LCMs适用的溶剂
LC-MS
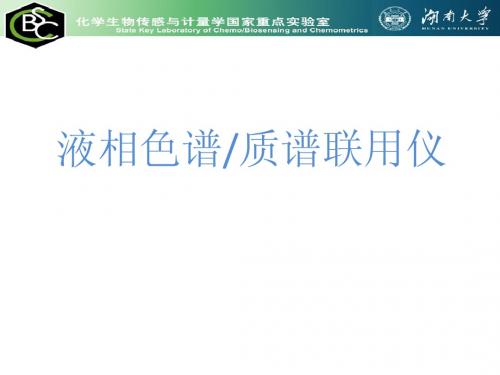
假设测定A和B
A的名义分子同位素分子量取整
1.样品处理
了解样品的结构极性情况,进而推测离子可 能的模式 A、B单标样本浓度都在10ppm左右 配好的样品用0.45μm的滤膜过滤到液质专用 小瓶 注:液质专用小瓶用溶剂超声洗涤
小结
开发定量方法逻辑顺序
子离子定量
1.全扫描scan,确认/得到化合物离子质量数 2.全扫描scan或sim,优化毛细管出口电压 (fragmentor),保证母离子的传输效率 3.子离子扫描daughter ion scan(使用已优化好的 fragmentor),选择定量/定性离子,优化碰撞 能量(CE),得到/优化子离子的响应 4.多反应监测MRM定量,使用已优化的 fragmentor和CE
母离子定量
1.全扫描scan,确认/得到化合物离子质量数 2.全扫描scan或sim,优化毛细管出口电压 (fragmentor),保证母离子的传输效率 3.选择离子监测sim定量,使用已优化的 fragmentor 一般建议:母离子定量也最好用MRM模式
3Q
3.MS条件
注意事项 K,Na外,其他金属离子不能进MS 离子源的选择
离子源的选择
ESI
离子在溶液中已生成 化合物无需具有挥发性 是分析热不稳定化合物的首选 除了生成单电荷离子之外还可以 生成多电荷离子
APCI
离子在气态条件中生成 化合物需具有一定的挥发性 化合物必需是热稳定的 只生成单电荷离子
Agilent QQQ系统
离子光学组件 Lens 1 Lens 2 Skimmer 毛细管
质量分析器
检测器 高能打拿极
八极杆
电子倍增器
全扫描(MS2 Scan)
羧甲基纤维素质谱lcms

羧甲基纤维素(Carboxymethyl Cellulose,简称CMC)是一种重要的纤维素衍生物,其钠盐(羧甲基纤维素钠)被广泛用作食品添加剂、乳化剂、增稠剂等。
质谱(LCMS)是一种分析化学技术,用于分析化合物。
在这里,LCMS被用于分析羧甲基纤维素的结构和性质。
LCMS分析羧甲基纤维素的过程主要包括以下步骤:
1. 样品制备:将羧甲基纤维素样品溶解在适当的溶剂中,通常为水或有机溶剂。
2. 色谱分离:通过液相色谱(LC)对羧甲基纤维素分子进行分离。
色谱柱通常选择合适的凝胶柱,如羟丙基-Sephadex G-75。
3. 质谱分析:通过串联质谱仪(MS)对经过色谱分离后的化合物进行质谱分析。
质谱分析可以帮助确定羧甲基纤维素的分子量、结构以及取代度等性质。
4. 数据处理:将质谱分析得到的数据进行处理,得到羧甲基纤维素的详细结构信息。
LCMS质谱高纯溶剂
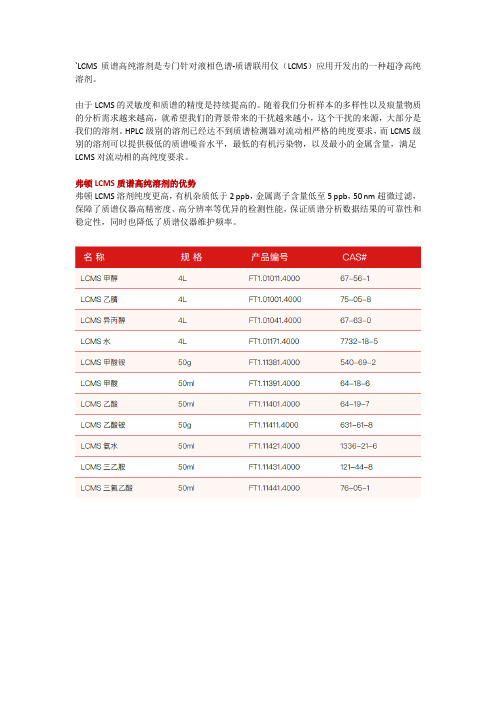
`LCMS质谱高纯溶剂是专门针对液相色谱-质谱联用仪(LCMS)应用开发出的一种超净高纯溶剂。
由于LCMS的灵敏度和质谱的精度是持续提高的。
随着我们分析样本的多样性以及痕量物质的分析需求越来越高,就希望我们的背景带来的干扰越来越小,这个干扰的来源,大部分是我们的溶剂。
HPLC级别的溶剂已经达不到质谱检测器对流动相严格的纯度要求,而LCMS级别的溶剂可以提供极低的质谱噪音水平,最低的有机污染物,以及最小的金属含量,满足LCMS对流动相的高纯度要求。
弗顿LCMS质谱高纯溶剂的优势
弗顿LCMS溶剂纯度更高,有机杂质低于2 ppb,金属离子含量低至5 ppb,50 nm超微过滤,保障了质谱仪器高精密度、高分辨率等优异的检测性能,保证质谱分析数据结果的可靠性和稳定性,同时也降低了质谱仪器维护频率。
常用溶剂在LCMS中的位置
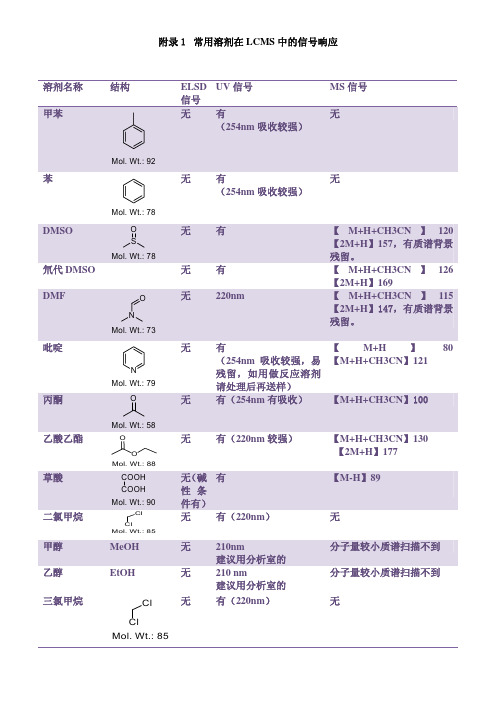
草酸
COOH COOH Mol. Wt.: 90
Cl Cl Mol. Wt.: 85
二氯甲烷 甲醇 乙醇 三氯甲烷
无 (碱 有 性 条 件有) 无 有(220nm) 无 无 210nm 建议用分析室的 210 nm 建议用分析室的 有(220nm)
无 分子量较小质谱扫描不到 分子量较小质谱扫描不到 无
有 【 M+H 】 (254nm 吸收较强,易 【M+H+CH3CN】121 残留,如用做反应溶剂 请处理后再送样) 有(254nm 有吸收) 【M+H+CH3CN】100
乙酸乙酯
O O Mol. Wt.: 88
无
有(220nm 较强)
【M+H+CH3CN】130 【+H】177 【M-H】89
MeOH EtOH
Cl Cl Mol. Wt.: 85
无
附录1 常用溶剂在 LCMS 中的信号响应
溶剂名称 甲苯
结构
ELSD UV 信号 信号 无 有 (254nm 吸收较强)
MS 信号 无
Mol. Wt.: 92
苯
无
有 (254nm 吸收较强)
无
Mol. Wt.: 78
DMSO
O S Mol. Wt.: 78
无
有
氘代 DMSO DMF
N Mol. Wt.: 73 O
无 无
有 220nm
【 M+H+CH3CN 】 120 【2M+H】157,有质谱背景 残留。 【 M+H+CH3CN 】 126 【2M+H】169 【 M+H+CH3CN 】 115 【2M+H】147,有质谱背景 残留。 80
阿拉丁(aladdin)LC-MS溶剂中文版目录

LC-MS 级溶剂1/2LC-MS 级溶剂LC-MS 级溶剂有着灵敏度更高的、使用更便捷的高效液相色谱质谱联用仪器(LC-MS)的问世,LC-MS 迅速地成为制药、生物化工、化学合成、环境保护和食品科学等领域的一种首选的分析工具。
提供一种绝对无杂质干扰的LC-MS 级溶剂,可确保LC-MS 分析达到最优化的条件和最优化的特性,同时可保证LC-MS 分析仪器和色谱柱有更长的的使用寿命。
阿拉丁的LC-MS 溶剂是是阿拉丁为拓展LC-MS 的应用而精心开发的最高级别的溶剂产品。
阿拉丁应用最先进的净化和控制技术,经过极其严格的脱杂处理制成。
阿拉丁使用严格的多层次质量控制和特别包装,可以保证每一批次阿拉丁LC-MS 溶剂的产品质量,确保其优异的批次稳定性。
阿拉丁LC-MS 溶剂是经过特别加工精制的溶剂,满足现代LC-MS 仪的需要,具有非常低的有机物杂质、金属杂质和微粒,具有极低的背景吸收,是您在使用LC-MS 和HPLC 时的最理想选择。
特点•低挥发物残留•低背景•低杂质•批次稳定性•低UV 吸收•低离子含量•通过梯度洗脱检测•经0.2µm 膜过滤•惰性气体封装应用适合≤ppb 级高灵敏度LC-MS 分析和其他需要极低污染和化学噪音的应用.LC-MS 级溶剂适合≤ppb 级高灵敏度LC-MS 分析和其他需要极低污染和化学噪音的应用.货号 品名 规格 CAS 包装2/2LC-MS 级溶剂A120771乙腈Acetonitrile LC-MS 75-05-81L, 6×1L 2.5L, 4×2.5L W119425水Water LC-MS 7732-18-51L4L ,4×4L M116125甲醇MethanolLC-MS 67-56-11L4L , 4×4L I112019异丙醇Isopropyl Alcohol LC-MS 67-63-01L 2.5L E116141乙酸乙酯Ethylacetate LC-MS 141-78-61L 2.5L H120773正己烷n-Hexane LC-MS 110-54-31L 2.5L H120772正庚烷HeptaneLC-MS142-82-51L 2.5LT103297三氟乙酸Trifluoroacetic acid LC-MS 76-05-110ml 1L F112037甲酸 Formate LC-MS 64-18-625ml 100ml A116168乙酸 AcetateLC-MS 64-19-7100mL 500mL A100186甲酸铵Ammonium formate LC-MS 540-69-250G A112060乙酸铵Ammonium acetate LC-MS 631-61-825G 100G A11208425%氨水溶液Ammonia solution LC-MS 1336-21-6100mL T103290三乙胺TriethylamineLC-MS121-44-850mL货号 品名 规格 CAS 包装LC-MS 流动相添加剂。
LCMSMS农药一齐分析法(Ⅱ)

1. 适用范围适用于LC/MS/MS农药的一齐分析法(Ⅱ)中农药的检测。
2. 编制依据依据日本厚生劳动省《LCMS农药一齐分析法Ⅱ》编制。
3. 方法原理水果、蔬菜、加工产品中的农药残留经乙腈提取,提取溶液经净化、浓缩用50%的甲醇定容后,LC/MS/MS测定,用外标法定量。
4. 试剂4.1 乙腈:色谱级4.2 丙酮:色谱级4.3 正己烷:色谱级4.4 乙酸乙酯:色谱级4.5 无水硫酸钠:分析纯4.6 甲苯:色谱级4.7甲醇:色谱级4.8 氯化钠:分析纯4.9 0.01M 盐酸:8.29ml浓盐酸定容至1000ml5. 仪器设备5.1 Waters UPLC TQD5.2 食品加工器5.3菜刀,案板5.4电子天平5.5均质机5.6布氏漏斗,分液漏斗,普通漏斗,滤纸5.7梨形瓶(250ml,100ml)5.8 锥形瓶(200ml,100ml)5.9 C18固相萃取柱(1G 6ml)5.10氨基复合柱(Carbon/NH2 500mg 6ml)5.11硅胶柱Silica (500mg 6ml)5.12旋转蒸发仪5.13 氮吹仪5.14 玻璃离心管(10mL)5.15 分液漏斗振荡器5.16 真空泵5.17 移液枪(1mL,200μL)6. 分析步骤6.1 试料的制备:(除不加样品外,试剂空白与样品同步处理)将送交实验室的不少于500克的样品于食品加工器搅细(对于大块状的样品可先用水果刀切成小块,再放于食品加工器中搅细)。
粉碎后的样品装于一次性的塑料袋中备用。
6.2 提取6.2.1 准确称取10.0g(精确到0.1g)搅细的样品(原料称取20.0g)于均质杯中,加入70ml乙腈,在高速组织捣碎机中均质5min。
6.2.2 将样品减压抽滤,用30ml乙腈冲洗均质杯,也过滤,合并滤液。
6.2.3 将滤液倒入装有7gNaCl的300ml分液漏斗中,用20mL0.01M 盐酸冲洗三角瓶,也倒入分液漏斗中,在分液漏斗震荡器上剧烈震荡10min,静置10min,使乙腈相和水相分层。
LCMS操作操作注意事项

LCMS操作操作注意事项LC/MS操作操作注意事项首先确定LC分析条件包括: mobile phase,gradient及column 等,并自备solvents,solvent/waste bottles,sample vials (1.8 ml).每次操作时: 分析前及完成后,需清洗管线至少半小时以上,确保无残留污染.MS分析条件已知或已找到相关参考资料.进入chemstation后,首先需check tune为pass才可进行以后之操作,否则立即通知仪器中心助理.条件设定,质谱解析及报告A. LC设定1. Column2. 液相条件 - 移动相的选择3. Set up Pump4. Set up Injector5. Set up DAD SignalsB. MS设定条件参考1.电喷雾(ESI)之3个基本步骤2.化学电离(APCI)之3个基本步骤:3.MS 雾化室(MSD spray chamber)4. Set up MSD signals 1- MSD Control (方框左方)5.Set up MSD Signals 2 - MSD Signal Settings (方框右方)6.选择离子检测(SlM)/ 扫描(scan)模式C. MS说明与解析:1.四极杆质量分析器有两种扫描方式:2.质谱常用术语3.质谱图相关概念4. LC-API/CID/MS5. MS解析规则与条件说明6. API电离步骤及过程:7. ESI 及APCI 最佳化条件8.将现有的LC方法改编为LC/API-MS方法9.萃取离子层析图(EIC)10. 建立校正表D. ReportA. LC设定1. Column :(1)通常填充物质的粒径3-10 μm的管柱是可用的.(2)大於10 μm的填充物不适合LC/MS分析,因为峰扩散很大.(3)粒径3-5 μm的填充物是较理想的.5 μm管柱可能更适合扫描模式,其峰宽度会比较小粒径的管柱宽.峰出现花费较少时间,可以有足够扫描速度和扫描整个峰.如果数据采集太快,损失谱图质量.3.5 μm管柱具有增加层析分辨率和灵敏度的优点.提高的分辨能力可更可靠地分离同分异构体.(4)根据实验需要选择Column的长度,对高流量简单分析用短Column.总分析时间会较短,当为复杂样品时,需要较长Column的分离能力.2. 液相条件 - 移动相的选择(1) 非挥发性的酸,碱,盐 => 挥发性的酸,碱,盐(2) Positive Ion ( pH 5.0 ; 9 preferred)●Acetic acid●Formic acid●Tri-fluoroacetic acid●Ammonium hydroxide●TEA(3) Post-column addition of acid or base may be used to adjust the pH if the chromatography won't work at the desired pH(4) 溶剂适用性Suitable for ES and APClSuitable for only APCIMethanoIAcetonitriIe ;*WaterEthanol ; Propanol ; lsopropanoI ; ButanoIDMF(1) ; DMSO(1)Acetic Acid ; Formic AcidAcetoneCH2C12 ; CHCI3THFTolueneBenzeneHydrocarbons (e.g Hexane)StyreneCCl4CS2Cyclic Hydrocarbons(e.g Cyclohexane)(5)许多常规的HPLC溶剂适合於API-MS.较好的电喷雾溶剂将在溶液中保持离子.因此,如甲苯这样的溶剂,在溶液中不会保持离子,不能用於电喷雾.虽然水是对离子极好的溶刹,但它的溶剂化能太高,去溶剂困难.(6)常用的LC/MS溶剂一般为高质量的甲醇,乙睛,水和挥发性添加剂,如甲酸,乙酸,甲酸铵和乙酸铵等.如果这些常规溶剂能满足分析的需要,可不考虑其他溶剂.(7)层析类型与离子源适用性ESIAPCIReversed phase*****Normal phase****p.s. Main compatibility problems: ions in solution are favorable for electrospray, but may lead to poor retention in reversed phase. High ionic strength and/or nonvolatile buffers may be hard to electrospray(8)ESI/APCI与HPLC的移动相之正反模式的相容性:(a)电喷雾有利於溶液中的离子.但是反相液相层析条件通常会抑制离子化,增加分子疏水性质.之后添加可用於克服这种类型的不相容性.样品通常挥发性低.(b)电喷雾对正相分离不是很有用,因为在正相移动相中通常不会形成离子.而APCI可处理非极性移动相,非极性样品通常较易挥发,因此正相层析可用於APCI.(c)以凝胶过滤形式的体积排阻层析与电喷雾相匹配.水相移动相和蛋白质样品是很好的匹配.凝胶过滤层析不适合APCI.通常样品不易挥发,而且缓冲液会引起APCI问题.然而,凝胶渗透层析由於移动相为有机性的,不太适合於电喷雾.3. Set up Pump(1)流速(flowrate) :流速就是溶剂沿著管柱流动的速度.保持流动速度为常数对确保精确的保留时间和峰面积测量是很重要的.(2)溶剂(solvent) :设定分析的溶剂组成.可选择一元分析或梯度流洗分析.设定通道B 的百分比为从0到100%的任何值.通道A为剩下的体积100-(%B).当运行反相层析分析时,建议把水相放在通道A.(3)压力界限(pressure limit) :设定压力限制的上限和下限,最高压力限是pump的自行停止的界限,保持分析系统压力避免过大.如果压力低於最低限以下,例如移动相走空以后,pump也将自动停止.(4)停止时间(stop time) :设定一个分析的时间.停止时间后,所有的梯度都会停止,并且pump 参教返回初始值.pump是一个完整分析系统停止时间的掌握者.(5)驻留时间(post time) :驻留时间是分析结束和下一个分析开始之间的最小时间间隔,可以利用驻留时间在改变移动相组成后平衡Column,例如梯度流洗以后.(6)时间表(time table) :时间表用於建立pump的梯度洗脱程序.pump时间表的值从时间表中定义的终值应随时间线性地变化.4. Set up Injector(1)标准的进样体积为0.1~100μL(标准配置).如果使用洗针功能,指定洗针所用的溶剂瓶号即可.其余参数一般不需要改变.(2)选择Use Injection programs可以按照使用者要求,安排程序进样.(3)如果希望两次进样取样间隔时间,可以选择Optimization选项:(a)Overlap Injection cycle:在前一针样品进行时,就提前把下一针的样品吸入定量瓶中,做好进样准备.要注意一定要改变后面的时间内定值,以保证前一针样品能够顺利进入液相系统,同时保证下一针样品的扩散最小.一般建议该时间选择在上一针样品进行停止之前1~2min.(b)Prefetch Sample Vial:在前一针样品进行时,就提前把下一针的样品抓取到针座上等候,做好进样准备.此时预约的时间要比上种选项要少,但没有样品扩散的风险.此时也要注意一定要改变后面的时间内定值,以保证前一针样品能顺利进入液相系统,一般建议该时间选择在上一针样品进行停止之前1~2min.(4)注意要保证Agilent 1200各个模式的Stop Time和Post Time的设定要一致,内定为As pump,即在pump的参数设定画面规定这两个时间即可.5. Set up DAD Signals(1) Signals :规定采集层析图所需的参数,要求样品的检测步长Wavelength(Sam.)设在化合物的最大吸收步长处,且大於溶剂的截止步长20nm以上.参考步长Wavelength(Ref)应设在样品没有吸收的地方,且越靠近样品的侦测步长越好.另外,要求BW(Ref)>BW(Sam.)>Slit,其中样品的谱带宽度Band Width(Sam.)应约等於其紫外吸收峰的半峰宽.注意记得保存所需通道的层析图,否则层析数据将不被保存.(2)Spectrum:规定采集吸收光谱时所需的参数.可保存所需张数的光谱图None, Apex + Baselines, Apex+Slopes+Baselines, Ail in Peak, Every 2nd Spectrum或All.若需要进行峰纯度检测或最佳化完全未知的样品检测条件,建议选择All模式.(3)Peakwidth:设定影响层析图的数据采集频率,Peakwidth应略小於层析上最窄的层析峰的半峰宽.数据采集速率太快,则层析图的毛刺过多,数据采集速率太慢,则所采集的数据点过少,造成层析峰变形,无法进行准确定量.B. MS设定条件参考1.电喷雾(ESI)之3个基本步骤: 喷雾及带电→去除溶剂→离子蒸发.ESI之雾化气压力,乾燥气流速及温度取决於流动相组成及流速,如流速越快,含水量越高即需越多乾燥气辅助去除液滴中溶剂.ESI需考虑(1)样品 :(a)在溶液中为离子态:儿茶酚胺,硫酸酯共轭物,丁基胺(b)有可诱导电离的化合物:甲醇(c)含杂原子的化合物:氨基甲酸酯类,苯并二氮杂原子类(含O, S, N)(d)溶液中带多电荷:蛋白质,多肽,低聚核甘酸(2)溶液化学参数 :(a)流速(b)样品的pKa,溶液pH(c)溶液导电性(3)应避免的样品 : 尤其非极性的样品PAHs,PCBs2.化学电离(APCI)之3个基本步骤: 雾化→蒸发液滴→气相电离.即蒸发后再进行离子化,用於可被蒸发之样品,且此过程只产生单电荷离子.APCI需考虑(1)样品(a)分子量和极性中等的化合物:PAHs, PCBs,脂肪酸,邻苯二甲酸酯类(b)不含酸性和碱性位点的化合物: 酮,酯,醇,醛,碳氢化合物(c)含有杂原子的化合物:脲,氨基甲酸酯等(d)对电喷雾响应不好的样品(2)溶液化学参数(a)较ES对溶液化学作用不灵敏(b)较ES更耐大的流速(c)适用ES不宜的一些溶剂(3)应避免的样品 : 在气化过程中热不稳定的化合物3.MS 雾化室(MSD spray chamber)(1) Method: API-ES ; APCI(a)乾燥气流速(drying gas flow, L/min) : 范围0-13即最高限值13 L/min.ESI通常为8-10 L/min;APCI通常为4.(b)喷雾气压力(Nebulizer pressure, psig) :APCI:60 psi;ESI与流速有关,最高限值60 psi.(c)乾燥气温度(drying gas temperature) :与流动相和流速有关,通常为300 ℃以上.水相比较多或高流速时要求较高的乾燥气温度.最高限为350 ℃(d)蒸发温度(Vaporizer temperature)(限於APCI) :与溶剂和流速有关,通常为350 ℃.水或高流速时要求较高的蒸发温度.最高限为500 ℃.(e)毛细管电压(Capillary voltage) : 最高限为6000 V正模式(V) 负模式(V)ESI 4000 3500APCI 4000 4000注: ESI模式负极性时,在高电压时全发生喷针放电.(f)电晕电流(Corona current, μA) (限於APCI) :对正极性: 通常为4 μA;对负极性: 通常为25 μA.(2) Method: MM-ES + APCI 复合源(a)注意charging voltage : 影响ESI区域离子化重要参数.4. Set up MSD signals 1- MSD Control (方框左方)(1) Use MSDMSD将被用作检测器.在standby状态,不会被作为检测器.(2)停止时间(Stop Time): 质谱停止采集的时间.(a)一般设为As pump:按照LC pump中所设的停止时间,(b)若使用者输入时间:当选到指定时间时,MSD数据采集将停止.(3)FlA状态: 选择FlA时,会显示FIA enabled.非FlA时,显示FIA disabled.(4)调整档案(tune file): 指定采集数据时MSD采用那个调整档案里的参数(5)峰宽度(Peak width): 输入预期的层析峰半宽度 (用分钟表示).典型的层析峰宽为0.05-0.15 min.不确定时使用较小的值.内定值为0.1 min.(6)循环时间(Cycle time): 完成一次所有活性信号扫描的时间(秒).也可想像层析图上一个采样点的时间.循环时间的计算与所输入峰宽有关.(7)快速扫描(Fast scan):(a)当设定的质谱信号参数(如峰宽,质量范围和步长)会导致采集数据点过少时,软体会提醒改变参数或选择快速扫描方式.(b)如果参数设定不需要快速扫描,即使选择快速扫描也不会被执行.在VL系统中没有这个选项.(c)在最佳化扫描速度时,会损失一些灵ㄧㄥㄣㄚㄚ陌性敏度和分辨率.因为四极柱的离子传输效率较低.Data reconstruction (selectable)可选,在此设定下采集的数据进行重组以提高电荷和多电荷质谱图的分辨率.某些需要快速扫描速度的应用,不一定要重组数据.对於这些应用,不选择数据重组.数据重组时应用一个移动平均过滤器以减少由快速扫描引起的质谱杂讯.(8)时间过滤器(Time Filter): 内定为选择时间过滤器.建议使用时间过滤器.(9)扫描数据存档(Scan Data Storage):(a)建议选择保存压缩数据(Condenced),即棒状质谱图,节省磁碟空间.(b)对多电荷样品使用完全数据 (连续质谱图),其它使用压缩数据.(c)Deconvolution只能在完全数据上进行.如果试图在压缩数据或SIM数据上使用Deconvolution,将会产生错误.5.Set up MSD Signals 2 - MSD Signal Settings (方框右方)(1)极性(Polarity):指定MSD检测离子的极性(positive/negative).必须先进行MSD tune.(2)模式(Mode): 采集质谱数据的方式.(3)扫描(Scan):从高到低指定的质量范围,并且以指定的间隔(步长)检测强度.在过程中这个程序不断重覆.适於定性分析,例如未知物的鉴定,峰纯度或多电荷样品分子量的确定.(4)碎裂器(ramp):适用於Scan模式和定义ramp.不同的质量数离子可应用不同的碎裂电压.(5)时间(Time): 进样后质谱参数开始进行生效的时间(min)如果第一行是非零时间,MSD不会采集,直到这个时间到达.质谱选择阀也会将LC流路送到废液瓶,直到开始数据采集.(6)质量范围(Mass range): MSD扫描的质量范围.范围越大,MSD须扫描得越快.要得到高品质数据,应扫描所需范围.(7)增益(Gain): 增益是MSD信号放大因子,将信号强度电子倍增器的电压相关联,增益5会得到增益为1时的信号强度的5倍.通常,检测器会在较低的增益值对工作即能产生合适的离子强度.高增益值同时会增加噪音,产生较差的信噪比.通过增加增益值来提高EMV电压会缩短电子倍增器的寿命.(8)碎裂器电压(Fragmentor voltage): 设定碎裂电压.当进行一个方法(Method)时,控制碎裂器电压优先是:(a)选择FIA时,包括碎裂电压的FIA表;(b)当选择碎裂器ramp时,碎裂器ramp表(FIA未选择);(c)如果以上的都没有选择时,参考下列信号设定表.(9)阀值(Threshold): 阀值是强度值.只有强度等於或大於这个值的数据点才被保留在每个扫描的质谱中.内定值150.(10)步长(Step size): 步长是设定扫描数据点之间步幅的大小.这个参数影响扫描速率.典型的值是0.1,范围为0.05到0.4.Fragmentor值与化合物性质有关: 不同化合物使用不同的Fragmemtor值CompoundMax. Mol. Ion2nd. M/z >50%Acid Red 4 (-) 100 130Alprazolam (+) 100 140Caffeine (+) 70 100Methamphetamine (+) <50 70Nitrophenol (-) 70 100Quinine (+) 90 130Quinine (++) 50 60Reserpine (+) 130 170Salbutamol (+) 200注: 最佳化碎裂电压: 表中数据表示不同化合物获得最强的准分子离子和第一个碎裂离子达到其50%强度的碎裂器电压值.每个化合物均通过FlA模式在碎裂电压50到200 V范围内进行分析的.6.选择离子检测(SlM)/ 扫描(scan)模式(1)Scan采集在特定范围中每个质量上的数据.对定性分析通常采用scan模式采集,例如确定样品的分子量或纯度.(2)SlM只采集预先设定离子的数据.此采集方式具灵敏度和专一性.定量分析通常选择SlM模式.Greater sensitivityBetter peak shapeTrace levels easily detected in complex matricesUsed for routine quantificationUseful when precise ion ratios are desired(a)SlM参数: SlM有三个可用自订的参数# of ionsm/z of each ionDwell time(b)每组可检测30个离子,每次采集可分50组.(c)每组检测最少数目的离子可获得最大的信噪比和准确度.(d)驻留时间(Dwell Time)是对每个离子检测的时间段.该时间取决於峰宽选择(17+2 cycles/peak).S/N正比於Dwell Time.(e)可以设定同组中每个离子驻留相同时间或设定每个离子相对驻留时间:*组内(group)为同时SIM的离子,组间为不同对SIM的离子.*组内SIM的离子越多,每个离子的驻留时间就越少.*操作者可根据层析峰的分离情况将不同离子设为组内,或组间SIM.*驻留时间是用於采集每个离子的时间.驻留时间与MSD参数对话框中设定的峰宽度值有关系.软体会根据峰宽度值,然后根据每个峰要有17+2个循环的需要以确定驻留时间.除非使用者指定相对的驻留时间,否则驻留时间会在组内的离子间平均分配.(3)选择 SlM 离子(a)定量离子:该离子的响应用於计算待测物的含量.(b)限定离子:该离子的响应值与定量离子响应值之比值用来确认待测物质化合物,以避免假检出.(c)选择相对强度较高的离子可以提高灵敏度.(d)选择质量数高的离子可以避免干扰.(e)选择唯一性高的离子.(f)尽量避免选择基质或背景中存在的离子.(g)如果可能,每个化合物选择多个离子.离子间的比例可以帮助判别化合物,特别是同位素族.(h)对於SIM定量,可以对每个层析峰选择一个定量离子.这个离子应该峰高且唯一.不要选择在基体或背景中相同的m/z.(i)为确定层析峰,选择一或两个限定离子.这些离子也应该峰高且唯一.在LC/MSD分析中,离子的出现和峰高度,缓冲液和碎裂器相关.(4)选择定量/限定离子(a)应用精确质量,不要用名义质量(例如195.2而不用195).(b)为得到最佳效果,用动态校正,即选择如括弧所示的样品质量(195.0,195.1,195.2,195.3,195.4)以选择响应值最佳的离子.(c)为长期的效果,检测前要对质量轴进行校正.(5)定量方法建立首先要扫描采样以确定在SIM分析中所要使用的离子.使用扫描采样数据,得到谱图列表展示有一位近似值的精确质量并输入到SIM表中.使用者应使用列表中的质量,例如195.2,而不是名义上的质量195.如果所选择的SlM质量偏差0.3daltons,则灵敏度会有70%的损失.另一方面,找到SlM分析的最佳m/z,要避免所有离子的SlM实验产生一位近似值.这就是所谓的动态调整.(6)动态SIM校正动态SIM校正可以获得最佳的SIM灵敏度.做SIM 试验:(a)以0.1 dalton为间隔采集多个质量例如:Caffeine 195.2 : 195.0 ; 195.1 ; 195.2 ; 195.3 ; 195.4(b)积分SIM数据并且选出响应值最大的离子作为定量离子(c)进行动态调整,此例子中,全扫描质谱图在195.2 daltons处有离子.(d)咖啡因的SIM实验将包括一组具有五个离子:195.0,195.1,195.2,195.3和195.4 daltons.评估各质量的离子EIC 积分结果,或直接看质谱图以找到最大强度的离子.这就是SIM定量的最佳离子.C. MS说明与解析:1.四极杆质量分析器有两种扫描方式:(1)全扫描方式(scan) : 主要用於定性分析,未知化合物的鉴定;(2)选择离子监测(SIM) : 主要用於已知化合物的定量分析.2.质谱常用术语1.分子离子(molecular ion)自由基 (radical)离子M.+.很活泼,易碎裂而产生广义的碎片离子.2.准分子离子*(quasi-molecular ion)由软电离技术产生的质子或其他阳离子加合离子以及去质子化或其他阴离子加合离子.3.碎片离子(fragment ion)电离后具有过剩内能的分子离子以多种方式裂解生成碎片离子.4.奇电子离子(odd-electron ion, OE);偶电子离子(even-eIectron ion, EE)OE含未配对电子,有较高的反应 (碎裂)活性,易生成碎片离子.5.多电荷离子(multiply-charged ion)6.同位素离子(isotopic ion)3.质谱图相关概念(1)总离子层析图(Total Ion Chromatogram, TIC)(2)质谱图 (Mass Spectrum, MS)(3)选择离子监测图 (Selected Ion Monitor, SlM)(4)萃取离子层析图 (Extract Ion Chromatogram, EIC)(1)总离子层析图(Total Ion Chromatogram, TIC)将质谱图上每一个离子的强度的总合对应扫描时间作图产生的.TIC层析图上每一个采样点都对应一张质谱图.(2)准分子离子当用大气压电离技术分析小分子时,通常在质谱中主要的峰是质子化准分子离子.例如苯基保泰松的实际质量308,在质谱图中其基峰即最大强度峰是准分子离子[M+H]+峰,m/z为309.(3)萃取离子层析图(EIC) : 可以(a)提高S/N去除背景(b)分离共流出峰(c)主要用於:寻找目标峰,准确定量注意:EIC是在数据采集后分析时生成的;而SIM是实际信号.4. LC-API/CID/MS :(1)通过调节毛细管出口电压,在毛细管出口处可发生碰撞诱导解离(Collision Induced Dissociation, CID)而产生碎片.利用碎片信息,有助於定性;利用碎片离子定量,可提高方法的专属性.(2)范例:甘油三酸脂的碎裂电压75V可得谱图中m/z 684.65呈现准分子离子峰[M+NH4]+.当碎裂电压升高到175V,可用碎片离子m/z 467或439表示.(3)API是一种相对的软电离技术,产生的主要是准分子离子.CID是用中性气体分子去碰撞离子产生碎片的过程,对定性分析和定量分析都很有用.(4)CID可以通过选择离子传输毛细管出口和第一级分离器之间的离子能量来控制.离子能量可以通过改变软体(chemstation)中所谓的碎裂参数来改变.5. MS解析规则与条件说明(1) [M+H]+离子的氮规则分子量为奇数[M+H]+ 为偶数的离子有奇数个N分子量为偶数[M+H]+ 为奇数的离子有偶数个N或不含N例1. m/z 300的[M+H] + 离子: mw = 299 ; 即化合物一定有奇数个氮例2. m/z 301的[M+H] + 离子: mw = 300 ; 即含有偶数个氮(0,2,4….)(2)API质谱图解释电喷雾和APCI是可提供分子量信息的软电离技术.检测到的离子种类与溶剂添加剂和分析所用的条件相关.正离子检测负离子检测-[M+H]+ 酸性条件-[M+Na]+ , [M+K]+ (有盐时)-[M+NH4]+ 有铵盐缓冲溶液-[M+X]+, x=溶剂或缓冲溶液中的阳离子-[2M+H]+在高浓度时形成的二聚体-[M+H+S]+ 溶剂添加剂-[M+H]- 碱性条件-[M+X]- , x=溶剂或缓冲溶液中的阴离子-[M-H+S]- 溶剂添加剂(3)样品的储存,准备,移动相和添加剂都将影响最后结果.当建立一个新方法时要注意这些因素.(4)CID (Collision-Induced Dissociation )通过与中性气体分子碰撞将能量传递给离子产生碎片的过程.能量传递足以导致断键和所选离子重排.对定性分析和定量分析都很有用.定性提供有关分子的结构信息.定量特性由限定离子的存在而增强.70年代初McLafferty (JACS,95,3886,1973)证明ClD使键断开和离子重排,产生离子代表中性分子的结构.结构解析:API过程中,准分子离子以偶电子离子形式出现.通过CID合产生碎片,碎裂过程如下:ABCD+→ ABC+ + D (中性碎片)电荷保留在质子亲和势较高的碎片上(5)ClD 质谱图解析: 常见中性损失( Even LOSS)(M+X)+-18水(M+X)+-H2O(M+X) +-20氟化氢(M+X)+-HF(M+X) +-28一氧化碳或乙烯(M+X)+-CO or (M+X) +-C2H4(M+X) +-30甲醛(M+X)+-H2CO(M+X) +-31甲胺(M+X)+-CH3NH2(M+X) +-32甲醇(M+X)+-CH3OH(M+X) +-36氯化氢(M+X)+-HCI(M+X) +-44二氧化碳(M+X)+-CO2(M+X) +-46二氧化氮(M+X)+-NO2(M+X) +-60乙酸(M+X)+-CH3CO2H(M+X) +-90硅醇(M+X)+-HO-Si-(CH3)36. API电离步骤及过程:(1)溶液中电离(样品pKa,溶液pH)→(2)喷雾(表面张力及黏度,气动辅助)→(3)去溶剂(乾燥气温度及流速,热容量Hvap)→(4)离子从溶液中解析(溶解能)→(5)气相中的离子反应(质子亲合力,电荷交换)(1)当分析物在溶液中以离子存在时得到最佳的电喷雾灵敏度对中性分子,离子相互作用比非离子相互作用大103至104倍(例如:凡得瓦力,氧键).因此分析物离子可以克服液滴的溶解能,从带电液滴中解析出来.(2)在溶液中如何产生离子(a)酸/碱化学性质: M - NH2 + 酸→ [M-NH3]+ +酸-M - COOH + 碱→ [MCOO]- + 碱+(b)螯合(对类似糖的中性物质): M.+ Na+ [M + Na]+ (碱金属,如20 M乙酸钠)(c)衍生化: 形成离子或酸/碱产物当分析物溶解在酸或碱之极性溶剂中时,可被离子化或具有强偶极距.对於电离的分析物,ESI通常简单且具有高灵敏度.不存在其它离子-离子相互作用干扰,离子在喷雾前已经存在於溶液中.在喷雾中这些离子易於从液滴中蒸发出来,得到较高的分析物离子强度.形成强偶极距但没有被电离的分析物也可分析.喷雾室中的强电极场驱动离子化过程.这些电场促使喷雾液滴带电荷,使液滴表面引起分析物分子离子化.通过使用特定的化学物质的交互作用,这些分析物也可被化学电离.(3)对电喷雾使用典型缓冲液产生离子的问题 - 离子对的形成(a)对正离子检测,由於离子对的形成,使溶液中或气相中的离子中和![M + H]+ + A- [M + H + A]o ;A = B, S, P : 有利於中性样品; A = 甲酸盐,乙酸盐: 有利於带电物质*离子对强度: B,S,P > 三氟乙酸>乙酸盐,甲酸盐 (B, S, P) = 硼酸盐,硫酸盐,磷酸盐(b)对负离子检测,由於离子对形成,使溶液或气相中离子的中和![M-H]- + C+ [M-H + C]0 ; C = Na,K,Li (中性产品); C = NH4+(带电物质)(4)气相中的离子反应(a)通过离子传输区域时从大气压喷雾室的反应中会产生质子转移和电荷交换反应.这个高压区允许发生1000次离子/分子反应.(b)质子转移:与HPLC添加剂相比,如:氨,三乙胺 (质子亲合力分别为206和232 kcal/mole),样品具有较低质子亲和力的会失去一个质子,变成中性或形成复合离子如[M+NH4]+.(c)溶液碱度在气相中会导致分析物离子的去质子化,致使分析物没有电喷雾信号.添加剂如三乙胺这样强气相碱的使用,会导致[M+H]+离子损失.添加剂(乙酸铵,甲酸铵,乙酸,甲酸和氢氧化氨)的使用会减少这些反应.(5)比较电喷雾电离源(ESI)大气压化学电离源(APCI)离子在溶液中已生成离子在气态条件中生成化合物无需具有挥发性化合物需具有一定的挥发性是分析热不稳定化合物的首选方法化合物必需是稳定的生成单电荷离子外亦可生成多电荷离子只生成单电荷离子7. ESI 及APCI 最佳化条件(1)APC-MS添加剂(a)调整PH : 使用乙酸,甲酸,TFA,氨氧化胺(b)一般的缓冲液/离子配对试剂 :乙酸铵/甲酸铵 ; 三氟乙酸(TFA) ; 七氟丁酸(HFBA) ;四乙基或四丁基氢氧化铵(TBAH)(c)阳离子化试剂 : 20-50 M的乙酸钠或乙酸钾(d)一般考虑:挥发性 (污染喷雾室,堵塞喷嘴)导电性 (对离子气化过程减少小液滴的形成)离子配对 (进入气相时中和预带电离子)(e)不能使用在HPLC中常使用的磷酸盐,硫酸盐或硼酸盐(f)在APl-ES中影响添加剂选择的主要因素是离子配对和挥发性.(g)对APCI添加剂选择的主要因素是挥发性.8. 将现有的LC方法改编为LC/API-MS方法(1)溶剂:(a)非挥发性缓冲盐 => 挥发性缓冲盐(b)浓度<10 mM(对於ES)或<100 mM(对於APCI)用醋酸鞍,甲酸,三氟乙酸 (TFA),七氟丁酸(TFBA),羟基四丁基胺取代磷酸盐,硫酸盐和硼酸盐.(Formic acid, acetic acid, TFA, ammonium hydroxide)如果必须使用非挥发性缓冲盐,使用仅阳离子或阴离子部分不挥发性缓冲盐,例如用醋酸钠而不用磷酸钠.(c)挥发性离子对试剂可以使用.(2)样品制备: 通常样品制备或没有样品制备会严重影响API-MS分析.很多情况下APl-MS技术的失败源於样品前处理.前处理不当会导致信号衰减或共同离子干扰.前处理需要考虑以下因素,其为分析成败关。
常用溶剂在LCMS中的信号响应
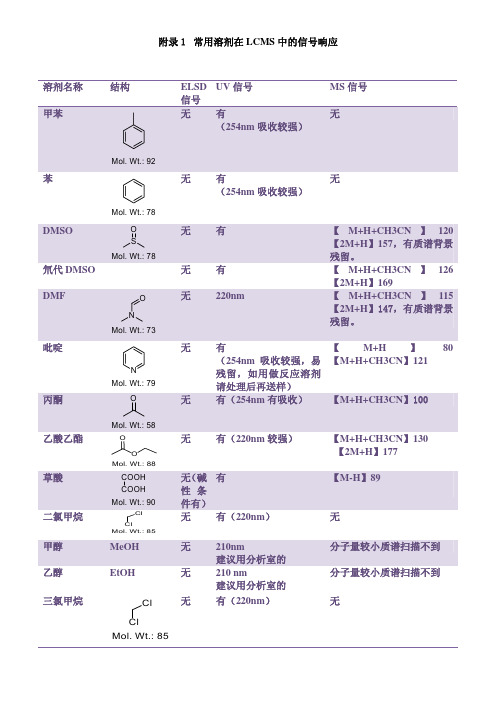
无 无
有 220nm
【 M+H+CH3CN 】 120 【2M+H】157,有质谱背景 残留。 【 M+H+CH3CN 】 126 【2M+H】169 【 M+H+CH3CN 】 115 【2M+H】147,有质谱背景 残留。 80
吡啶
N Mol. Wt.: 79
无
丙酮
O Mol. Wt.: 58
无
附录1 常用溶剂在 LCMS 中的信号响应
溶剂名称 甲苯
结构
ELSD UV 信号 信号 无 有 (254nm 吸收较强)
MS 信号 无
Mol. Wt.: 92
苯
无
有 (254nm 吸收较强)
无
Mol. Wt.: 78
DMSO
O S Mol. Wt.: 78无有来自氘代 DMSO DMF
N Mol. Wt.: 73 O
有 【 M+H 】 (254nm 吸收较强,易 【M+H+CH3CN】121 残留,如用做反应溶剂 请处理后再送样) 有(254nm 有吸收) 【M+H+CH3CN】100
乙酸乙酯
O O Mol. Wt.: 88
无
有(220nm 较强)
【M+H+CH3CN】130 【2M+H】177 【M-H】89
草酸
COOH COOH Mol. Wt.: 90
Cl Cl Mol. Wt.: 85
二氯甲烷 甲醇 乙醇 三氯甲烷
无 (碱 有 性 条 件有) 无 有(220nm) 无 无 210nm 建议用分析室的 210 nm 建议用分析室的 有(220nm)
lcms操作规程

lcms操作规程
LCMS的操作规程如下:
1.打开LCMS仪器,确保仪器处于正常工作状态。
2.准备样品,确保样品浓度适中,一般为0.1mg/ml左右,并用适量的
甲醇溶解。
如果样品不溶于甲醇,可以考虑使用少量的DMSO进行溶解,然后再稀释到所需的溶剂中。
样品瓶要确保无尘。
3.在LCMS系统中设定并下载之前设定的MethodFile,即将仪器参数从
程序配置到仪器中。
4.如果仪器刚刚开机,需要等待各个部件预热完成后再进行操作。
5.在操作过程中,要确保按照仪器规定的步骤进行,不得随意更改操作
程序或跳过某些步骤。
6.在完成样品分析后,关闭LCMS仪器,并按照规定进行清理和维护。
以上就是LCMS的操作规程,供您参考。
具体操作可能因仪器型号和操作环境的不同而有所差异,建议参考仪器说明书或咨询专业技术人员。
LCMS(ESI)的基本原理及应用

D r y i n g g a s : 3 5 0 癈 , 1 2 l / m i n
2 . 9 6 4 507.1 485.1
4 0 0 6 0 0 8 0 0
150.12.
1 0 0 0
液滴产生噪音更大的谱图
适合电喷雾的样品
含杂原子的样品 通过引入可接受一个电荷的化合物 在溶液中是多电荷的样品 在溶液中是离子的化合物 适用于热喷雾的样品
质谱常用术语
1、分子离子(molecular ion) 自由基(radical)离子M•+。很活泼,易碎裂而产生广义的碎片离子 2、准分子离子(quasi-moleculanr ion) 由软电离技术产生的质子或其他阳离子加合离子以及去质子化或其他阴 离子加合离子。 3、碎片离子(fragment ion) 电离后具有过剩内能的分子离子能以多种方式裂解生成碎片离子。 4、多电荷离子(multiply-charged ion) 5、同位素离子(isotopic ion)
ESI的电离模式的选择原则
ESI(+): - 适应于碱性样品,含有仲氨或叔氨基时可优先考虑 - 适合酸性系统: 容易加合质子
ESI(-): - 适应于酸性样品,含氯、含溴和多个羟基时可尝试使用 - 在 碱性系统 中:易失去质子 对于本身 离子流较弱的样品建议 - 在酸性系统中做ESI(+)
ESI正、负离子模式常见离子:
正离子模式:
[M+H]+
[M+ Na]+ [M+K]+ [M+NH4]+ [2 M+H]+
[M+1]+
[M+23]+ [M+39]+ [2M+18]+ [2M+1]+
岛津LCMS-2020操作规程

岛津LCMS-2020操作规程岛津LCMS 2020 操作规程一、目的1.1 制定标准的岛津LCMS 2020操作规程。
1.2 确保操作人员规范操作,减少操作上的误差。
二、范围2.1 本规程适用于岛津LCMS 2020。
可定量和定性分析,此仪器暂适用于定性测试。
三、EHS3.1 高效液相色谱质谱联用仪应置于稳定的工作台上,避免震动、阳光直射和气流。
3.2 机械泵要防止共振现象,泵油要定时观察,防止泵油变黑或者降到最低刻线下。
低于刻度下限也要换油,注意要用同型号的油。
3.3 温度10-30℃(应恒定):相对湿度25%-75%;电源电压220±20V。
(通风良好,附近无明显的震动源。
断电对仪器的损害最大,应配备应急电源。
使用UPS电源断电时间不得超过6小时。
3.3 补充液氮应佩戴防冻手套及防护面罩,挂上醒目警示标牌。
对于液氮有泄漏现象要想安全部门说明。
如果液氮达不到所需要的压强,能够适当开启液氮增压阀。
(液氮压强不得超过1.5Mpa)。
3.4 样品管中含有易挥发性、具放射性有机溶剂,样品架及溶剂应密封、存放于通风柜内。
3.5 液质联用做样品途中(质谱仪HV绿灯亮),离质谱仪一米远。
3.6 质谱仪清洗前至少冷却0.5t。
四、名词解释调谐五、样品制备样品能够经过固液萃取,液液萃取(除非挥发性盐)。
样品的最低限度在10ppm,样品不要过浓,选用适当溶剂对其溶解(二氯甲烷,三氯甲烷,正己烷、二甲亚砜、苯、二甲基甲酰胺、四氢呋喃、乙醚等溶剂在流动相中的比例低于10%),流动相需经0.45um滤膜过滤,存在不溶物的样品需经0.22um滤膜过滤,流动相需经脱气处理,应符合紫外分光光度法度溶剂的要求。
六、仪器操作6.1 打开质谱仪,按下真空泵开关,抽真空6个小时以上直至[status]键常亮。
6.2 待抽完真空,根据实验目的,安装适宜的色谱柱。
打开LC 系统电源(A泵、B泵、UV检测器),待泵和检测器自检结束后,用乙腈/水体系平衡色谱柱30min, 更换所需流动相并排气泡(日常维护:用10%的异丙醇溶液冲洗柱塞杆,进样前清洗进样口和注射器)。
液质联用(LCMS)原理简析

液质联用(LCMS)原理简析1.质谱法质谱分析是先将物质离子化,按离子的质荷比分离,然后测量各种离子谱峰的强度而实现分析目的的一种分析方法。
质谱的样品一般要汽化,再离子化。
不纯的样品要用色谱和质谱联用仪,是通过色谱进样。
即色谱分离,质谱是色谱的检测器。
离子在电场和磁场的综合作用下,按照其质量数m和电荷数Z的比值(m/z,质荷比)大小依次排列成谱被记录下来,以检测器检测到的离子信号强度为纵坐标,离子质荷比为横坐标所作的条状图就是我们常见的质谱图。
2.质谱仪质谱仪由以下几部分组成数据及供电系统┏━━━━┳━━━━━╋━━━━━━┓进样系统离子源质量分析器检测接收器┗━━━━━╋━━━━━━┛真空系统质谱仪一般由进样系统、离子源、分析器、检测器组成。
还包括真空系统、电气系统和数据处理系统等辅助设备。
(1)离子源:使样品产生离子的装置叫离子源。
液质的离子源有ESI,APCI,APPI,统称大气压电离(API)源,实验室常用液质的离子源为ESI源。
电喷雾(ESI)的特点通常小分子得到[M+H]+ ]+,[M+Na]+ 或[M-H]-单电荷离子,生物大分子产生多电荷离子。
电喷雾电离是最软的电离技术,通常只产生分子离子峰,因此可直接测定混合物,并可测定热不稳定的极性化合物;其易形成多电荷离子的特性可分析蛋白质和DNA等生物大分子;通过调节离子源电压控制离子的碎裂(源内CID)得到化合物的部分结构。
(2)质量分析器: 由它将离子源产生的离子按m/z分开。
离子通过分析器后,按不同质荷比(M/Z)分开,将相同的M/Z离子聚焦在一起,组成质谱。
质量分析器有:磁场和电场、四极杆、离子阱、飞行时间质谱、傅立叶变换离子回旋共振等。
实验室目前液质的质量分析器类型:三重四极杆(QqQ):离子源→第一分析器→碰撞室→第二分析器→接收器MS1 MS2Q1 q2 Q3QqQ仪器可以方便的改变离子的动能,因此扫描速度快,体积小,常作为台式进入常规实验室,缺点是质量范围及分辨率有限,不能进行高分辨测定,只能做到单位质量分辨。
LCMS 简易操作流程

LCMS 簡易操作流程(一) 為提升分析數據品質,請依下列方式進行樣品前處理1、純化。
鹽類會干擾質譜訊號,故樣品含有鹽類請先去除。
2、樣品應適用溶於水、Methanol及Acetonitrile等溶劑之高極性樣品,並控制濃度為0.5-10μM。
(1m g樣品以20-60μl相容溶劑溶解後以50%ACN +0.1%FA水溶液稀釋50×300倍) 。
3、無法全溶且有懸浮固體時應過濾或離心取澄清液以免阻塞管路。
(二) 測樣流程1、以syringe取100μl 80%ACN手動推針沖洗管路。
再以syringe取150μl配樣溶液並將syringe固定在樣品推送台上。
2、質譜儀參數設定。
(1) Capillary voltages: 2.5-3.5kV。
(2) Cone voltage:10-60V。
3、開氮氣,啟動氮氣供應系統(選按Gas),穩定後按操作按鈕(operate)。
4、設定注射速度(50μl/min)啟動樣品注射(syringe pump),推針50μl後將流速緩降至10μl後按(Acqu)。
5、設定存檔參數:file name、tex t、range) ,按(Start)開始測background。
6、樣品測定時可即時監看。
測樣完成按(Standby)並停止樣品注射。
7、取150μl濃度0.5-10μM之樣品。
質量監測得穩定peak或推針50μl後將流速緩降至10μl,按下(Acqu) 設定存檔參數後按(Start)。
8、測樣完成後取150μl配樣溶液沖洗管路至無樣品殘留。
9、降溫至100℃以下時按停氣體供應系統,關氮氣,簽使用記錄本。
10、印出質譜圖。
關螢幕。
LCMS使用注意事项

LCMS使用注意事项LC-MS添加剂:酸:不能使用无机酸,否则会腐蚀离子源;可以推荐使用甲酸或乙酸,在阳离子模式下可以作为质子供体;碱:不能使用碱金属类强碱,否则会腐蚀离子源;推荐使用氨水,在阴离子模式下作为质子受体;表面活性剂:洗涤剂或其他表面活性剂会抑制离子化,不建议使用;三氟(氯)乙酸:可以提高色谱分辨率,但在质谱中对正负离子模式都有一定的离子抑制效应,用量应该小于0.1%(v/v);异丙醇(Isopropyl Alcohol):适合负离子模式,可以增强阴离子的形成,加入有机相(B)的量应该在10%左右;三乙胺(TEA)和三甲胺(TMA):适合负离子模式,加入可以增强阴离子的形成;难挥发性盐:HPLC中也尽量避免难挥发性盐的加入,如碱金属磷酸盐、硼酸盐、柠檬酸盐等;缓冲溶液:缓冲液浓度应低于20mM,尽量使用易挥发性盐如醋酸铵、甲酸铵;使用缓冲液时应该勤清洗ESI的加热毛细管以及API的stack;LC-MS不应该使用tris缓冲液,否则蛋白(多肽)质谱信号会受到tris盐的影响而使样品质谱信号降低,tris的质谱峰为m/z 122,243,265和327。
.单独做RP-HPLC可以允许蛋白样品中含有少量DMSO或者Tris。
常用的LC-MS溶剂:甲醇、乙腈、水、异丙醇、二氯甲烷、氯仿、己烷。
ESI中不同溶剂系统LC-MS的作用不同,产生的离子化总数不同:50/50 ACN/H2O 0.1% NH4OH50/50 MeOH/H2O50/50 ACN/H2O100 H2O100 MeOH100 ACN50/50 MeOH/H2O 1% Acetic50/50 MeOH/H2O 0.1% Formic 50/50 ACN/H2O 1% Acetic50/50 ACN/H2O 0.1% Formic50/50 MeOH/H2O 5mM NH4OAc 50/50 MeOH/H2O 10mM NH4OAc 50/50 MeOH/H2O 0.1% TFA50/50 MeOH/H2O 0.05% TFA50/50 MeOH/H2O 0.02% TFA50/50 ACN/H2O 0.1% TFA50/50 ACN/H2O 0.05% TFA50/50 ACN/H2O 0.02% TFA50/50 MeOH/H2O 0.1% NH4OH S o l v e n t S y s t e mCounts (protonated ion species)。
LCMS基础

LCMS基础LCMS是有机合成中重要的分析工具,解析LCMS谱图也是一项基本技能。
LCMS基本原理和特性1)LCMS的特性:是HPLC和MS的结合,有两者的功能,有没有两者精确。
2)流动相方法:常见0-30,0-60,10-80,30-90四种方法,0,10,30都是指乙腈的含量,乙腈含量越大,流动相极性越小,出峰越靠前。
3)正离子源适用于碱性化合物:含氮化合物更容易粘附氢正离子,在正离子源中容易出分子离子峰。
负离子源适合酸性化合物:酸性化合物更容易轰击掉氢正离子,如酸,酚类化合物。
看LCMS步骤1)先看MS部分, 看有没有所要离子峰, 并且要看清楚该化合物是否有MS信号, 是否掩盖周围的峰。
2)再看HPLC部分, 看含量有多少, 并且要看清楚该化合物是否有强的HPLC信号, 是否掩盖周围的峰。
3) 两者结合起来看, 推测反应进行的程度和反应产生的杂质。
常见加合离子峰[M+Na]+ = [M+23]+ 加钠离子;[M+K]+ = [M+39]+加钾离子;[M+NH4]+ = [M+18]+加铵离子;[M +H +H2O]+ = [M+19]+加水;[M+X]+ 这里X 是指溶剂缓冲液中的阳离子;[M+H+Solvent]+溶剂加合峰,如[M+H+CH3CN]+ =[M+42 ]+ 是CH3CN加合离子,[M+H+CH3OH]+ = [M+33 ]+ 是CH3OH加合离子;减峰:M-56(脱叔丁基)和M-100(脱Boc),M-16(脱NH3)和M-17(脱水)以及M+2/2(比较常见),其他少见。
同位素峰特别注意精确分子量和摩尔分子量的区别常见氯和溴同位素的表现: 一个氯峰高比M+2/M=1:3;一个溴为峰高比M+2/M=1:1;多个同位素的表现可以用Chemdraw精确模拟。
注意事项1.在LCMS报告中,MS响应强的组分有可能掩盖MS响应弱的组分,可通过提取离子流或扣除背景等方式进行判断,LCMS报告必须将LC和MS两部分结合,相互佐证。
LCMS及制备HPLC使用教程

Zewei Wang
流动相的准备 样品的准备
日常维护
常规LCMS操作 数据分析
注意事项
流动相的准备
常用流动相:水、乙腈、甲醇 常用添加剂:甲酸、乙酸、三氟乙酸;醋酸铵;氨水
溶剂必须为HPLC级,超纯水(18 Ω.m);普通有机溶剂须过滤 添加剂须为HPLC级,一般添加0.1%
checktune: MSD Tune >>> OK >>> Tune >>> checktune
常规LCMS操作
单个样品时,选方法 5-100% Common.M >>> RunControl >>> Sample Info,填好后待压 力平稳再点击开始。初始压力100Bar左右,当100% MeCN时,压力50Bar左右。
Dimer 2M-H [-1] 2M+Cl- [35.37] 2M+HCOO- [45] 2M+TFA- [113]
注意事项
• 样品一定要用滤膜过滤,此外样品中最好不要有三乙胺!(三乙胺在LCMS中 残留严重),如果含有三乙胺,请先用酸处理下样品再送样。 • LCMS中的方法请不要随意更改!
• 液相小瓶盖用过2-3次后及时更换新的。样品瓶请勿用洗衣粉、洗洁精等表面
2)用滴管蘸取 一下(不是吸取 一滴),残留在 管壁上的用溶剂 稀释至0.5 mL
PS: 如果样品不溶于MeOH或 MeCN,可用DMSO溶解,但尽 量不用DMSO
日常维护
• 流动相须勤换,2-3天换一次,否则放置过久,水相易长菌,乙腈易聚合。 可分别在周一、三、五更换。 • 每周清洗一次喷雾室,可在周一更换流动相的间隙进行 • 每周做一次checktune,校准MS。可在清洗完MS后进行。
LCMs适用的溶剂

LCMS适用的溶剂通常根据目标化合物的溶解性和与LCMS中使用的各种电离技术的兼容性选择溶剂。
在ESI和其它常压电离技术中,溶剂的挥发性和给质子的能力很重要。
使用的主要质子溶剂像甲醇和其与水的混合物,比如1:1的甲醇水,或1:1的乙腈水(甲醇水混合物增加的粘度超过了纯净的水或甲醇,因为发生了放热反应)。
当使用100%的水时,水相对低的蒸汽压可能对灵敏度不利。
通过添加挥发性有机溶剂,降低表面张力,能提高灵敏度。
表面活性剂,虽然能增加从喷雾液滴中释放出离子,但因其较高的质子亲和力,可能降低灵敏度。
质子惰性的共溶剂,像10%DMSO水溶液和异丙醇,对一些化合物,能提高溶解度。
在确保被测物比溶剂更偏碱性的前提下,甲酸通常以较低的水平(0.1%)添加,便于电离。
一些酸,即使是很少量,像TFA,也可能限制灵敏度,但对增加一些化合物的溶解度可能是必需的。
在ESI电离模式中,缓冲液和盐(Na+,k+和磷酸盐)可降低蒸汽压,导致信号减弱。
液滴的表面张力增加,挥发性降低,可用相对更易挥发的缓冲液,像醋酸铵,形成弱酸-碱对,进行补救。
选择溶剂需要考虑的问题- 对于比溶剂更偏碱性的分子,气相中的溶剂将限制ESI电离。
光电离除外(不是酸碱电离),但受溶剂调节。
- 从电离区域去除溶剂和水蒸气,增加在大气压下电离化合物的种类。
- 相对于样品或溶解在液体中的目标被测物减少液体体积,将提高ESI的性能(如,使用较低流速)。
- 有用的溶剂- 可接受的添加物- 非挥发性盐(磷酸盐,硼酸盐,柠檬酸盐等等)会在离子源沉积,阻塞毛细管,因此需要更多的清洗和维护操作。
现代离子源设计,相比以往的设计,能较好地处理非挥发性物质。
- 表面活性试剂(表面活性剂/去垢剂)抑制电喷雾电离的效率- 无机酸具有腐蚀性- 三氟乙酸(TFA)超过0.01%的水平时,会在一定程度上抑制阳离子电喷雾。
大大的抑制了阴离子电喷雾。
- 三乙基胺(TEA)高PA(232千卡/摩尔)在m/z102处,产生强[M+H]离子。
LC MS 溶剂 高纯度溶剂 说明书

产品资料用于LC/MS分析的溶剂和溶剂混合物J.T.Baker®高纯度LC/MS溶剂产品系列日益扩大,不断满足您的需求。
三十多年来,Mallinckrodt Baker公司生成的高纯度溶剂已经成为准确性高、重现性好、高分辨分析效果产品的标准。
我们将蒸馏、溶剂混合、提纯、质量控制、分析监测以及包装方面的专知结合起来,生产用于LC/UV法和LC/MS法用途优化和功能检测的溶剂。
一致性高、重现性好的性能可供选择的高品质J.T.Baker®溶剂和溶剂混合物: • 批次间一致性 • 严格的UV技术规范 • 功能检测 LC/UV和LC/MS用 • 采用0.2μm过滤器过滤除去颗粒杂质。
• 便捷的包装方式经济有效、节省时间凭借下列优点可提高您实验室操作效率: • 无需清洗玻璃器具 • 无需对腐蚀性酸进行检测 • 无需采购过量试剂 • 色谱柱平均寿命更长减少污染风险用于LC/UV 和LC/MS开发和检测产品的特性: • 蒸发后残渣最少 • 金属加合物生成最少 • 有机污染最少 • 离子抑制作用最小LC/MS 溶高纯度溶9829-029829-039828-039830-029830-039827-039831-029831-03LC/M S LC/M S LC/M S LC/M S LC/M S LC/M S LC/M S LC/M S1L 4L 4L 1L 4L 4L 1L 4L9832-029832-039835-029835-039837-029837-039839-029839-039838-029838-039840-029840-039834-029834-039836-029836-03C/M S C/M S LC/M S LC/M S C/M S C/M S LC/M S LC/M S C/M S C/M S LC/M S LC/M S C/M S C/M S LC/M S LC/M S1L 4L 1L 4L 1L 4L 1L 4L 1L 4L 1L 4L 1L 4L 1L 4L乙腈乙腈乙酸乙酯甲醇甲醇2-丙醇水水乙腈-0.1%甲酸乙腈-0.1%甲酸乙腈-0.1%三氟乙酸乙腈-0.1%三氟乙酸水-0.5%甲酸水-0.5%甲酸水-0.5%三氟乙酸水-0.5%三氟乙酸水-0.15%甲酸水-0.15%甲酸水-0.15%三氟乙酸水-0.15%三氟乙酸水-0.1%甲酸水-0.1%甲酸水-0.1%三氟乙酸水-0.1%三氟乙酸用LC/MS分析的溶剂和溶剂混合物除非另有说明,本目录中商标所有权均归Mallinckrodt Baker公司所有。
LCMS抽提protocol

编号步骤说明1 采集样品注意采样时间和采样位置乙酸乙酯一定要没过样品,最多不能超过三角瓶或试管2/3 2 果实:称取10-11g切碎后放入100ml三角瓶,加入50ml乙酸乙酯。
叶片:200mg, 加入20ml乙酸乙酯。
3 果实用玻璃棒压碎样品,尽量压碎;叶片用液氮研磨至粉末4 上超声破碎仪,10min一次,共3次。
每次结束后再次用玻璃棒压碎。
若3次之后还不够碎,可以再10min过程中用塞子塞好三角瓶或试管5 用滤斗进行过滤6 使用旋蒸仪蒸干液体。
使用19/26的50ml旋蒸瓶,和29/33的中继头。
7 用1ml注射器吸取1ml甲醇,加入50ml旋蒸瓶,通过超声波使其充分溶解。
8 用1ml注射器吸取已溶解好的液体,通过0.22um滤膜进行过滤,直接入样品瓶。
将样品瓶盖好盖子。
9 萃取好的样品可以在4℃进行保存注:除了对温度进行具体说明外,未具体说明温度部分,都为常温。
编号步骤说明1 检查顶部瓶内液体是否足够,需要及时补充,补充的液体需要经0.22um的滤膜过滤。
2 开启机器,分子量在104,电流在0.1时,仪器比较干净,不容易有误差。
TraceFinder:通常国家质检需要的,不能修改参数Xcalibur:实验室通常选用,可以修改参数3 洗管子100% —50%(1-2min)—10%(用于平衡压力,非常重要)这里师兄建议选用A、D通道。
这两个通道使用甲醇洗管压力稳定在150左右比较好。
(中间箱子指示灯绿)4 打开XApps中的sequence setup开始录入并摆放样品Sample type:选用了unknowFilename:输入样品名和编号Path:储存路径Inst meth:使用方法Position:对应盘的坐标位置。
A1,B1,C2之类的。
红色开头R,蓝色B,以此类推。
Inj vol:上样量。
默认为20ul。
后面参数默认。
注:1.为防止堵管,在摆放样品时,应摇晃并对光查看是否有沉淀。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
LCMS适用的溶剂
通常根据目标化合物的溶解性和与LCMS中使用的各种电离技术的兼容性选择溶剂。
在ESI和其它常压电离技术中,溶剂的挥发性和给质子的能力很重要。
使用的主要质子溶剂像甲醇和其与水的混合物,比如1:1的甲醇水,或1:1的乙腈水(甲醇水混合物增加的粘度超过了纯净的水或甲醇,因为发生了放热反应)。
当使用100%的水时,水相对低的蒸汽压可能对灵敏度不利。
通过添加挥发性有机溶剂,降低表面张力,能提高灵敏度。
表面活性剂,虽然能增加从喷雾液滴中释放出离子,但因其较高的质子亲和力,可能降低灵敏度。
质子惰性的共溶剂,像10%DMSO水溶液和异丙醇,对一些化合物,能提高溶解度。
在确保被测物比溶剂更偏碱性的前提下,甲酸通常以较低的水平(0.1%)添加,便于电离。
一些酸,即使是很少量,像TFA,也可能限制灵敏度,但对增加一些化合物的溶解度可能是必需的。
在ESI电离模式中,缓冲液和盐(Na+,k+和磷酸盐)可降低蒸汽压,导致信号减弱。
液滴的表面张力增加,挥发性降低,可用相对更易挥发的缓冲液,像醋酸铵,形成弱酸-碱对,进行补救。
选择溶剂需要考虑的问题
- 对于比溶剂更偏碱性的分子,气相中的溶剂将限制ESI电离。
光电离除外(不是酸碱电离),但受溶剂调节。
- 从电离区域去除溶剂和水蒸气,增加在大气压下电离化合物的种类。
- 相对于样品或溶解在液体中的目标被测物减少液体体积,将提高ESI的性能(如,使用较低流速)。
- 有用的溶剂
- 可接受的添加物
- 非挥发性盐(磷酸盐,硼酸盐,柠檬酸盐等等)
会在离子源沉积,阻塞毛细管,因此需要更多的清洗和维护操作。
现代离子源设计,相比以往的设计,能较好地处理非挥发性物质。
- 表面活性试剂(表面活性剂/去垢剂)抑制电喷雾电离的效率
- 无机酸具有腐蚀性
- 三氟乙酸(TFA)
超过0.01%的水平时,会在一定程度上抑制阳离子电喷雾。
大大的抑制了阴离子电喷雾。
- 三乙基胺(TEA)
高PA(232千卡/摩尔)在m/z102处,产生强[M+H]离子。
抑制弱碱性化合物阳离子的电喷雾。
- 四氢呋喃(THF)
100%的THF具有高可燃性,因此APCI和绝大多数接口技术使用氮气作为喷雾气。
(使用空气可能引起爆炸危险。
)会与PEEK®管反应。
离子抑制
离子抑制是质谱学家使用ESI作为电离方式时面对的比较多的具体问题之一。
2001年,美国食品药品管理局(美国FDA)出版了工业生物分析有效方法指南(联邦注册号,66,100,28526),表明确保分析质量的要求是不能妥协的。
该条款指明了可用于评估离子抑制是否存在的几个实验方案。
将基质提取后加标样品中的被测物的多反应监测(MRM)响应(峰面积或峰高),与直接溶于纯流动相的被测物的多反应监测响应进行对比。
基质中被测物的信号比在纯溶剂中的地,表明基质中存在干扰物质。
C.Mallet等发表的文章表明,在色谱图中被测物(和内标物)基质效应的存在。
试验人员使用三通装置,将含有目标被测物及其内标物的溶液以连续进样方式引入质谱,将空白基质样品抽取物通过LC系统自动进样引入质谱后,连续的基线出现下降,表明连续进样的被测物的电离受到抑制,因为基质中有干扰物质存在。
柱化学杂交柱化学和直径低于2微米的高选择性颗粒的使用,是色谱柱技术的一项革命性进步。
这种杂交化学性质不依赖于可能引起离子抑制的流动相的改性,并且增加了颗粒的选择性。
超高压力LC与传统HPLC
通常称为UHPLC(超高压液相色谱),J.Jorgenson教授(北卡罗来纳大学)的工作近来实现了这项技术的商业化,UHPLC为增加常规LCMS分析的信息量提供了可能。
Waters公司对这项技术进行了商业化,称为UPLC技术,或超高效液相色谱,与HPLC相比,UPLC的峰容量增加,对HPLC中形成较宽峰的共流出物,可以在UPLC能够实现分离。
将色谱峰形(通常条件下)浓缩成2秒或更短的谱带,为灵敏度的提高提供了可
能,有利于质谱响应,改善信噪比。
UPLC技术的概念改变了传统LC分离实践中建立起来的一些熟知的参数,比如流速、颗粒大小,甚至对范第姆特曲线的理解。
其工作压力从大约2000psi增加到高达20000psi,固定相颗粒直径小于2μm,接近1969年John Knox在其"Knox方程式"中理论极限。
一些伴随出现的问题,象增大机械压力和过大的热效应等,促进了MS性能的提高,也稍微偏离了对理论结果的直觉。
图20:由范第姆特曲线描述的线速度的变化,导致分离效能变化的趋势。
由图可知,1.7μm直径颗粒色谱柱效能更好,且不随流速变化而改变。
虽然所有色谱柱的证据表明在极端低线速度下会降低柱效,但是对HPLC我们所熟知的一个事实是,填料直径越小分离性能越好,并且随着线速度的增加,性能较少受到影响。
在现在被称为‘传统'HPLC分离与UPLC分离的比较中,可以认为是一个关于技术怎样重新定义实验设计方法的事例。
不但在原理上重新定义分离技术(速度快近4倍),而且增强了选择性,揭示了一些常规HPLC无法看清的细节,比如在图中的咪达唑仑的代谢物。
提高的分离度显示出葡萄糖苷酸的二次级代谢物,m/z=548.125。
图21:技术的提高通常能揭示更多未知细节,比如单一葡萄糖苷酸代谢物的色谱峰。
咪达唑仑的氧化代谢是由肝脏细胞色素P450蛋白催化的。
在上述药物结构式中,代谢氧化[羟化]最可能发生的分子内主要位点已使用红星标出。
1对咪达唑仑在胆汁中的代谢物,使用HPLC/MS和UPLC/MS 比较分析,发现在HPLC上有一名义m/z为548的色谱峰。
但在UPLC/MS中,则分离出一对色谱峰,每个的准确质量值相同,m/z为548.1248。
实测裂解确认这两个代谢物均为葡萄糖苷酸化代谢物。
作者给出了完全
分离的两种物质准确质量对应的经验分子式,表明咪达唑仑在标示的位点发生双羟化,然后可能在一个位点发生O-甲基化,另一个与葡萄糖醛酸结合,反之亦然。