手把手教你构建系统发育树
系统发育树构建教程(PHYLIP)

系统发育树构建教程(PHYLIP)PHYLIP网址:/phylip.html(一)序列的前期准备1.用ENTREZ或SRS搜索同源DNA/蛋白质序列(same sequence in different organisms) 2.用CLUSTALX进行多条序列比对,在output format option选定PHY格式,构建进化树需要这个phy文件。
Figure 4.1 用clustalx进行多条序列比对3.解压缩phylip-3.68.exe,得到三个文件夹,doc文件夹里是关于所有PHYLIP子程序的使用说明,exe文件夹里是直接可以使用的各个子程序,src文件夹里是所有程序的源文件。
4.打开exe文件夹,双击SEQBOOTt子程序(SEQBOOT是一个利用bootstrap方法产生伪样本的程序),输入刚刚生成的phy文件的路径,点击enter。
5.所有PHYLIP程序默认的输入文件名为infile, 输出文件名为outfile。
如果在exe文件夹里找不到默认的输入文件,会提示can’t find input file “infile”。
Figure 4.2 seqboot程序起始界面6.进入程序参数选择页面(Figure 4.3)。
第一列中的D、J、%、B、R、W、C、S等代表可选的参数。
想改变哪个参数,就键入此参数对应的字母,并点击回车键,对应参数将会发生改变。
当我们设置好所有参数后,(这里我们可以不做任何修改),键入Y,按回车。
此时程序询问“random numbe r seed? <must be odd>”,这是询问生成随机数的种子是多少,输入一个4N+1的数,点击回车程序开始运行,输出结果到文件outfile,保存在当前文件夹里。
.Figure 4.3 seqboot程序参数选择页面主要参数解释:D: 数据类型,有Molecular sequence、discrete morphology、restriction sites和gene frequencies4个选项。
系统发育树构建PPT(共10张PPT)

系统发育树构建软件 窗Mo口de上lt面es的t&下Mr拉M菜od单elt可es让t(碱你基选替择换传模统型多筛重选比软对件和)轮廓比对需要的所有选项。
• BEAST 名CLUS称T:ALX-Un是cuCltLuUreSdTAbaLc多te重riu序m列cl比on对e 程YU序2的01WH1in0dows版本。
• 序列长度:353 • 相 似 比: 99%
• 核酸序列 • 分类地位
打开软件clustalx
• CLUSTALX-是CLUSTAL多重序列比对程序的Windows 版本。Clustal X为进行多重序列和轮廓比对和分析结果提 供一个整体的环境。 序列将显示屏幕的窗口中。采用多色彩的模式可以在比 对中加亮保守区的特征。窗口上面的下拉菜单可让你选 择传统多重比对和轮廓比对需要的所有选项。
D并is用ta系nc统e-进ba化se树d来m概eth括od生s物基间于的距这离种的亲方缘法关系。
• Figtree (树形显示软件) C窗lu口st上al面x比的对下结拉果菜是单构可建让系你统选发择育传树统的多前重提比对和轮廓比对需要的所有选项。
名Maxim称u:m paUrnscimulotunrye(dMbPa)最cte大riu简m约c法lone YU201H10
3
系统进化树
结点:表示一个分类单元。
进ቤተ መጻሕፍቲ ባይዱ支:两种以上生物(DNA序列 )及其祖先组成的树枝。
进化分支长度:用数值表示的进化枝 的变化程度(遗传距离) 距离标尺:生物体或序列之间差异的的 数字尺度。
根:所有分类的共同祖先。
外群:一个或多个无可争议的同源物 种,与分析序列相关且具有适当的亲缘 根 关系
MEGA 软件——系统发育树构建方法

• 双击图标
,
• 下载下来的序列片段保存文件为FASTA格式,打开方式为TXT格式。 • 将Blast对比后所Download的序列筛选后,构建系统发育进化树。 • 构建系统发育树需要测序所得序列1个,Blast对比得出序列5-10个, 构建出的为单枝系统发育进化树。通过这个对比可以确定出所测 定的序列最相似物种,当相似度为99%甚至100%时,基本可以确 定所测定的基因序列所属物种。
ME待测PCR产物送测序后,一个星期左右会得到生物 公司发的邮件,里面包含测序结果的附件,下载后得到文件压缩 包 ,将文件包解压,可以看到文件夹里有文件
• 测序公司会提供一款解读软件Chromas,免安装类型,能够直接 打开测序结果并通过软件直接进入NCBI数据库进行Blast搜索。
• 可培养真菌可以选定对比物种种属后构建大型系统发育进化树, 不可培养真菌则可直接构建系统发育进化树。
• 双击
后打开MEGA软件
手把手教你构建系统进化树(2021年)

97 NR 116489.1 Pseudomonas stutzeri strain VKM B-975 16S ribosomal RNA partial sequence NR 113652.1 Pseudomonas stutzeri strain NBRC 14165 16S ribosomal RNA partial sequence
进化分析流程
测序组装
• 将克隆扩增测序得到的基因进行测序。
Blast
• 比对找到相似度最高的几个基因,将这几个基因的 序列(Fasta格式文件)下载下来,整合在一个*.txt 文档中。
比对序列
• 用Mega 7.0的ClustalW做多序列联配,比对结果用*.meg格式 保存。或者用Clustal X软件进行比对,比对结果保存为*.aln, 再用Mega 转化为*.meg格式。
DNA→ DNA
ezbiocloud https:///identify
cDNA→蛋 白质
蛋白质 →cDNA
蛋白质→蛋白 质
NCBI
输入测序组装后的序列
ezbiocloud
输入序列名称 输入测序组装后的序列
比对序列
MEGA可识别fasta格式文件比对前将xxx.txt 重命名为xxx.fasta
构建系统进化树
1) 在构建系统树时,使用了Bootstrap法进行检验。在做Bootstrap时,以原序列为蓝本随机重组生成新的序列, 重复估算模型。如果原序列计算得到的分枝在新Bootstrap中依然频繁出现,则该分枝的可信度高。分枝在 Bootstrap中出现的频率就是表征分枝可信度的参数。 2) Original Tree是应用估算模型形成的最优系统树。在Original Tree上有计算得到的距离数据,可以表征两个基 因的亲缘远近;MEGA形成的Original Tree上也有频率参数,实际来自Bootstrap Consensus Tree的对应分枝。 3) Bootstrap Consensus Tree 是很多次Bootstrap得到的平均结果,它不包含进化距离信息(在设置View时无法 调用,也没有意义),分枝上的数字代表该分枝的频率参数。另外,它的拓扑结构也可能与Original Tree很不相同。
手把手教你构建系统发育树.

Extraction of chromosomal DNA PCR amplification of the16S rRNA gene
Assmbly of 16S rRNA gene fragments
Obtain closely related sequences
TOOLS
NCBI RDP / http://www.bacterio.cict.fr/
EzTaxon http://147.47.212.35:8080/
NCBI homepage /
YOU NEED A REGISTRATION NUMBER TO LOG IN THIS PAGE FIRST
Nocardiopsis kunsanensis DSM 44524 AF195412
99 17 27 80 81
Nocardiopsis aegyptia DSM 44442T AJ539401 Nocardiopsis lucentensis DSM 44048T X97888 Nocardiopsis dassonvillei subsp. dassonvillei DSM 43111T X97886 Nocardiopsis dassonvillei subsp. albirubida DSM 40465T X97882 Nocardiopsis synnemataformans DSM 44143T Y13593 Nocardiopsis quinghaiensis DSM 44739T EF597511
Phylogenetic analysis & Bootstrap analysis
TOOLS
初二生物系统发生树构建方法

初二生物系统发生树构建方法生物系统发生树是一种用来描述生物进化关系的图形工具,它可以帮助我们理解不同物种之间的亲缘关系和进化历史。
构建生物系统发生树需要收集大量的生物学数据,并运用一系列的分析方法。
本文将介绍初二生物系统发生树构建的方法。
一、确定研究对象要构建生物系统发生树,首先需要确定研究对象。
可以选择一组具有亲缘关系的物种作为研究对象,比如不同种类的昆虫、鱼类或者植物。
在选择研究对象时,需要考虑它们的进化关系已经被广泛研究并且有可靠的分类信息可供参考。
二、收集分类信息为了构建生物系统发生树,我们需要收集各个物种的分类信息。
这包括它们的科、属、种等分类级别。
分类信息可以通过查阅专业的生物学书籍和数据库获得。
三、收集形态和分子数据除了分类信息,形态和分子数据是构建生物系统发生树的重要依据。
通过观察物种的外部形态特征,比如体型、花朵形状、翅膀结构等,可以推测它们的亲缘关系。
此外,通过分析物种的DNA序列,尤其是核酸或蛋白质序列,可以揭示它们的遗传关系。
因此,我们需要收集和记录物种的形态和分子数据。
四、进行系统发生树分析系统发生学是构建生物系统发生树的主要工具。
它利用形态和分子数据进行计算,通过比较不同物种之间的相似性和差异性,推断它们之间的进化关系。
常用的系统发生学方法包括距离法、最大简约法和最大似然法等。
在进行系统发生树分析前,需要选择合适的方法,并准备好相关的计算软件。
五、树构建在进行系统发生树分析后,我们可以通过计算机软件生成生物系统发生树。
树的构建过程会考虑到物种之间的进化距离和分支长度等因素。
树的构建可以使用专业的系统发生学软件,如MEGA和PHYLIP 等。
生成的树结构可以通过图像输出和基因组数据展示。
六、树解读和分析生成生物系统发生树后,我们需要对树进行解读和分析。
树的分枝结构和分枝长度等特征可以帮助我们理解物种之间的进化关系。
同时,树还可以用来预测物种的共同祖先和演化路径等信息。
通过对树的分析,我们可以深入理解生物的进化历史和多样性。
系统发育树构建的三种方法

系统发育树构建的三种方法
1. 距离法(Distance Method):该方法将各个物种之间的差异转化为距离值,并根据这些距离值构建系统发育树。
距离可以基于基因序列或形态特征等进行计算。
该方法不考虑进化模式和序列的进化过程,仅提供基于相似性的分支结构。
2. 最大简约法(Maximum Parsimony):该方法基于最小进化原则,即最可能的树是具有最少次数的进化事件的树。
它寻求在进化树上使得进化事件(如插入、缺失、突变)的次数最少的树。
该方法是需要较多计算的方法,但树的建立结果更加准确。
3. 最大似然法(Maximum Likelihood):该方法也是基于最小进化原则,但它考虑进化模式和序列的进化过程,并将最可能的进化树视为产生的序列数据的最大概率估计。
该方法需要更复杂的计算,但对于数据信息的准确推断较好。
叙述系统发育树的构建过程

叙述系统发育树的构建过程嘿,咱今儿就来讲讲系统发育树的构建过程,这可有意思啦!你看啊,系统发育树就像是一棵大树,它的枝桠代表着各种生物之间的关系。
那怎么把这棵大树给“种”出来呢?首先得有一堆生物的数据呀,就像盖房子得有砖头一样。
这些数据可以是各种各样的,比如基因序列啦、形态特征啦等等。
然后呢,就开始比对这些数据,这就好比把不同的砖头摆在一起,看看哪些相似,哪些不同。
接着,就根据这些比对的结果来确定它们之间的亲缘关系。
这就好像在给砖头们找它们的“家族”一样,哪些是近亲,哪些是远亲。
这可不是一件容易的事儿啊,得非常仔细地去分析。
然后呢,把这些亲缘关系用一种特别的方式表示出来,就像把砖头们按照一定的规律摆好,形成一个结构。
这个结构慢慢就变成了系统发育树的雏形。
这时候,就像是在给大树修剪枝叶一样,要对这个雏形进行调整和优化。
要确保每个部分都放对了位置,不能有差错。
最后,一棵完整的系统发育树就出来啦!哇塞,你想想看,通过这么多复杂的步骤,终于把生物之间的关系给清楚地呈现出来了,这难道不神奇吗?你说,这系统发育树构建的过程,像不像一个艺术家在精心雕琢一件作品?每一个细节都要处理好,才能呈现出完美的结果。
而且啊,这可不是一次性就能完成的事儿,得反复地去研究、去调整。
你再想想,要是没有系统发育树,我们怎么能知道各种生物之间有着这样那样的联系呢?我们怎么能更好地理解生命的奥秘呢?所以啊,这个构建过程虽然复杂,但真的超级重要呢!咱平时生活中也有类似的情况呀,比如说搭积木,不也是一块一块地搭起来,最后形成一个完整的造型嘛。
这和构建系统发育树不是有点像嘛!总之呢,系统发育树的构建过程就是这么神奇又有趣,它让我们对生物的世界有了更深的了解和认识。
这可真是一项伟大的工作啊!你难道不这么觉得吗?。
构建系统发育树的方法

构建系统发育树的方法
构建系统发育树的方法有多种,其中常用的方法包括:
1. 形态学方法:该方法主要利用物种形态特征的相似性进行分类和构建系统发育树。
通过比较物种的形态特征,如外部形态、骨骼结构等,可以确定物种间的相似程度,并将相似的物种进行分类和构建系统发育树。
2. 分子生物学方法:该方法利用物种的遗传信息进行分类和构建系统发育树。
通过分析物种的DNA序列或蛋白质序列,可以确定物种间的遗传关系,并将不同的物种进行分类和构建系统发育树。
3. 同工酶分析:该方法利用物种的同工酶差异进行分类和构建系统发育树。
同工酶是由不同基因或等位基因编码的酶,通过对物种中同工酶的电泳分析,可以确定物种间的同工酶差异,从而进行分类和构建系统发育树。
4. 基因组学方法:该方法利用物种的整个基因组信息进行分类和构建系统发育树。
通过对物种的基因组序列进行比较和分析,可以确定物种间的遗传关系,并将不同物种进行分类和构建系统发育树。
以上方法通常会结合使用,以获得更准确和可靠的系统发育树。
此外,还有其他一些辅助分析方法,如模型选择和统计分析等,可以进一步优化和验证系统发育
树的构建结果。
系统发育树构建

系统发育树的主要目的是揭示物种的进化历程,帮助科学家理解生物多样性的 起源、物种的演化路径以及生物进化的规律。
系统发育树的基本组成
01
02
03
04
节点
代表物种或共同祖先,节点间 的连线表示物种间的亲缘关系
。
分支
连接节点间的线段,代表物种 间的进化关系。
叶节点
代表可观测的物种,是系统发 育树的末端节点。
WENKU DESIGN
树的解读与注释
根部的位置
系统发育树的根部通常代表进化关系中最为原始的物种。
分支长度
分支长度可以反映物种之间的进化距离,较长的分支表示较大的 进化距离。
节点注释
节点注释包括该节点的物种名称、化石记录等信息,有助于理解 该节点在进化历史中的位置。
系统发育关系推断
同源性分析
通过比较不同物种的基因或蛋白质序 列,确定它们之间的同源性,进而推 断它们之间的进化关系。
03
通过比较不同物种在特定环境下的适应性特征,可以分析这些
特征的进化起源和演化过程。
PART 05
系统发育树的应用
REPORTING
WENKU DESIGN
物种分类与系统发生学研究
物种鉴定
系统发育树可以帮助确定物种间的亲缘关系,从而对未知物种进 行鉴定和分类。
生物多样性研究
通过构建系统发育树,可以了解生物多样性的起源、演化和分布, 为保护和利用生物资源提供科学依据。
分子钟假设
基于分子钟假设,通过比较不同物种 基因或蛋白质序列的进化速率,可以 推断它们之间的相对进化时间。
物种进化历史分析
物种起源与分化
01
系统发育树揭示了物种的起源和分化过程,有助于理解物种多
MEGA 系列软件系统发育树构建方法

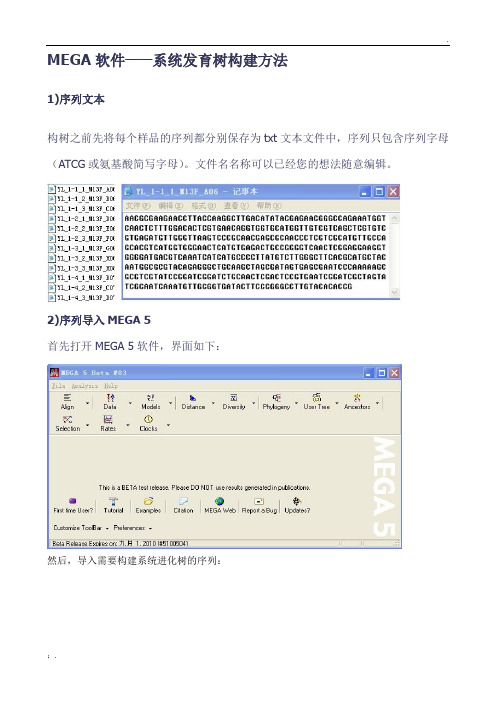
MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都保存在同一个txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑(不能有中文)。
保存为fasta格式2)右键点击fasta文件,打开方式,mega3、全选,点击alignment,algin by culstx(按钮W),OK4、关闭此窗口,点击Yes保存5、再次点击Yes保存,6、点击cancel取消7、选择是否为编码蛋白质的核酸序列8、选择是否用mega打开文件9、点击YES,激活mega,此时mega的菜单栏与刚开始打开的菜单栏有区别。
10、系统发育树构建原理不讲了,此处以构建NJ树为例。
点击工具栏上的phylogeny,construct phylogeny,neighbor joining (NJ).出现如下界面(注意几个绿颜色的小方块):点击第一个小绿方块,选择,小绿方块会变成四个点的省略号,再点击出现如下页面:选择Bootstrap,后面的replication改为1000,点击对勾。
然后点击第三个小绿方块,这个时候对于蛋白质序列以及DNA序列,两者模型的选择是不同的。
对于蛋白质的序列,多选择Poisson Correction (泊松修正)这一模型。
而对于核酸序列,多选择Kimura 2-parameter (Kimura-2参数) 模型。
所有设置完毕之后,点击compute,雏形的树就出来了:可以对此树做出一些修改,比如线条粗细,树的形状等等,此处自己多试试。
6)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键,就可以对文字的字体大小,倾斜等做出修饰。
见下图:PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
系统发育树的详细构建方法

构建系统发育树需要注意的几个问题1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。
2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。
3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。
4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。
5 枝长可以用来表示类间的真实进化距离。
6 重要的是理解系统发育分析中的计算能力的限制。
任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。
7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。
然而,有些方法可以检测系统发育树检测的可靠性。
第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。
分子进化研究的基本方法对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。
表型的(phenetic)和遗传的(cladistic)数据有着明显差异。
Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。
这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。
表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。
进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。
文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。
系统进化树分有根(rooted)和无根(unrooted)树。
系统发育进化树构建

系统发育进化树构建系统发育进化树(Phylogenetic tree)是一种用于描述物种或群体之间进化关系的图形表示。
通过构建系统发育进化树,我们可以了解不同物种之间的亲缘关系,以及它们的共同祖先。
本文将介绍系统发育进化树的构建方法和其在生物学领域中的应用。
一、系统发育进化树的构建方法1. 选择合适的基因或序列:构建系统发育进化树需要选择适当的基因或序列进行分析。
常用的基因包括核糖体RNA(rRNA)和线粒体DNA(mtDNA)等。
2. 收集物种样本:从不同物种中收集样本,并提取相应的基因或序列。
3. 序列比对:将收集到的序列进行比对,找出它们之间的相同和差异。
4. 构建进化模型:根据序列比对的结果,选择适当的进化模型,如最大似然法或贝叶斯推断等。
5. 构建进化树:利用选定的进化模型,根据序列的相似性和差异性,构建系统发育进化树。
二、系统发育进化树的应用1. 物种分类:系统发育进化树可用于物种分类,帮助我们理解不同物种之间的亲缘关系。
通过比较进化树上的分支长度和节点位置,我们可以判断物种之间的相似性和差异性。
2. 进化研究:系统发育进化树可用于研究物种的进化历史和进化速率。
通过比较不同物种之间的进化树,我们可以了解它们的共同祖先以及它们之间的演化路径。
3. 分子演化研究:系统发育进化树在分子演化研究中起着重要的作用。
通过比较不同物种的基因或序列,我们可以推断它们的演化历史和演化速率。
4. 物种保护:系统发育进化树可用于指导物种保护工作。
通过研究物种的进化关系,我们可以了解哪些物种是濒危物种或有特殊保护需求的物种。
5. 药物开发:系统发育进化树可用于药物开发。
通过比较不同物种的基因或序列,我们可以了解它们之间的差异,并找到可能具有药用潜力的物种。
总结:系统发育进化树是一种重要的工具,用于描述物种或群体之间的进化关系。
通过构建系统发育进化树,我们可以了解不同物种之间的亲缘关系,以及它们的共同祖先。
系统发育进化树在物种分类、进化研究、分子演化研究、物种保护和药物开发等领域都有着广泛的应用。
MEGA软件——系统发育树构建方法
MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都分别保存为txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑。
2)序列导入MEGA 5首先打开MEGA 5软件,界面如下:然后,导入需要构建系统进化树的序列:点击OK出现新的对话框,创建新的数据文件导入成功3)序列比对分析点击W,开始比对。
比对完成后删除序列两端不能完全对其的碱基。
系统分析然后,关闭该窗口,在弹出的对话框中选择保存文件,文件名随便去,比如保存为1。
4)系统发育树构建以NJ为例Bootstrap选择1000,点Computer,开始计算计算完毕后,生成系统发育树。
以下“系统发育树树的修饰”方法沿用斑竹brightfuture01的方法5)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键-编辑图片,就可以对文字的字体大小,倾斜等做出修饰。
见下图:这个时候可以通过Adobe professional 对其进行图像导出:先将此word文档打印成PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
手把手教你构建系统发育树

Extraction of chromosomal DNA PCR amplification of the16S rRNA gene
Assmbly of 16S rRNA gene fragments
Obtain closely related sequences
0.01
Example2
PHYLIP---NJ
BIOEDIT SEQBOOT DNADIST NEIGHBOR CONSENSE TREVIEW
A number= 4n+1>0
SEQBOOT.EXE
Outfile
infile
DNADIST.EXE
Outfile
infile
NEIGHBOR.EXE
TOOLS
NCBI RDP / http://www.bacterio.cict.fr/
EzTaxon http://147.47.212.35:8080/
NCBI homepage /
YOU NEED A REGISTRATION NUMBER TO LOG IN THIS PAGE FIRST
38 89 40
Nocardiopsis listeri DSM 40297T X97887 Nocardiopsis alba DSM 43377T X97883 Nocardiopsis tropica DSM 44381T AF105971
99
Nocardiopsis umidischolae DSM 44362T AY036001
57
Nocardiopsis halotolerans DSM 44410T AJ290448 Nocardiopsis trehalosi DSM 44380T AF105972
系统发育进化树构建

系统发育进化树构建
系统发育进化树是一种用来表示生物种类和它们之间进化关系的图表。
它基于共有衍征和演化关系的分析,可以帮助我们理解物种的起源和演化过程。
下面是一个示例的系统发育进化树构建过程:
1. 收集数据:我们需要收集关于不同物种的特征和遗传信息的数据。
这些数据可以包括形态特征、分子序列等。
2. 数据处理:接下来,将收集到的数据进行处理,例如进行序列比对、计算相似性指数等。
这些处理会将数据转化为可以进行系统发育树分析的形式。
3. 构建系统发育树:通过使用系统发育树构建软件,如MEGA、PHYLIP等,我们可以利用处理后的数据构建系统发育树。
这些软件通常使用一些统计模型和算法来计算物种之间的相似性和进化关系。
4. 评估树的可靠性:构建系统发育树后,还需要对树的可靠性进行评估。
这可以通过计算支持值或进行自举分析等方法来实现。
支持值表示构建树的数据集中的信息支持树的某个分支。
5. 进行树的修正:如果评估发现树的某些分支的可靠性较低,我们可以根据需要进行进一步的分析,例如添加更多的数据或调整分析的参数。
6. 结果解读:在构建了系统发育树之后,可以通过对树的结构和分支进行解读,了解物种的起源和演化过程。
树的结构可以显示物种之间的近缘关系和进化路径。
注意:以上只是一个概括的系统发育进化树构建过程,具体的步骤和方法可能会因不同的研究目的和数据类型而有所不同。
在实际研究中,还需要根据具体情况选择适合的分析方法和工具。
手把手教你构建系统进化树

3、比对序列,比对结果转化为*.meg格式
用 Mega 6.0 的 ClustalW 做多序列联配,比对结果用 *.meg格式保存。或者用Clustal X软件进行比对,比对结果 保存为*.aln,再用Mega 6.0转化为*.meg格式。
4、构建系统进化树
打开保存的*.meg格式文件,选择邻接法构建系统发育 进化树。
以外米缀蛾的cds为例,点击cds,出现下图。
点击FASTA,出现下图。
该图为外米缀蛾的 FASTA格式,如何保 存见下图
一般情况下点 击该页的右上 角有send 图标, 选择后点击 create file 即 可下载。Txt可 以打开。 该图显示的是 序列全长的 FASTA格式下 载。
因为我采取基于氨 基酸序列比对,所 以选择coding sequences和fasta protein,下载编码 区氨基酸序列。
文件名未下载时不要更改,下下来之后再更改
MEGA6可以识别fasta格式文件。如图,将全 部-基因.txt重命名为全部-基因.fasta
•选择打开方式为MEGA6,打开全部-基因.fasta,自动跳出序列窗口 •用ClustalW做多序列联配
如何构建系统进化树
YZU.TRY
系统发生树(英文: Phylogenetic tree ) 又称为演化树( evolutionary tree ),是 表明被认为具有共同祖先的各物种间演化关 系的树。是一种亲缘分支分类方法 ( cladogram )。在树中,每个节点代表其 各分支的最近共同祖先,而节点间的线段长 度对应演化距离(如估计的演名称要么全部 斜体,要么全部不斜体,无法只让拉丁文斜体
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Phylogenetic analysis & Bootstrap analysis
METHODS
Distance-based method Maximum parsimony method (MP) Maximum likehood method(ML)
Unweightedpair group method with arithmetic mean(UPGMA) Neighbor joining(NJ) Minimum evolution(ME)
335
Nocardiopsis arabia DSM 45083T EF095149 Actinomadura echinospora DSM 43163T AJ420135
Example3 PHYLIP-----MP
BIOEDIT SEQBOOT DNAPARS CONSENSE TREEVIEW
Nocardiopsis kunsanensis DSM 44524 AF195412
99 17 27 80 81
Nocardiopsis aegyptia DSM 44442T AJ539401 Nocardiopsis lucentensis DSM 44048T X97888 Nocardiopsis dassonvillei subsp. dassonvillei DSM 43111T X97886 Nocardiopsis dassonvillei subsp. albirubida DSM 40465T X97882 Nocardiopsis synnemataformans DSM 44143T Y13593 Nocardiopsis quinghaiensis DSM 44739T EF597511
1000 278
Nocardiopsis umidischolae DSM 44362T AY036001 Nocardiopsis litoralis DSM 45168T EU583726
1000 917 607 962
Nocardiopsis kunsanensis DSM 44524T AF195412 Nocardiopsis xinjiangensis DSM 44589T AF251709 Nocardiopsis salina KCTC 19003T AY373031
TOOLS
NCBI RDP / http://www.bacterio.cict.fr/
EzTaxon http://147.47.212.35:8080/
NCBI homepage /
YOU NEED A REGISTRATION NUMBER TO LOG IN THIS PAGE FIRST
Construction of Phylogenetic Tree
Extraction of chromosomal DNA PCR amplification of the16S rRNA gene
Assmbly of 16S rRNA gene fragments
Obtain closely related sequences
102 264 408
323
Nocardiopsis 996 Nocardiopsis Nocardiopsis 482 Nocardiopsis 588 62 Nocardiopsis
Nocardiopsis lucentensis DSM 44048T X97888 Nocardiopsis aegyptia DSM 44442T AJ539401 Nocardiopsis quinghaiensis DSM 44739T EF597511 526 Nocardiopsis halotolerans DSM 44410T AJ290448 Nocardiopsis 997 Nocardiopsis 961 Nocardiopsis 913 Nocardiopsis 611 YIM 90008 Nocardiopsis 926 Nocardiopsis 851 Nocardiopsis 515 Nocardiopsis 628 Nocardiopsis 422 Nocardiopsis 981 Nocardiopsis 308 988 Nocardiopsis 542 Nocardiopsis litoralis DSM 45168T EU583726 kunsanensis DSM 44524T AF195412 xinjiangensis DSM 44589T AF251709 salina KCTC 19003T AY373031 halophila DSM 44494T AJ421018 baichengensis DSM 44845T AY619716 chromatogenes DSM 44844T AY619715 potens DSM 45234T FM253114 composta DSM 44551T AF360734 rhodophaea DSM 44843T AY619714 rosea DSM 44842T AY619713 gilva DSM 44841T AY619712 arabia DSM 45083T EF095149
EzTaxon http://147.47.212.35:8080/
OR
RDP(RIBOSOMAL DATABASE PROJECT) homepage http://www.bacterio.cict.fr/
Multiple-sequences alignments
TOOL---CLUSTAL_X
Multiple alignments
Phylogenetic analysis
Bootstrap analysis
Assmbly of 16S rRNA gene
TOOL ---SeqMan(DNASTAR)
Select your sequences here
R
A
Delete it
Obtain closely related sequences
74
31
35
49
Nocardiopsis composta DSM 44551T AF360734
Nocardiopsis potens DSM 45234T FM253114
42 99 91
Nocardiopsis chromatogenes DSM 44844T AY619715
Nocardiopsis baichengensis DSM 44845T AY619716 Nocardiopsis halophila DSM 44494T AJ421018 Actinomadura echinospora M 43163T AJ420135
290
YIM 90008
Nocardiopsis dassonvillei subsp. albirubida DSM 40465T X97882
811 815 995 258 169
Nocardiopsis synnemataformans DSM 44143T Y13593 Nocardiopsis dassonvillei subsp. dassonvillei DSM 43111T X97886 Nocardiopsis lucentensis DSM 44048T X97888 Nocardiopsis aegyptia DSM 44442T AJ539401 Nocardiopsis quinghaiensis DSM 44739T EF597511
0.01
Example2
PHYLIP---NJ
BIOEDIT SEQBOOT DNADIST NEIGHBOR CONSENSE TREVIEW
A number= 4n+1>0
SEQBOOT.EXE
Outfile
infile
DNADIST.EXE
Outfile
infile
NEIGHBOR.EXE
CONSENSE.EXE
DNAPARS.EXE TREEVIEW.EXE
MP TREE
Nocardiopsis 866 Nocardiopsis 554 550 Nocardiopsis Nocardiopsis 787 581
exhalans DSM 44407T AY028325 valliformis DSM 45023T AY336503 metallicus DSM 44598T AJ420769 ganjiahuensis DSM 45031T AY336513
Outtree
intree
CONSENSE.EXE
NJ TREE
631 364 417
Nocardiopsis valliformis DSM 45023T AY336503
994 695 875
Nocardiopsis exhalans DSM 44407T AY028325 Nocardiopsis metallicus DSM 44598T AJ420769 Nocardiopsis ganjiahuensis DSM 45031T AY336513
Phylogenetic analysis & Bootstrap analysis
TOOLS
MEGA-----UPGMA\NJ\MP\ME
PHYLIP----NJ\MP\ML
Example 1 MEGA--------NJ
Example MEGA ---------NJ
99 68 88 65
568
Nocardiopsis halotolerans DSM 44410T AJ290448 Nocardiopsis rosea DSM 44842T AY619713
1000
973 995 446 994