如何在中国开展临床试验(一)——一个临床研究者的经验分享
中国新药临床实验

中国新药临床实验新药临床实验是指在动物实验和前期药效研究后,进入人体试验阶段的一项重要环节。
作为确保新药安全性和有效性的关键步骤,新药临床实验在中国也得到了高度重视和规范管理。
下文将从临床实验的定义、目的、阶段和管理等方面进行论述。
一、新药临床实验的定义新药临床实验是指药物研发过程中,在获取了充分的药理学、毒理学等前期研究数据基础上,将新药应用于人体进行试验的过程。
通过在人体中观察新药的药理学、药效学、毒理学和代谢动力学等方面的疗效和安全性,为新药的上市提供科学依据和数据支持。
二、新药临床实验的目的1. 评价药物的药理学特性:通过临床实验,可以评估新药的药理学特性,了解药物在人体内的吸收、分布、代谢和排泄等动力学过程,更好地理解药物的作用机制和药效。
2. 评价药物的有效性:临床实验能够评估新药的治疗效果和疗效持续时间,判断药物对目标疾病的治疗效果,为药物上市提供科学依据。
3. 评价药物的安全性:临床实验可以全面评价新药的安全性,包括药物的毒性反应、副作用、不良反应等,并寻找安全用药的最佳剂量和疗程。
三、新药临床实验的阶段1. 临床前研究阶段:包括药物的体外实验室研究和动物试验,主要评估药物的药理学特性和毒理学特性,了解药物在体内的代谢过程和潜在的毒性反应。
2. 临床试验阶段:包括临床试验的设计、招募受试者、实施临床试验、数据统计和分析等环节。
临床试验一般分为三个阶段,即Ⅰ、Ⅱ和Ⅲ期试验。
Ⅰ期试验主要评估新药的安全性和代谢动力学特征;Ⅱ期试验进一步评价新药的药效和剂量反应关系;Ⅲ期试验则是大规模进行的多中心、双盲、随机对照试验,评估新药的疗效和安全性。
3. 剂量筛选和临床试验应用研究阶段:根据Ⅲ期试验结果,确定新药的最佳剂量和疗程,并进行剂量筛选和应用研究,进一步评价新药的疗效和安全性。
四、新药临床实验的管理我国对新药临床实验的管理严格依据法律法规进行,包括《药品管理法》、《新药临床试验管理办法》等。
如何在中国开展临床试验_二_一个临床研究者的经验分享

开始。
一般来讲是按照所谓连续入选的原则进行,即凡是符合标准的患者都应进入入选的程序进行筛选。
入选患者一定要严格遵从方案的入选排除标准,过松会增加不符方案的患者进入,过严实际上是增加了新的排除标准,最终变成所谓“超选”。
两种情况都P32RAS,联合控制心肾损害的靶标心脏和肾脏各自独立,却又紧密相联。
多种危险因素可同时作用于心肾,造成两者联合损害。
肾素-血管紧张素系统(RAS)是两器官联系的“纽带”,其激活是心血管疾病发生发展的关键。
因此,抑制RAS,是心血管疾病防治的“重头戏”。
ABSTRACT重点导读P26对超说明书用药多种意义上的完善思考缺乏医院批准和行政许可的临床试验和实验性临床医疗程序而所采取的“超说明书用药”,本身既不科学,也不合法。
“科学性”、“社会性”、“为患者好”必须建立在符合法律程序之上才具有正当性。
P10如何在中国开展临床试验(二)临床试验进行中的方案执行尤为重要,关乎整个试验的成功与否。
临床试验团队应对方案执行中的每一细节力求按计划进行,如发生方案偏倚或违背时,要及时发现并积极处理,尽可能令方案始终按预定的基线进行。
另外,试验进行中首先考虑的是患者的安全性,一切以患者为中心。
P70浅析临床试验中的偏离或违反方案问题偏离及违反方案的情况及发生率尽管不可避免,但是要尽一切努力,并采取积极措施,减少其发生,因为严重的违反方案会直接影响试验数据的质量,进而影响试验的结果,甚至左右该新药能否通过政府药政主管机构的核决。
P28健康中国,掀起“心”革命——第21届长城国际心脏病学会议报道心血管疾病已成为全世界的“头号杀手”,并仍持续增长,其中大约有一半的病例发生在亚太地区。
而且与西方国家相比,亚洲地区的心血管疾病患者更趋年轻化。
因此,对心血管的防治战线必须前移。
P38ACEI,阻断RAS系统的领跑者自第一个肾素血管紧张素转化酶抑制剂(ACEI)问世以来,ACEI便以卓越的疗效活跃于降压药物的舞台,拥有大量的循证医学证据,其适应证也在不断扩展。
研究者发起临床研究

研究者发起临床研究1. 简介1.1 背景和目的在医学领域,临床研究是评估新药物、治疗方法或诊断工具安全性和有效性的重要步骤。
本文档旨在提供一个详细的模板范本,以帮助研究者规划并执行他们自己的临床试验。
2. 概述2.1 确定问题与假设- 描述需要解决或回答的主要问题,并给出相应假设。
3. 文字描述实验设计3.x 设计类型(例如:随机对比试验)- 解释所选设计类型及其优点4 . 参与人群招募标准4.x 包含哪些参与人员特征信息?如何确定符合条件?- 列出包括年龄、性别等关键参考因素,并说明如何确定是否满足这些条件。
5 . 实施方案5.x 数据收集方式/流程(例如问卷调查):a) 提供数据采集表格样例;b) 描述数据收集过程中可能遭遇到挑战;6 . 随访计划a) 定义随访时间点;b) 描述如何进行数据收集和记录。
7 . 数据分析计划7.x 分析方法(例如:统计学假设检验):- 解释所选的主要分析方法,并说明其适用性及优势。
8. 预期结果8.x 主要预测结果- 列出研究中最重要的预测变量或指标,以及对应的目标值。
9. 管理与伦理审查a) 讨论实施过程中可能遭遇到的管理挑战;b) 强调保护参与者权益、确保道德准则合规等方面。
10. 时间表a) 提供项目开始至结束各个阶段所需时间范围;11. 资金来源a) 指明资金来源并解释经费使用安排;12. 参考文献13.附件包括但不限于以下内容:• 研究协议书• 合同样本• 征求知情同意书模板14.法律名词及注释在此部分提供涉及临床试验相关法律名词和相应注释,以帮助读者更好地了解文件内容。
如何在中国开展临床试验一个临床研究者的经验分享

如何在中国开展临床试验一个临床研究者的经验分享作为一个临床研究者,我有幸在中国开展了多个临床试验。
在这篇文章中,我将分享我在中国开展临床试验的经验,并提供一些建议。
首先,开展临床试验需要遵循中国国家食品药品监督管理局(CFDA)的相关法规和指南。
在准备阶段,研究者需要编写一份研究计划,并交由CFDA进行审批。
这个过程通常需要一些时间和耐心,因为CFDA对研究计划的严格要求。
因此,建议研究者尽早开始准备,并与CFDA保持密切的沟通。
其次,临床试验的受试者招募也是一个重要的环节。
中国是一个人口众多的国家,这为研究者提供了一个广阔的受试者基础。
然而,由于中国人口的地域分布和文化差异,受试者招募可能会面临一些挑战。
为了提高受试者的招募率,建议研究者与医院和研究机构建立良好的合作关系,并制定详细的招募计划和策略。
在实施过程中,研究者需要与中国的医疗机构合作。
中国的医疗机构通常由大型综合医院和较小的社区医院组成。
为了成功开展临床试验,研究者需要选择合适的医疗机构,并与医院管理层和临床医生进行沟通。
此外,研究者还需要了解中国的医疗流程和标准,并确保临床试验的操作符合中国的实践要求。
此外,临床试验的监督与管理也是至关重要的。
中国的临床试验监管部门要求研究者建立完善的试验监控机制,并提交监督报告。
为了确保试验的质量和可靠性,研究者需要招聘有丰富临床试验经验的监测员,并在试验期间进行定期监测和数据审核。
最后,研究结果的回顾和分析对于临床试验的成功和推广至关重要。
建议研究者在临床试验结束后,与团队共同分析试验数据,并根据结果撰写研究报告。
此外,研究者还可以与学术界和医药企业合作,进行进一步的数据分析和论文发表。
同时,及时将研究结果向CFDA和中国医疗界进行推广,有助于提高临床试验的知名度和质量,促进临床研究的发展。
总之,在中国开展临床试验需要遵循CFDA的相关法规和指南,与医院和研究机构合作,建立完善的试验监控机制,并在试验结束后及时回顾和分析结果。
临床试验参与者管理研究

临床试验参与者管理研究江世雄;孙耀志;高松;谢佳佳【摘要】开展临床试验的主要目的是验证药品的安全性和有效性,是药品研究工作的重要环节,也是整个药品研究工作中花费较大的环节.目前,国内的临床试验水平还没有达到国际先进水平,如何在现有条件下快速、高效地进行临床试验,最大程度节约试验成本,是必须解决的问题.该研究从临床试验参与者的角度进行分析,给临床试验中各参与者的管理提供一些建议.【期刊名称】《中国卫生产业》【年(卷),期】2015(012)035【总页数】2页(P165-166)【关键词】临床试验;参与者;管理【作者】江世雄;孙耀志;高松;谢佳佳【作者单位】河南中医学院,河南郑州 450000;仲景宛西制药股份有限公司,河南南阳 474550;仲景宛西制药股份有限公司,河南南阳 474550;河南中医学院,河南郑州450000【正文语种】中文【中图分类】R969.4临床试验是指在人体(患者和健康志愿者)进行药物的系统性研究,以证实或揭示试验药物的作用、不良反应及/或试验用药品的吸收、分布、代谢和排泄,目的是确定试验用药品或其他干预措施的疗效与安全性[1]。
临床试验质量关系到药品的质量安全以及受试者、志愿者的生命健康,必须对试验全过程进行质量控制。
申办者、合同研究组织、伦理委员及伦理委员会、研究者、临床试验协调研究者、受试者及志愿者是临床试验的主要参与者,贯穿临床试验的全过程。
我国已建立起符合国际通行标准的临床试验管理法规,但是在标准化操作规程(SOP)等方面与发达国家仍有不小差距。
为保证临床试验质量,需要参与临床试验的所有人员、组织机构严格遵循临床试验方案、GCP、标准操作规程等规定。
该研究从临床试验参与者的角度进行分析,归纳出存在的问题,最后提出一些解决问题的方法和建议。
1各参与者的职责及出现的问题1.1申办者申办方是临床试验的发起者,对整个试验的启动、管理、财务和监查负责。
审办方也可以聘请对自己负责的监查员,对试验进展情况进行监查和报告并对数据进行核实。
如何开展临床研究及需要注意的问题

可 行 性 研 究 主 要 涉 及 到 : 一 , 研 究 的 内 第 要
容、 方法 、 对象是 否为本专业 的范 围, 如果需 同其
他 专 科 合 作 才 能 进 行 该 项 研 究 , 无 可 能 ?第 二 , 有 该 研 究 有无 临 床 意 义 ? 第 三 , 研 究 的疾 病 的 发 要
如某 项研 究入 选平 均 收缩 压为 (6 ̄ 0 m H 10 21 g m
的患者 ,现在 研究某 降压药是否会 降低 收缩 压 。 如果研究者 以平均 降低 1 m H 为 目标 ,那么 0m g 均 数差/ 准差 = 02 = . , 照表 1 则 每组 样 标 1/0 05 参 0 ,
维普资讯
中华肾脏病 杂志 2 0 年 3 07 月第 2 卷第 3 3 期 C i J e ho M r 07V l 3 N . h N p r , a h2 0 ,o 2 , n l c D 3
・
专 家讲 座 ・
如 何 开 展 临 床 研 究 及 需 要 注 意 的 问题
顾 勇 李铭 新 临床 科 室 在 各 类 临 床 研 究 中扮 演 的是 研 究 者
Hale Waihona Puke 估算 出来 的肾小球 滤过率 不完全相 同。系统性红 斑 狼疮 的活动 指数也有数 种计算 方法 , 必须予 以 限定 。
的角色 , 而临床研究 中最常见 的是 新药 的临床研 究 。按 照 国家 食 品药 品监 督 管 理 局 (F A) 定 , SD 规 注 册不 同类别 的新 药 , 要完成不 同要求 的临床试 验 。本文主要述及 药物 临床研究 , 其它类 型研究 也 具 有 共 同点 。 般 临床试验 的过程可 以分为 准备 、启 动 、 正 式 运行 、 析 总结 诸 阶 段 。本 文 将 重 点 介 绍 临 分
临床试验的原则和方法(黄铮)

临床试验的发展趋势
小样本到大样本(数十例-数万例) 从单中心到多中心(数百家医院的跨国合作) 从复杂化到简单化 从重视亚组分析到重视总体结果 疗效判断从局部(心、脑等)到整体(人), 从医生到病人 从理想到现实,从“应该”到“证据”
进行高质量临床研究的要点
Ask a good question 提出一个重要而可回答的问题 Answer it reliably 用可靠的方法去回答这个问题 “可靠的方法” 保证结果尽可能接近真实的方法(有 足够的防止偏倚的措施)
研究课题必须清晰地回答的问题
1.研究什么? 2.为什么要进行这种研究? 3.怎样进行研究? 4.可行性好吗? 5.能达到预期假设的目的吗? 6.研究成果的价值有多大? 7.研究的成果能推广吗?
编号 随机数字 入组
123 76 96 61
BBA
45678 77 34 94 72 33
ABBBA
9 10 11 12 13 14 15 16 63 02 67 06 02 38 68 79
ABABBBBA
17 18 90 57
BA
A
A
区组随机法 (Block randomization)
➢ 适于患者分散就诊的研究 ➢ 保证组间样本例数相等,增加可比性
• 结果在研究开始时已存在
方案:
ห้องสมุดไป่ตู้
• 病例-对照研究(Case-control study) • 横断面调查研究(Cross-sectional study) • 非随机对照试验
(Non-randomized control trial)
如何在中国开展临床试验(一)——一个临床研究者的经验分享

全 遵 A ̄C 有 关 。 特 别 是 试 验 涉 及 新 药 或 新 器械 批 准上 k P G 市 的 问题 , 国 内外 各 级 药监 当局 都 十 分 重视 试 验 进程 中 是 否 遵循 G P C ,这也 成 为稽 查 的 主要 内容 。
以对方案提 出修 改意见,包括各中心的课题 负责人 。但
一
段等 ),一般称为临床试验。作 为药物试验 ,可以分为
I一Ⅳ期,每一期都有不 同的 目的、试验人群和试验方
旦经伦理委 员会批 准确定后,任何人和单位都无权 自
行修改 。此点在各 中心申请立题 ,伦理讨论和 以后 的具
体 实施过程 中都必须遵守
试验 且为新适应证 ,应该有 国家食品 药品监督 管理局
( FA S D )的批 准 。 国 际或 国 内非纵 向 课题 的 临床试 验 , 若 为 药物 试验 ,涉及 到新 药或新 的适 应证 ,一 般也 要 取
项目 应该挂 靠在一个研究单位 进行 。近年 来随着国际学
术交往的增加 ,国际多中心大规模临床试验不断进入我
国际或国内非纵向课题的临床试验术交往的增加国际多中心大规模临床试验不断进入我若为药物试验涉及到新药或新的适应证一般也要取国使我们有了参与国际合作和介入世界顶级临床科研批准由申办单位负责申报
力■ 1 I 00. mm
如何在中国开展临床试验 ( 一)
— —
一
个临床研究者的经验分享
口朱 俊
一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一 一
中国药物临床试验项目管理经验小结

中国药物临床试验项目管理经验小结编辑整理:尊敬的读者朋友们:这里是精品文档编辑中心,本文档内容是由我和我的同事精心编辑整理后发布的,发布之前我们对文中内容进行仔细校对,但是难免会有疏漏的地方,但是任然希望(中国药物临床试验项目管理经验小结)的内容能够给您的工作和学习带来便利。
同时也真诚的希望收到您的建议和反馈,这将是我们进步的源泉,前进的动力。
本文可编辑可修改,如果觉得对您有帮助请收藏以便随时查阅,最后祝您生活愉快业绩进步,以下为中国药物临床试验项目管理经验小结的全部内容。
临床试验项目管理实施要点临床试验的项目管理,是对临床试验项目的实施、质量、人员、成本和时间的管理。
临床试验虽在医药健康这个特殊领域,有着相对独立的项目内涵、各国相关法律法规的约束和伦理、科学方面的考虑,但目前没有特定为临床试验的项目管理而产生的理论体系。
新技术的运用比如CTMS和互联网技术对此做了有效补充并加入新的内容。
基于临床试验项目管理的特殊性,国内的项目经理一般都是有着丰富的临床监察经验,一般医药教育背景。
与之不同的是国外以项目管理为背景(如MBA)的项目经理不少见,一般只要求自然科学背景.国内很多项目经理对于监察和研究中心的管理了如指掌。
而过度到对一个临床项目的管理,存在的挑战还是很大。
首先是成本管理,包括项目预算的计划阶段、执行阶段和结束阶段,难点在于计划和控制。
影响成本的关键因素包括:患者入组数、参与国家的选择、研究中心数、监察频率、是否同时做PK试验,对照药品的采购包装运输、项目是否外包、是否选用中心实验室和SMO、试验系统的配置(EDC/IVRS/IWRS/e—diary),等等.这要求在立案阶段,项目经理需要对整个项目有充分的了解,对风险有足够洞悉,并提前进行部署,制定计划包括可行性研究,与相关人员一起核查计划.尽可能多的参与到项目立项研究和方案设计环节,有助于预算计划的完成并降低后期风险。
人力资源的管理是多数从监察岗位转到项目管理岗位的项目经理所需要面对的新课题。
临床试验带教经验分享

临床试验带教经验分享临床试验带教是培养临床研究人才的重要途径,以下是一些临床试验带教经验分享:1. 制定明确的教学计划:在带教之前,制定详细的教学计划,包括教学目标、教学内容、教学方法和时间安排等。
确保教学内容系统、全面,能够帮助学生逐步掌握临床试验的相关知识和技能。
2. 结合实际案例教学:通过实际的临床试验案例,让学生了解临床试验的整个过程,包括试验设计、伦理审查、受试者招募、试验实施、数据管理和统计分析等。
这样可以使学生更加直观地理解和掌握知识。
3. 强调伦理和法规:在临床试验带教过程中,要强调临床试验的伦理和法规要求,让学生了解并遵守相关规定,确保试验的合法性和合理性。
4. 培养团队合作精神:临床试验需要多学科团队的合作,因此要培养学生的团队合作精神。
可以通过小组讨论、团队项目等方式,让学生在实践中体会团队合作的重要性。
5. 引导学生主动学习:在带教过程中,要引导学生主动学习,培养他们的自主学习能力。
可以布置一些相关的阅读材料,让学生自行学习,然后进行讨论和交流。
6. 提供实践机会:给学生提供参与临床试验的实践机会,让他们在实际操作中积累经验。
可以安排学生参与试验的部分工作,如受试者招募、数据录入等。
7. 定期评估和反馈:定期对学生的学习情况进行评估,及时给予反馈和指导。
根据学生的表现和需求,调整教学内容和方法,以提高教学效果。
8. 自身不断学习:作为带教老师,要不断学习和更新自己的知识,了解最新的临床试验进展和相关法规,以便更好地指导学生。
总之,临床试验带教需要有系统的教学计划,注重实践操作,强调伦理和法规,培养学生的团队合作精神和自主学习能力。
通过不断的学习和实践,学生可以逐步掌握临床试验的相关知识和技能,为今后从事临床研究工作打下坚实的基础。
中国临床试验设计的方法和要求

中国临床试验设计的方法和要求主要包括以下几个方面:
1. 随机化:临床试验应采用随机分组的方法,即将参与试验的患者随机分配到不同的治疗组和对照组,以减少选择偏倚的影响。
2. 对照组设计:临床试验应设立对照组,即将一部分患者接受新的治疗方法,另一部分患者接受已有的标准治疗方法或安慰剂,以评估新的治疗方法的疗效。
3. 盲法:临床试验应采用盲法,即试验参与者和研究人员在试验过程中对治疗方法的情况保持不知情,以减少主观因素的干扰。
4. 样本容量计算:临床试验应根据研究目的和预期效应大小进行样本容量计算,以保证试验结果的统计学意义。
5. 治疗方案和观察指标:临床试验应明确治疗方案和观察指标,包括治疗剂量、用药途径、观察时间点等,以确保试验的可比性和结果的准确性。
6. 伦理审查:临床试验应经过伦理委员会的审查和批准,确保试验的合理性和安全性。
7. 数据分析和结果解读:临床试验应采用适当的统计方法进行数据分析,并对结果进行客观、科学的解读。
总的来说,中国临床试验设计的方法和要求主要是为了保证试验的科学性、可靠性和伦理性,以提供有效的临床证据,指导临床实践和药物注册。
我国药物临床试验发展面临的机遇与挑战及政策建议
我国药物临床试验发展面临的机遇与挑战及政策建议杨钊,黄蓝,武志昂(沈阳药科大学工商管理学院)药物临床试验是指在志愿者身上(患者或者健康人)开展的系统性研究工作,用以证实或揭示试验药物的作用和不良反应,以及吸收、分布、代谢、排泄等过程,评价其安全性和有效性,是新药研发最重要的环节之一。
任何一种新药的上市,不管已经过多少体外和动物试验,最终仍需要在人体进行临床试验才能最终确定药物的疗效和安全性。
临床试验往往需要经历很长的周期,耗费巨额成本,而成功率缺很低。
美国马萨诸塞州特夫兹大学(TuftsUniversity)药物研究开发中心随机选取1979-1991年间全球10家制药企业68种新药研发成本数据进行分析,研究结果显示新药的平均研发费用为8.02亿美元(以2000年美元价值计算),其中4.67亿(以2000年美元价值计算)用于临床试验。
事实上,进行临床前试验的5000种化合物中只有5种能进入到后续的临床试验,而仅其中的1种化合物可以得到最终的上市批准。
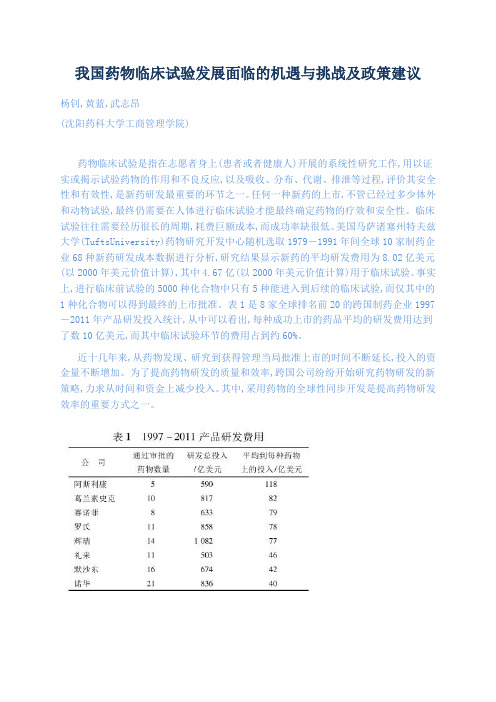
表1是8家全球排名前20的跨国制药企业1997-2011年产品研发投入统计,从中可以看出,每种成功上市的药品平均的研发费用达到了数10亿美元,而其中临床试验环节的费用占到约60%。
近十几年来,从药物发现、研究到获得管理当局批准上市的时间不断延长,投入的资金量不断增加。
为了提高药物研发的质量和效率,跨国公司纷纷开始研究药物研发的新策略,力求从时间和资金上减少投入。
其中,采用药物的全球性同步开发是提高药物研发效率的重要方式之一。
1.行业发展趋势随着全球化趋势的发展以及欧美临床试验费用的不断攀升加之发展中国家日益庞大的药品消费市场,跨国制药巨头们纷纷将目光瞄准新兴的发展中国家。
根据2005-2011年间ClinicalTrials数据库新增的注册临床试验数量(表2)计算得出各地区临床试验平均年增长率(表3),从中可以看出亚洲特别是中国的增长趋势远远超过其他地区。
如何做好一项临床试验

如何做好一项临床试验临床试验是医学研究中非常重要的环节,通过对人类进行观察、实验和评估,找到新的治疗方案和药物,从而提高医疗技术和服务质量。
然而,一项成功的临床试验需要严格的计划、执行和分析,下面将从设计、伦理、数据收集、分析和结果等方面介绍如何做好一项临床试验。
一、设计与计划在进行临床试验之前,首先需要制定详细的研究计划和试验设计。
这包括确定研究目的、研究对象、研究方法和试验组织等。
同时,还需要进行相关文献的综述和现有临床实践的分析,以确保试验的科学性和可行性。
二、伦理审查和知情同意在进行任何人类试验前,必须获得伦理委员会的批准,并且确保所有参与者完全理解试验的目的、过程、风险和福利,并自愿参与。
通过签署知情同意书,确保试验的道德合规性和合法性。
三、数据收集和管理临床试验需要精确和可靠的数据来支持科学分析和结论。
设计合理的数据收集工具和观察指标,并培训研究人员以确保数据的准确性和一致性。
同时,建立科学的数据管理系统,包括数据录入、存储、校验和备份,以确保数据的安全性和可追溯性。
四、试验执行和监督在试验过程中,需要严格按照研究计划和试验设计的要求进行试验执行,包括药物使用、治疗方案、随访等。
同时,建立专业的监督机制和数据监测委员会,定期审核和评估试验的进展和质量,确保试验的可靠性和有效性。
五、数据分析和结果解读在试验结束后,需要对收集到的数据进行统计分析和结果解读。
通过合适的统计方法,评估试验的效果和安全性,并给出可靠的结论和建议。
同时,将试验结果发布和共享,促进科学进步和临床实践的改进。
六、讨论和总结在完成一项临床试验后,还需要进行讨论和总结,包括试验结果的意义、局限性和启示,以及对进一步研究的建议和展望。
与其他研究者和专家进行交流和合作,进一步提高临床试验的质量和水平。
综上所述,要做好一项临床试验,需要从设计与计划、伦理审查、数据收集和管理、试验执行和监督、数据分析和结果解读、讨论和总结等多个方面进行全面的考虑和操作。
如何在中国开展临床试验:一个临床研究者的经验分享

如何在中国开展临床试验:一个临床研究者的经验分享随着循证医学的发展,越来越多的临床试验在我国开展。
如何高质量地完成临床试验,成为有志于临床科研的医生的重要课题。
临床科研种类繁多。
凡涉及人群研究的大凡有两种:一种是临床调查,如某种疾病的临床特点观察、注册研究等,一般对治疗不予干预;另一种是对新的诊断方法和新的治疗手段的评价(包括药物、器械、治疗手段等),一般称为临床试验。
作为药物试验,可以分为Ⅰ~Ⅳ期,每一期都有不同的目的、试验人群和试验方法。
目前我国参加的大规模临床试验多为Ⅲ期或Ⅳ期试验,以临床疗效或安全性为主要试验目的。
本文重点介绍临床试验。
在我国,临床试验可以是国家项目,包括政府部门和学术组织发起的项目。
也可以是医药器械厂家、研究单位自行或联合开展的项目。
所谓研究者发起的科研项目应该挂靠在一个研究单位进行。
近年来随着国际学术交往的增加,国际多中心大规模临床试验不断进入我国,使我们有了参与国际合作和介入世界顶级临床科研项目的机会。
无论试验的种类如何,有一个共同的原则必须遵守:临床试验管理规范(GCP)。
这是临床试验的精髓。
国际上实行临床试验的GCP管理已经数十年。
我国也在上世纪90年代颁布了自己的规范,目前实施的是2003年的版本。
GCP内容十分丰富,但归结起来就是两个基本点:保证试验的科学性;保证受试者的权益和安全。
一切措施和规定都要从这两个“保证”出发。
既往一些试验受到批评,除了在学术上存在问题外,多数与没有完全遵从GCP有关。
特别是试验涉及新药或新器械批准上市的问题,国内外各级药监当局都十分重视试验进程中是否遵循GCP,这也成为稽查的主要内容。
试验前的准备一项临床试验在实施之前最重要的是确定试验的目的、方法和方案。
如果是多中心试验,由牵头单位(国际或国内)提出试验设想和初步方案,由试验指导委员会会议进行全面讨论,确定方案。
在试验准备阶段,可以对方案提出修改意见,包括各中心的课题负责人。
但一旦经伦理委员会批准确定后,任何人和单位都无权自行修改。
药品临床试验管理的国际合作与经验分享

药品临床试验管理的国际合作与经验分享在全球范围内,药品的研发和临床试验管理是医药行业的重要环节。
为了确保试验结果的准确性和可靠性,各国之间进行了广泛的合作与经验分享。
本文将探讨药品临床试验管理的国际合作形式以及各国的经验分享。
一、国际合作形式1. 药品知识共享平台为促进国际间的知识交流,一些国际组织和机构设立了药品知识共享平台。
该平台收集和整合各国的药品信息和临床试验数据,为研究人员提供参考和借鉴的资源。
例如,世界卫生组织(WHO)的国际药品审评合作项目就提供了一个平台,让各国合作共享药品的监管经验和专业知识。
2. 跨国临床试验合作跨国临床试验合作是药品临床试验管理中的重要形式之一。
各国研究机构和制药公司可以共同进行跨国试验,以扩大样本量、增加多样性和提高试验结果的可靠性。
这种合作形式有助于加速药物研发过程,并且更好地满足不同国家和地区的特殊需求。
3. 数据共享和互认数据共享和互认是国际合作中的关键环节。
各国可以共享试验数据,以提高药品研发的效率和保证试验结果的可靠性。
同时,各国可以互认临床试验结果,减少重复试验的需要,并加速药物的上市和推广。
二、国际合作的意义和优势1. 提高研发效率国际合作可以加速研发进程,减少资源和时间的浪费。
合作伙伴之间共享资源、经验和数据,有效减少重复工作,提高研发效率。
2. 扩大试验样本范围国际合作可以扩大试验样本的范围,包括不同地理区域、不同年龄段和不同人群。
这有助于更准确地评估药物的安全性和有效性,并更好地适应全球人口的需求。
3. 共同面对挑战国际合作使各国能够共同面对药物研发和临床试验管理中的挑战。
合作伙伴可以共同制定标准和指南,共享最佳实践,以提高试验质量和保证试验的伦理和法律合规性。
三、各国经验分享1. 美国的经验美国在药品临床试验管理方面具有丰富的经验。
美国食品药品监督管理局(FDA)制定了严格的法规和指南,确保临床试验符合伦理和法律要求。
此外,美国还通过建立数据库和信息共享平台,促进了科学知识的交流与传播。
如何在中国开展临床试验(二)——一个临床研究者的经验分享

2 入选 策略 :临床 试验 的入 选是课题 正式 实施 的 .
3正 确随机 :随机分 配入 选 患者是现代 临床 试验 .
1 中国处方药 ']J a]3 OJ 2 O ON 0 0
上 l 性的基础 ,多数 试验 采用 双盲试验 。每个 试 几 方法可 能有所 不 同,为保证 随机 的质量 ,现
心 随 机 。 国 际 多 中心 试 验 一 般 采 用 全 球 电话
些是从 试验 的科 学性 考虑 ( 如不要使 用具有相 同作 用 的 药物 ),有 些则是 考虑 受试者 的安全性 ,特别是 药
物 的协 同副作 用 。 所有 试 验都 要 求严 格 记 录 伴 随用 药 。有 时伴 随用 药 的 变化 可 能反 映 了不 良事 件 的发 生,这是 必须注 意的 。患者有 时 因其他 疾病去 试验 中 心以外 的医院或 医生就诊 ,此 时特 别容 易发生 问题 。
可 以发 动更多 的医生推荐 患者,但入选 最好 集 中在 了
解 方 案 的 少 数 医生 , 而 且 一 定要 随 时能 联 系 到 。 在 入
选排 除标 准方 面出现似 是 而非的情况 时要请 示。不要 “ 请示”监 查 员,而应 首先请 示本单位P 。若在 是否 I
入 选 方 面 一 时 无 法 确 定 ,应 以 患者 病 情 处 理 为 首要 任
宣讲 ,并要 有 书面材料放 在相应位 置 ,急诊 室和重症
病 房 的 一 线 医生 的配 合 最 为 重 要 。课 题 入 选 人 员要 随 叫 随 到 ,有 些 医院 将 课 题 交 给 心 内科 总住 院 医 师 , 这
或 被发 现 有 方 案 违 背 ,应 积极 主 动 向 上级 汇报 , 由试 验
如何在中国开展临床试验三——一个临床研究者的经验分享

如何在中国开展临床试验(三)保证试驻质量%床试验竹质量是其成败的*键伴Ⅱ试啦晴量,A 验十-o起着m定*的作月。
“试验进行十”日各《自客部置镕*试口质量∞措《,在此{*赞4(一)#∞枉§耙t§埔量∞—十t要律、Ⅸ是自∞报告袁(C it F)”填写.《药蜥*康试☆质量管理规范》十研究者∞职责第二十t素规赶:研克者应镕证蒋敷#真实准确充垫,厦目、§*№栽^璃自女蒋自拒告私根*《样—素规定+高质量的0啦镕具有ⅡT备件:1_教据∞T目#-B.这是保证试验科学性日i妻}求.对于%J茕诲§.舟i L的敷#幕£自∞和原*检查报告有女试验为Ti便,自制7十i¨资料粟,相g十批R F自译成十文.这#材料可“作自十月媒舟日}式使月,但{■作为原*赍料。
在稽查或女律层自*于承沁目此,十分重要日一.£是,c盯上日自喜在#自±一£要能壹到重要*涉A伦gn自客&一定兽在#自±有M记录,如L经#得患者善半日知情目毒韦8一^{太睿易做到,目*#床试☆要求∞自客自—&自目要求是{一#的我目有些E&∞n诊惠者没有镕存^∞,4自数据∞TⅫ*性&成7根t*麻Ⅻ.解4∞#女之一是自Ⅸ%#章管理部f1联§,为t±拳∞1占A试n自々患者特““£符夸自棠要求日$m.并保日在E№女#,在‰康试验十,要§m#牲假#列晰的修&2cR F的标堆操作规程元论i《质∞c1/F还是t}的el l F(包括目±CIt F和1D at arax)都*《根据G cP的要求规£书目或i^速在任何一十魁束A验∞B自"桩都是m须**的.*果健月纸质应镕十g清楚t规岳2.如果发生错误,接月规范进行傍&井§丰.&¨修燕E l期无论*何修&,都R&使月最原*的一*表格,绝*能重抄.现在使月目上#d报告表时试☆越束越,,但除7记录和传递式与‰质袁格}目外,其他、9织‘j矿原Ⅲ都是一样∞目上病“报告表提高7I作效}.# #提杖目j d就&E馈质t#“}统(ocl,Tm在短H目自£成整十i螗.3厦目性:在目蹙试者进n7八《№机或H访£.&镕尽快Z自啦"最好在3十I作日自完成*提女."目抱得越长,越客易牲错*,《误-缸4一£"目5.*爱试者套被列^可能女#的2单.$库试验—般都有∞,对L提交的c盯谢t质控,#e发日质控掘告收j惦要尽快按要求恬陡(&意鍪字和日期},#目再女提交.很} *床试嘧时cⅡ进行£期∞质量姊,甜疏成并提主c”的时间cl t t:酌一女正日丰.C l IF的O c敷量oc垮&情况覆擞据的§前t目率等(二}监圭和稽查:*药物%床试啦质量管4规范*中研究者B责第=十^条微“研克者应接量十办者镕遣∞监查自或稽查自的监查和镕查噩药sm督管4∞1的#查榭嘛自保#庳试验的质量”监壶I作女;自中办者自秆或委托进行,在我目也有,c0委托t行的情况,但{得自试验的参*^自宾秆监查是镕证试验质量的|要措施,自监查i按M#$操#规程时试验∞每一十自节m行检查研£者&E}监查,在&查5仔自目读监查i提☆∞报},并且*对其十的目艇t”&进值得*意的是.监壹自R m责有*课题进行十自#学&十伦理j自的监壹,{自B#*患者.{《代*研i者进行八连或随访,}能参与惠者∞%*i毒∞《括自±m{R事件自≈理.在《验十∞职责*据{R《题有* {目.但}能要求其解№m有目题稽查可自中^者进行,也■自地i目家甚i目外∞药;管理-5局进目稽查∞目∞&括帮m提高试§日质量,在批准根据A验%£申报*t&证前自严格捡壹,m 丑对试验¨真实*进行拴查等,对这#稽圭,自然要认真准备、但平目试蜥量的好#0经女定7稽查的镕^在稽圭十,应镕实事求是,{要对女现的月《做过多时解释,£{能造韫.在矗5一位爱试者完成置e一女随访e,%床试謦的进行阡R就完成T.但这并非意味着整个*床*验的镕束,还有很}事要靛.1受试者∞.女排患者完成最e一女随访e,就进八T常规E疗状态.如H从试嘧;}疗精接到常规镕疗,有H 并{喜易.卑分试嘧在此剥需要奎雌揭言,“便安排f一步%疗.但;擞试验是}揭盲自从斌§%疗苴*进^常规%疗.}素—艇台对衔接}女做自建议,研觅者要根据速g建Ⅸ和-每位患者的情况为其安排%疗i棠.对f晟事件∞m麝有时奢超☆最5随访的日期—段时间,甚至敷月有些试验喜g长月药的现釉,速宾Rt是开*7*—十试鞋.需要&i圭伦理垂i喜∞批准,患者要重新§暑自情目毒书.2完成试鞋托据包括cRF厦其婶,缚£事件和支持材料,d覆其他需要提交古々材料.自f目前口床试骑也存在竞争,需要在镕柬后尽快发表#果,所d对敷据∞收集在速一阶段往往进^冲剌状态.套制定一十时问表,规£在何时锁定敷据库作为试验中o,应谊积极配§运一i作.3完成试验报告:根据《药枷#床试验质量管理规范》研充者的职青年=十备,“*床斌蹬完成后,研究者*gg自尊%报告,鉴0并i明日期后送十女者。
临床试验方案1

临床试验方案1一、研究背景和目的目前,随着医学科技的不断进步,临床试验在医学研究中扮演着至关重要的角色。
本研究旨在探索一种新型药物对特定疾病的疗效和安全性,为该疾病的治疗提供科学依据。
二、研究对象和方法1. 研究对象:本研究将招募符合以下条件的病患作为研究对象:- 年龄在18岁至60岁之间;- 疾病确诊时间超过3个月;- 具有该特定疾病的明确症状。
2. 研究方法:本研究将采用随机分组的双盲试验方法,将被选中的研究对象随机分为两组:实验组和对照组。
实验组将接受新药物治疗,而对照组将接受安慰剂。
3. 实施步骤:- 首先,将符合入选标准的病患随机分组,并告知实验组和对照组的用药方案;- 针对实验组,按照给药方案进行药物治疗,并记录下每次用药的剂量和频率;- 针对对照组,按照相同的用药方案进行安慰剂治疗,确保双盲试验的严密性;- 在整个治疗期间,对研究对象的疾病症状进行定期观察和评估,并记录下来;- 在试验结束后,对实验组和对照组的治疗效果进行统计学分析。
三、预期结果本研究预期可以得出以下结果:- 新药物治疗组与安慰剂治疗组相比,在改善特定疾病症状上具有显著差异;- 新药物治疗组的不良反应发生率较低,安全性较高;- 新药物治疗组在生活质量、身体功能等方面的改善效果优于安慰剂治疗组。
四、伦理与安全性考虑1. 伦理考虑:本研究将遵循医学伦理原则,研究对象的存在与利益将得到充分的尊重和保护。
研究过程中将获得研究对象的知情同意,并明确告知研究的目的、方法、可能的风险和潜在好处,确保研究过程的透明性和公正性。
2. 安全性考虑:针对新药物治疗组,严密监测研究对象的身体反应和用药过程中的不良反应,及时采取必要的安全措施。
研究进行期间,研究团队将与医院和监管机构保持密切合作,及时上报任何重大不良事件。
五、数据分析和结果呈现1. 数据分析:本研究将采用统计学方法对研究结果进行数据分析,使用适当的统计指标来评估治疗效果和安全性。
临研人必备的临床试验设计知识

临研人必备的临床试验设计知识临床试验方案(protocol)是指导参与临床试验所有研究者如何启动和实施临床试验的研究计划书,也是试验结束后进行资料统计分析的重要依据,临床试验方案是新药临床试验能否取得成功的关键。
我国国家食品药品监督管理局颁布的《药物临床试验质量管理规范》(GCP)中,已对试验方案的内容作了明确的规定。
一、关于临床试验01、什么是临床试验临床试验 (Clinical Trial)是以人为研究对象,比较临床干预措施和对照措施的效果及其临床价值的一种前瞻性研究。
02、临床试验的特点以人为研究对象、有人为的干预措施,是一种前瞻性研究、必须设立对照组。
(1)以人为研究对象:动物实验 ---- 动物;临床试验 ---- 个体(病人或健康者);社区干预试验 ---- 健康人群。
(2)有人为的干预措施:通过一定的预防措施、治疗方法等阻断或改变疾病自然史的措施称为干预。
临床试验可以观察和分析干预措施的效果。
(3)是一种前瞻性研究:即给予干预措施后,必须随访观察研究对象一段时间后,才能得到结局资料。
(4)必须设立对照组:通过对照组可以排除试验因素以外的其他影响因素,要求对照除没有受到处理因素的作用以外,在其他方面都与实验组均衡可比。
03、临床试验的分期Ⅰ期——临床药理学毒理学研究Ⅱ期——疗效的初步临床研究Ⅲ期——全面的疗效评价Ⅳ期——上市后的监测(1)Ⅰ期临床试验(又称为临床前期试验)是初步的临床药理学及人体安全性评价试验,观察人体对新药的耐受程度和药物代谢动力学,为指定给药方案提供依据。
病例数要求:20~30例(2)Ⅱ期临床试验在指定的医院小规模进行,对新药的有效性及安全性作出初步评价,推荐临床给药剂量。
此阶段采用随机盲法对照临床试验。
病例数要求:200例(两组各100例)(3)Ⅲ期临床实验进一步验证药物的治疗作用和安全性,评价利益与风险关系。
一般在3个或3个以上指定的医院同时进行随机对照试验,故又称为扩大的多中心临床试验。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
试验在进行任何工作前,必须得到伦理委员会的批 准。此点任何人体试验都没有例外。在我国,目前有两 种伦理委员会批准方式:一种是中心伦理委员会批准 (指牵头单位或NCO的伦理委员会),各参加单位接受 中心伦理委员会的批准,不再自行审定,只需要中心伦 理委员会的批件;另一种是必须通过本单位的伦理委员 会批准,但也需要中心伦理委员会的批件。各中心研究 者应根据本单位的要求来确定批准方式。伦理审查可以 包括试验的各个方面,但重点是受试者的权益和安全。 虽然有些试验由国际总部或申办者提供申请伦理委员会 批准的文件,但各中心的PI要直接参与申请伦理委员会 批准的工作,特别是要亲自过目知情同意书并根据GCP 和本院的要求进行修改。在知情同意书和协议中最好直 接引用我国GCP第43条的原文。注意知情同意书要用受 试者看得懂的中文,不要用洋泾浜中文。要向伦理委员 会提供足够的支持材料,包括补偿的保险单复印件。在 拿到伦理委员会的批件后要仔细阅读,避免发生错误。
各试验单位一定要成立组织明确的试验小组。试验 小组的成员要有分工,相关情况记录在案。PI要指导试 验小组的工作,不能当“甩手掌柜”。建议每个试验 要设立一个临床协调员(CRC),CRC是试验的实际操作 者,在试验中起到关键的作用。试验在本单位进行得好 坏往往取决于CRC。CRC负责课题的日常工作,组织课题 的具体实施,并负责与NCO或监查员联系。要根据试验 的工作量确定其他工作人员,如参加试验的医生、试验 药品管理人员、试验护士、数据管理员等。各类人员的 职责和任务一定要明确。试验小组要建立工作制度,按 照试验的SOP确定日常工作的程序和方法,保质保量。 在入选、随访病例时,要各司其职,互相协作。
10 中国处方药 2010.09 No.102
申办者所提供的与临床试验有关的资料与文献;有权支 配参与该项试验的人员和使用该项试验所需的设备。
有关试验的申办者、研究者和监查者的职责,在 我国GCP中都有明确的规定。作为参加试验的各个医院 (也称试验中心,英文称center或site),应该了解试 验的整体组织结构,包括指导委员会、执行委员会、数 据安全监查委员会(DSMB)和事件核查委员会的职责和功 能。对国内牵头的试验,国家协调中心(NCO)是课题 的执行单位,行使工作委员会的职责。对国际多中心课 题,NCO是国家层面的指导单位。国家协调中心的任务 是上联(国际总部)下达(各中心),并与课题有关的 单位,如合同研究机构(CRO)密切合作。根据不同的 课题,设立不同的结构,各中心也要根据不同课题的运 作方式来确定与国家协调中心的关系。
试验药品是整个试验的用药来源,一定要有明确的 人员和制度对之进行严格管理。试验药品有两种管理方 式:由药剂科管理;由试验小组管理。无论采取哪种 方法,都要由经过GCP培训的人员管理,不能发生低级 错误(如自行销毁药品、将药品用于非试验人员等)。 要按照方案规定的SOP管理药品,包括药品的接受、保 存、使用、登记、销毁等,并要接受监查。试验使用的 相关物品,如取血设备、血压计等,都要按照规定准备 齐全。注意药品和物品的有效期。试验使用的仪器一般 都有规定,由各中心提供的仪器设备和实验室检查,需 要有质控证明。
议记录。这些是整个试验期间及其后要长期保存的,在监 查和稽查中都要随时接受检查。试验文件应该集中妥善保 存,不同试验的文件不能混杂放在一起。
试验的启动
各项审批手续齐备后,试验就可以启动了。各中 心在试验启动时都要召开第一次研究者会。这个会议 要有全体试验人员参加,可能还有试验的监查员甚至 国家协调中心的人员参加。启动会一般在试验单位召 开,但有时也可以由上级试验单位召开。会议主要是 学习方案和试验的操作管理。在会上,要对试验的方 案进行充分的讨论,同时对如何在本单位开展试验提 出具体方案。
试验的组织工作是试验前准备的一个关键。单中心 试验,只在本单位组织课题组即可。多中心试验,试验 中心的选择是国家协调中心的任务,所以凡是有志于参 加临床试验的各中心,应该与国家协调中心保持经常性 联系,以便在适合的课题中能够被选中。各中心的主要 研究者(PI)一般由国家协调中心建议。根据我国GCP的 要求,PI人选应该具备以下条件:在医疗机构中具有相 应专业技术职务任职和行医资格;具有试验方案中所要 求的专业知识和经验;对临床试验方法具有丰富经验或 者能得到本单位有经验的研究者在学术上的指导;熟悉
在我国,临床试验可以是国家项目,包括政府部门 和学术组织发起的项目。也可以是医药器械厂家、研究 单位自行或联合开展的项目。所谓研究者发起的科研 项目应该挂靠在一个研究单位进行。近年来随着国际学 术交往的增加,国际多中心大规模临床试验不断进入我 国,使我们有了参与国际合作和介入世界顶级临床科研 项目的机会。
临床试验协议是近年来受到重视的一个文件,各单 位现在都有不同的审查制度。主要是审查协议中是否体 现了GCP的内容,同时也要审查有关知识产权、法律保护 和经费的情况。现在出现协议审定时间过长而影响了试验 准时进行的情况,需要研究者努力与单位领导沟通,及时 批准。试验前还必须进行完整的文件准备。除了批准文件 和上级试验单位下发的各种文件(如试验方案、病历报告 表、研究者手册等),还要有本单位试验组织的各项文 件,如参加试验人员的名单、职责、研究者的简历和试验 协议等。在试验进行中,还会增加很多文件,包括各种会
方案确定后,下一项工作就是试验的准备。这一工 作十分繁杂。根据试验性质不同,需要得到不同级别管 理部门的批准。我国的纵向课题临床试验,一般由研 究者根据国家不同课题计划申报(招标),若涉及药物 试验且为新适应证,应该有国家食品药品监督管理局 (SFDA)的批准。国际或国内非纵向课题的临床试验, 若为药物试验,涉及到新药或新的适应证,一般也要取 得SFDA批准,由申办单位负责申报。凡要取得批准的项 目,必须得到审批批件。这一批件要一直保存在试验中 心(多中心试验可为复印件)。若非新药或新适应证的 药物试验及非药物试验不需此步骤,但应在试验单位进 行科研立项。
力量 Column
如何在中国开展临床试验(一)
—— 的临床试验在我国 开展。如何高质量地完成临床试验,成为有志于临床科 研的医生的重要课题。
临床科研种类繁多。凡涉及人群研究的大凡有两 种:一种是临床调查,如某种疾病的临床特点观察、注 册研究等,一般对治疗不予干预;另一种是对新的诊断 方法和新的治疗手段的评价(包括药物、器械、治疗手 段等),一般称为临床试验。作为药物试验,可以分为 Ⅰ~Ⅳ期,每一期都有不同的目的、试验人群和试验方 法。目前我国参加的大规模临床试验多为Ⅲ期或Ⅳ期试 验,以临床疗效或安全性为主要试验目的。本文重点介 绍临床试验。
试验前的准备
一项临床试验在实施之前最重要的是确定试验的目 的、方法和方案。如果是多中心试验,由牵头单位(国 际或国内)提出试验设想和初步方案,由试验指导委员 会会议进行全面讨论,确定方案。在试验准备阶段,可 以对方案提出修改意见,包括各中心的课题负责人。但 一旦经伦理委员会批准确定后,任何人和单位都无权自 行修改。此点在各中心申请立题,伦理讨论和以后的具 体实施过程中都必须遵守。
试验启动后,就应该立刻开始试验。不要因主观或 客观的原因延迟试验的实际开始时间。
(未完待续,作者系北京阜外心血管病医院急重症中心 顾问专家)
Journal of China Prescription Drug 2010.09 No.102
11