MEGA构建系统进化树的步骤(以MEGA7为例)教学文案
怎样使用MEGA建立进化树

如何使用建立进化树1、首先是双击软件打开如下图所示
2、现在是处于DNA序列,而我们要做蛋白质的进化树的话,就如下操作
3、接下来我们要进行序列的输入,点击左边那个红箭头,则出现下面的窗口;
4、然后右击sequence 1,修改名字,如改成DPV
5、然后从Word 里复制蛋白质序列,然后在下面的位置粘贴
6、则可出现如下图的序列了
7、然后点击窗口上的保存图标保存
8、重复从3开始,直到你的序列输入完
9、序列输入完后进行最后的保存,方法如下:
要输入ul7两次保存名字—然后关闭这个窗口; 接下来打开
出现下面这个窗口
接下来就可以建立各种样式的进化树
嗯,只是把过程写出来,方便大家建立进化树,不足的地方,大家补充好。
Mega的使用以及进化树的绘制

1.MEGA构建系统进化树的步骤2.CLUSTALX进行序列比对1.MEGA构建系统进化树的步骤1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
如图:2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图:。
3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,单击右键,选择delete即可,如图:。
4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。
根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。
最后出现一个对话框询问是否打开,选择Yes,如图:。
5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,如图:6. 最后,使用TreeExplorer窗口中提供的一些功能可以对生成的系统进化树进行调整和美化。
MEGA构建系统进化树的步骤(以MEGA7为例)教学文案

M E G A构建系统进化树的步骤(以M E G A7为例)MEGA构建系统进化树的步骤(以MEGA7为例)本文是看中国慕课山东大学生物信息学课程总结出来的分子进化的研究对象是核酸和蛋白质序列。
研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA序列。
因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。
所以这种情况下应该选用DNA序列,而不选蛋白质序列。
2)如果DNA 序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。
1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致 ( 5’-3’)。
想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。
所以我们以后者为例。
2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta 文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。
如果是比对好的多序列比对可以直接选择“Analyze”。
3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这里选择熟悉的ClustalW),弹出窗口询问“Nothing selected for alignment,Selectall?”选择“OK”。
4. 之后,弹出多序列比对参数设置窗口。
这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。
MEGA 软件——系统发育树构建方法

• 双击图标
,
• 下载下来的序列片段保存文件为FASTA格式,打开方式为TXT格式。 • 将Blast对比后所Download的序列筛选后,构建系统发育进化树。 • 构建系统发育树需要测序所得序列1个,Blast对比得出序列5-10个, 构建出的为单枝系统发育进化树。通过这个对比可以确定出所测 定的序列最相似物种,当相似度为99%甚至100%时,基本可以确 定所测定的基因序列所属物种。
ME待测PCR产物送测序后,一个星期左右会得到生物 公司发的邮件,里面包含测序结果的附件,下载后得到文件压缩 包 ,将文件包解压,可以看到文件夹里有文件
• 测序公司会提供一款解读软件Chromas,免安装类型,能够直接 打开测序结果并通过软件直接进入NCBI数据库进行Blast搜索。
• 可培养真菌可以选定对比物种种属后构建大型系统发育进化树, 不可培养真菌则可直接构建系统发育进化树。
• 双击
后打开MEGA软件
手把手教你构建系统进化树(2021年)

97 NR 116489.1 Pseudomonas stutzeri strain VKM B-975 16S ribosomal RNA partial sequence NR 113652.1 Pseudomonas stutzeri strain NBRC 14165 16S ribosomal RNA partial sequence
进化分析流程
测序组装
• 将克隆扩增测序得到的基因进行测序。
Blast
• 比对找到相似度最高的几个基因,将这几个基因的 序列(Fasta格式文件)下载下来,整合在一个*.txt 文档中。
比对序列
• 用Mega 7.0的ClustalW做多序列联配,比对结果用*.meg格式 保存。或者用Clustal X软件进行比对,比对结果保存为*.aln, 再用Mega 转化为*.meg格式。
DNA→ DNA
ezbiocloud https:///identify
cDNA→蛋 白质
蛋白质 →cDNA
蛋白质→蛋白 质
NCBI
输入测序组装后的序列
ezbiocloud
输入序列名称 输入测序组装后的序列
比对序列
MEGA可识别fasta格式文件比对前将xxx.txt 重命名为xxx.fasta
构建系统进化树
1) 在构建系统树时,使用了Bootstrap法进行检验。在做Bootstrap时,以原序列为蓝本随机重组生成新的序列, 重复估算模型。如果原序列计算得到的分枝在新Bootstrap中依然频繁出现,则该分枝的可信度高。分枝在 Bootstrap中出现的频率就是表征分枝可信度的参数。 2) Original Tree是应用估算模型形成的最优系统树。在Original Tree上有计算得到的距离数据,可以表征两个基 因的亲缘远近;MEGA形成的Original Tree上也有频率参数,实际来自Bootstrap Consensus Tree的对应分枝。 3) Bootstrap Consensus Tree 是很多次Bootstrap得到的平均结果,它不包含进化距离信息(在设置View时无法 调用,也没有意义),分枝上的数字代表该分枝的频率参数。另外,它的拓扑结构也可能与Original Tree很不相同。
用MEGA构建进化树

如何用MEGA构建进化树是一个关于序列分析以及比较统计的工具包,其中包括有距离建树法和MP建树法;可自动或手动进行序列比对,推断进化树,估算分子进化率,进行进化假设测验,还能联机的Web 数据库检索;下载后可直接使用,主要包括几个方面的功能软件:iDNA和蛋白质序列数据的分析软件;ii序列数据转变成距离数据后,对距离数据分析的软件; iii对基因频率和连续的元素分析的软件;iv把序列的每个碱基/氨基酸独立看待碱基/氨基酸只有0和1的状态时,对序列进行分析的软件;v绘制和修改进化树的软件,进行网上blast搜索;用MEGA构建进化树有以下步骤:1. 16S rDNA测序和参考序列选取从环境中分离到单克隆,去重复后扩增16S rDNA序列并测序,然后与数据库比对,找到相似度最高的几个序列,确定一下你分离的细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定你分离得到的就是Blast到的那个,然后找一到两个同科的,再找一到两个同目的,再找一到两个同纲的细菌,把序列全部下下来,以FSATA形式整合在TXT文档中,如>TS1GCAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAA CACGTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCG GA TAGGACCTCGGGA TGCA TGTTCCGGGGTGGAAAGGTTTTCCGGTGCAGGATGGGCC>gi|6|gb|| Rhodococcus sp. Atl25 16S ribosomal RNA gene, partial sequence CGATTAGAGTTTGA TCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAA GTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAACAC GTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAA TACCGGA T>TS2TGCAAGTCGAGCGAATGGA TTAAGAGCTTGCTCTTA TGAAGTTAGCGGCGGACGGGTG AGTAACACGTGGGTAACCTGCCCA TAAGACTGGGATAACTCCGGGAAACCGGGGCTAA TACCGGATAACA TTTTGAACTGCATGGTTCGAAA TTGAAAGGCGGCTTCGGCTGTCACT>gi||emb|| Bacillus cereus partial 16S rRNA gene, strain TMWGA TGAACGCTGGCGGCGTGCCTAA TACATGCAAGTCGAGCGAA TGGATTAAGAGCTTG CTCTTA TGAAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCCATAAGAC TGGGATAACTCCGGGAAACCGGGGCTAATACCGGATAACATTTTGAACYGCATGGTTC ………………………….………………………….参考序列选择有几个原则:a,不选非培养unclutured微生物为参比;b,所选参考序列要正确,里面无错误碱基;c,在保证同属的前提下,优先选择16S rDNA全长测序或全基因组测序的种;d,每个种属选择一个参考序列,如果自己的序列中同一属的较多,可适当选择两个参考序列;2. 序列比对将整理好的序列导入,如图接着程序自动运行,得出结果,自动输出.aln和.dnd 为后缀的两个文件; 序列比对也可以直接用MEGA来做;3. 打开程序MEGA,如下图所示:4. 只能打开meg格式的文件,但是它可以把其他格式的多序列比对文件转换过来,用.aln格式Clustal的输出文件转换.meg文件;点File:Convert to MEGA Format,打开转换文件对话框,从目的文件夹中选中Clustal 对比分析后所产生的.aln文件,点击打开;5. 转换好的meg文件,会弹出一个提示信息,点击ok;查看meg序列文件最后是否正常,若存在clustal. 行,即可删除;点存盘保存meg文件,meg文件会和aln文件保存在同一个目录;6. 关闭转换窗口,回到主窗口,现在点面板上的“Click me to activate a data file”打开刚才的meg文件;如果为蛋白质序列,选择“protein sequence”,电击“OK”,得到以下图示,数据输入之后的样子,窗口下面有序列文件名和类型;而在另外一个窗口内,出现以下数据文件点击选择和编辑数据分类图标, 可对所选择的序列进行编辑,完成后点击close即可;序列编辑完成后,可进行保存,点击保存后出现以下界面,点击ok即可;7. 构建进化树的算法主要分为两类:独立元素法discrete character methods和距离依靠法distance methods;所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了;而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的;进化树枝条的长度代表着进化距离;独立元素法包括最大简约性法Maximum Parsimony methods和最大可能性法Maximum Likelihood methods;距离依靠法包括除权配对法UPGMAM和邻位相连法Neighbor-joining;1 phylogeny→UPGMA2用Bootstrap构建进化树,MEGA的主要功能就是做Bootstrap验证的进化树分析,Bootstrap 验证是对进化树进行统计验证的一种方法,可以作为进化树可靠性的一个度量;各种算法虽然不同,但是操作方法基本一致;进化树的构建是一个统计学问题;我们所构建出来的进化树只是对真实的进化关系的评估或者模拟;如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”;模拟的进化树需要一种数学方法来对其进行评估;不同的算法有不同的适用目标;一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序列上的每一个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多大于几千个碱基;用最大可能性法分析序列则不需以上的诸多条件,但是此种方法计算极其耗时;如果分析的序列较多,有可能要花上几天的时间才能计算完毕;过程如下①参数的设置:phylogeny→bootstrap test of phylogeny→NJ②系统进化树的测试方法,可以选择用Bootstrap,也可以选择不进行测试;重复次数Replications通常设定至少要大于100比较好,随机数种子可以自己随意设定,不会影响计算结果;一般选择500或1000;有许多Model供选择,默认为Kimura 2-paramete r,不同的Model有不同的算法,具体请参考专业的生物信息学书籍;设定完成,点compute,开始计算;②结果输出:这个过程所耗时间和序列的数量和长短成正比,程序就会产生这么一个树,该窗口中有两个属性页,一个是原始树,一个是bootstrap验证过的一致树;树枝上的数字表示bootstrap验证中该树枝可信度的百分比; 结果如下:8. 进化树的优化:1利用该软件可得到不同树型,如下图所示:除此之外,还可以有多种树型,根据需要来选择; 2显示建树的相关信息:点击图标i;3点击优化图标,可进行各项优化:Tree栏中,可以进行树型选择:rectangular tree/circle tree/radiation tree;每种树都可以进行长度,宽度或角度等的设定Branch:可对树枝上的信息进行修改;Lable:可对树枝的名字进行修改;Scale:标尺设置Cutoff:cut off for consensus tree;一般为50%;9、进化树的分类优化Place root on branch:可以来回转换;Flip subtree:180度翻转分枝,名字翻转180度;Swab subtree:交换分枝,名字不翻转;Compress/expand subtree与Set divergent time:可以把同一分枝的基因压缩或扩展;点击Compress/expand subtree后,在要压缩的分枝处点击,出现以下界面,在name/caption 中输入文件名例如w,其他还有很多的选项,设置好了,点击OK;所得到的结果,可以在压缩和扩展之间转换;10. 调整进化树根据所的进化树的效果,要进行调整,包括多余序列删除、不足序列添加、种属名称标注等等,还要根据投稿杂志要求在PHOTOSHOP 中修改等;完成后的进化树应包含充足的信息;本人所做进化树完成图如下:。
使用mega构建进化树的流程

使用mega构建进化树的流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!使用 MEGA 构建进化树的流程如下:1. 数据准备:收集需要构建进化树的序列数据,可以是 DNA 序列或蛋白质序列。
mega操作过程-多序列比对、进化树PPT幻灯片课件

信
(neighbour joining)、phylip、dist
息
学
CORRECT DIST:决定是否做距离修正。对于小的序列歧异(<
及
10%),选择与否不会产生差异;对于大的序列歧异,需做出
应
修正。因为观察到的距离要比真实的进化距离低。
用
IGNORE GAPS:选择on,序列中的任何空位将被忽视。
Extremely slow computation.
20
Progressive Alignment Method
DbClustal:
http://igbmc.u-strasbg.fr:8080/DbClustal/dbclustal.html
基
础 Poa (Partial order alignments):
第二节 多序列比对程序及应用
基 础
Progressive Alignment Method
生
物
Iterative Alignment
信
息 学
Block-Based Alignment
及 应
DNASTAR
用
DNAMAN
12
1、Progressive Alignment Method
学
A distance matrix is built to derive a guide tree, which is
及
then used to direct a full multiple alignment using the
应
progressive approach.
用
Outperforms Clustal when aligning moderately divergent
mega操作过程多序列比对进化树ppt文档

1 2 3 4 5 6 7 8 91 ⅠY D G G A V - E AL ⅡY D G G - - - E AL ⅢF E G G I L V E AL
ⅣF D - G I L V Q AV ⅤY E G G A V V Q AL
表1 多序列比对的定义 表示五个短序列(I-V)的比对结果。通过插入空位,使5个序列中 大多数相同或相似残基放入同一列,并保持每个序列残基顺序不变
➢ 在NCBI/EBI的FTP服务器上可以找到下载的软件包。 ClustalW 程序用选项单逐步指导用户进行操作,用户 可根据需要选择打分矩阵、设置空位罚分等。
ftp:///pub/software/
➢ EBI的主页还提供了基于Web的ClustalW服务,用户可以 把序列和各种要求通过表单提交到服务器上,服务器 把计算的结果用Email返回用户(或在线交互使用)。
计算机程序自动比对 ➢ 通过特定的算法(如穷举法,启发式算法等),由计算机程 序自动搜索最佳的多序列比对状态。
穷举法
穷举法(exhaustive alignment method)
➢ 将序列两两比对时的二维动态规划矩阵扩展到多维矩阵。即用 矩阵的维数来反映比对的序列数目。这种方法的计算量很大, 对于计算机系统的资源要求比较高,一般只有在进行少数的较 短的序列的比对的时候才会用到这个方法
概念 多序列比对的意义 多序列比对的打分函数 多序列比对的方法
1、概念
多序列比对(Multiple sequence alignment) ➢ align multiple related sequences to achieve optimal matching of the sequences.
mega操作过程多序列比对进化树
MEGA 系列软件系统发育树构建方法

MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都保存在同一个txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑(不能有中文)。
保存为fasta格式2)右键点击fasta文件,打开方式,mega3、全选,点击alignment,algin by culstx(按钮W),OK4、关闭此窗口,点击Yes保存5、再次点击Yes保存,6、点击cancel取消7、选择是否为编码蛋白质的核酸序列8、选择是否用mega打开文件9、点击YES,激活mega,此时mega的菜单栏与刚开始打开的菜单栏有区别。
10、系统发育树构建原理不讲了,此处以构建NJ树为例。
点击工具栏上的phylogeny,construct phylogeny,neighbor joining (NJ).出现如下界面(注意几个绿颜色的小方块):点击第一个小绿方块,选择,小绿方块会变成四个点的省略号,再点击出现如下页面:选择Bootstrap,后面的replication改为1000,点击对勾。
然后点击第三个小绿方块,这个时候对于蛋白质序列以及DNA序列,两者模型的选择是不同的。
对于蛋白质的序列,多选择Poisson Correction (泊松修正)这一模型。
而对于核酸序列,多选择Kimura 2-parameter (Kimura-2参数) 模型。
所有设置完毕之后,点击compute,雏形的树就出来了:可以对此树做出一些修改,比如线条粗细,树的形状等等,此处自己多试试。
6)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键,就可以对文字的字体大小,倾斜等做出修饰。
见下图:PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
MEGA构建系统进化树的步骤(以MEGA7为例)

MEGA构建系统进化树的步骤(以MEGA7为例)本文是看中国慕课山东大学生物信息学课程总结出来的分子进化的研究对象是核酸和蛋白质序列。
研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA 序列。
因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。
所以这种情况下应该选用DNA序列,而不选蛋白质序列。
2)如果DNA序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。
1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。
所以我们以后者为例。
2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。
如果是比对好的多序列比对可以直接选择“Analyze”。
3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这里选择熟悉的ClustalW),弹出窗口询问“Nothing selected for alignment,Select all?”选择“OK”。
4. 之后,弹出多序列比对参数设置窗口。
这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。
构建系统进化树的详细步骤

构建系统进化树的详细步骤1. 建树前的准备工作1.1 相似序列的获得——BLASTBLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool 的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。
国际著名生物信息中心都提供基于Web的BLAST服务器。
BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为核向两端延伸,以找出尽可能长的相似序列片段。
首先登录到提供BLAST服务的常用,比如国的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。
这些提供的BLAST服务在界面上差不多,但所用的程序有所差异。
它们都有一个大的文本框,用于粘贴需要搜索的序列。
把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。
如果是DNA序列,一般选择BLASTN搜索DNA数据库。
这里以NCBI为例。
登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。
BLASTN结果如何分析(参数意义):>gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, completesequenceScore = 2020 bits (1019), Expect = 0.0Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / PlusQuery: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60|||||||||||||||||||||||||||||||||||||||||| ||||||||| ||||| Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120|| ||||||||||||||||||||||||||||||| | |||||| ||||||||||||| Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118Score :指的是提交的序列和搜索出的序列之间的分值,越高说明越相似; Expect:比对的期望值。
用MEGA如何画进化树?
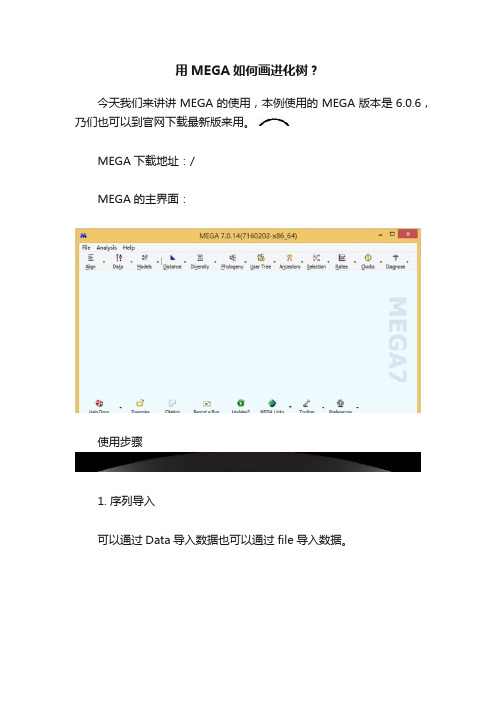
用MEGA如何画进化树?今天我们来讲讲MEGA的使用,本例使用的MEGA版本是6.0.6,乃们也可以到官网下载最新版来用。
MEGA下载地址:/MEGA的主界面:使用步骤1. 序列导入可以通过Data导入数据也可以通过file导入数据。
如果打开的文件是比对结果,选择Analyze;如果打开的文件是序列文件,选择Align。
另外双击这些后缀名文件即可自动导入序列,导入后会弹出MEGA比对界面。
如果fasta 序列导入报错,多是因为序列长度不同导致:如果序列长度不同,可以采用新建文件,将序列文件导入的方法。
步骤:Align → Edit/Build Alignment → create a new alignment → Data → open → Retrieve sequences from File将复制输入的序列另存输出看看。
步骤:data → Export alignment → fasta format序列长度都被用横线补齐了。
2. 多序列比对选择muscle或者clustalw进行比对:clustalw 一般用于DNA ,muscle多用于蛋白。
在比对之前需先选中要进行比对的序列(Shift),还可以对序列或者序列名进行编辑(双击)。
比对参数选择:保存比对文件,进化树分析提供数据。
一般导出的比对结果保存为fasta格式,或者直接点击保存按钮将结果,保存为二进制的mas或meg文件。
3. 构建进化树导入数据:将刚刚另存的meg 文件重新导入到mega程序中(直接拖入工作界面),并选择构建进化树。
参数选择:参数设置,Bootstrap method一般选择1000~1500;第一次绘图时建议选择500,这样运行速度会比价快,结果合适再调至1000重新进行进化分析。
描述:进化树可视化:View→Tree/Branch style选择树的模式,也可以通过右图菜单进行选择。
发散树环状树进化树简单美化:可视化文件的保存:•Newick——标准树文件,用于下游可视化软件的导入•png——压缩图像文件•pdf——矢量图像文件保存为png/pdf,显示不全怎么办?——将图和图注复制到WORD中保存即可(Image Copy to Clipboard 粘贴到WORD)。
系统发育树构建构建步骤

• 两个临近的分支的连接处称为节点 (node),表示推断祖先的现存类群在树最 底部的分支点成为根节(root node), 一 个单一的共同的祖先被定义为进化支 (clade)或者单源群(monophyletic group)
• 树的分支模式被成为拓扑结构(tree topology)
系统发育树建立方法
Mega下载地址/
作业: 1.使用entrez获取登录号为P26374的蛋白序列,然后 通过blastp,搜索nr库中最相似的10个序列,记录登 录号,用Mega软件进行系统发育树构建(要求用两 种以上方法)。
打开软件clustalx
• CLUSTALX-是CLUSTAL多重序列比对程 序的Windows版本。Clustal X为进行多重 序列和轮廓比对和分析结果提供一个整体 的环境。 序列将显示屏幕的窗口中。采用多色彩的 模式可以在比对中加亮保守区的特征。窗 口上面的下拉菜单可让你选择传统多重比 对和轮廓比对需要的所有选项。
?了解分子进化及系统发育分析?掌握多序列比对方法?熟悉系统发育树建立方法?掌握用mega软件进行构建系统发育树?系统发生phylogeny是指生物形成或进化的历史?系统发生学phylogeictree描述物种之间进化关系?系统发育树phylogenetictree又称称evolutionarytree进化树就是描述这一群有机体发生或进化顺序的拓扑结构
2.获取P03958序列,进行psi-blast搜索,看看结果 和blastp搜索有什么不同。
• 系统发育树(phylogenetic tree)——描 述物种之间进化关系
• 系统发育树(phylogenetic tree)——又 称evolutionary tree进化树)就是描述这一 群有机体发生或进化顺序的拓扑结构。它 可以用来研究不同物种间的进化关系,这 一直是生物学的研究热点。
手把手教你构建系统进化树

3、比对序列,比对结果转化为*.meg格式
用 Mega 6.0 的 ClustalW 做多序列联配,比对结果用 *.meg格式保存。或者用Clustal X软件进行比对,比对结果 保存为*.aln,再用Mega 6.0转化为*.meg格式。
4、构建系统进化树
打开保存的*.meg格式文件,选择邻接法构建系统发育 进化树。
以外米缀蛾的cds为例,点击cds,出现下图。
点击FASTA,出现下图。
该图为外米缀蛾的 FASTA格式,如何保 存见下图
一般情况下点 击该页的右上 角有send 图标, 选择后点击 create file 即 可下载。Txt可 以打开。 该图显示的是 序列全长的 FASTA格式下 载。
因为我采取基于氨 基酸序列比对,所 以选择coding sequences和fasta protein,下载编码 区氨基酸序列。
文件名未下载时不要更改,下下来之后再更改
MEGA6可以识别fasta格式文件。如图,将全 部-基因.txt重命名为全部-基因.fasta
•选择打开方式为MEGA6,打开全部-基因.fasta,自动跳出序列窗口 •用ClustalW做多序列联配
如何构建系统进化树
YZU.TRY
系统发生树(英文: Phylogenetic tree ) 又称为演化树( evolutionary tree ),是 表明被认为具有共同祖先的各物种间演化关 系的树。是一种亲缘分支分类方法 ( cladogram )。在树中,每个节点代表其 各分支的最近共同祖先,而节点间的线段长 度对应演化距离(如估计的演名称要么全部 斜体,要么全部不斜体,无法只让拉丁文斜体
怎样使用MEGA建立进化树

如何使用MEGA4.0建立进化树1、首先是双击软件打开如下图所示
2、现在是处于DNA序列,而我们要做蛋白质的进化树的话,就如下操作
3、接下来我们要进行序列的输入,点击左边那个红箭头,则出现下面的窗口;
4、然后右击sequence 1,修改名字,如改成DPV
5、然后从Word 里复制蛋白质序列,然后在下面的位置粘贴
6、则可出现如下图的序列了
7、然后点击窗口上的保存图标保存
8、重复从3开始,直到你的序列输入完
9、序列输入完后进行最后的保存,方法如下:
要输入ul7两次保存名字—然后关闭这个窗口。
接下来打开
出现下面这个窗口!
接下来就可以建立各种样式的进化树!
嗯,只是把过程写出来,方便大家建立进化树,不足的地方,大家补充好!
(注:可编辑下载,若有不当之处,请指正,谢谢!)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
M E G A构建系统进化树的步骤(以M E G A7为例)MEGA构建系统进化树的步骤(以MEGA7为例)本文是看中国慕课山东大学生物信息学课程总结出来的分子进化的研究对象是核酸和蛋白质序列。
研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA序列。
因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。
所以这种情况下应该选用DNA序列,而不选蛋白质序列。
2)如果DNA 序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。
1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致 ( 5’-3’)。
想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。
所以我们以后者为例。
2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta 文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。
如果是比对好的多序列比对可以直接选择“Analyze”。
3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这里选择熟悉的ClustalW),弹出窗口询问“Nothing selected for alignment,Selectall?”选择“OK”。
4. 之后,弹出多序列比对参数设置窗口。
这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。
MEGA的所有默认参数都是经过反复考量设置的,这保证了MEGA傻瓜机全自动档的品质,所以当你无从下手,或者没有什么特别要求的时候,直接点击“OK”,接受这些默认参数,开始多序列比对。
了解两个参数:①替换记分矩阵,替换记分矩阵是反映残基之间相互替换率的矩阵,也就是说,它描述了残基两两相似的量化关系。
DNA 序列有 DNA 序列的替换记分矩阵,蛋白质序列有蛋白质序列的替换记分矩阵,两者不可混用。
DNA 序列的替换记分矩阵主要有三种:1)等价矩阵。
相同核苷酸得分为1,不同核苷酸间的替换得分为 0。
由于不含碱基的理化信息和不区别对待不同的替换,一般只用于理论计算。
2)转换-颠换矩阵。
转换:DNA分子中的嘌呤被嘌呤或嘧啶被嘧啶替换。
颠换:DNA分子中的嘌呤被嘧啶或嘧啶被嘌呤替换。
在进化过程中,转换发生的频率远比颠换高。
为了反映这一情况,转换-颠换矩阵中,转换的得分比颠换要高为-1 分,而颠换的得分为-5 分。
3)BLAST 矩阵。
经过大量实际比对发现,如果令被比对的两个核苷酸相同时得分为+5 分,不相同为-4 分,这时比对效果最好。
这个矩阵广泛地被 DNA 序列比较所采用。
没有为什么,就是好,实践经验所得。
因为这个矩阵最早应用于 BLAST 工具,因此得名 BLAST 矩阵。
蛋白质的替换记分矩阵要比核酸的复杂一些: 1)等价矩阵。
相同得 1 分,不同得 0 分。
2)PAM矩阵。
基础的 PAM-1矩阵反应的是进化产生的每一百个氨基酸平均发生一个突变的量值,是基于相似度>85%的序列产由统计方法计算得到的。
由PAM-1 自乘 n 次可以外推得到 PAM-n ,表示发生了更多次突变。
如果序列亲缘关系远,也就是说序列间会有很多突变,那就选 PAM 后面跟一个大数字的矩阵;如果亲缘关系近,也就是突变比较少,序列间大多数地方都是一样的,那就选 PAM 后面跟一个小数字的矩阵。
3)BLOSUM矩阵。
后面也有一个编号,是通过对大量符合特定要求的序列计算而来的。
比如BLOSUM62是指这个矩阵是由一致度≥62%的序列计算得到的。
如果序列亲缘关系远,序列相似度低,那就选BLOSUM 后面跟一个小数字的矩阵;如果序列亲缘关系近,序列相似度高,那就选BLOSUM 后面跟一个大数字的矩阵。
总结,亲缘关系较近的序列之间的比较,用 PAM 数小的矩阵或BLOSUM 数大的矩阵;而亲缘关系较远的序列之间的比较,用 PAM 数大的矩阵或 BLOSUM 数小的矩阵。
对于关系较远的序列之间的比较,由于 PAM250 是通过矩阵自乘推算而来的,所以其准确度受到一定限制。
相比之下BLOSUM 矩阵更具优势。
对于关系较近的序列之间的比较,用 PAM 或 BLOSUM 矩阵做出的比对结果,差别不大。
如果关于要比较的序列不知道亲缘关系远近,那么就闭着眼睛用BLOSUM62 吧!如果你记不住或者听不懂上面讲的种种,那就记住 BLOSUM62 这个名字,也可以走遍天下全不怕!图1:氨基酸差异与矩阵编号对②空位罚分包括两种: gap 开头(gap open)和gap延长(gap extend)。
默认gap开头罚分高,gap延长罚分低,这样得出的结果gap很集中,有很多长串出现的gap,这可以比对两条很相似的序列--同源序列;相反,如果gap开头罚分少,gap延长罚分高,比对结果gap就比较分散,极少出现连续长串的gap(可以想象其中的原因,总是要保证得分高),这可以比对两条绝大部分序列都很相似,但其中一条的一个功能区在另一条序列中是缺失的两条序列,可以找出这个功能区。
5. 比对过程是先进行双序列比对,在进行多序列比对,最后会出现一个多序列比对结果。
将之作为中间结果保存下来。
在Alignment Explorer窗口中选择“Data”→“Export Alignment”→“MEGA Format”。
这里一定选择MEGA format以方便MEGA后续分析(其他格式适用于其他软件的分析),MEGA自动赋予“.meg”后缀名,保存后,弹出窗口,“为这组数据命名”,自己看得懂知道就可以,我这里命名为“il1r2 alignment”。
6. 生成的“.meg”文件可以双击直接导入MEGA。
也可以将其拖入MEGA主窗口中。
拖入后主窗口增加了一个“TA”按钮,点击弹出新窗口“Sequence Data Explorer”,其是多序列比对结果。
再点击“Sequence Data Explorer”上的“TA”按钮,点击后多序列最上面增加了一行,这一行是根据多序列比对结果分析得出的共有序列(consensus sequence),也就是一列里出现次数最多的字母。
多序列比对中每一列里的字母如果和共有序列相同则打点,不同则标出不同的字母,空位还是空位。
如果还想进一步了解序列的保守程度,可以点击“C”按钮,以黄色标记保守序列;或者点击“V”按钮,以黄色标记不保守序列。
通过进一步的分析,可以淘汰掉一些序列,比如海选的的序列里有一些不合群的序列,就可以把他们去掉,不让他们参与建树,以免影响建树质量。
此外,还可以对这些序列进行分组标记。
点击分组按钮,点击“加号”按钮,更改组名,然后按住Ctrl键同时选中Ungrouped Taxa 列表中的要放入这个组的序列,选中后点击“箭头”按钮,即可将序列放入分组。
同理,可以创建其他分组。
当序列数量较多时,人为分组,可以从树上更加清晰的看出组内哪些成员叛逃了去了别的组。
此外,输入序列的名字较长,作为构建的系统发生树上叶子的名字,会破坏树的外观也不利于信息的解读。
因此,需要人为修改一下序列的名字。
选中序列后点击,把名字改为能区分彼此的关键词,全部改好之后点击“save”按钮,准备工作全部完成。
7. 开始建树。
点击MEGA主窗口上的Phylogeny下拉菜单,选择Neighbor Joining(最近邻居法)。
弹出窗口询问是否使用当前 .meg里面的数据,选Yes。
接下来,弹出参数设置窗口(Analysis Preferences)。
参数设置对构建的系统发生树的准确程度非常重要。
在树构建好之后,还经常需要根据树的具体情况,重新设置参数,并重新建树,如此反复,纸质结果令人满意为止。
同样的如果对参数设置摸不着头脑,就接受默认设置,也能做出基本满意的系统发生树。
至少应该掌握其中三个参数的设置:① Test of Phylogeny(建树的检验方法),是用来检验建树的质量的。
默认的检验方法是Bootstrp method (步长检验)。
步长检验需要设定检验次数,通常为100的倍数,默认设置为500。
步长检验是根据所选择的建树方法,计算并绘制指定次数株系统发生树。
因为大多数建树的方法的核心算法都是统计概率模型,所以每次计算出来的树都会有所差别。
而剑豪的系统发生树上每个节点上都会标有一个数字,它代表了指定次数次计算所得出的系统发生树中有百分之多少的树都含有这一节点。
一般来讲,绝大多数节点上的数值都大于70%的树才可信。
个别低于70%的节点可以暂且容忍,或通过添加、山间序列来改善质量。
② Substitution Model。
是选择计算遗传距离时使用的计算模型。
理论上应该尝试各种模型,根据检验结果选择最合适的模型计算。
但在实际操作中,可先尝试选用较简单的距离模型,比如p-distance。
③ Gap/Missing Data Treatment,大多数建树方法会要求删除多序列比对中含有空位较多的列。
但是根据遗传距离度量方法的不同,删除原则也不同。
如果是以序列间不同残基的个数来度量遗传距离的话,这里需要选择 Complete deletion(全部删除)。
如果是其他方法,比如这里选用的 NJ 方法,可以选择 Partial deletion(部分删除)。
删除程度定在 50%,即,保留一半含有空位的列。
8. 按照以上方案参数设置后,点击“Compute”按钮,开始构建系统发生树。
经过一番计算之后,新窗口 Tree Explorer 里展示的就是创建好的系统发生树。
这个窗口里有两个标签页。
第一个是 Original Tree(原始树),第二个是 Bootstrap consensus tree(步长检验合并出来的树)。
Bootstrap consensus tree 上,节点处的数字表示,经步长检验有百分之几的树具有这根树枝,即,反应了该树枝的可信度。
当前构建的这株系统发生树中,绝大多数节点处的数值都是≥70 的话,这株树整体上就是可信的。
Original Tree 是步长检验构建的 500 株树中的一株,未经过多棵树合并,所以树枝的长短可以精确代表遗传距离。