The Gene Ontology Annotation (GOA) Database--an integrated resource of GO annotations to the UniProt
如何使用Gene

網址:/
在首頁的GO website 底下的 在首頁的 ontology files 點進去
在GO format 底下四個資料夾 function 、process、comnpent、defs 、 、 按右鍵另存新檔下載
程式使用順序
GO_格式化
Step1:run GO的格式化.cpp(把<字元取代成%字元) Input file: 下載下來的process.ontology.txt檔 output file:定為000 Step2 runGO_id 中的go.cpp(擷取2個欄位) Input file: 下載下來的gene_association.goa_human output file: 定為001 Step3:run 刪除多餘的GO.cpp(刪除兩列或多列相同的字串) Input file: 上步驟的 001 output file: 定為002
二、下載GO 對應的Swiss-Prot
在首頁的GO website 底下的 在首頁的 annotations點進去 點進去
找到Homo sapiens 找到 GO Annotations 下 載 按 右 鍵 另 存 新 檔
以下是程式使用說明 依照step逐一執行 紅色 表示功能及目的 棕色 表示要執行的程式檔 藍色 表示是輸入的程式檔 藍色 表示是輸出的程式檔
-
Step4:run GO_term中的GOid的F&P&C的term.cpp (擷取出term term關鍵字和goid關鍵字 ) tmponent.ontology.txt 裡面的GOID先截取 出來,再加上 下載下來的GO.defs檔 output file: 定為003 Step5:run GO_statement中的: GO的功能.cpp (印出Swissprot 和GO的id關鍵字以及%開頭的字串) Swissprot Input file:用step3的002 以及下載下來的function.ontology output file: 定為004
gene ontology(GO基因注释)

GO(gene ontology)是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的,堆积因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准.GO是多种生物本体语言中的一种,提供了三层结构的系统定义方式,用于描述基因产物的功能.基因本体论(gene ontology)的建立现今的生物学家们浪费了太多的时间和精力在搜寻生物信息上。
这种情况归结为生物学上定义混乱的原因:不光是精确的计算机难以搜寻到这些随时间和人为多重因素而随机改变的定义,即使是完全由人手动处理也无法完成。
举个例子来说,如果需要找到一个用于制抗生素的药物靶点,你可能想找到所有的和细菌蛋白质合成相关的基因产物,特别是那些和人中蛋白质合成组分显著不同的。
但如果一个数据库描述这些基因产物为“翻译类”,而另一个描述其为“蛋白质合成类”,那么这无疑对于计算机来说是难以区分这两个在字面上相差甚远却在功能上相一致的定义。
Gene Ontology (GO)项目正是为了能够使对各种数据库中基因产物功能描述相一致的努力结果。
这个项目最初是由1988年对三个模式生物数据库的整合开始:: FlyBase (果蝇数据库Drosophila),t Saccharomyces Genome Database (酵母基因组数据库SGD) and the Mouse Genome Database(小鼠基因组数据库MGD)。
从那开始,GO不断发展扩大,现在已包含数十个动物、植物、微生物的数据库。
GO的定义法则已经在多个合作的数据库中使用,这使在这些数据库中的查询具有极高的一致性。
这种定义语言具有多重结构,因此在各种程度上都能进行查询。
举例来说,GO可以被用来在小鼠基因组中查询和信号转导相关的基因产物,也可以进一步找到各种生物地受体酪氨酸激酶。
这种结构允许在各种水平添加对此基因产物特性的认识。
基因功能注释

基因功能注释
基因功能注释是利用不同策略,来确定一个基因的蛋白质产物。
解析基因的功能有助于理解生物体健康和疾病状况,帮助用户分析和保存基因的功能及其相关联的健康风险。
这里推荐一款基因功能注释的软件——Gene Ontology Annotation。
Gene Ontology Annotation(GOA)是一个基于Web的数据库,用于基因组数据注释,主要用于内在属性的分类和分析。
GOA拥有丰富的信息,如基因和基因组概念、分子功能,以及各种细胞类型和发育阶段的功能注释。
GOA的优点:首先,对于对大规模数据进行基因功能注释,GOA提供了针对这类数据的“网状注释”(Web-based annotation)方法。
它是一种快速的整体搜索策略,使用自定义的网状模式(web-based pattern)进行基因注释。
而且,GOA 拥有丰富的信息以及强大的数据库,它可以在短时间内将大量数据转换为可使用的信息,并可以实现复杂的基因功能注释,便于研究者理解不同基因及其细胞类型和发育阶段的功能。
另外,GOA符合人义,它涵盖了各种功能及注释,不仅数量庞大,而且内容丰富,包括超过17万个有用的条目,涵盖细胞生物学、分子生物学、发育学、免疫学、比较基因组学等研究领域等。
用户可以利用它,快速地看到基因的功能,准确地了解不同基因的作用,保存及分析这些相关的信息,一般来说,GOA的操作比较简单,而且搜索结果也很准确。
总之,Gene Ontology Annotation是一个帮助研究者更好地了解基因功能的非常有用的软件,它强大的数据库与丰富的信息,使用起来较为方便,能够有效地帮助研究者细致地研究基因的功能,为生物学研究提供了重要的参考资料,强烈推荐使用它!。
基因本体论(go)功能注释 gene ontology annotation

基因本体论(go)功能注释 gene ontologyannotation基因本体论(Gene Ontology,简称GO)是一种用来描述基因功能的标准化系统。
GO的功能注释则是使用GO术语为基因或蛋白质序列进行注释,帮助科学家理解生物体内基因的功能和相互关系。
本文将介绍基因本体论(GO)的概念和作用,以及基因本体论功能注释的流程和应用。
一、基因本体论(GO)的概念和作用基因本体论(GO)是一种标准化的词汇系统,用于描述基因和蛋白质的功能、过程和组件。
GO包含三个主要的本体:分子功能(Molecular Function)、生物过程(Biological Process)和细胞组件(Cellular Component)。
每个本体都包含一系列术语和相应的定义,科学家可以根据这些术语和定义来描述基因的功能。
基因本体论的作用是帮助科学家对基因和蛋白质进行分类和理解。
通过将基因和蛋白质注释到GO术语上,科学家可以更准确地了解它们的功能、参与的生物过程以及位于细胞的哪个组件。
这对于研究基因的功能以及疾病的发生和发展有着至关重要的意义。
二、基因本体论功能注释的流程基因本体论功能注释是指将基因或蛋白质序列与基因本体论术语进行关联的过程。
下面是一般的基因本体论功能注释流程:1.数据预处理:获取待注释基因或蛋白质的序列数据,排除冗余数据和噪音数据。
2.基因本体论术语获取:从基因本体论数据库中获取相应的术语,包括分子功能、生物过程和细胞组件。
3.序列比对:将待注释的基因或蛋白质序列与已知序列进行比对,找出相似序列。
4.注释:根据序列比对的结果,将相似序列的注释信息转移到待注释序列上。
5.术语关联:根据注释信息,将待注释基因或蛋白质与相应的基因本体论术语进行关联。
6.结果验证:对注释结果进行验证和统计分析,评估注释的准确性和可靠性。
三、基因本体论功能注释的应用基因本体论功能注释在生命科学研究中有着广泛的应用。
以下是一些常见的应用领域:1.基因功能研究:通过注释基因的功能,科学家可以更好地理解基因在细胞中的作用,从而揭示生物体内复杂的生物过程。
Gene Ontology(GO)简介与使用介绍

5.GO 文件格式
GO 的所有数据都是免费获得的。GO 数据有三种格式:flat(每日更新)、XML(每月更新)和 MySQL(每月更 新)。 这些数据格式都可以在 GO ftp 的站点上下载。XML 和 MySQL 文件是被储存于独立的 GO 数据库中。
如果需要找到与某一个 GO 术语相关的基因或基因产物,可以找到一个相应表格,搜寻到这种注解的编号,并且 可以链接到与之对应的位于不同数据库的基因相关文件。
如图为 DAG-Edit 的界面,可以分为四个部分:
1) 定义编辑面板(term editor panel)
显示当下的本体论。也是主要的编辑本体论结构的工具,可以通过点击和拖动术语来修改本体论的从属关系。
2) 文本编辑面板(text editor panel)
修改术语中的内容。在修改多个术语时,会出现一个选择菜单,可以选中后逐个修改。
8.GO 的应用
GO 的局限性
1). GO 不是基因序列或基因产物数据库,相反的,GO 强调基因产物在细胞中的功能。
2). GO 不是整合数据库的一种方式(如联邦式整合数据库),它并不能做到这点是因为:
4)如何得到由 GO 术语注解的蛋白序列?
在 GO 网页上选择能查询到所有数据库的 Amigo 浏览器,键入 GO 术语(如“线粒体”),在结果中显示了被注 释的基因。然后选择你所需基因,在网页的最低端把选项拖至“get fasta sequence”区域,再确定即可。 5)如何能够找到所有和一个特定的 GO 术语相关的人类基因呢?
GO 对基因和蛋白的注释阐明了基因产物和用于定义他们的 GO 术语之间的关系。基因产物指一个基因编码的 RNA 或蛋白产物。因为一个基因可能编码多个具有很不相同性质的产物,所以 GO 推荐的注释是针对基因产物的而不 是基因的。一个基因是和所有适用于它的术语联系在一起的。
gene?Ontology?(基因本体论)

gene Ontology (基因本体论)gene ontology为了查找某个研究领域的相关信息,生物学家往往要花费大量的时间,更糟糕的是,不同的生物学数据库可能会使用不同的术语,好比是一些方言一样,这让信息查找更加麻烦,尤其是使得机器查找无章可循。
Gene Ontology就是为了解决这种问题而发起的一个项目。
Gene Ontology中最基本的概念是term。
GO里面的每一个entry都有一个唯一的数字标记,形如GO:nnnnnnn,还有一个term 名,比如"cell", "fibroblast growth factor receptor binding",或者"signal transduction"。
每个term都属于一个ontology,总共有三个ontology,它们分别是molecular function, cellular component 和biological process。
一个基因product可能会出现在不止一个cellular component里面,也可能会在很多biological process里面起作用,并且在其中发挥不同的molecular function。
比如,基因product "cytochrome c" 用molecular function term描述是"oxidoreductase activity",而用biological process term描述就是"oxidative phosphorylation"和"induction of cell death",最后,它的celluar component term是"mitochondrial matrix"和"mitochondrial inner membrane"。
网络生物分析 GENE ONTOLOGY

Biological Process
regulation of gluconeogenesis 糖质调节
Biological Process
limb development 肢体发展
Ontology Structure
• Terms are linked by two relationships
内膜 外部膜
嵴 细胞
Cellular Component
亲水区
醣蛋白脂类结合成磷脂横跨膜的Cellular Component
• Enzyme complexes in the component ontology refer to places, not activities.
Molecular Function
• terms are related within a hierarchy
GO structure
gene A
GO structure
• This means genes can be grouped according to userdefined levels
• Allows broad overview of gene set or genome
What is the Gene Ontology?
• Genes are linked, or associated, with GO terms by trained curators at genome databases
– known as ‘gene associations’ or GO annotations
Ontology Structure
cell
Directed Acyclic Graph (DAG) - multiple parentage allowed
差异基因go注释

差异基因go注释差异基因是指在不同生物体或者同一生物体不同组织、不同发育时期以及在不同条件下表达水平发生明显变化的基因。
这些差异基因的研究对于深入理解生物体的发生发展、适应环境变化以及疾病的发生机制具有重要意义。
为了更好地理解差异基因的功能,科研人员常常对差异基因进行GO(Gene Ontology)注释。
GO注释是一种常见的功能注释方法,它根据基因的功能特征将其分类并进行注释。
GO注释基于GO数据库,该数据库将基因功能划分为三个主要方面:生物学过程(Biological Process)、分子功能(Molecular Function)和细胞组分(Cellular Component)。
通过GO注释,可以为差异基因的功能特征提供详细的描述和分类,从而为后续的生物信息分析和研究提供基础。
在进行差异基因GO注释之前,首先需要进行差异分析。
差异分析是通过比较不同样本间基因表达水平的差异,筛选出差异显著的基因。
差异分析常用的方法包括t检验、方差分析和基因表达差异倍数筛选等。
这些方法可以帮助我们确定哪些基因是差异表达的,为后续的功能注释提供基础。
差异基因GO注释的流程通常包括以下几个步骤:1. 数据准备:准备进行差异基因GO注释所需的数据,包括差异基因表达矩阵和差异基因列表。
2. GO数据库下载:从相应的数据库中下载最新的GO注释文件,常用的数据库包括Gene Ontology Consortium、UniProt和NCBI等。
3. 数据筛选和预处理:根据差异基因列表,筛选出在GO数据库中存在注释信息的基因。
4. GO注释:将差异基因与GO数据库中的功能项进行匹配,得到差异基因的功能注释信息。
5. GO富集分析:对差异基因的功能注释信息进行统计分析,找出在某个功能项上显著富集的基因集合。
6. 结果展示:将GO注释和GO富集分析的结果进行整理和可视化展示,方便研究人员进行进一步的分析和解读。
当然,差异基因GO注释的具体方法和流程还需要根据实际研究的需求和数据情况进行调整和优化。
生物信息学中的基因功能注释方法解析

生物信息学中的基因功能注释方法解析随着基因组学技术的快速发展,我们已经进入了一个大数据时代,生物信息学的重要性日益突显。
在基因组学研究中,了解基因的功能是至关重要的一步。
基因功能注释是指根据已有的研究和数据库,对基因进行功能预测和解释。
本文将详细介绍生物信息学中常用的基因功能注释方法。
1. 基因本体注释基因本体注释是一种基于知识库的方法,通过将基因与生物过程、分子功能和细胞组成等术语进行关联,从而预测基因的功能。
最著名的基因本体知识库是基因本体组织(Gene Ontology, GO)。
GO分类了三个方面的术语:分子功能、生物过程和细胞组成。
基因本体注释可以通过比对基因序列与已知基因的相似性来实现。
比对结果可以通过统计学方法来确定注释结果的可靠性。
2. 基于序列相似性的注释基于序列相似性的注释是最常用的注释方法之一。
该方法根据已知的基因序列、蛋白序列或基因家族,来推断未知基因的功能。
通过使用比对算法,如BLAST,可以在数据库中搜索与目标基因序列具有相似序列的已知基因。
根据相似性,可以预测目标基因的功能。
这种方法的优点是简单快速,但是也存在一些限制,比如只能预测已知的功能。
3. 基于域的注释基于域的注释是在基因序列中寻找特定的保守域来推断基因的功能。
保守域是指在进化过程中高度保守的序列片段,对蛋白质的功能至关重要。
有多种工具可以用来识别和注释保守域,如Pfam和InterPro。
通过比对目标基因序列与保守域数据库中的已知域,可以推断出目标基因的功能。
4. 基于组学数据的注释随着高通量技术的不断发展和研究成果的积累,大量的组学数据可用于基因功能注释。
这些数据包括转录组学、蛋白质组学和代谢组学等。
通过分析这些数据,可以识别基因表达模式、蛋白质互作网络和代谢途径等信息,从而预测基因的功能。
一些常用的基因功能注释工具,如DAVID和Enrichr,可以利用这些组学数据进行功能注释。
5. 基于机器学习的注释随着机器学习算法的进展,基于机器学习的基因功能注释方法也越来越受到关注。
go基因注释与功能分类

背景
随着后基因组 (post-genomics) 时代的来临,基因组学的研究 重心开始从阐明所有遗传信息转移到在整体分子水平对功能进行 研究。这种转变的一个重要标志是产生了功能基因组学 (functional genomics)。
任务
功能基因组学的主要任务之一是进行基因组功能注释 (genome annotation),了解基因的功能,认识基因与疾病的关 系,掌握基因的产物及其在生命活动中的作用等。
举例
这里以检索神经源性分化因子6(NEUROD6)为例。在检索框 中输入“NEUROD6”并勾选“gene and proteins”和“exact match”,运行后所得基因产物检索结果如图所示。
此图显示了该基因产物的基本信息,包括类型、物种、 别名来源和序列
人民卫生出版社8年制及7年制临床医学等专业用《生物信息学》
KEGG数据库的改进与更新
KEGG PATHWAY 还存储了一些人类疾病通路数据,这些 疾病通路被分为六个子类:癌症、免疫系统疾病、神经退行 性疾病、循环系统疾病、代谢障碍、传染病循环系统疾病。 KEGG DRUG数据库也在不断地完善,其中的药物数据几乎 涵盖了日本的所有非处方药和美国的大部分处方药品 。 DRUG 是一个以存储结构为基础的数据库,每条记录都包含 唯一的化学结构以及该药物的标准名称,以及药物的药效、 靶点信息、类别信息等。药物的靶点通过KEGG PATHWAY 查询,药物的分类信息是KEGG BRITE数据库的一部分,通 过药物的标准名称可以找到该药物的商品名,还可以找到药 物销售的标签信息。此外,DRUG还包括一些天然的药物和 中药的信息,有些药物被日本药典所收录。
富集分析中常用的统计方法有累计超几何 分布、Fisher精确检验等。
go基因注释与功能分类
其中排在第一位的是人类 基因“PGM1”的相关信息, 点击该条目进入到详细信 息页面。 该页面以表格的形式列出 了该基因有关的详细信息, 包括基因编号,基因的详 细定义,所编码的酶的编 号,基因所在通路,以及 序列的编码信息。同时, 在页面的右侧还提供了该 基因在其他分子生物学数 据库的链接,如OMIM、 NCBI、GenBank等。
KEGG存储内容
KEGG目前共包含了19个子数据库,它们被分类成系统信息、 基因组信息和化学信息三个类别 。
基因组信息存储在GENES数据库里,包括全部完整的基因组序列和部 分测序的基因组序列,并伴有实时更新的基因相关功能的注释。 KEGG中化学信息的 6个数据库被称为 KEGG LIGAND数据库,包含 化学物质、酶分子、酶化反应等信息。KEGG BRITE数据库是一个包 含多个生物学对象的基于功能进行等级划分的本体论数据库,它包括 分子、细胞、物种、疾病、药物、以及它们之间的关系。
一些小的通路模块被存储在MODULE数据库中,该数据库还存储了其 他的一些相关功能的模块以及化合物信息。
KEGG DRUG数据库存储了目前在日本所有非处方药和美国的大部分 处方药品。 KEGG DISEASE是一个存储疾病基因、通路、药物、以及疾病诊断标 记等信息的新型数据库。
KEGG数据库的注释与检索
一、富集分析算法
富集分析方法通常是分析一组基因在某个功能结点上是否 过出现(over-presentation)。这个原理可以由单个基因的注 释分析发展到大基因集合的成组分析。 由于分析的结论是基于一组相关的基因,而不是根据单个 基因,所以富集分析方法增加了研究的可靠性,同时也能 够识别出与生物现象最相关的生物过程。
GeneOntology(GO)分析
Gene Ontology
现今的生物学家们浪费了太多的时间和精力在搜寻生物信息上。这种情况归结为生物学上定 义混乱的原因:不光是精确的计算机难以搜寻到这些随时间和人为多重因素而随机改变的定 义,即使是完全由人手动处理也无法完成。举个例子来说,如果需要找到一个用于制抗生素 的药物靶点,你可能想找到所有的和细菌蛋白质合成相关的基因产物,特别是那些和人中蛋 白质合成组分显著不同的。但如果一个数据库描述这些基因产物为“翻译类”,而另一个描述 其为“蛋白质合成类”,那么这无疑对于计算机来说是难以区分这两个在字面上相差甚远却在 功能上相一致的定义。 Gene Ontology (GO)项目正是为了能够使对各种数据库中基因产物功能描述相一致的努力结 果。这个项目最初是由 1988 年对三个模式生物数据库的整合开始:: FlyBase (果蝇数据库 Drosophila),t Saccharomyces Genome Database (酵母基因组数据库 SGD) and the Mouse Genome Database (小鼠基因组数据库 MGD)。从那开始,GO 不断发展扩大,现在已包含数 十个动物、植物、微生物的数据库。 GO 的定义法则已经在多个合作的数据库中使用,这使在这些数据库中的查询具有极高的一 致性。这种定义语言具有多重结构,因此在各种程度上都能进行查询。举例来说,GO 可以 被用来在小鼠基因组中查询和信号转导相关的基因产物,也可以进一步找到各种生物地受体 酪氨酸激酶。这种结构允许在各种水平添加对此基因产物特性的认识。 GO 的结构包括三个方面:分子生物学上的功能、生物学途径和在细胞中的组件作用。当然, 它们可能在每一个方面都有多种性质。如细胞色素 C,在分子功能上体现为电子传递活性, 在生物学途径中与氧化磷酸化和细胞凋亡有关,在细胞中存在于线粒体质中和线粒体内膜 上。下面,将进一步的分别说明 GO 的具体定义情况。 基因产物 基因产物和其生物功能常常被我们混淆。例如,“乙醇脱氢酶”既可以指放在 Eppendorf 管里 的基因产物,也表明了它的功能。但是这之间其实是存在差别的,一个基因产物可以拥有多 种分子功能,多种基因产物也可以行使同一种分子功能。比如还是“乙醇脱氢酶”,其实多种 基因产物都具有这种功能,而并不是所有的这些酶都是由乙醇脱氢酶基因编码的。一个基因 产物可以同时具有“乙醇脱氢酶”和“乙醛歧化酶”两种功能,甚至更多。所以,在 GO 中,很 重要的一点在于,当使用“乙醇脱氢酶活性”这种术语时,所指的是功能,并不是基因产物。 许多基因产物会形成复合物后执行功能。这些“基因复合物”有些非常简单(如血红蛋白由血
Gene Ontology分析

Gene OntologyGO分析Gene Ontology可分为分子功能Molecular Function生物过程biological process和细胞组成cellular component三个部分。
蛋白质或者基因可以通过ID 对应或者序列注释的方法找到与之对应的GO号而GO号可对于到Term即功能类别或者细胞定位。
参考网站 功能富集分析功能富集需要有一个参考数据集通过该项分析可以找出在统计上显著富集的GO Term。
功能或者定位有可能与研究的目前有关。
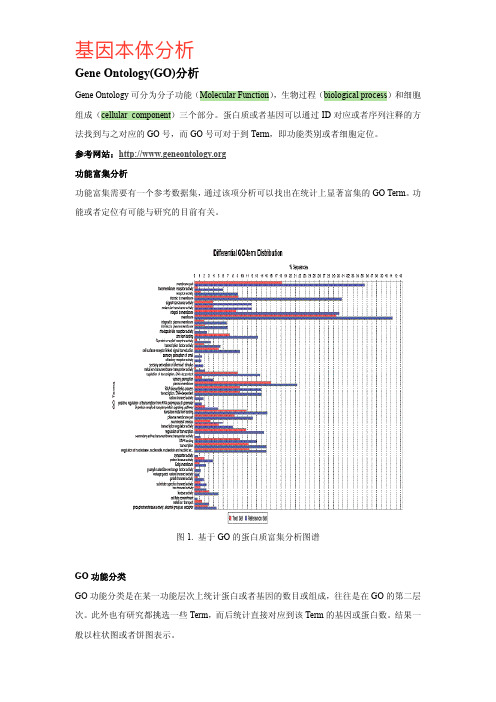
图1. 基于GO的蛋白质富集分析图谱GO功能分类GO功能分类是在某一功能层次上统计蛋白或者基因的数目或组成往往是在GO的第二层次。
此外也有研究都挑选一些Term 而后统计直接对应到该Term的基因或蛋白数。
结果一般以柱状图或者饼图表示。
1.GO分析根据挑选出的差异基因计算这些差异基因同GO 分类中某几个特定的分支的超几何分布关系GO 分析会对每个有差异基因存在的GO 返回一个p-value小的p 值表示差异基因在该GO 中出现了富集。
GO 分析对实验结果有提示的作用通过差异基因的GO 分析可以找到富集差异基因的GO分类条目寻找不同样品的差异基因可能和哪些基因功能的改变有关。
2.Pathway分析根据挑选出的差异基因计算这些差异基因同Pathway 的超几何分布关系Pathway 分析会对每个有差异基因存在的pathway 返回一个p-value小的p 值表示差异基因在该pathway 中出现了富集。
Pathway 分析对实验结果有提示的作用通过差异基因的Pathway 分析可以找到富集差异基因的Pathway 条目寻找不同样品的差异基因可能和哪些细胞通路的改变有关。
与GO 分析不同pathway 分析的结果更显得间接这是因为pathway 是蛋白质之间的相互作用pathway 的变化可以由参与这条pathway 途径的蛋白的表达量或者蛋白的活性改变而引起。
Annotating genomes using ontologies

GPCR activity
receptor activity
is_a
Combining with an extracellular or intracellular messenger to initiate a change in cell activity
transmembrane receptor activity
Molecular Function
insulin binding GO:0005009 insulin receptor activity
Molecular Function
GO:0015238 drug transporter activity
Molecular Function
[Term] id: GO:0015238 name: drug transporter activity namespace: molecular_function def: "Enables the directed movement of a drug into, out of, within or between cells. A drug is any naturally occurring or synthetic substance, other than a nutrient, that, when administered or applied to an organism, affects the structure or functioning of the organism; in particular, any such substance used in the diagnosis, prevention, or treatment of disease." [ISBN:0198506732] is_a: GO:0005215 ! transporter activity
go基因注释与功能分类
意义
快速有效的基因注释对进一步识别基因,研究基因的表达调 控机制,研究基因在生物体代谢途径中的地位,分析基因、基因 产物之间的相互作用关系,预测和发现蛋白质功能,揭示生命的 起源和进化等具有重要的意义。
第二节 基因注释数据库
Gene Annotation Database
基因注释数据库产生的原因
KEGG 通 常 被 看 作是生物系统的计算 机表示,它囊括了生 物系统中的各个对象 与对象之间的关系。 在分子层面、细胞层 面、组织层面都可以 对数据库进行检索。 每个数据库中的检索 条目按照一定规律被 赋予一个检索号,也 就是 ID 。表中列出了 KEGG的13个核心数据 库的检索号。
另外一种化学注释的方法是 以小分子化学结构的生物学 意义为特征来实现的。 在 KEGG 数据库中,酶与酶 之间的反应信息以及相关的 化学结构信息分别存储在 KEGG REACTION 数据库和 KEGG REPAIR数据库中。 每个化合物的化学结构都被 转 化 为 RDM (atom type changes at R:reaction center D:diffevent atom M:matched atom )模式。
KEGG数据库的改进与更新
为了满足日益增长的科学研究需求,KEGG数据库在最近 几 年 里 不 断 扩 充 , 新 增 加 的 50 多 个 通 路 使 KEGG PATHWAY数据库更加完善。这50多个新增加的通路包括 信号传导通路、细胞生物过程通路和人类疾病通路等。 KEGG对通路数据新增了两个补充内容:第一个补充是一 张全局通路图,这张全局通路图是通过手工拼接 KEGG的 120 多个现存通路图生成的,存储为 SVG 文件。另一个补 充内容是 KEGG MODULE 数据库,这是一个收集了通路 模块以及其他一些功能单元的新型数据库,功能模块是在 KEGG子通路中被定义为一些小的片段,通常包括几个连 续的反应步骤、操纵子、调控单元,以及通过基因组比对 得到的系统发生单元和分子的复合物等。
一种基于生物数据的多层关联规则挖掘算法_硕士学位论文

挖掘算法A Thesis Submitted in fulfillment of the Requirements forthe Degree of Master of EngineeringAn Algorithm for Mining Biological DataMultilevel Association RulesCandidate : Zhang PingMajor : Computer Software and TheorySupervisor : Prof. Lu YanshengHuazhong University of Science & TechnologyWhuhan 430074, P.R.ChinaJune, 2007独创性声明本人声明所呈交的学位论文是我个人在导师指导下进行的研究工作及取得的研究成果。
尽我所知,除文中已经标明引用的内容外,本论文不包含任何其他个人或集体已经发表或撰写过的研究成果。
对本文的研究做出贡献的个人和集体,均已在文中以明确方式标明。
本人完全意识到,本声明的法律结果由本人承担。
学位论文作者签名:日期:年月日学位论文版权使用授权书本学位论文作者完全了解学校有关保留、使用学位论文的规定,即:学校有权保留并向国家有关部门或机构送交论文的复印件和电子版,允许论文被查阅和借阅。
本人授权华中科技大学可以将本学位论文的全部或部分内容编入有关数据库进行检索,可以采用影印、缩印或扫描等复制手段保存和汇编本学位论文。
保密□ ,在_____年解密后适用本授权书。
本论文属于不保密□。
(请在以上方框内打“√”)学位论文作者签名:指导教师签名:日期:年月日日期:年月日华中科技大学硕士学位论文摘要数据挖掘是从大量数据中发现潜在的、有趣的知识的过程,是解决“数据丰富,知识贫乏”状况的有效方法。
关联规则挖掘用于从大量数据中揭示项集之间的有趣关联或相关联系,是数据挖掘的一项重要研究内容,在现实生活中有着广泛的应用。
gene annotation analysis -回复

gene annotation analysis -回复gene annotation analysis是一项基因组学领域的重要研究方法,它通过对基因组中已知和未知基因的功能进行注释,从而揭示基因在细胞过程和生物学功能中的作用。
本文将逐步介绍gene annotation analysis的原理、方法和现实应用,并通过具体案例解释其在基因研究领域的重要性。
首先,我们来了解gene annotation analysis的原理。
基因组是一个由DNA序列组成的巨大网络,其中包含了成千上万个基因。
然而,对于大多数基因来说,我们对其功能和作用仍知之甚少。
基因的annotation就是为了解决这一问题而进行的研究。
基因的annotation可以分为三个层次:DNA层面的序列注释、RNA转录层面的注释和蛋白质层面的注释。
其中,DNA序列注释是指对基因组中的DNA区域进行分类和注释,将其与已知的基因数据进行比对,确定其可能的功能和特征。
RNA转录注释是指对基因组中的RNA序列进行分析和注释,确定其是否转录成mRNA并参与蛋白质合成。
而蛋白质注释则是指对基因组中已知的蛋白质序列和结构进行比对和分析,确定基因可能编码的蛋白质的功能和特征。
接下来,我们将介绍gene annotation analysis的方法。
gene annotation analysis涉及到很多基因组学的工具和数据库,其中一些常用的工具包括:BLAST、Gene Ontology (GO)、Kyoto Encyclopedia of Genes and Genomes (KEGG)等。
BLAST是一种基于序列比对的工具,它可以将未知序列与已知序列比对,从而确定其可能的功能。
例如,我们可以将一条未知的DNA序列与已知的基因组中的DNA序列进行BLAST 比对,找出与其相似的序列,并据此推测其功能和特征。
GO是一个用于描述基因和基因组功能的分类系统,它将基因的功能分为不同的层次和分类,并提供了一系列标准化的注释术语。
gene_ontology(GO基因注释)

GO(gene ontology)是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的,堆积因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准.GO是多种生物本体语言中的一种,提供了三层结构的系统定义方式,用于描述基因产物的功能.基因本体论(gene ontology)的建立现今的生物学家们浪费了太多的时间和精力在搜寻生物信息上。
这种情况归结为生物学上定义混乱的原因:不光是精确的计算机难以搜寻到这些随时间和人为多重因素而随机改变的定义,即使是完全由人手动处理也无法完成。
举个例子来说,如果需要找到一个用于制抗生素的药物靶点,你可能想找到所有的和细菌蛋白质合成相关的基因产物,特别是那些和人中蛋白质合成组分显著不同的。
但如果一个数据库描述这些基因产物为“翻译类”,而另一个描述其为“蛋白质合成类”,那么这无疑对于计算机来说是难以区分这两个在字面上相差甚远却在功能上相一致的定义。
Gene Ontology (GO)项目正是为了能够使对各种数据库中基因产物功能描述相一致的努力结果。
这个项目最初是由1988年对三个模式生物数据库的整合开始:: FlyBase (果蝇数据库Drosophila),t Saccharomyces Genome Database (酵母基因组数据库SGD) and the Mouse Genome Database(小鼠基因组数据库MGD)。
从那开始,GO不断发展扩大,现在已包含数十个动物、植物、微生物的数据库。
GO的定义法则已经在多个合作的数据库中使用,这使在这些数据库中的查询具有极高的一致性。
这种定义语言具有多重结构,因此在各种程度上都能进行查询。
举例来说,GO可以被用来在小鼠基因组中查询和信号转导相关的基因产物,也可以进一步找到各种生物地受体酪氨酸激酶。
这种结构允许在各种水平添加对此基因产物特性的认识。
GeneOntology教程

24th Feb 2006
Jane Lomax
GO tools
• Many tools exist that use GO to find common biological functions from a list of genes:
/GO.tools.microarray.shtml
24th Feb 2006
Jane Lomax
What is the Gene Ontology?
• Set of biological phrases (terms) which are applied to genes:
– protein kinase – apoptosis – membrane
Cellular Component
• where a gene product acts
24th Feb 2006
Jane Lomax
Cellular Component
24th Feb 2006
Jane Lomax
Cellular Component
24th Feb 2006
Jane Lomax
cell
is-a part-of
membrane
chloroplast
mitochondrial membrane
24th Feb 2006 Jane Lomax
chloroplast membrane
Ontology Structure
• Ontologies are structured as a hierarchical directed acyclic graph (DAG) • Terms can have more than one parent and zero, one or more children
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
In Silico Biology 4 (2004) 5–6IOS PressElectronic publication can be found in In Silico Biol. 4, 0002 <http://www.bioinfo.de/isb/2003/04/0002/>, 1 December 2003. 1386-6338/04/$17.00 © 2004 – IOS Press, Bioinformation Systems e.V. and the authors. All rights reserved 5Short CommunicationThe Gene Ontology Annotation (GOA) Database – An Integrated Resource of GO Annotations to the UniProt KnowledgebaseEvelyn Camon*, Daniel Barrell, Vivian Lee, Emily Dimmer and Rolf ApweilerEuropean Bioinformatics Institute (EBI), Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, UKEdited by E. Wingender; received 17 September 2003; accepted 30 September 2003; published 1 December 2003 This short communication is provided to accompany a lecture given at the '‘Workshop on Ontologies and Data Integration for Biology’', May 2003, Japan.Gene Ontology (GO) [1] is a well-established structured vocabulary, which has been successfully used for 6 years in protein annotation. The vocabulary was designed by biologists to replace the multiple no-menclatures used by specialised and large knowledge bases that can hinder data integration. Currently GO consists of over 16,000 terms, distributed over three ontologies and these describe what a protein does (molecular function), how it does it (biological process) and where it performs this task in a generic cell (cellular component). The GO Consortium is successful because it limited its scope to these three ontolo-gies from the outset and has involved the biological community throughout its evolution. In making GO immediately available for biological annotation some errors were inevitable, however the Consortium is committed to its maintenance and upkeep as new genomic databases get involved. As additional ontolo-gies to GO are needed to model biology and experimentation, the GO Consortium has created the Open Biological Ontologies (OBO) web site to encourage communication and the creation of other standard vocabularies that could be freely used in tandem with GO.At the European Bioinformatic Institute there are two main GO activities, it houses the GO editorial of-fice (edit GO ontologies) headed by Michael Ashburner and Midori Harris and the GO annotation team (annotate GO to gene products) headed by Rolf Apweiler and Evelyn Camon. The rest of this lecture will describe the Gene Ontology Annotation (GOA) database [2].The GOA database (/GOA) uses the GO vocabulary to provide high quality elec-tronic and manual annotations to gene products contained in UniProt Knowledgebase (Swiss-Prot, __________________________________________________*Corresponding author. Tel.: +44 1223 494465; Fax: +44 1223 494468; E-mail: goa@.E. Camon et al. / The Gene Ontology Annotation (GOA) Database6TrEMBL, PIR-PSD) [3]. As a supplementary archive of GO annotation, GOA promotes a high level ofintegration of the knowledge represented in Swiss-Prot with other databases. This is achieved by convert-ing Swiss-Prot annotation into a recognised computational format. GOA provides annotated entries for nearly 60,000 species (GOA-SPTR) and is the largest and most comprehensive open-source contributorof annotations to the GO Consortium annotation effort. By integrating GO annotations from other model organism groups (FlyBase, SGD, MGD), GOA consolidates specialised knowledge and expertise to en-sure the data remains a key reference for current biological knowledge. Furthermore, the GOA database fully supports the Human Proteomics Initiative (HPI) [4] by fast-tracking the annotation of proteins likelyto benefit human health and disease. In addition to a non-redundant set of annotations to the human pro-teome (GOA-Human) and monthly releases of its GO annotation for all species (GOA-SPTR), a series of GO mapping files (Swiss-Prot keyword to GO, InterPro to GO), and specific cross-references in other databases are also regularly distributed. GOA can be queried through a simple user-friendly web interface via our QuickGO browser or downloaded in a parsable format via the EBI (ftp:///pub/databases/GO/goa/) and GO FTP sites (ftp:///pub/go/gene-associations/). The GOA dataset can be used to enhance the annotation of particular model organism or gene expression datasets (via slimmed down version of GO, GO-slim, ftp:///pub/go/GO_slims/), although increasingly it has been used to evaluate GO pre-dictions generated from text mining or protein interaction experiments. In 2004, the GOA team will buildon its success and will continue to supplement the functional annotation of UniProt, improve recall and searching with GO and try to facilitate access to all available biological information.Researchers wishingto feedback to the GOA project are encouraged to e-mail: goa@.The GOA project is grateful for the support of Grants QRLT-2001-00015 and QLRI-2000-00981 of the European Commission and a supplementary NIH grant, 1R01HGO2273-01.KEYWORDS: Gene Ontology, annotationREFERENCES[1]Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K.,Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M. and Sherlock, G. (2000). Gene Ontology: tool for the unifica-tion of biology. Nature Genet. 25, 25-29.[2]Camon, E., Magrane, M., Barrell, D., Binns, D., Fleischmann, W., Kersey, P., Mulder, N., Oinn, T., Maslen,J., Cox, A. and Apweiler, R. (2003). The Gene Ontology Annotation (GOA) project: implementation of GO in Swiss-Prot, TrEMBL and InterPro. Genome Res. 13, 662-672.[3]Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., Martin, M. J., Mi-choud, K., O'Donovan, C., Phan, I., Pilbout, S. and Schneider, M. (2003). The SWISS-PROT protein know-ledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365-370.[4]O'Donovan, C., Apweiler, R. and Bairoch, A. (2001). The human proteomics initiative (HPI). Trends Bio-technol. 19, 178-181.。