,换成5ml全培养基第四天第五天:1、9:00与17:00各收取一次5ml培养液,共20ml(-80℃保存)2、过滤:用孔径为0、45mm得过滤器除去上清中得293T细胞3、加入5ml 5XPEG8000/NaCl溶液,每30min-1h上下摇匀一次4、4℃过夜第六天1、4℃,3500rpm,20min2、弃上清,倒扣纸上静置1-2min,吸干残余液体3、加入120—150μlPBS,缓慢吹打,以防形成气溶胶4、50μl分装,—80℃保存。
慢病毒转染步骤

转染法步骤:
1分至适宜浓度的细胞到六孔板中,次日细胞应30%~50%融合
2.第二天,吸掉板中的生长培养基并加入新的含聚凝胺的培养基,加入适当数量的病毒于细胞中(见“决定慢病毒效价”部分),每孔溶液终体积为3ml
注意:考虑到细胞在自旋过程中不一定能完全被培养基覆盖,不推崇减少六孔板中的溶液终体积。
调整孔中聚凝胺的终浓度为8mg/ml.通过涡旋六孔板轻轻地混合培养基,病毒以及聚凝胺。
3.为减少自旋-转染过程中悬浮物质的形成以及孔与孔之间液体接触的情况发生,用无菌微孔密封膜封闭六孔板(直接盖在板上),封闭好后,再在密封膜上加上一层未经灭菌的密封箔,最后盖上六孔板盖。
4.在标准的组织培养离心机中,37°C,2250rpm(1200g)条件下自旋-转染细胞60min.
5.一自旋-转染后就移走密封膜和密封箔,将孔中原有的培养基换为新鲜的生长培养基(每孔2ml就足够)
6.转染24h后,吸掉孔中的培养基并每孔加入新鲜的含有嘌呤霉素的2ml生长培养基。
注意:嘌呤霉素的浓度应被每个细胞株充分利用,一般规定在1-5μg/mL
7.进一步的培养时间主要取决于细胞株的类型以及转染后的实验分析。
通常,嘌呤霉素筛选细胞需要大约48h。
下面推荐的是结合给定的细胞株和分析实验来得到的嘌呤霉素筛选
时间
>转染后实验分析嘌呤霉素筛选细胞时间> mRNA knockdown (qPCR) 2+ days
> Protein knockdown (Western) 3+ days
> Phenotypic assay 表型分析3+ days
8.检测目的感染细胞的表面分子。
整理)慢病毒稳转细胞株步骤

稳转慢病毒一、所需试剂1、慢病毒载体(详细信息见附录及《质粒的扩增提取》)(大肠杆菌-80℃保存2-3年,质粒-20℃保存2-3年,病毒液-80℃保存1年)(1)载体质粒:两端的LTR、剪切位点、包装信号Ψ以及抗性或荧光基因、gag基因5′端350bp的序列及位于env序列中的RRE,含宿主RNA聚合酶识别部分(2)包装质粒(psPAX2):包含了pol、gag包装成分(3)包膜质粒(pMD2.G):用其他病毒的包膜蛋白代替了env基因.三种质粒共同转染产生不具有自我复制能力的病毒载体。
2、包装细胞:293T细胞3、菌株:大肠杆菌,用于提取质粒4、转染试剂:XTREME-GENE(-20℃保存,不可分装),一种脂质与其他组份构成的混合物5、浓缩试剂(配好后4℃保存,原材料室温保存):5X PEG8000/NaCl溶液(聚乙二醇):NaCl 8.766 g; PEG8000 50g溶解在200ml Milli-Q纯水中,高压蒸汽灭菌**也可直接从公司买来病毒液(-80℃封口膜封口冻存管保存,4℃保存3天):滴度一般为108TU/ml6、10mg/ml polybrene(-20℃分装保存):溴化己二甲铵。
是带正电的小分子,与细胞表面的阴离子结合,提高慢病毒对细胞的感染效率,通常加入polybrene 能提高感染效率2~10 倍。
有一定细胞毒性,需要摸索浓度(1~10μg/ml)7、无血清培养基:optimen8、贴壁细胞(复苏后3代以上的细胞)9、puromycin:嘌呤霉素,用于筛选稳转细胞二、具体步骤<一>病毒包装与收集(中皿,转染步骤类似于瞬转)第一天1、种板,10×105个293T细胞,加入全培养基双抗DMEM 4-5ml,过夜2、配制5X PEG8000/NaCl溶液称取NaCl 8.766 g; PEG8000 50g溶解在200ml Milli-Q纯水中;121摄氏度30min 湿热灭绝30min;保存在4℃第二天2、加入2ml全培养基DMEM3、将1加入2,孵育10h,换成5ml全培养基第四天第五天:1、9:00和17:00各收取一次5ml培养液,共20ml(-80℃保存)2、过滤:用孔径为0.45mm的过滤器除去上清中的293T细胞3、加入5ml 5XPEG8000/NaCl溶液,每30min-1h上下摇匀一次4、4℃过夜第六天1、4℃,3500rpm,20min2、弃上清,倒扣纸上静置1-2min,吸干残余液体3、加入120-150μl PBS,缓慢吹打,以防形成气溶胶4、50μl分装,-80℃保存。
贴壁细胞转染条件

贴壁细胞转染条件一、前言贴壁细胞是指生长在培养皿上的细胞,常用于细胞培养和转染实验。
转染是指将外源性DNA或RNA导入到目标细胞中,使其表达特定的基因或蛋白。
贴壁细胞转染条件影响着转染效率和细胞生长状态,因此需要进行优化。
二、影响贴壁细胞转染效率的因素1. 细胞密度:过低的密度会导致转染效率低下,而过高的密度则会影响细胞生长状态。
一般来说,应该选择处于对数生长期的细胞进行转染。
2. 转染剂:常用的转染剂有磷脂体、聚乙烯酰胺(PEI)等。
不同的转染剂对不同类型的细胞有不同的适用性,需要根据实验需求进行选择。
3. 转染时间:过短或过长的时间都会影响转染效率。
一般来说,应该根据实验需求和具体情况确定最佳转染时间。
4. 转染浓度:过高或过低的浓度都会影响转染效率。
一般来说,应该根据实验需求和具体情况确定最佳转染浓度。
5. 细胞状态:细胞处于不同的生长状态时,对转染的敏感度也不同。
一般来说,处于对数生长期的细胞对转染更为敏感。
三、常用的贴壁细胞转染方法1. 磷脂体介导法:将DNA或RNA与磷脂体混合后加入培养皿中进行转染。
优点是操作简单,适用于大多数贴壁细胞;缺点是转染效率较低。
2. 电穿孔法:利用高压电脉冲使细胞膜发生短暂的孔洞,从而导入外源性DNA或RNA。
优点是转染效率高;缺点是操作复杂且易造成细胞损伤。
3. 聚乙烯酰胺(PEI)介导法:将DNA或RNA与PEI混合后加入培养皿中进行转染。
优点是适用范围广,转染效率高;缺点是有毒性且易形成颗粒堵塞孔径。
四、常用的贴壁细胞转染条件1. 细胞密度:一般来说,应该选择处于对数生长期的细胞进行转染,细胞密度应该控制在70%左右。
2. 转染剂:常用的转染剂有磷脂体、PEI等。
具体选择应根据实验需求和细胞类型进行。
3. 转染时间:一般来说,转染时间应该控制在4-6小时左右。
4. 转染浓度:具体选择应根据实验需求和细胞类型进行。
5. 细胞状态:处于对数生长期的细胞对转染更为敏感。
实用指导(二):慢病毒感染贴壁细胞实验步骤

实用指导(二):慢病毒感染贴壁细胞实验步骤和元生物|2016-03-1513:51慢病毒慢病毒(Lentivirus)是一类改造自人免疫缺陷病毒(HIV)的病毒载体,基因组为RNA,对分裂细胞和非分裂细胞均具有感染能力。
慢病毒基因组进入细胞后,在细胞浆中反转录为DNA,形成DNA整合前复合体,进入细胞核后,DNA整合到细胞基因组中。
整合后的DNA转录成mRNA,回到细胞浆中,表达目的蛋白;或产生小RNA。
慢病毒介导的基因表达或小RNA干扰作用持续且稳定,并随细胞基因组的分裂而分裂。
以24孔为例:第一天:准备细胞将状态良好的目的细胞接种到24孔板中,细胞计数后,进行铺板,每孔加入500ul培养基。
保证第二天感染病毒时融合效率达到30-40%。
通常,24孔板内以5-8*10^4cells/孔的密度铺板。
第二天:病毒感染,换液1计算病毒加药量选择合适MOI值,进行病毒感染。
加药量计算方法:(细胞数×MOI值/病毒原液滴度)×10^3=病毒加药量(μl)。
2弃去部分培养基将每组中加入的病毒及感染增强剂的体积去掉,保证加入病毒和感染增强剂后,每孔中仍保持500ul的液体量。
3加入慢病毒加药量计算方法(细胞数×MOI值/病毒原液滴度)×10^3=病毒加药量(μl),培养基的总量必须充分覆盖住细胞。
4添加感染增强剂polybrene,保证终浓度为5ug/mlPolybrene是一种聚阳离子,可以中和电荷以促进假病毒外壳与细胞膜的作用。
Polybrene 最佳浓度因不同细胞株而异并应根据经验而定(通常范围为2-10μg/ml)。
过长时间暴露于Polybrene(>12小时)可对某些细胞产生毒性作用。
5病毒感染8~12小时后,观察细胞状态。
如细胞状态差,尽快换新鲜培养基;如细胞状态良好,可以在24小时内换液,但不宜超过24小时。
弃去培养基后每孔加入500μl新鲜的完全培养基。
慢病毒转染实验流程

慢病毒转染实验流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!慢病毒转染实验流程。
1. 病毒液与靶细胞准备。
将慢病毒液从-80℃冰箱中取出,置于冰上融化。
慢病毒转染原理

慢病毒转染原理慢病毒转染是一种常用的基因转染方法,适用于长期表达外源基因的研究。
慢病毒转染原理主要是利用慢病毒载体将外源基因导入宿主细胞,并将其整合入宿主细胞基因组中,从而实现外源基因的稳定表达。
本文将从慢病毒转染的原理、步骤和应用等方面进行介绍。
慢病毒转染原理。
慢病毒转染的原理主要包括以下几个步骤,首先,将外源基因插入慢病毒载体中,构建成慢病毒表达载体;然后,将慢病毒表达载体转染至包装细胞中,利用包装细胞的辅助病毒蛋白包装慢病毒颗粒;最后,将包装好的慢病毒颗粒用于感染目标细胞,外源基因被整合入宿主细胞基因组中,实现稳定表达。
慢病毒转染步骤。
慢病毒转染的步骤包括慢病毒载体构建、包装细胞培养、病毒颗粒包装和目标细胞感染等。
首先,构建慢病毒表达载体时,需要选择合适的慢病毒载体和适当的启动子,将外源基因插入载体中,并经过序列分析和酶切验证。
其次,包装细胞的培养和病毒颗粒包装是慢病毒转染的关键步骤,需要选择合适的包装细胞系,转染慢病毒表达载体并培养包装细胞,最终收集包装好的慢病毒颗粒。
最后,利用包装好的慢病毒颗粒感染目标细胞,实现外源基因的稳定表达。
慢病毒转染应用。
慢病毒转染在基因功能研究、基因治疗和细胞工程等领域具有广泛的应用。
在基因功能研究中,可以利用慢病毒转染实现基因敲除、过表达和靶向修饰等,进而研究基因在生理和病理过程中的功能。
在基因治疗中,慢病毒转染可以用于治疗遗传性疾病、肿瘤和传染病等,为基因治疗提供了重要的技术支持。
在细胞工程中,慢病毒转染可以用于改良细胞系,提高细胞的表达稳定性和产量,满足生物制药和工业生产的需要。
总结。
慢病毒转染是一种重要的基因转染方法,具有稳定、高效、广泛应用等特点。
通过慢病毒转染,可以实现外源基因在宿主细胞中的稳定表达,为基因功能研究、基因治疗和细胞工程等领域提供了重要的技术支持。
因此,深入理解慢病毒转染的原理和步骤,对于开展相关研究具有重要的意义。
慢病毒转染操作

一、慢病毒转染贴壁细胞实验方法
1、慢病毒转染前18-24小时,将贴壁细胞以1×10^5/孔铺到24孔板中。
使细胞在慢病毒转染时的数量为2×10^5/孔左右。
2、第二天,用含有6 μg/ml polybrene的2ml新鲜培养基替换原培养基,加入适量病毒悬液。
37℃孵育。
3、(对polybrene毒性敏感的细胞选作此步骤)4小时后加入2ml新鲜培养基以稀释polybrene。
4、继续培养24小时,用新鲜培养基替换含有病毒的培养基。
5、继续培养。
如果慢病毒含有荧光蛋白,一般转染48小时后可见明显荧光表达,72小时后更加明显。
如需FACS检测转染效率,可在转染后72-96小时进行。
如果慢病毒含有抗性基因并且需要加药筛选,可以在转染3-4天后开始加药。
二、慢病毒转染悬浮细胞实验方法
1、在2×10^5/ml悬浮细胞中加入polybrene至6 μg/ml和适量病毒,充分混匀。
37℃孵育。
或者150g室温离心4小时(选作,部分难转染的细胞系采用此步骤可以提高转染效率)。
2、(对polybrene毒性敏感的细胞选作此步骤)4小时后(或离心结束后)加入等体积新鲜培养基以稀释polybrene。
3、继续培养3-4天。
中间视细胞生长情况可传代或换液。
慢病毒感染protocol

慢病毒载体感染流程(带嘌呤霉素筛选标签)方案1.取对数期生长的待感染细胞铺入24孔板,分别设置空白对照孔、对照慢病毒感染孔、目的基因慢病毒感染孔,37℃孵箱培养至细胞密度为50% -60% ;培养时间在24h之内要达到,否则重新铺3细胞。
技巧,在你算好的细胞体积,按70% 100%(你的计算细胞浓度和体积) 130% 铺三种浓度,一般过夜后至少有一组将满足你的要求,省时、省力。
2.取对照慢病毒和目的基因慢病毒,置于冰盒融化,用含10%胎牛血清的培养液+(24孔板每孔约500μl)稀释病毒原液成所需浓度(最佳MOI 值,因细胞而异),同时加入Polybrene 5μg/m l(增加病毒感染效率),轻轻混匀后,分别加入对照慢病毒感染孔、目的基因慢病毒感染孔,空白对照孔只加入含10%胎牛血清的培养液。
3.轻轻混匀后,37℃细胞培养箱培养12-16小时,使病毒感染细胞;4.移除含病毒的培养液,更换含10%胎牛血清的培养液+青链霉素,继续培养48-72小时至细胞密度为90%左右;5.不带EDTA的胰酶消化细胞下来以后,直接加温浴过的10%胎牛血清的培养液+青链霉素至24孔板中,连同胰酶一起转移到6孔板,根据细胞类型,贴壁时间不一样,经过3-10h不等,细胞贴壁后移去带胰酶的培养基,加10%胎牛血清的培养液+青链霉素,培养48-72h后。
如果你的慢病毒是带有嘌呤霉素筛选标志,执行以下方案培养分别在空白对照孔、对照慢病毒感染孔、目的基因慢病毒感染孔加入合适浓度的嘌呤霉素(1ug-2ug/ml终浓度)筛选7-10天后(视情况换液或传代),可见空白对照孔的细胞均已死亡;对照慢病毒感染孔和目的基因慢病毒感染孔仍有一定比例的细胞存活,这些存活的细胞即为稳定感染株。
用含1μg/m l嘌呤霉素(维持筛选压力)的培养液扩增稳定感染株细胞,冻存或用于后续研究。
附:嘌呤霉素筛选浓度的获得1、取对数生长期的待感染细胞铺入96孔板,设置复孔;2、用含10%胎牛血清的培养液稀释嘌呤霉素成不同浓度,如1μg/m l、1.5μg/m l、2μg/m l、2.5μg/m l、3μg/m l;3、将含不同浓度嘌呤霉素的培养液,加入96孔板;4、37℃细胞培养箱培养5-7天后,视情况换液;5、观察96孔板中细胞的存活情况,以能杀死孔板中所有细胞的最低嘌呤霉素浓度为最终筛选浓度。
慢病毒感染细胞具体方法及详细步骤

慢病毒感染细胞具体方法及详细步骤
材料和试剂
1. 6 cm细胞培养皿(Fiosher).
2. 人类或小鼠细胞系,细胞试验所需要的生长培养基。
3. 凝聚胺(海美溴铵; Sigma H 9268)
4. 合适的选择性抗生素。
仪器
1. 细胞培养箱
步骤
慢病毒感染方法应该根据不同的细胞系和以细胞为基础的试验来进行优化。
例如,下列的参数应该在大规模的感染之前进行优化,以确定一个实验的最适条件:
1) 细胞种植密度
2) 慢病毒的数量
3) 嘌呤霉素的浓度
4) 感染时间
2. 在6cm培养皿中种植适当密度的细胞,每孔体积6ml。
1) 贴壁细胞:转染前1天种植细胞
2) 悬浮细胞:转染当天种植细胞,培养基中需要含有凝聚胺。
3. 细胞中加入病毒:
1) (贴壁细胞):弃掉培养基,加入新鲜的含有凝聚胺的培养基。
或者,弃掉部分培养基并且补充含有凝聚胺的培养基。
调整体积和凝聚胺的浓度,使得凝聚胺的终浓度为8ug/ml。
4. 病毒感染:
1) 孵育细胞过夜。
2) 感染后24h更换培养基。
弃掉培养基,换入6ml新鲜的培养基。
如果需要抗生素选择,则使用含有抗生素的新鲜培养基。
注意:嘌呤霉素的浓度应该根据每种细胞系来进行优化;常用的浓度范围为:2-5 μg/mL.
5. 孵育细胞,根据需要每隔几天更换培养基(如果需要,则要含有抗生素) 。
孵育时间的长短主要依赖于感染后的试验。
慢病毒转染PROTOCOL

慢病毒转染PROTOCOL第一天病毒感染前24 小时将细胞置于12 孔板。
加入 1 ml 适当的完全培养基(含有血清和抗生素)孵育过夜。
在传染当天细胞应该达到约50%平铺(Day 2)。
注意:也可用其它型号培养板转导。
这样需要根据孔径或培养板调节细胞量。
第二天准备完全培养基和Polybrene? (sc-134220) 混合物,Polybrene 终浓度为:5 μg/ml。
移去培养基并添加1 ml Polybrene/培养基混合物于每孔中(适用于12 孔板)。
注意:Polybrene 是一种聚阳离子,可以中和电荷以促进假病毒外壳与细胞膜的作用。
Polybrene 最佳浓度因不同细胞株而异并应根据经验而定(通常范围为2-10 μg/ml)。
过长时间暴露于Polybrene (> 12 小时)可对某些细胞产生毒性作用。
在室温下溶化慢病毒颗粒,使用前轻轻混匀。
培养液中加入shRNA 慢病毒颗粒以感染细胞。
轻轻振动培养板使其混匀并孵育过夜。
病毒颗粒使用量因细胞株的特点而有较大不同。
注意:溶化的shRNA 慢病毒颗粒置于冰上。
反复冻溶或长时间将病毒颗粒暴露于常温可使病毒效价降低。
注意:当你是第一次转导shRNA 慢病毒结构进入细胞,我们建议你用几个不同剂量的shRNA 慢病毒颗粒。
此外,我们建议你用一孔shRNA 慢病毒颗粒转导的细胞作对照(sc-108080)。
注意:用copGFP 对照慢病毒颗粒:sc-108084 观察转导效率。
第三天移去培养液并添加1 ml 完全培养基(不含Polybrene)。
孵育细胞过夜。
第四天欲选择稳定表达shRNA 的克隆,根据细胞类型不同将其分成1:3 到1:5 并继续在完全培养基中孵育24-48 小时。
第五-六天及以后用Puromycin dihydrochloriede (sc-108071) 筛选稳定表达shRNA 的克隆。
Puromycin 筛选法:用足够剂量的Puromycin 杀死非转导的细胞。
siRNA转染步骤—贴壁细胞

siRNA转染步骤—贴壁细胞
示例:使用Lipofectamine RNAiMAX和HeLa cell 用于siRNA 的在6孔板上进行的转染实验。
注:请实验前确认实验使用的细胞系是否适合转染试剂。
1、转染前一天,在6孔板中每个孔内播种 3.0 x 10^5 HeLa cells,每孔放2.5mL不含抗生素的生长培养基。
这样在转染时会有50-60%的融合率。
2、从细胞中去除生长培养基。
3、加入1.5 mL新鲜的不含血清的生长培养基。
4、对于每个转染的孔,按照如下方法准备siRNA 和Lipofectamine RNAiMAX 混合物。
4-1、在250μl的无血清生长培养基中加入100pmole siRNA,柔和混匀。
4-2、Lipofectamine RNAiMAX使用前要先混匀,然后在250μl 生长培养基中加入5μl的Lipofectamine RNAiMAX进行稀释,室温下温育5分钟。
4-3、混合稀释好的siRNA和Lipofectamine RNAiMAX,室温下温育20分钟。
5、加入混合物到含有HeLa cells的6孔板中,每孔终体积为2ML,并轻晃混匀。
6、在37℃的二氧化碳培养箱中培养细胞 5-6小时。
7、更换含有血清的培养基并培养细胞24-48小时,直到进行基因敲除实验。
整理)慢病毒稳转细胞株步骤
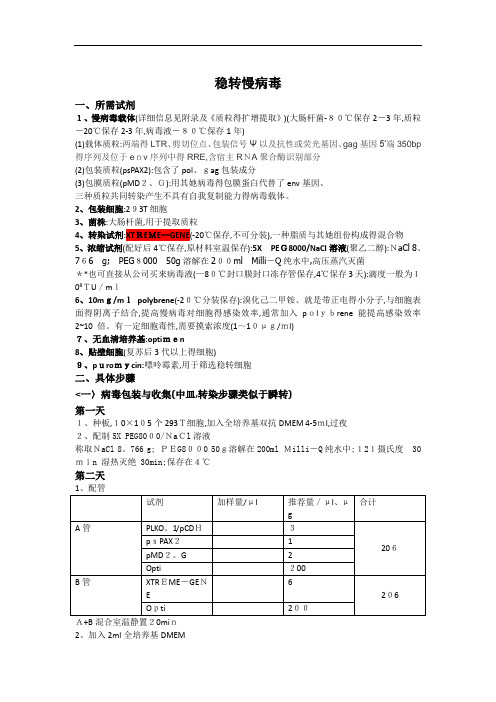
稳转慢病毒一、所需试剂1、慢病毒载体(详细信息见附录及《质粒得扩增提取》)(大肠杆菌-80℃保存2-3年,质粒-20℃保存2-3年,病毒液-80℃保存1年)(1)载体质粒:两端得LTR、剪切位点、包装信号Ψ以及抗性或荧光基因、gag基因5′端350bp 得序列及位于env序列中得RRE,含宿主RNA聚合酶识别部分(2)包装质粒(psPAX2):包含了pol、gag包装成分(3)包膜质粒(pMD2、G):用其她病毒得包膜蛋白代替了env基因、三种质粒共同转染产生不具有自我复制能力得病毒载体。
2、包装细胞:293T细胞3、菌株:大肠杆菌,用于提取质粒4、转染试剂:XTREME—GENE(-20℃保存,不可分装),一种脂质与其她组份构成得混合物5、浓缩试剂(配好后4℃保存,原材料室温保存):5X PEG8000/NaCl溶液(聚乙二醇):NaCl 8、766g;PEG800050g溶解在200ml Milli-Q纯水中,高压蒸汽灭菌**也可直接从公司买来病毒液(—80℃封口膜封口冻存管保存,4℃保存3天):滴度一般为108TU/ml6、10mg/mlpolybrene(-20℃分装保存):溴化己二甲铵、就是带正电得小分子,与细胞表面得阴离子结合,提高慢病毒对细胞得感染效率,通常加入polybrene 能提高感染效率2~10 倍。
有一定细胞毒性,需要摸索浓度(1~10μg/ml)7、无血清培养基:optimen8、贴壁细胞(复苏后3代以上得细胞)9、puromycin:嘌呤霉素,用于筛选稳转细胞二、具体步骤<一〉病毒包装与收集(中皿,转染步骤类似于瞬转)第一天1、种板,10×105个293T细胞,加入全培养基双抗DMEM 4-5ml,过夜2、配制5X PEG8000/NaCl溶液称取NaCl 8。
766 g; PEG8000 50g溶解在200ml Milli-Q纯水中;121摄氏度30min 湿热灭绝 30min;保存在4℃第二天A+B混合室温静置20min2、加入2ml全培养基DMEM3、将1加入2,孵育10h,换成5ml全培养基第四天第五天:1、9:00与17:00各收取一次5ml培养液,共20ml(—80℃保存)2、过滤:用孔径为0、45mm得过滤器除去上清中得293T细胞3、加入5ml 5XPEG8000/NaCl溶液,每30min-1h上下摇匀一次4、4℃过夜第六天1、4℃,3500rpm,20min2、弃上清,倒扣纸上静置1-2min,吸干残余液体3、加入120-150μl PBS,缓慢吹打,以防形成气溶胶4、50μl分装,-80℃保存。
整理)慢病毒稳转细胞株步骤

稳转慢病毒一、所需试剂1、慢病毒载体(详细信息见附录及《质粒的扩增提取》)(大肠杆菌-80℃保存2-3年,质粒-20℃保存2-3年,病毒液-80℃保存1年)(1)载体质粒:两端的LTR、剪切位点、包装信号Ψ以及抗性或荧光基因、gag基因5′端350bp的序列及位于env序列中的RRE,含宿主RNA聚合酶识别部分(2)包装质粒(psPAX2):包含了pol、gag包装成分(3)包膜质粒(pMD2.G):用其他病毒的包膜蛋白代替了env基因.三种质粒共同转染产生不具有自我复制能力的病毒载体。
2、包装细胞:293T细胞3、菌株:大肠杆菌,用于提取质粒4、转染试剂:XTREME-GENE(-20℃保存,不可分装),一种脂质与其他组份构成的混合物5、浓缩试剂(配好后4℃保存,原材料室温保存):5X PEG8000/NaCl溶液(聚乙二醇):NaCl 8.766 g; PEG8000 50g溶解在200ml Milli-Q纯水中,高压蒸汽灭菌**也可直接从公司买来病毒液(-80℃封口膜封口冻存管保存,4℃保存3天):滴度一般为108TU/ml6、10mg/ml polybrene(-20℃分装保存):溴化己二甲铵。
是带正电的小分子,与细胞表面的阴离子结合,提高慢病毒对细胞的感染效率,通常加入polybrene 能提高感染效率2~10 倍。
有一定细胞毒性,需要摸索浓度(1~10μg/ml)7、无血清培养基:optimen8、贴壁细胞(复苏后3代以上的细胞)9、puromycin:嘌呤霉素,用于筛选稳转细胞二、具体步骤<一>病毒包装与收集(中皿,转染步骤类似于瞬转)第一天1、种板,10×105个293T细胞,加入全培养基双抗DMEM 4-5ml,过夜2、配制5X PEG8000/NaCl溶液称取NaCl 8.766 g; PEG8000 50g溶解在200ml Milli-Q纯水中;121摄氏度 30min 湿热灭绝 30min;保存在4℃第二天2、加入2ml全培养基DMEM3、将1加入2,孵育10h,换成5ml全培养基第四天第五天:1、9:00和17:00各收取一次5ml培养液,共20ml(-80℃保存)2、过滤:用孔径为0.45mm的过滤器除去上清中的293T细胞3、加入5ml 5XPEG8000/NaCl溶液,每30min-1h上下摇匀一次4、4℃过夜第六天1、4℃,3500rpm,20min2、弃上清,倒扣纸上静置1-2min,吸干残余液体3、加入120-150μl PBS,缓慢吹打,以防形成气溶胶4、50μl分装,-80℃保存。
病毒转染原理及步骤

病毒转染原理及步调之羊若含玉创作在细胞相关的实验操纵中,对于一些按通例办法难以转染甚至无法转染的细胞,通过病毒介导的实验可以或许大大提高基因的转导效率,以达到目标基因的高效瞬时表达.病毒转染包含以下步调:1构建载体2包装提纯病毒3沾染靶细胞.以慢病毒为例.慢病毒(Lentivirus)载体是以HIV-1(人类免疫缺陷I型病毒)为基本成长起来的基因治疗载体.区别一般的逆转录病毒载体,它对决裂细胞和非决裂细胞均具有沾染才能.慢病毒载体的研究成长得很快,研究的也异常深入.该载体可以将外源基因有效地整合到宿主染色体上,从而达到持久性表达.在沾染才能方面可有效地沾染神经元细胞、肝细胞、心肌细胞、肿瘤细胞、内皮细胞、干细胞等多种类型的细胞,从而达到优越的的基因治疗效果,在美国已经开展了临床研究,效果异常幻想,因此具有辽阔的应用前景.一、慢病毒载体构建原理:慢病毒载体的包装系统一般由两部分组成,即包装成分和载体成分.包装成分由HIV-1基因组去除了包装、逆转录和整合所需的顺式作用序列而构建,可以或许反式提供产生病毒颗粒所必须的蛋白;载体成分则与包装成分互补,即含有包装、逆转录和整合所需的顺式作用序列,同时具有异源启动子掌握下的多克隆位点及在此位点拔出的目标基因.将包装成分与载体成分的多个质粒共转染包装细胞,即可在细胞上清中收获携带目标基因的复制缺陷型慢病毒载体颗粒.慢病毒表达载体包含了包装、转染、稳定整合所需要的遗传信息.慢病毒包装质粒可提供所有的转录并包装RNA 到重组的假病毒载体所需要的所有帮助蛋白.为产生高滴度的病毒颗粒,需要应用表达载体和包装质粒同时共转染细胞,在细胞中进行病毒的包装,包装好的假病毒颗粒排泄到细胞外的造就基中,离心取得上清液后,可以直接用于宿主细胞的沾染,目标基因进入到宿主细胞之后,经由反转录,整合到基因组,从而高水平的表达效应分子.对于一些较难转染的细胞,如原代细胞、干细胞、不分化的细胞等,能大大提高目标基因转导效率,并且目标基因整合到宿主细胞基因组的几率大大增加,这就为RNAi,cDNA克隆以及陈述基因的研究提供了一个有利的途径.对进行稳转细胞株的筛选,为活体动物模子实验提供高质量的包含目标基因的病毒液提供基本.二、慢病毒载体构建及包装流程:(一)实验流程(1和2为并列步调)1.慢病毒过表达质粒载体的构扶植计上下游特异性扩增引物,同时引入酶切位点,PCR(采取高保真KOD酶,3K 内突变率为0%)从模板中(CDNA质粒或者文库)调取目标基因CDS区(coding sequence)连入T载体.将CDS区从T载体上切下,装入慢病毒过表达质粒载体.2.慢病毒干扰质粒载体的构建合成siRNA对应的DNA颈环构造,退火后连入慢病毒干扰质粒载体3. 慢病毒载体的包装与浓缩纯化制备慢病毒穿梭质粒及其帮助包装原件载体质粒,三种质粒载体分离进行高纯度无内毒素抽提,共转染293T细胞,转染后6 h 改换为完全造就基,造就24和48h后,分离收集富含慢病毒颗粒的细胞上清液,病毒上清液通过超离心浓缩病毒.(二)实验资料:慢病毒载体、包装细胞和菌株该病毒包装系统为三质粒系统,组成为pspax2, pMD2G, pLVX-IRES-ZsGreen1/pLVX-shRNA2.其中质粒上的ZsGreen1表达框能表达绿色荧光蛋白(GFP).细胞株293T,慢病毒的包装细胞,为贴壁依赖型成上皮样细胞,生长造就基为DMEM(含10% FBS).贴壁细胞经造就生长增殖形成单层细胞.菌株大肠杆菌菌株DH5α.用于扩增慢病毒载体和帮助包装载体质粒.三、慢病毒转染细胞实验办法(一)慢病毒转染贴壁细胞实验办法1、慢病毒转染前18-24小时,将贴壁细胞以1×105/孔铺到24孔板中.使细胞在慢病毒转染时的数量为2×105/孔左右.2、第二天,用含有6 μg/ml polybrene的2ml新鲜造就基替换原造就基,参加适量病毒悬液.37℃孵育.3、(对polybrene毒性敏感的细胞选作此步调)4小时后参加2ml新鲜造就基以稀释polybrene.4、持续造就24小时,用新鲜造就基替换含有病毒的造就基.5、持续造就.如果慢病毒含有荧光蛋白,一般转染48小时后可见显著荧光表达,72小时后加倍显著.如需FACS检测转染效率,可在转染后72-96小时进行.如果慢病毒含有抗性基因并且需要加药筛选,可以在转染3-4天后开端加药.(二)慢病毒转染悬浮细胞实验办法1、在2×105/ml悬浮细胞中参加polybrene至6 μg/ml和适量病毒,充分混匀.37℃孵育.或者150g室温离心4小时(选作,部分难转染的细胞系采取此步调可以提高转染效率).2、(对polybrene毒性敏感的细胞选作此步调)4小时后(或离心停止后)参加等体积新鲜造就基以稀释polybrene.3、持续造就3-4天.中间视细胞生长情况可传代或换液.病毒沾染细胞原本效率就较低,一定要注意以下几点:1.沾染时的造就基尽量少些;2. 每1ml的造就基中参加5-10ul的Polybrene(储存液浓度为2mg/ml ),可以提高效率约3-5倍.不过病毒沾染效率的问题都是相对的,达到自己的研究目标即可.北京英格恩生物公司最新研发合成一种新型纳米聚合物病毒沾染增强试剂(病毒转染试剂)EnvirusTM,具有对病毒吸赞同独有的浓缩功效.EnvirusTM通过物理作用将病毒大量富集在细胞概况,大大增加病毒与细胞的接触,最大限度促进病毒沾染细胞,可大大提高腺病毒,慢病毒,腺相关病毒,逆转录病毒等病毒的沾染效率,并且细胞毒性很小.通常情况下,比不必EnvirusTM增强剂,提高沾染效率20-50倍.病毒沾染细胞原本效率就较低,整个进程相对庞杂难操纵,一般一次消费周期至少两三个月,多则几个月到十几个月不等,经费也是几万到十几万.万一实验失败需要重复操纵会消费更多的时间和开支.所以有效提高病毒转染效率是病毒转染细胞的症结.。
慢病毒使用操作手册

慢病毒使用操作手册简介:慢病毒(Lentivirus)是一种常用于生命科学研究中的病毒载体,其具有较强的基因转染能力和稳定的基因表达特性。
本操作手册旨在向研究者提供一个详细的慢病毒使用指南,帮助他们顺利进行慢病毒基因转染实验并获得准确可靠的研究结果。
一、慢病毒基本原理慢病毒属于反转录病毒,其基因组为单链RNA。
慢病毒在寄主细胞内通过逆转录过程将RNA转录为DNA,随后将DNA插入宿主基因组中。
这使得慢病毒成为将外源基因稳定集成到宿主基因组中的理想工具。
二、慢病毒使用前准备1. 实验室条件准备:确保工作台面干净整洁,并准备好所需的培养物、培养器具和试剂。
2. 慢病毒载体制备:根据实验需要,选择合适的慢病毒载体,并通过慢病毒包装系统将目标基因插入载体中。
三、慢病毒转染实验步骤1. 细胞培养:将目标细胞接种在培养皿中,并选择合适的培养基进行细胞培养。
2. 慢病毒感染:将预制的慢病毒悬液加入培养皿中,控制感染浓度和时间,以实现最佳的感染效果。
3. 筛选标记:根据实验需要,在感染后适当的时间点添加筛选标记物,如抗生素或荧光标记剂。
4. 选择和扩增:将受筛选标记影响的细胞单克隆分离,扩增和保存。
5. 验证表达:使用合适的实验方法,如western blot或PCR等,来确认目标基因的表达情况。
6. 结果分析:对实验结果进行统计学分析,并绘制适当的图表。
四、注意事项和常见问题解决方案1. 实验前应认真阅读文献,了解慢病毒的基本原理和实验操作流程。
2. 制备慢病毒载体时,应仔细验证目标基因的序列和正确插入。
3. 慢病毒感染时应注意控制感染浓度和时间,避免细胞毒性和非特异性感染。
4. 筛选标记物的选择应根据实验需要和细胞类型进行合理选择。
5. 实验过程中,注意严格遵守实验室安全和生物安全操作规范。
6. 常见问题解决方案:如遇到感染效率低或细胞毒性问题,可以尝试优化感染条件或调整细胞培养条件。
如果基因表达不稳定,可以尝试选择合适的筛选标记物或优化基因载体。
慢病毒转染原理

慢病毒转染原理
慢病毒转染是一种常用的基因传递技术,它利用慢病毒病毒粒子作为载体将目标基因导入靶细胞。
慢病毒是一类特殊的病毒,具有较高的感染效率和稳定的基因表达能力,因此被广泛应用于基因转染领域。
慢病毒转染的基本原理可以分为以下几个步骤:
1. 构建慢病毒载体:首先,将目标基因插入到慢病毒载体的适当位点上。
慢病毒载体通常包括了病毒的基因组结构,例如表达型蛋白、包装型蛋白等。
2. 包装慢病毒:将构建好的慢病毒载体导入特定细胞系,并通过培养和传代扩增病毒。
这些细胞会产生足够多的慢病毒病毒颗粒。
3. 收集病毒颗粒:从培养基中收集慢病毒病毒颗粒。
通常,病毒颗粒可以通过浓缩和超速离心等方法获得。
4. 转染靶细胞:将收集到的病毒颗粒加入待转染的靶细胞培养基中。
病毒颗粒会与细胞表面的受体结合,进入细胞。
5. 病毒基因组整合到靶细胞基因组:一旦进入细胞内,慢病毒会释放出自己的遗传物质,包括病毒基因组RNA和逆转录酶。
逆转录酶能够将病毒基因组的RNA逆转录成DNA,并将其整合到宿主细胞的染色体上。
从而实现病毒基因的稳定表达。
综上所述,慢病毒转染是一种利用慢病毒病毒粒子作为载体将基因导入到靶细胞的技术。
通过包装慢病毒病毒颗粒,并与靶细胞进行转染,实现了基因的稳定表达。
这种技术在基因治疗、基因功能研究等领域具有重要的应用价值。
慢病毒转染实验流程

慢病毒转染实验流程英文回答:Lentiviral Transduction Protocol.Materials:Lentiviral vector.Target cells.Transfection reagent (e.g., Polybrene)。
Culture medium.Selection reagent (optional)。
Procedure:1. Prepare virus and target cells:Thaw the lentiviral vector and aliquot it into appropriate volumes for your experiment.Count and prepare the target cells at a suitable density for transduction.2. Add the transfection reagent:Add the transfection reagent (e.g., Polybrene) to the virus-containing solution according to the manufacturer's instructions. Incubate for the recommended time.3. Incubate virus with cells:Add the virus solution to the target cells and incubate for 24-72 hours. The optimal incubation time may vary depending on the target cell type and lentiviral vector used.4. Remove virus and select for transduced cells:Remove the virus-containing medium and wash the cells with fresh culture medium.If desired, select for transduced cells using an appropriate selection reagent (e.g., antibiotic resistance marker or fluorescent protein).5. Culture and analyze transduced cells:Culture the transduced cells in the appropriate medium and conditions.Analyze the cells for transduction efficiency and transgene expression using methods such as flow cytometry, microscopy, or Western blotting.Additional Notes:Optimization of the transduction protocol may be necessary for different target cell types and lentiviral vectors.Safety precautions should be taken when handling lentiviral vectors, as they contain potentially infectious material.The use of a spin infection protocol can enhance transduction efficiency.Transduction can be scaled up or down as needed by adjusting the volumes used.中文回答:慢病毒转染实验流程。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
贴壁细胞慢病毒转染步骤及方法
一、细胞培养Panc-1 细胞,常规培养使用含10% FBS (GIBCO) 的DMEM 培养基(含1.5 mg/L-Glutamine,100 U/mL penicillin,100 μg/mL Streptomycin) 中,37ºC 5% CO2饱和湿度培养箱中培养。
二、病毒侵染实验材料及试剂DMEM培养基+ 10% FBSPBS(Life Science Products&Services)Trypsin-EDTA Solution (Gibco)24孔板(Corning)Lentivirus-NC 病毒液(GenePharma,1×109 TU/mL)
三、步骤
1. 将状态良好的Panc-1 消化后重悬,取适量细胞接种至24 孔板中,37°C培养箱中过夜;
2. 取阴性对照病毒,按1:10、1:100、1:1000 与DMEM培养基混合稀释,总体积约500 μL,并加入终浓度5 μg/mL Polybrene;
3. 吸去24 孔板中原培养基,换入阴性对照病毒梯度稀释液,37°C培养箱中培养;
4. 24h 后吸去阴性对照病毒稀释液,换入500 μL新鲜培养基,37°C培养箱中培养;
5. 48-96h 于荧光倒置显微镜下观察并记录结果。