分子图形软件网络教程精编
神经网络软件操作手册-Neurosolutions0

1、打开Neurosolutions,进入以下界面值点击NS Excel '按钮,在Excel 加载项中出现Neurosolutions 项。
者文件封 硝如 祖眼⑵ 插入⑴ 梏式约 工具。
的据此 窗口口 增助如 咯1171t :加口与0昌国导融X 。
息-W g 》包£1曲鼬?宋^2、标记数据选定指标数据列(x1 x5),点击Neurosolutions 菜单选择Tag Data 下Column(s) As Input 选 项,将(x1 x5)标记为输入。
选定指标数据列y,点击Tag Data 下Column(s) As Desired 选 项,将y 标记为输出。
选定第1到53个样本所在的行,点击Tag Data 下Row(s) As Training 选项,将其标记为训练集。
最后选定第53到58个样本所在的行,点击Tag Data 下Row(s) As Testing 选项,将其标记为测试集。
标记完成后界面如下。
力文杵0 ««'E:'福图也人工I 格加0 IM'I)她存艰 营口的 也助如 乐u&LW.a 口片& 寻&晶怪五,U M 唾些 I :**磔3、预处理数据点击Neurosolutions 菜单,选择Preprocess Data 下Randomize Rows 选项,完成数据处理, Excel 出现 sheet1Randomize.亘]交件区i锚强£J次困以置入UJ格式®।IflCU 窈竟亚।宙口-J黜助皿Heur由IxLi皿□N kl 目鼻咎EL占心T ,他E TIMIB鄱B:^:、B c0E F:H I■2 4U71334QB95q 4峥95 4U6928 4069T7S40G939 4067&IO4Ci?1711407051245r o 913iUB&il1440&E21540719IE4U70617口 ,一16:'■''1940BBD如4涮521406B32240679Z3 4Q&7B.:;4U71D2E-40705EE 407 IS274067E.ME4、建立BP神经网络模型选择Excel的sheetl工作表界面,点击Neurosolutions菜单,选择Create/Open Network下的New Custom Network选项,出现以下界面。
分子模拟软件简介

分子模拟软件简介3D分子图形显示工具 (RasMol and OpenRasMol)(免费)AMBER (分子力学力场模拟程序)autodock (分子对接软件)(免费)GROMACS (分子动力学软件)(免费)GULP (General Utility Lattice Program)(免费)NIH分子模拟中心的化学软件资源导航(Research Tools on the Web) X-PLOR (大分子X光晶体衍射、核磁共振NMR的3D结构解析)(免费)高通量筛选软件PowerMV (统计分析、分子显示、相似性搜索等)(免费)化合物活性预测程序PASS(部分免费)计算材料科学MathubC4:Cabrillo学院化学可视化项目以及相关软件(免费)Databases and Tools for 3-D Protein Structure Comparison and Alignment(三维蛋白质结构对比)(免费)Democritus (分子动力学原理演示软件)DPD应用软件cerius2(免费)EMSL Computational Results DataBase (CRDB)MARVIN'S PROGRAM (表面与界面模拟)(免费)XLOGP(计算有机小分子的脂水分配系数)(免费)量子化学软件中文网美国斯克利普斯研究院:金属蛋白质结构和设计项目(免费) /doc/1f7136708.html,/(免费)3D Molecular Designs (蛋白质及其他3D分子物理模型快速成型技术)3D-Dock Suite Incorporating FTDock, RPScore and MultiDock (3D分子对接)(免费)AMSOL (半经验量子化学计算)(免费)Amsterdam Density Functional (ADF, 第一原理电子结构计算) Bilbao晶体学服务器(免费)BOSS (蒙特卡罗模拟软件)CADPAC (剑桥量子化学计算软件)(免费)Car-Parrinello分子动力学(CPMD, ab-initio分子动力学计算软件)(免费)CHARMM (大分子分子力学模拟计算软件)(部分免费)Chem2Pac package (A computational Chemistry Integrator)(免费) ChemTK Lite (QSAR软件)(免费)Chemweb计算化学在线工具:GAMESS(免费)Clebsch-O-Matic (在线计算器)(免费)Collaborative Computational Projects (协同计算计划) COLUMBUS (量化从头计算分子电子结构程序集)(免费) CrystalMaker Software (晶体结构可视化软件)DL_POLY (分子动力学模拟软件)(免费)DockVision (分子对接程序)(部分免费)DPMTA (分子动力学并行模拟软件)(免费)Dr. Pablo Wessig 研究小组开发的计算化学软件(免费)eHiTS: Electronic High Throughput ScreeningEMSL Gaussian Basis Set Order Form(免费)GAMESS-UK (分子电子结构计算软件)GAMESS: The General Atomic and Molecular Electronic Structure System(免费)Genebrowser (生物技术、基因治疗资源导航)Glide (分子对接程序)GROMOS (通用分子动力学软件包)(部分免费)HyperChem (分子模拟)Interprobe Chemical Services (分子模型化软件)Jmol (分子可视化软件)(免费)List of Computationally Sick Species (ab initio计算出现问题的物质、方法)MacroModel (分子力学计算程序)MARDIGRAS和CORMA (弛豫矩阵分析)(免费)MCPRO (用于蛋白质和核酸的蒙特卡罗模拟软件)MDRANGE (分子动力学计算ion ranges)(免费)MDynaMix (分子动力学计算软件)(免费)MidasPlus (分子建模软件, 美国加利福尼亚大学旧金山分校计算机图形实验室开发)MOE(分子模拟应用环境、方法开发平台)MOLMOL (生物大分子3D结构分析和显示、NMR结构解析)(免费) MolPOV 2.0.8 (一个将PDB文件转为POV-Ray文件的软件)(免费) MOLPRO 量子化学软件包(用量化从头方法计算分子电子结构)(免费) Mopac 2002 (通用半经验量子力学程序)NAMD (并行分子动力学计算软件)(免费)Norgwyn Montgomery (化学软件公司)NWChem (计算化学软件包)OpenEye (快速计算分子的静电性质、形状)ORAC (用于模拟溶剂化生物分子的分子动力学计算程序, 意大利佛罗伦萨大学)(免费)ORTEP-III (美国橡树岭国家实验室晶体结构可视化--热椭圆体绘图程序)(免费)OSIRIS Property Explorer (LogP, 溶解度、成药可能性预测)(免费) PAPA (计算粒状物料的三维并行分子动力学计算程序)(免费) PETRA (反应性参数预测,包括生成焓、键离解能等)(部分免费) PharmTree (3D药效团生成和化合物分类)Pipeline Pilot (药物发现集成平台)PMDS (并行分子动力学模板库)(免费)PreADMET (ADMET预测)Protein Domain Motion Analysis Software: DynDom (蛋白质分析软件)(免费)ProtoMol (分子动力学并行计算软件)(免费)PSI3量子化学软件包(量化从头计算)(免费)Q-Chem (量子化学计算软件包)SGI应用于化学、生物信息学的软件、硬件产品SIGMA (分子动力学相关软件)SimBioSys (药物设计软件SPROUT)SMILECAS 数据库 (描述分子结构、子结构和复合结构的线性编码系统)SURFNET (量子化学计算程序)(免费)Sweet (依据标准命名方法和分子顺序建立糖类三维模型)Swiss PDB Viewer (PDB蛋白质结构可视化软件)SYBYL/Base(分子模拟和药物发现平台)TINKER (分子设计软件)(免费)UCSF Chimera (可扩展的, 交互式分子图形程序)(免费)VAMP/VASP (采用从头计算量子力学的分子力学)(免费)VHMPT (螺旋膜蛋白拓扑结构显示与编辑程序)(免费)Virtual Molecular Dynamics Laboratory (分子动力学软件)(免费) voidoo(空腔探测软件)(免费)WAM: Web Antibody Modeling (抗体模型构建)(部分免费) WebMO (基于3W界面的计算化学软件包)(免费)并行分子动力学模拟软件DoD-TBMD(免费)大分子对接程序Hex(免费)大规模原子(分子)并行模拟器 LAMMPS(免费)单晶和粉末衍射合作计算项目开发的免费软件(CCP14)(免费)蛋白质分子动力学模拟软件:CONCOORD(免费)蛋白质模拟资源导航,美国橡树岭国家实验室ORNL分子的物理化学性质在线计算(用在基于结构的药物设计, 可计算logP, PSA,等)(免费)化合物3D结构VRML文件的自动生成(免费)化学中的常用计算机软件与资源(免费)化学资源工具箱(免费)计算机模拟的分子运动图像集(DSMM)(免费)可下载的教学软件(伦敦大学玛丽女王学院化学系提供)(免费)量子化学网美国华盛顿州立大学化学系:无机化学教学资源美国加州大学圣地亚哥分校所开发的化学软件(化学反应计算、分子建模和可视化)美国康奈尔大学理论中心计算生物服务单元提供的免费软件(计算生物与化学方面)(免费)美国马里兰大学:生物技术研究所Gilson研究小组美国能源部Ames实验室:经典分子动力学软件AL_CMD(免费) 免费远程计算:贵州大学GHPCC量子化学从头计算系统(免费)模拟蛋白质的并行分子动力学计算程序EGO(免费)牛津大学:抗癌药物分布式计算项目 Screensaver Lifesaver欧洲科学基金资助项目:分子模拟所面临的挑战(Simu: Challenges in Molecular Simulations)生物大分子结构分析和确认系列软件(美国加州大学圣地亚哥分校大分子结构计算研究中心开发)(免费)牙买加西印度大学Mona分校化学系所开发的免费软件(免费)应用于双原子分子的数值Hartree-Fock程序(免费)原子轨道3维模拟演示。
中科大MS培训教程画物质XRD图谱

数据处理
对收集到的数据进行处理,包 括背景扣除、平滑处理等。
数据收集与分析
数据收集
在实验过程中,需要实时记录衍射角度和对应的 强度数据。
图谱分析
根据处理后的数据,绘制XRD图谱,并进行物 相分析、晶体结构分析等。
ABCD
数据处理
对收集到的数据进行处理,包括背景扣除、平滑 处理等,以获得更准确的衍射图谱。
峰位
指X射线衍射峰的位置,通常以角 度表示。通过分析峰位,可以确 定晶体的晶格常数、晶面间距等 参数。
解析方法
根据布拉格方程(nλ=2dsinθ)和 已知的λ值,计算出晶面间距d;根 据晶格常数和晶体类型,进一步确 定物质成分。
峰强解析
峰强
指X射线衍射峰的强度,通常以相对 强度或积分强度表示。通过分析峰强 ,可以确定晶体结晶度、杂质含量等 信息。
在结晶度分析中,需要注意衍射峰的 宽度和峰高比值。峰宽越窄,表明晶 体结构越完整;峰高比值越大,表明 结晶度越高。这些信息有助于了解物 质的结构和性能。
感谢您的观看
THANKS
结果解读
根据分析结果,解读样品的物相组成、晶体结构 等信息。
03
XRD图谱绘制
数据处理
数据清洗
去除异常值、填补缺失值、平滑噪声等,确保数 据质量。
归一化
将数据范围调整到同一尺度,以便更好地比较和 识别模式。
滤波和降噪
通过平滑技术减少数据中的随机噪声,提高图谱 的清晰度。
图谱绘制软件
01
02
03
添加图例和标签
为图谱添加图例和标签,说明不同颜 色或标记的含义,提高图谱的可读性。
调整线条和标记样式
分子原子原胞晶体显示软件Visual Molecular Dynamics 使用说明

分子原子原胞晶体显示软件Visual Molecular Dynamics使用说明本文档旨在为用户提供使用分子原子原胞晶体显示软件Visual Molecular Dynamics(VMD)的详细说明。
本文档将从安装步骤、界面介绍、基本操作、高级功能等方面进行阐述。
一、安装步骤1.VMD软件安装包。
2.运行安装包,并按照提示完成安装过程。
3.启动VMD软件。
二、界面介绍1.主窗口:展示主要的分子模型和操作界面。
2.工具栏:提供常用的快捷操作按钮。
3.菜单栏:包含各种功能选项和设置项。
4.侧边栏:显示当前分子的相关信息和属性。
5.3D显示区域:展示分子结构的三维模型。
三、基本操作1.导入分子模型:通过菜单栏中的导入选项或拖放文件的方式导入分子模型。
2.移动和旋转:通过鼠标拖动实现分子模型的移动和旋转。
3.缩放和平移:通过鼠标滚轮调整分子模型的缩放比例,并通过鼠标拖动实现分子模型的平移。
4.选择和编辑:通过选择工具选择分子的特定部分,并通过编辑工具进行编辑。
5.显示模式:通过菜单栏中的显示选项,切换分子的显示模式,如线框模式、球棍模式等。
四、高级功能1.分子动力学模拟:通过设置分子的初始参数和模拟条件,进行分子动力学模拟。
2.路径绘制:根据分子模型的路径,绘制出分子的运动轨迹。
3.能量计算:对分子进行能量计算,并可显示计算结果。
4.分子表面绘制:绘制分子的表面结构,提供更详细的分子信息。
附件:1.VMD安装包()法律名词及注释:1.分子:由原子组成的稳定结构体。
2.晶体:具有排列有序的原子、分子或离子的固体物质。
3.原子:构成分子和晶体的最基本的微观粒子。
高斯软件-基础教程
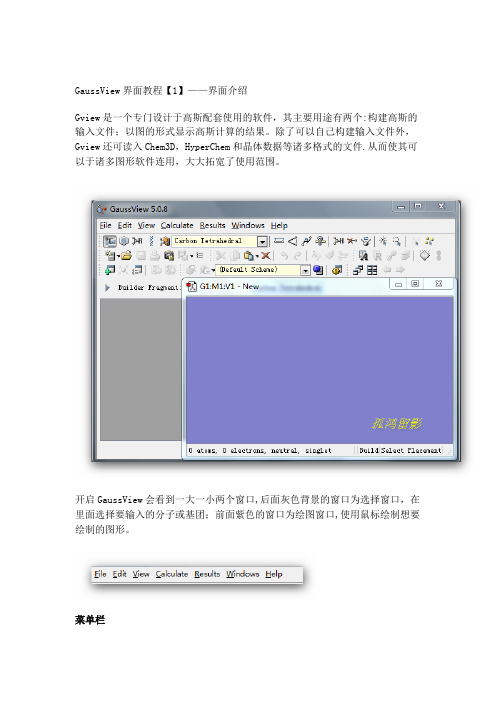
GaussView界面教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件.从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到.3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】-—构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
(完整word版)10分钟教你掌握分子对接模拟软件(医药向)

首先介绍一下自己吧,本人毕业于南方某知名211大学药学系,目前于澳门科技大学攻读硕士研究生.从本科开始自己就在接触CADD(计算机辅助药物设计)方面的软件知识,在此将分享一些自己的纯干货!下面将以一个实例操作带大家迅速认识和掌握分子模拟对接,希望给各位从事医药行业和药物化学合成的同学带来帮助。
话不多说,下面进入正题。
首先我们搞清楚一个概念:什么是分子模拟对接.分子模拟对接简单来说就是利用电脑软件将受体蛋白与配体分子进行模拟对接,计算它们的结合能(KJ/MOL)大小来判断结合是否紧密,若结合效果比较理想,那么该蛋白受体或配体则是我们理想的分子,可以进一步进行实验室操作,避免盲目实验带来的人力经济损失.接下来我将介绍一下本篇文章的主角,也是我们所要用到的软件PyRx、Chemdraw、AutodockTools 以及PyMol.为了便于理解,简要概括之:Chemdraw为化合物分子绘图软件;PyRx为Autodock Vina 算法搭载软件,能够调用其算法直接进行模拟对接;AutodockTools是PyMol为对接结果成像软件,可以进一步分析其结构.下面正式进入正题,我将大致分为三个板块来进行推进:受体配体的准备;分子对接;结果分析.研究类型为:已知若干配体分子结构,通过受体蛋白测试配体分子活性。
本次筛选意在以COMT酶为受体,从20种与常见氨基酸形成环二肽的目标化合物中筛选出与COMT 酶受体结合最为紧密的一种环二肽结构,大大减少了随机筛选的盲目性,有利于进一步研究该类化合物分子的生物学活性与改造成抗帕金森疾病前药的可能。
图1展示了20种不同环二肽结构物质的统一结构,随着R基团的不同,所对应的氨基酸也不同。
而表1则展示了20种不同环二肽的分子式。
图1 Cycol[DOPA(6—NO2)-AA]表1 待筛选的20种配体分子配体名称分子量Cycol[DOPA(6-NO2)—Ala] 307。
079 Cycol[DOPA(6—NO2)—Arg]351.089Cycol[DOPA(6—NO2)-Asn]350.104 Cycol[DOPA(6-NO2)-Asp]351.089 Cycol[DOPA(6-NO2)-Cys]339.145 Cycol[DOPA(6—NO2)-Gln]364.131 Cycol[DOPA(6-NO2)-Glu]365.116 Cycol[DOPA(6-NO2)—Gly]293.052 Cycol[DOPA(6—NO2)—His]373.141 Cycol[DOPA(6—NO2)—Ile]349。
介绍一款优秀的分子图形软件Chimera

口
墓丝 功能
对 于蛋 白质 和 D A, 以进 行 多 序 列 比对 。使 N 可
更为可贵的是 , 这些命 令可以组合在一起 , 做成
一
用其 中的 M lln u ag 查看器可以方便 的查看。通过此 ti 功能可以进行分子的叠合 。另外通过 M t M kr a h ae 工 c 具可以进行大分子的叠合 。另外该软件还有 V l e om u 查看器 , 用来显示和分析体积数据 , 其可 以支 持多数 mp a 类型文件。更多的功能在此不赘述。
教学方面 , 尤其是多媒体课件的制作上有意想不到的 用途和其他软件无法 比拟的优势。但该软件在 国内 科研机构 中应用较少 , 其具有的其他类似软件所不具 备 的功能和特点仍鲜为人知 , 故在此分 以下几个方面
进 行介 绍 。
球棍、 球形 、 线性等 。对于大分子来说 , 可以很方便的 显示各种模型 。例如对 于蛋 白质 分子来说 , J 只需 要点击鼠标就可以让分子只显示骨架 , 或者只显示螺 旋、 折叠等, 也可以只显示某一种氨基酸 , 或者只显示 分子的某一部分 。如果为了强调效果 , 亦可以采用改
变颜 色或者 改变 显 示 模 式 等 加 以 突 出。类 似 可 以显
示的大分子还有 D A、 N 聚合物 , 甚至病毒外壳等。
日 委 佥
频繁使用 鼠标 是一件很辛苦的事情 , C i e 但 h r m a
Ⅱ 壬缉 亘 塑
分子的可视化是 C ie h r m a的核心部分 , 它做到 了
[ ] G dadTD, un F rnTE Sf a x ni s 2 odr H agcc, er . ow r et s n i t e e o
t S h me a f r it r cie vs aiai n o ag - o UC F c i r o n e a t iu l t fl e mo v z o r
梦龙软件网络图编辑操作教程

通过输入具体数值或选择预设的粗细选项,可以调整连线的粗细。较粗
的连线可以强调重要的关系,而较细的连线则可用于表示次要关系。
插入图片和文本框美化网络图
插入图片
在网络图中插入相关图片可以增强视觉效果和表达力。选择需要插入的图片文件,调整其大小和位置,使其 与网络图内容相协调。
添加文本框
文本框可用于添加说明性文字或标注重要信息。在梦龙软件中,可以创建文本框并输入所需文字,然后调整 文本框的位置和大小以适应网络图布局。
行业应用
梦龙软件在各行各业中都有广泛的应用,如 IT、教育、科研、金融、制造等行业。在IT 行业中,梦龙软件可用于软件开发过程中的 流程图设计、系统架构图绘制等;在教育行 业中,梦龙软件可用于课件制作、教学计划 安排等;在科研行业中,梦龙软件可用于实
验数据分析、科研论文插图绘制等。
02
梦龙软件界面与基础操作
学员互动交流环节
环节一
学员提问与答疑
01
环节二
经验分享与交流
03
环节三
案例分析与讨论
05
02
说明
在此环节,学员可以提出自己在使用梦龙软 件过程中遇到的问题或困惑,老师和其他学 员会积极解答和提供帮助。
04
说明
鼓励学员分享自己在使用梦龙软件过 程中的实用经验和技巧,促进学员之 间的交流和学习。
06
说明
批量处理节点属性方法
选择多个节点
按住鼠标左键并拖动,可以框选 多个节点。也可以通过按住Ctrl 键并单击需要选择的节点进行多
选。
应用批量处理
右键单击选中的任何一个节点, 在弹出的菜单中选择“批量处理
”选项,打开批量处理窗口。
打开批量处理窗口
(2024年)《完整的visio教程》ppt课件

1 2
无法连接到团队协作服务
检查网络连接、确认服务器地址和端口号是否正 确、更新Visio版本等。
版本控制出现错误
检查文件是否被其他程序占用、确认文件路径和 名称是否正确、重新启动Visio等。
3
团队协作效率低下
优化网络连接、减少文件大小、提高计算机性能 等。
2024/3/26
26
06
高级功能应用与技巧分享
19
在Visio中创建数据可视化图表
将图表形状拖拽到画布中,并根据需 要调整大小和位置。
根据数据自动生成图表,并可通过右 侧属性面板调整图表样式和格式。
2024/3/26
双击图表形状打开“数据”窗口,在 此处输入或导入数据。
20
报表生成方法
报表生成器介绍:Visio内置的报表生成器可快速创建基于数据的报表。
连接形状并设置连接线 格式。
使用文本工具添加和编 辑文本。
应用主题和样式统一图 形外观。
导出图形为PDF、图片 等格式。
2024/3/26
7
02
绘制流程图与组织结构图
2024/3/26
8
流程图基本概念及符号含义
流程图基本概念
流程图是一种用图形符号表示系统或它的组成部分和各类人员之间相互联系、 相互作用情况的图。它可以描述系统的工作过程和逻辑功能。
绘制组织结构框架
使用Visio中的“组织结构图”模板,选择合适的图形和 符号,搭建组织结构的框架。
添加职位和人员信息
在组织结构图中添加各个职位和人员的信息,包括姓名、 职位、联系方式等。
2024/3/26
调整和优化
根据需要调整和优化组织结构图的布局和细节,使其更加 清晰易读。同时,可以使用不同的颜色和线条来区分不同 的部门和职位,提高可读性。
Discovery Studio官方教程(Help-Tutorials) 可视化分子对接非键作用

Discovery Studio Analyze Ligand Poses教程介绍分子之间的非键相互作用是生命体系中分子识别的基础。
在基于结构的药物设计过程中,识别和优化配体分子和受体分子间的相互作用是一个基本过程,因此,全面细致的分析非键相互作用非常关键。
本教程将采用热稳定的腺苷A2A受体和反相激动剂ZM241385的复合物,其中A2A受体是G-蛋白偶联受体,是帕金森氏症的药物靶标,其拮抗剂Preladenant正处于临床阶段。
本教程将分析对接至A2A受体的配体分子构象。
执行计算并分析结果在文件浏览器(Files Explorer)中,双击打开Samples|Tutorials|Receptor-Ligand Interactions|3pwh_dockedposes.dsv。
打开一个分子对接结果,包含3pwh蛋白分子和多个小分子的对接构象。
(图1)说明:对接分子共有50个,具有一定多样性,其中20个为参照分子,带有CHEMBL前缀,30个为诱饵分子,带有ZINC前缀。
这50个分子基于Dock Ligands (CDOCKER)模块对接至已经预处理过的腺苷A2A受体分子,最终得到3pwh_dockedposes.dsv文件中的对接结果。
本教程通过对该对接结果的分析来获取重要的非键相互作用信息,从而进一步优化虚筛流程。
图1 3pwh对接结果在工具浏览器(Tools Explorers)中,展开Receptor-Ligand Interactions |View Interactions,点击Analyze Ligand Poses。
相应的参数出现在参数浏览器中。
点击Input Ligands参数项,下拉列表中选择3pwh_dockedposes:All,选中3pwh_dockedposes 窗口中所有配体构象。
其余参数保留默认设置,点击Run运行任务。
(图2)点击Background让任务后台运行,等待任务完成。
2023年中级软考《信息安全工程师》考试全真模拟易错、难点精编⑴(答案参考)试卷号:10

2023年中级软考《信息安全工程师》考试全真模拟易错、难点精编⑴(答案参考)(图片大小可自由调整)一.全考点综合测验(共50题)1.【单选题】数字信封技术能够()A.对发送者和接收者的身份进行认证B.保证数据在传输过程中的安全性C.防止交易中的抵赖发送D.隐藏发送者的身份正确答案:B2.【单选题】防火墙作为一种被广泛使用的网络安全防御技术,其自身有一些限制,它不能阻止()A.内部威胁和病毒威胁B.外部攻击C.外部攻击、外部威胁和病毒威胁D.外部攻击和外部威胁正确答案:A3.【单选题】数字水印技术通过在数字化的多媒体数据中嵌入隐蔽的水印标记,可以有效地对数字多媒体数据的版权保护等功能。
以下各项工,不属于数字水印在数字版权保护必须满足的基本应用需求的是()A.安全性B.隐蔽性C.鲁棒性D.可见性正确答案:D4.【单选题】关于C2 等级安全性的描述中,错误的是()A.用户与数据分离B.安全性高于C1C.存取控制的单位是用户D.具有托管访问控制正确答案:D本题解析:解析:C2 等级具有受控的访问控制,存取控制以用户为单位,用户与数据分离,安全性高于C1。
B1 是标记安全保护,除了C2 级的安全要求外,增加安全策略模型,数据标号(安全和属性),托管访问控制等。
根据解析, D 选项错误,故选择 D 选项。
5.【单选题】()是企业在信息时代市场竞争中生存和立足的根本。
A.人才优势B.原材料优势C.经营式优势D.信息优势正确答案:D6.【单选题】扫描技术()A.只能作为攻击工具B.只能作为防御工具C.只能作为检查系统漏洞的工具D.既可以作为工具,也可以作为防御工具正确答案:D7.【单选题】以下关于IPSec 协议的叙述中,正确的是()A.IPSec 协议是解决IP 协议安全问题的一B.IPSec 协议不能提供完整性C.IPSec 协议不能提供机密性保护D.IPSec 协议不能提供认证功能正确答案:A8.【单选题】许多黑客利用软件实现中的缓冲区溢出漏洞进行攻击,对于这一威胁,最可靠的解决方案是()。
2024年VISIO培训教程

VISIO2024培训教程一、概述Visio2024是一款功能强大的图形绘制软件,由微软公司推出。
它广泛应用于各种场合,如流程图、网络图、组织结构图等。
本教程旨在帮助用户快速掌握Visio2024的基本功能和操作技巧,提高绘图效率。
二、界面及功能介绍1.菜单栏:包含文件、编辑、视图、插入、格式、数据、审阅等菜单,用户可以通过这些菜单执行相应操作。
2.工具栏:包含常用的绘图工具和快捷按钮,方便用户快速操作。
3.绘图区域:用于绘制图形和图表。
4.形状窗口:提供丰富的形状和模板,用户可以拖拽到绘图区域使用。
5.属性窗口:显示选中对象的属性,如大小、颜色、线条等,用户可以在此修改属性。
6.任务窗格:包含绘图资源、图层、图例等,方便用户管理绘图内容。
三、基本操作1.创建绘图:启动Visio2024,选择“新建”-“绘图”,在弹出的对话框中选择合适的模板,即可创建一个新的绘图文件。
2.选择对象:鼠标左键选择单个对象,按住Shift键选择多个对象。
3.移动对象:选中对象后,按住鼠标左键拖动,即可移动对象。
4.复制和粘贴:选中对象,鼠标右键,选择“复制”,在绘图区域鼠标右键,选择“粘贴”,即可复制和粘贴对象。
5.旋转对象:选中对象,鼠标右键,选择“旋转”,输入旋转角度,即可旋转对象。
6.对齐和分布:使用工具栏中的对齐和分布按钮,可以快速对齐和分布对象。
7.组合和拆分:选中多个对象,鼠标右键,选择“组合”,即可将多个对象组合为一个整体。
选中组合对象,鼠标右键,选择“拆分”,即可拆分组合对象。
四、绘图技巧1.使用模具:Visio2024提供了丰富的模具,用户可以根据需求选择合适的模具进行绘图。
2.使用图层:通过图层,用户可以更好地组织和管理绘图内容。
新建图层,设置图层属性,如颜色、线条等,然后选择对象,将其移动到指定图层。
3.连接线:使用连接线功能,可以快速连接两个对象。
选择连接线工具,起点对象,拖动到终点对象,释放鼠标左键,即可完成连接。
人教版四年级数学下册易错题精编讲义第16讲平移(附答案)

第16讲平移(讲义)(知识梳理+易错汇总+易错精讲+易错专练)1、在方格中画简单图形平移后的图形的方法。
(1)选点。
在原图形上选几个能决定图形形状和大小的点;(2)描点。
按要求把所选的点向规定的方向平移规定的格数;(3)连线。
把平移后的点连点成形。
2、平移的应用。
应用图形的平移可以将不规则图形转化成规则图形,进而解决问题。
1、图形平移的距离是指对应点之间的距离,而不是指两个图形之间空格的距离。
2、在对图形进行两次平移时,一定要正确理解题意,明确平移的顺序、方向和距离。
3、把不规则图形转化为规则图形的方法往往不止一种。
【易错一】下面()组的两个图形经过平移能够完全重合。
A.B.C.D.【分析】平移:在平面内,将一个图形上的所有点都按照某个方向作相同距离移动的图形运动。
平移后图形的位置改变,形状、大小、方向不变。
【解答】解:的两个图形经过平移能够完全重合。
故选:B。
【点评】此题考查了平移的意义及在实际当中的运用。
【易错二】移一移,说一说。
图①向平移了格;图②向平移了格;图③向平移了格。
【分析】根据图中两图的相对距离及箭头指向即可确定平移的方向和距离。
【解答】解:根据平移定义可知,图①向上平移了2格;图②向左平移了4格;图③向右平移了6格。
故答案为:上;2;左;4;右;6。
【点评】本题主要考查了平移,解题的关键是确定平移的方向和距离。
【易错三】如图所示:(1)图形①平移到图形②的位置,可以先向平移格,再向平移格.(2)画出中间图形的另一半,使它成为轴对称图形.(3)画出右边图形的全部对称轴.【分析】(1)根据平移的性质分别数出图形①向右和向下平移到图形②的距离即可求解;(2)先根据对称轴,分别找出中间图形的对应点,再依次连接即可;(3)如果一个图形沿着一条直线对折后,直线两旁的部分完全重合,这样的图形叫做轴对称图形,依据定义即可作出所给图形的对称轴.【解答】解:(1)图形①平移到图形②的位置,可以先向右平移4格,再向下平移5格;(2)、(3)解答如图:故答案为:右,4,下,5.【点评】考查了平移,将简单图形旋转一定的度数,确定轴对称图形的对称轴条数及位置,本题综合性较强,但难度不大.【易错四】描述与画图。
《分子模拟教程》课件

人工智能和机器学习技术将在分子模拟中发挥越 来越重要的作用,例如用于优化模拟参数、预测 性质等。
多尺度模拟
目前分子模拟主要集中在原子或分子级别,未来 将进一步发展多尺度模拟方法,将微观尺度和宏 观尺度相结合,以更全面地理解物质性质和行为 。
跨学科融合
分子模拟将与生物学、医学、材料科学等更多学 科领域进行交叉融合,为解决实际问题提供更多 可能性。
环境科学
在环境科学领域,分子模拟可用于研究污 染物在环境中的迁移转化机制,为环境保 护提供理论依据。
THANKS.
分子动力学模拟的常见算法
Verlet算法
一种基于离散时间步长的算法,用于计算分子位置和速度。
leapfrog算法
一种常用的分子动力学模拟算法,具有数值稳定性和计算效率高的特 点。
Parrinello-Rahman算法
一种基于分子力场的算法,可以用于模拟大尺度分子体系的运动。
Langevin动力学算法
材料科学
通过模拟材料中分子的运动和相互作 用,可以研究材料的力学、热学和电 学等性质,为材料设计和优化提供依 据。
03
Monte Carlo模拟
Monte Carlo模拟的基本概念
随机抽样
Monte Carlo模拟基于随 机抽样的方法,通过大量 随机样本的统计结果来逼 近真实结果。
概率模型
Monte Carlo模拟建立概 率模型,模拟系统的状态 变化和行为。
通过模拟药物分子与靶点分子的相互作用,预测 药物活性并优化药物设计。
材料科学
研究材料中分子的结构和性质,预测材料的物理 和化学性质。
生物大分子模拟
模拟生物大分子的结构和动力学行为,如蛋白质 、核酸等,有助于理解其功能和性质。
化学结构式编辑软件ChemDraw课程

目录第一章 chemdraw基础知识 (1)1.1、工作环境 (2)1.1.1工作环境综述 (2)1.1.2 图形工具板 (2)1.2 chemdraw的基本操作 (4)1.1.1 chemdraw文件的建立 (4)1.1.2 打开chemdraw文件 (4)1.1.3 chemdraw文件的存储 (5)1.1.4 关闭chemdraw文件 (6)1.1.5 退出chemdraw (6)第二章实例指导 (7)2.1指导实例1 反应方程式 (7)2.1.1 绘制前的操作准备 (7)2.1.1 绘制实例 (8)2.2 指导实例2 绘制中间体结构 (14)2.3 指导实例3 复杂环化合物 (17)2.4 指导实例4 Fischer葡萄糖结构图 (20)2.5 指导实例5 绘制透视图形 (22)2.6 指导实例5 Newman结构 (25)练习: (28)第一章chemdraw基础知识计算机作为一种化学学习和研究的工具有着不可替代的作用。
它不仅能够帮助我们进行文字及图形处理等文书工作,而且可以在化学学习与研究的各个方面协助我们更快、更好的工作。
本章介绍一些常用的能在PC机上使用的化学类软件,以期能帮助读者在自己的学习和研究中做出有效、快速的选择。
有关化学结构式编辑的软件市面上非常之多,它们各有所长。
既有商品的,亦有对教育界及家用免费的。
其功能主要是描绘化合物的结构式、化学反应方程式、化工流程图、简单的实验装置图等化学常用的平面图形的绘制。
常见的这类软件有:ChemDraw, ChemWindow, ISIS Draw, ChemSketch等。
前两个为商业软件,有关它们的资料可以查阅各自的网站/ 和/。
后两个对教育界及家用为免费软件,可以在它们各自的网站/和/上下载。
ChemDraw是世界上使用最多的大型软件包ChemOffice中的一个组件,其它两个组件为Chem 3D(分子结构模型)和ChemFinder(化学数据库信息),其最新版本是Chem Office 2004(ChemDraw Pro 8.0、Chem3D Ultra8.0、ChemFinder Ultra8.0) 。
用XP软件画图(晶体结构解析用)

椭圆球图形的画法:1.将后缀为res的文件(比如为50710c.res)复制到硬盘根目录下(比如E:盘)2.启动单晶分析软件XP3.XP>>read e:\ 50710c ↵4.XP>>fmol ↵5.XP>> kill $q ↵6.XP>>proj ↵出现画图框,你可以按右上的按钮旋转或显示所有的原子。
将分子结构旋转到适当位置,也就是旋转的尽可能使每个原子都不被挡住。
当旋转到适当位置后按右上角的exit退出7.XP>>labl 1 450 ↵8.XP>>telp 0-30 ↵回车至现plorfile:的提示符9.plotfile:mol ↵mol是为你所要画的分子结构图起的名字,你也可以起别的名字。
注意:当你按了回车后会进入画图界面,这时候鼠标已经变成了一个矩形框。
将矩形框移动到显示的原子边上合适位置后点击鼠标左键,就对该原子标注了它的顺序。
鼠标在你标注了第一个原子后会自动移到下一个要标注的原子上或旁边,你仍将矩形框移动到合适位置后按鼠标左键进行标注。
重复以上的操作,直至将全部原子标注完毕。
然后按键盘上的b键保存后自动退出到XP程序的dos操作界面10.XP>>draw mol↵mol还是刚才的文件名11.当你上步操作按完回车后会出现一句话,可能的意思是:将该图形保存成什么格式的文件,有几个选项。
请选择按键盘上的a键(可能是保存成可用acrobat打开的文件),然后回车12.为你的图形取一个名字,你可以仍用mol13.上步回车后又会出现一句话,意思是要保存为黑白图形还是彩色图形。
按回车即保存成黑白图形,请按回车。
以上就画好了分子结构图。
你可以在这时候推出XP,即在XP>>提示符后输入quit或exit。
你也可以继续画堆积图。
14.XP>>cell ↵回车后会出现5个数字,前3个数字依次表示的是a、b、c轴。
Discovery Studio Visualizer简易教程

PDB: 3K9F
哈尔滨医科大学 解鸿波
工具栏
工具导航栏 主窗口
Ctrl+H
氨基酸残基
Ctrl+T
氨基酸相应性质
Ctrl+D
编辑原子
必须先 选择原 子
线型显示 棍型显示 球棍显示
编辑蛋白
必须先 选择原 子
线型显示 棍型显示 线型条带
No.2
索拉菲尼(Sorafenib)
HMG-CoA还原酶 PDB code: 1HWI
No.3
氟伐他汀(Fluvastatin)
神经氨酸酶 PDB code: 4KS1
No.4
达菲(Tamiflu)
青霉素结合蛋白 PDB Code: 3ITA
No.5
氨苄西林(Ampicillin)
Create Pharmacophore Manully工具 (根据已知分子构建药效团,但不实用)
写字板打开*.pdb文 件查看信息,内附 原子坐标,原子类 型及氨基酸序号等 信息。
HIV-1蛋白酶 PDB code: 3OXC
No.1
沙奎那韦(Saquinavir)
p38 MAP激酶 PDB code: 3GCS
3k9f工具导航栏工具栏主窗口ctrlhctrlt氨基酸残基氨基酸相应性质ctrld编辑原子线型显示棍型显示球棍显示必须先选择原编辑蛋白线型显示棍型显示线型条带必须先选择原条带改变背景颜色viewviewview画分子工具不推荐使用蛋白或dna等大分子配体分子其他分子例如水等按字母顺序排列不同部分字母重新循环代表晶格pdb编号侧链编号点加号查看侧链所含有氨基酸残基氨基酸及编号点加号查看氨基酸所含有原子去除对勾表示隐藏仅显示配体分子并双击选中分子显示为黄色
ISISDraw2.2的基本使用

ISISDraw2.2的基本使用ISIS/Draw2.2的基本使用万法及技巧ISIS/Draw2.2的程序界面是标准的Window窗口,在窗口中点击右键,可以击活快捷工具(早期ISIS/Draw1.0无此功能)。
上方是常用的官能团与分子模板工具条,左键点击选取后,直接在窗口中点击即可绘出结构。
左边为垂直工具条,依次排有各种工具按钮,选取、3D旋转、橡皮、绘制原子、化学键、链、箭头、括号、文本、线条、多边形等等,有的工具按钮上有“向右的小三角”图标,表明有多重选项,按住左键向右拖,置黑所需工具按钮即可选中。
最为可贵的是,ISIS/Draw2.2提供了即时帮助功能,只要你击活任一工具按钮,在按钮下方就会即时显示操作方法和工具按钮的作用,一目了然,无需你在浩瀚的帮助文件中费神地寻找,这充分显示了当今软件“界面友好”的流行趋势。
l、分子式绘制基本步骤:(l)在模板工具条或模板上选取相应的官能团,绘出基本框架;(2)在框架上进行原子、键及分子编辑,绘制出目标结构式;(3)运行“Chem Inspector”,进行结构式检查,确保所绘结构式的正确性(可选)。
A、运用模板画结构式:模板可以从窗口上方模板工具条选取,也可点击菜单栏上“ T emplate”项,菜单下列有程序自带的数十类几百个模板,从芳环、多元环、羰基化合物到糖、氨基酸等等应有尽有,使用十分方便。
点击选取后直接在窗口中欲绘制处点击左键即可。
同时窗口上的模板工具条也可根据日常研究工作的需要进行定制。
方法是在工具条右边点击右键,击活快捷工具栏,选择“Customize Menu And T o ol”,弹出定制对话框,将所需官能团移至工具条上点击“OK”。
B、键、链、原子基团的绘制:单击左边垂直工具条上“Bond”或“Chain”工具按钮,选取单、双、叁键或链,在绘图区单击鼠标左键或按住左键拖动鼠标即可绘制键或链。
单击左边垂直工具条上“Atom”工具按钮,在欲绘部位单击左键,即出现文本输入框,可以直接从键盘输人或从下拉菜单中选取欲输人的原子基因。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
分子图形软件网络教程精编Document number:WTT-LKK-GBB-08921-EIGG-22986DS ViewerProDS ViewerPro 是基于 PC 机硬件平台的显示效果最佳的软件之一,它具有构建、显示、分析和测定分子性质等多种功能:( 1 )使用 DS ViewerPro 的绘图工具,用户可在屏幕的工作区内绘制二维分子的骨架,软件内存储了原子的成键化学数据,可进一步构建三维分子结构模型。
DS ViewerPro 可以方便地通过坐标变化构建同分异构体,也可以通过扭角变化构建同分异构体。
构建的分子模型可在三维空间中任意旋转、平移、缩放,可方便地从任何角度观看分子模型的全部或局部。
用户还可以根据自己的意愿对原子和化学键进行着色或标定,以增强显示效果。
( 2 )具有多种分子结构的显示模式:线形、棒型、球棒、比例球棒、电子云空间填充、多面体等。
DS ViewerPro 提供了蛋白质、 DNA 等生物大分子的带状显示模式,它还可显示晶胞和分子表面图形,除了显示有机分子模型外,对于许多聚合物材料和无机材料也有特殊的显示模式。
( 3 )分子中不同化学键和原子可以通过不同颜色和大小进行区分,显示顔色丰富多彩,显示效果良好。
用户可任意调整显示质量和打印质量,高质量的显示和打印明显地增加了图形外沿的平滑,可以方便地将高分辨率的分子结构图形进行排版和印刷。
( 4 )用户可以将分子的结构性质参数拷贝到 Excel 兼容的电子表格,也可以将分子模型直接拷贝并嵌入到Microsoft Office 文件中。
在 Office 文件中,通过双击这些嵌入分子图形,可调用 DS ViewerPro 程序,直接进行编辑处理。
( 5 ) DS ViewerPro 带有较强的计算功能,可方便地进行分子结构几何参数测定,显示出键长、键角、二面角、非键距离和其他立体化学信息,这些数据会随着分子结构的变动和调整进行动态地修改。
DS ViewerPro 可提供两个窗口,以纵向平铺方式显示结构层次窗口和 3D 窗口。
( 6 ) DS ViewerPro 可以读入多种分子图形软件的格式文件,它可以将 ISIS/Draw 绘制的二维结构的格式文件自动转换成三维分子结构模型,还可以读入晶体衍射数据。
DS ViewerPro 可输出多种格式分子图形文件,最具有特色的是它可以输出 VRML World 格式的文件( .wrl ),而不必使用其它的转换程序。
DS ViewerPro 最新版本是版, Accelrys 公司提供 DS ViewerPro Suit 的 30 天试用版免费软件,它除了不具备建立分子模型等少数功能之外,其他功能与 DSViewerPro 基本相同。
本节说明 DS ViewerPro 的使用方法。
DS ViewerPro 的程序界面由下拉式菜单、水平工具栏、垂直工具栏和工作区等组成,如图 9-7 所示。
在工作区中根据不同的工作目的,可以显示分子模型的 3D 窗口,或者显示结构数据的数据表,或者显示分子层次等级的Hierarchy 窗口。
3D 窗口可用于显示分子模型,并通过3D 工具对窗口中的模型进行旋转、平移和缩放,还可以通过绘制工具构建分子模型,并进行文本注释等。
1 .菜单选项介绍DS ViewerPro 基本命令是通过选择下拉式菜单中的命令选项实现的,程序提供以下 7 个下拉菜单:( 1 ) File (文件)菜单有: New (建立新文件)、Open (打开已有文件)、 Open Location (在互联网上打开文件)、 Close (关闭文件)、 Save (存盘)、Save as (将文件另存为)、 Print (打印)、 Print preview (打印预览)、 Print setup (打印设置)、Send (通过 E-mail 发送)、 Exit (退出程序)等选项。
图 9-7 DS ViewerPro 的程序界面( 2 ) Edit (编辑)菜单有: Undo (取消上一个操作)、 Redo (重复上一个操作)、 Cut (剪切)、Copy (复制)、 Paste (粘贴)、 Delete (删除)、Paste from (从指定位置粘贴)、 Select all (全部选择)、 Select (选择)、 Group (将选定的内容组合成组)和 Properties (性质)等选项。
( 3 ) View (显示)菜单有: Display Style (显示模式)、 Color (颜色)、 Reset Rotation (恢复旋转前状态)、 Fit to Screen (在窗口合适位置显示模型)、 Center (在窗口中间显示模型)、Animate (动画显示)、 Spin (自动旋转显示)、 Full Screen (全屏显示)、 Show (显示)、 Hide (隐藏)、 Show All (显示窗口中的全部内容)、 Show Only (仅显示选定内容)、 Tool Bar (工具栏显示)和 Option (参数选项)等选项。
( 4 ) Tools (工具)菜单有: Hydrogens (氢原子)、 Labels (标识符号)、 Monitors (结构参数测定)、 Surfaces (分子表面)、 Query Feature (疑问性质)、 Markers (原子标识)、 Crystal Cell (晶胞参数)、 Element Properties (元素性质)、Calculate Properties (计算性质)、 Enter Command (运行命令)、 Play Script (运行命令文件)等选项。
( 5 ) Modify (修改)菜单有: Element (原子元素符号)、 Charge (电荷状态)、 Bond (化学键类型)、 Hybridization (杂化方式)、 Stereochemistry (手性立体结构)、 Insert Atom (插入原子)、Contract Bond (缩减键)、 Invert Center (构型倒置)、 Fuse (融合)、 Align Structure (排列结构)、 Torsion Kick (通过扭角变化构建同分异构体)、 Coordinate Kick (通过坐标变化构建同分异构体)、 Clean Structure (构建三维分子结构)、 Add on Molecules (能量优化)等选项。
( 6 ) Window (窗口)菜单有: New 3D Window (新建三维分子模型窗口)、 New Stereo 3D Window (新建三维立体分子模型窗口)、 New Hierarchy Window (新建层次结构窗口)、 New Data Table (新建数据表窗口)、 Cascade (叠层显示窗口)、 Tile Horizontal (水平平铺显示窗口)、 Tile Vertical (垂直平铺显示窗口)、 Title Molecules in Windows (将窗口中分子平铺显示)、 Arrange Icons (重排图标)、 Close All (关闭所有窗口)等选项。
( 7 ) Help (帮助)菜单有: Contents (目录)、Index (索引)、 Tip of the Day (每日一题)、About ViewerPro (关于信息)等选项。
2. 工具栏介绍除了上述菜单指令以外, DS ViewerPro 还提供了各种工具图标。
在菜单栏下方有水平工具栏,水平工具栏共有22 个工具图标。
按从左至右的顺序分别是: New (建立新文件)、 Open (打开已有文件)、 Save (存盘)、Print (打印)、 Cut (剪切)、 Copy (复制)、Paste (粘贴)、 Fit to Screen (在窗口合适位置显示模型)、 Display Style (显示模式)、 Measure (结构数据)、 Calculate Properties (计算分子性质)、Add Hydrogens (加氢)、 Hide Hydrogens (隐藏氢原子)、 Single Bond (单键工具)、 Double Bond (双键工具)、 Aromatic Bond (芳香键工具)、 Triple Bond (三键工具)、 Change Element (改变元素)、Clean Structure (构建结构模型)、 Coordinate Kick (通过坐标变化构建同分异构体)、 Dreiding (能量优化)、 Torsion Kick (通过扭角变化构建同分异构体)。
上述工具栏图标是系统安装后的默认图标,用户可以使用 View / Toolbars 命令增减其它工具图标的显示,也可以通过 Customize 选项定制自己个性化的工具栏图标。
在主窗口的左侧还有垂直工具栏,垂直工具栏共有 13 图标,用于屏幕显示、选择、控制和建立分子模型。
按从上至下顺序分别是: Select (选择, F5 )、 Rotate (旋转, F6 )、 Translate (平移, F7 )、 Zoom (尺寸缩放, F8 )、 Torsion (旋转扭角)、 Sketch (绘制结构)、 Ring (绘制环状结构)、 Chain (绘制链状结构)、 Annotation (注释文本)、 Text Box (带框注释文本)、 Text Callout (带指向框注释文本)等。
最后二个工具图标只有在使用层次结构窗口才可使用,它们是: Select (选择)、 Display (显示)。
在点击某一工具图标之后,在屏幕最下方的状态栏会显示该图标的操作帮助提示。
使用垂直工具栏的工具时,还要注意以下要点:( 1 )如果要将“ Select ”(选择)工具的选择方式由“ Lasso ”(套索)改为“ Rubber band box ”(矩形框)可使用 View / Options / Tool 命令更改。
( 2 )完成了一个选择操作后,如要进行多个对象的同时选择,可按下“ Shift ”键,再进行另一个选择。
( 3 )使用“ Torsion ”(旋转扭角)工具时,先要点击构成扭角的中间化学键,再进行拖拉旋转。
( 4 )如果工作区中存在多个分子或对象,要对某一个分子或对象进行旋转或平移操作,可先选择该分子或对象,按“ Ctrl ”键,再进行“ Rotate ”(旋转)或“ Translate ”(平移)操作。
( 5 )如要选择某一分子,可双击分子中任一个原子或键。