Site Evaluation and RFI spectrum measurements in Portugal at the frequency range 0.408-10 G
紫外分光光度法在保健食品检测中的应用

食品科技紫外分光光度法在保健食品检测中的应用罗婷婷(广西华测检测认证有限公司,广西南宁 530000)摘 要:紫外分光光度法是一种常用的光谱分析技术,可以用于分析物质的结构、浓度、纯度等参数。
在保健食品的检测中,紫外分光光度法有着广泛的应用,可以用于检测保健食品的成分含量、质量和安全性等。
本研究简单分析了紫外分光光度法在保健食品检测中的应用,讨论了紫外分光光度法在保健食品检测中的必要性,探讨了紫外分光光度法在保健食品测定中的应用,旨在为我国未来保健食品的生产与管理提供帮助与参考。
关键词:紫外分光光度法;保健食品;检测分析;食品安全管理Application of Ultraviolet Spectrophotometry in the Detectionof Health FoodLUO Tingting(Guangxi Huace Testing and Certification Co., Ltd., Nanning 530000, China) Abstract: Ultraviolet spectrophotometry is a commonly used spectral analysis technique that can be used to analyze parameters such as the structure, concentration, and purity of substances. In the detection of health food, ultraviolet spectrophotometry is also widely used, which can be used to detect the content, quality, and safety of ingredients in health food. This study briefly analyzed the application of ultraviolet spectrophotometry in the detection of health food, discussed the necessity of ultraviolet spectrophotometry in the detection of health food, and discussed the application of ultraviolet spectrophotometry in the determination of health food, aiming to provide assistance and reference for the future production and management of health food in China.Keywords: ultraviolet spectrophotometry; health food; detection analysis; food safety management紫外分光光度法是一种常用的光谱分析技术,利用化合物分子在紫外区或可见光区的吸收特性,通过检测样品溶液中不同波长光线的吸收强度,确定分子的结构、浓度、纯度等参数[1]。
美国FDA食品接触材料相关检测标准

美国FDA食品接触材料相关检测标准
随着食品生产技术的日益多元化,食品安全问题已经不仅仅限于食品本身,还包括与食品直接或间接接触的材料。
这些材料包括食品容器、包装材料、餐厨具等,统称为食品接触材料。
由其导致的食品安全问题越来越受到社会各界的关注。
食品接触材料的测试也称为“食品级测试”。
欧盟和美国对食品接触材料管控非常严格,要求出口到欧盟及美国的产品必须通过相应的测试认证。
我国对不同的食品接触材料也有着严格的卫生要求。
因此,无论是出口或是在国内销售的食品接触材料,都必须通过相应的质量安全检测。
莱德凭借专业的技术人才及实验室,针对不同的产品类型、出口国家等,为客户提供全面的检测、认证及咨询服务。
在食品接触材料方面,我们能够为您提供以下FDA检测标准要求:
美国要求
欧盟要求
中国要求。
国外主要无损检测标准含中英文名称对照

ASTM A 754/A 754M-1996X射线荧光法测量涂层厚度的试验方法Test Method for Coating Thickness by X-Ray FluorescenceASTM B567-1998用B射线背散射法测量涂层厚度的试验方法Test Method for Measurement of Coating Thickness by the Beta Backscatter MethodASTM B568-1998X射线光谱仪法测量涂层厚度的试验方法Test Method for Measurement of Coating Thickness by X-Ray SpectrometryASTM C637-1998辐射屏蔽混凝土用集料的标准规范Standard Specification for Aggregates for Radiation-Shielding ConcreteASTM C638-1992辐射屏蔽混凝土集料组分的描述术语Descriptive Nomenclature of Constituents of Aggregates for Radiation-Shielding Concrete ASTM C1455-2000用Y射线谱法无损检定仍然有效特殊核材料指南ASTM D2599-1987X射线光谱法测定汽油含铅量的试验方法(05.02)Test Method for Lead in Gasoline by X-Ray Spectrometry (05.02)ASTM D4294-1998用非色散X射线荧光光谱法测定石油产品中含硫量试验方法Sulfur in Petroleum Products by Non-Dispersive X-Ray Fluorescence Spectrometry, Method of Test for (05.02)ASTM D4452-1985 土壤样品的X 射线照相法X-Ray Radiography of Soil SamplesASTM D5059-1998X-射线光谱法测定汽油含铅量的试验方法Test Method for Lead in Gasoline by X-Ray Spectroscopy (05.03)ASTM D5187-1991X射线衍射法测定煅烧石油焦炭中结晶尺寸(LC)的试验方法Test Method for Crystallite Size (LC) of Calcined Petroleum Coke by X-Ray Diffraction (05.03) ASTM D6247-1998X射线荧光光谱法分析聚烯烃中元素含量的试验方法Test Method for Analysis of 日emental Content in Polyolefins by X-Ray Fluorescence SpectrometryASTM E94-2004(2010)射线照相检验标准指南Standard Guide for Radiographic Examination ASTM E142-1996 射线照相检测的质量控制方法Method for Controlling Quality of Radiographic TestingASTM E155-2010铝镁合金铸件射线照相检验标准参考照片Standard Reference Radiographs for Inspection of Aluminum and Magnesium CastingsASTM E170-1999有关辐射测量和剂量测定的术语ASTM E181-1998放射性核素探测器的校准和分析的一般方法General Methods for Detector Calibration and Analysis of RadionuclidesASTM E186-2010厚壁(50.8-114mm)钢铸件标准参考射线照片Standard Reference Radiographs for Heavy-Walled (2 to 4 /2-In./50.8 to 114-mm) Steel Castings ASTM E192-2004(2010)e1宇航用熔模钢铸件标准参考射线照片Standard Reference Radiographs for Investment Steel Castings of AerospaceApplicationsASTM E242-2001(2010)某些参数改变时射线照相图象显示的标准参考射线照片Standard Reference Radiographs for Appearances of Radiographic Images asCertain Parameters Are ChangedASTM E272-2010高强度铜基及银铜合金铸件的标准参考射线照片Standard Reference Radiographs for High-Strength Copper-Base andNickel-Copper Alloy CastingsASTM E280-2010厚壁(114-305mm)铸钢件标准参考射线照片Standard Reference Radiographs for Heavy-Walled (4 R to 12-in. (114 to305-mm)) Steel CastingsASTM E310-2010锡青铜铸件标准参考射线照片Standard Reference Radiographs for Tin Bronze CastingsASTM E390-2011钢熔焊焊缝标准参考射线照片Standard Reference Radiographs for Steel Fusion WeldsASTM E431-96(2011)半导体和相关器件射线照片判读指南Standard Guide to Interpretation of Radiographs of Semiconductors andRelated DevicesASTM E446-2010厚度至50.8mm钢铸件的标准参考射线照片Standard Reference Radiographs for Steel Castings up to 2 in. (50.8 mm) inThicknessASTM E505-2001(2011)铝和镁压铸件检验的标准参考射线照片Standard Reference Radiographs for Inspection of Aluminum and Magnesium Die Castings ASTM E545-2005(2010)确定直接热中子射线照相检验成象质量的标准试验方法Standard Test Method for Determining Image Quality in Direct ThermalNeutron Radiographic ExaminationASTM E586-88Y与X射线照相检测的术语定义ASTM E592-1999(2009)e16〜51mm厚钢板X射线照相检验和25〜152mm厚钢板钻60照相检验获得ASTM当量穿透灵敏度的标准指南Standard Guide to Obtainable ASTM Equivalent Penetrameter Sensitivity forRadiography of Steel Plates 14 to 2 in. (6 to 51 mm) Thick with X Rays and 1 to 6 in. (25 to 152 mm) Thick with Cobalt-60ASTM E665-1994测量暴露在X闪光射线机的X射线照射下的材料中相对深度的吸收剂量Determining Absorbed Dose Versus Depth in Materials Exposed to the X-RayOutput of Flash X-Ray MachinesASTM E666-1997Y 或X 射线剂量吸收的计算Calculating Absorbed Dose from Gamma or X RadiationASTM E689-2010球墨铸铁标准参考射线照片Standard Reference Radiographs for Ductile Iron CastingsASTM E746-2007测定工业射线照相成像系统相关图象质量响应的标准方法Standard Practice for Determining Relative Image Quality Response ofIndustrial Radiographic Imaging SystemsASTM E747-2004(2010)射线照相用线型象质计(IQI)的设计、制造及材料组分类的标准方法Standard Practice for Design, Manufacture, and Material GroupingClassification of Wire Image Quality Indicators (Iqi) Used for RadiologyASTM E748-2002(2008)材料热中子射线照相标准方法Standard Practices for Thermal Neutron Radiography of MaterialsASTM E801-2006(2011)电子装置射线照相检验的质量控制标准方法Standard Practice for Controlling Quality of Radiological Examination of Electronic Devices ASTM E802-1995(2010)厚度至114mm的灰口铸铁标准参考射线照片Standard Reference Radiographs for Gray Iron Castings up to 4 1 in. (114 mm) in ThicknessASTM E803-1991(2008)确定中子射线透照束长径比的标准方法Standard Method for Determining the L/D Ratio of Neutron Radiography BeamsASTM E915-1996残余应力测量用X射线衍射仪校准检定的试验方法Test Method for Verifying the Alignment of X-Ray DiffractionInstrumentation for Residual Stress MeasurementASTM E999-2010工业射线照相胶片处理的质量控制标准指南Standard Guide for Controlling the Quality of Industrial Radiographic FilmProcessingASTM E1000-98(2009)射线照相检测标准指南Standard Guide for RadioscopyASTM E1025-2011射线照相检测用孔型象质计设计、制造和材料组分类的标准方法Standard Practice for Design, Manufacture, and Material GroupingClassification of Hole-Type Image Quality Indicators(IQI) Used forRadiographyASTM E1030-2005(2011)金属铸件射线照相检验的标准试验方法Standard Test Method for Radiographic Examination of Metallic CastingsASTM E1032-2012焊缝射线照相检验的标准试验方法Standard Test Method for Radiographic Examination of WeldmentsASTM E1079-2010透射密度计校准的标准方法Standard Practice for Calibration of Transmission DensitometersASTM E1114-2009e1测定铱192工业射线照相源尺寸的标准试验方法Standard Test Method for Determining the Size of Iridium -192 IndustrialRadiopraphic SourcesASTM E1161-2009半导体和电子元件射线检验的标准方法Standard Practice for Radiologic Examination of Semiconductors andElectronic ComponentsASTM E1165-2004(2010)用针孔成象法测量工业X射线管焦点的标准试验方法Standard Test Method for Measurement of Focal Spots of Industrial X-RayTubes by Pinhole ImagingASTM E1168-1995 核设施工人辐射防护训练Radiological Protection Training for Nuclear Facility WorkersASTM E1254-2008射线照片及未曝光工业射线照相胶片储藏的标准指南Standard Guide for Storage of Radiographs and Unexposed IndustrialRadiographic FilmsASTM E1255-2009射线透视检验标准方法Standard Practice for RadioscopyASTM E1320-2010钛铸件标准参考射线照片Standard Reference Radiographs for Titanium CastingsASTM E1390-2012工业射线照相观片灯标准规范Standard Specification for Illuminators Used for Viewing IndustrialRadiographsASTM E1400-1995高剂量辐射量测定校准实验室的特性和性能规程Characterization and Performance of a High-Dose Radiation DosimetryCalibration Laboratory, Practice for (12.02)ASTM E1411-2009射线照相系统鉴定的标准方法Standard Practice for Qualification of Radioscopic SystemsASTM E1416-2009 焊缝射线检验的标准试验方法Standard Test Method for Radioscopic Examination of WeldmentsASTM E1441-2011计算机层析(CT)成像的标准指南Standard Guide for Computed Tomography (CT) ImagingASTM E1441-2000 计算机层析成像(CT)指南Guide for Computed Tomography (CT) Imaging ASTM E1453-2009含模拟或数字射线照相数据的磁带媒体存储标准指南Standard Guide for Storage of Magnetic Tape Media that Contains Analog orDigital Radioscopic DataASTM E1475-2002(2008)数字射线照相检验数据计算机化传输的数据区标准指南Standard Guide for Data Fields for Computerized Transfer of DigitalRadiological Examination DataASTM E1496-2005(2010)中子射线照相尺寸测量的标准试验方法Standard Test Method for Neutron Radiographic DimensionalMeasurements(With drawn 2012)ASTM E1570-2011计算机层析(CT)检验标准方法Standard Practice for Computed Tomographic (CT) ExaminationASTM E1647-2009确定射线照相检测对比度灵敏度的标准方法Standard Practice for Determining Contrast Sensitivity in RadiologyASTM E1648-1995(2011)铝熔焊焊缝检验标准参考射线照片Standard Reference Radiographs for Examination of Aluminum Fusion WeldsASTM E1672-2006选择计算机层析(CT)系统的标准指南Standard Guide for Computed Tomography (Ct) System SelectionASTM E1695-1995(2006)e1计算机层析(CT)系统性能测量的标准试验方法Standard Test Method for Measurement of Computed Tomography (Ct) System PerformanceASTM E1734-2009铸件射线照相检验标准方法Standard Practice for Radioscopic Examination of CastingsASTM E1735-2007确定经4-25MV X射线曝光的工业射线胶片相关成像质量的标准试验方法Standard Test Method for Determining Relative Image Quality of IndustrialRadiographic Film Exposed to X-Radiation from 4 to 25 MVASTM E174〃E1742M-2011射线照相检验标准方法Standard Practice for Radiographic ExaminationASTM E1814-1996(2007)铸件计算机层析(CT)检验标准方法Standard Practice for Computed Tomographic (CT) Examination of CastingsASTM E1815-2008工业射线照相胶片系统分类的标准试验方法Standard Test Method for Classification of Film Systems for IndustrialRadiographyASTM E1817-2008使用典型象质计(RQIs)控制射线检验质量的标准方法Standard Practice for Controlling Quality of Radiological Examination byUsing Representative Quality Indicators(RQI-s)ASTM E1894-1997选择脉冲X射线源用的剂量测定系统的标准指南Standard Guide for Selecting Dosimetry Systems for Application in PulsedX-Ray SourcesASTM E1931-2009X射线康普顿散射层析技术标准指南Standard Guide for X-ray Compton Scatter TomographyASTM E1935-1997(2008)校准和测量计算机层析(CT)密度的标准试验方法Standard Test Method for Calibrating and Meausring CT DensityASTM E1936-2003(2011)评估射线照相数字化系统性能的标准参考射线照片Standard Reference Radiograph for Evaluating the Performance ofRadiographic Digitization SystemsASTM E1955-2004(2009)与美国材料与试验协会ASTM E 390参考射线照片等级比较钢中焊缝完善性的标准射线检验Standard Radiographic Examination for Soundness of Welds in Steel byComparison to Graded ASTM E390 Reference RadiographsASTM E2002-1998(2009)测定射线照相图象总不清晰度的标准方法Standard Practice for Determining Total Image Unsharpness in RadiologyASTM E2003-2010中子射线照相波束纯度指示计制作的标准方法Standard Practice for Fabrication of the Neutron Radiographic Beam PurityIndicators [Metric]ASTM E2007-2010计算机射线照相标准指南(用于CR的标准指南)(可激射线发光[PSL]法)Standard Guide for Computed Radiology (Photostimulable Luminescence (PSL)Method)ASTM E2023-2010制作中子射线照相灵敏度指示计的标准方法Standard Practice for Fabrication of Neutron Radiographic SensitivityIndicatorASTM E2033-1999(2006)计算机射线照相的标准方法(用于CR的标准实施方法)(可激射线发光晓1]法)Standard Practice for Computed Radiology (Photostimulable LuminescenceMethod)ASTM E2104-2009优质航空与涡轮材料和构件射线照相检验的标准方法Standard Practice for Radiographic Examination of Advanced Aero andTurbine Materials and ComponentsASTM E2120-2000便携式X射线荧光光谱仪测量涂膜中铅含量的性能评估规程Practice for the Performance Evaluation of the Portable X-Ray FluorescenceSpectrometer for the Measurement of Lead in Paint FilmsASTM E2339-2004无损评价中的数字成像和通讯Digital Imaging and Communication in NDE(DICONDE)ASTM E2422-2011铝铸件标准参考数字射线图像(钛和钢铸件也适用)Standard Digital Reference Images for Al. Casting(Titanium & steel Casting also available) ASTM E2445-2005(2010)计算机射线照相系统的长期稳定性与鉴定的标准方法(用于CR系统的质量认定和长期稳定性的标准实施方法)Standard Practice for Qualification and Long-Term Stability of ComputedRadiology SystemsASTM E2446-2005(2010)计算机射线照相系统分类的标准方法(用于CR系统分类的标准实施方法)Standard Practice for Classification of Computed Radiology SystemsASTM E2597-2007e1数字探测器阵列制造特性的标准规程Standard Practice for Manufacturing Characterization of Digital DetectorArraysASTM E2660-2011航空用优质钢铸件标准参考数字射线图像Standard Digital Reference Images for Investment Steel Castings forAerospace ApplicationsASTM E2662-2009航空用平面与夹芯复合材料射线照相检验的标准方法Standard Practice for Radiologic Examination of Flat Panel Composites andSandwich Core Materials Used in Aerospace ApplicationsASTM E2669-2011数字射线照相(DR)检测方法的数字图像与通信无损评价(DICONDE)的标准方法Standard Practice for Digital Imaging and Communication in NondestructiveEvaluation (DICONDE) for Digital Radiographic (DR) Test MethodsASTM E2698-2010使用数字探测器阵列的射线照相检验标准方法Standard Practice for Radiological Examination Using Digital DetectorArraysASTM E2736-2010数字探测器阵列射线照相检测标准指南Standard Guide for Digital Detector Array RadiologyASTM E2737-2010评价数字探测器阵列性能和长期稳定性的标准方法Standard Practice for Digital Detector Array Performance Evaluation andLong-Term StabilityASTM E2738-2011使用计算机射线照相(CR)检测方法的数字图像与通讯无损评价(DICONDE) 的标准方法Standard Practice for Digital Imaging and Communication NondestructiveEvaluation (DICONDE) for Computed Radiography (CR) Test MethodsASTM E2767-2011使用X射线计算机层析(CT)检测方法的数字图像与通讯无损评价(DICONDE)的标准方法Standard Practice for Digital Imaging and Communication in NondestructiveEvaluation (DICONDE) for X-ray Computed Tomography (CT) Test MethodsASTM E2861-2011测量中子辐射束发散与校准的标准试验方法Standard Test Method for Measurement of Beam Divergence and Alignment inNeutron Radiologic BeamsASTM F629-1997铸造金属外科手术植入物射线照相检查实施方法(14)ASTM F727-1981透明照相干版透光度测量的试验方法Test Method for Measuring Transmittance of See-Through PhotoplateASTM F784-1982校准放射性同位素密封测试仪的试验方法Test Method for Calibrating Radioisotope Hermetic Test ApparatusASTM F864-1984 硬表面玻璃照相干板的检验Inspection of Hard-Surface Glass Photoplates ASTM F947-1985测定照相胶片低能级X射线辐射灵敏度的试验方法Test Method for Determining Low-Level X-Radiation Sensitivity ofPhotographic FilmsASTM F1035-1991使用橡胶帘布圆盘验证轮胎X射线成象系统的辩别能力Use of Rubber-Cord Pie Disk to Demonstrate the Discernment Capability of aTire X-Ray Imaging SystemASTM F1039-1987X射线安全屏系统中测量低剂量X辐射的试验方法Test Method for Measurement of Low Level X-Radiation Used in X-RaySecurity Screening SystemsASTM F1467-1999微电子装置电离辐射效应中X射线测试仪(近似等于10keV辐射量子)的使用Use of an X-Ray Tester (is Approximately Equal to 10 keV Photons) inIonizing Radiation Effects Testing of Microelectronic DevicesASTM PS95-1998便携式X射线荧光(XRF)装置现场测定涂料或其它涂层含铅量的质量体系的标准临时操作规程Standard Provisional Practice for Quality Systems for Conducting In SituMeasurements of Lead Content in Paint or Other Coatings usingField-Portable X-Ray Fluorescence (XRF) DevicesASTM PS 116-1999测量涂膜含铅量用的便携式X射线荧光光谱仪性能评价的临时操作规程Provisional Practice for the Performance Evaluation of the Portable X-RayFluorescence Spectrometer for the Measurement of Lead in Paint FilmsANSI/ANS6.1.1-1991中子及r射线对剂量因素的影响Neutron and Gamma-Ray Fluence-to-DoseFactorsANSI/IEEE 309-1970 盖革-弥勒计数器的试验程序Geiger-Muller Counters, Test Procedure for ANSI IT9.2-1991成象介质-已处理的照相胶片、平板和相纸-归档盒及储存箱Imaging Media - Photographic Processed Films, Plates and Papers - FilingEnclosures and Storage ContainersANSI IT9.8-1989成象介质-照相胶片耐折强度的测定Imaging Media - Photographic Film - Determination of Folding EnduranceANSI N13.2-1969 辐射监测的管理规程指南Administrative Practices in Radiation Monitoring, Guide toANSI N13.5-1972直读和非直读式袖珍X和Y射线辐射剂量仪的性能Direct Reading and Indirect Reading Pocket Dosimeters for X- and GammaRadiation, PerformanceANSI N13.7-1983辐射防护照相胶片剂量仪性能标准Radiation Protection - Photographic Film Dosimeters - Criteria forPerformanceANSI N13.11-2001 个人剂量测定的试验标准Personnel Dosimetry Performance, Criteria for TestingANSI N13.27-1981袖珍式报警辐射剂量仪和报警记数率计的性能要求Performance Requirements for Pocket-Sized Alarm Dosimeters and AlarmRatemetersANSI N15.36-1994核材料无损化验测量的控制和保证Nuclear materials - Nondestructive assay measurement control and assuranceANSI N15.37-1981核材料控制的自动无损化验系统指南Automation of Nondestructive Assay Systems for Nuclear Materials Control,Guide toANSI N42.16-1986用于液体闪烁计数器的密封放射检查源的规范Specifications for sealed radioactive check sources used in liquid-scintillation countersANSI N42.20-1995 个人辐射监视仪的性能标准Performance criteria foractive personnel radiation monitorsANSI N42.26-1995辐射防护仪器监测设备X和Y辐射个人报警装置Radiation Protection Instrumentation - Monitoring Equipment - PersonalWarning Devices for X and Gamma RadiationsANSI N43.3-1993通用辐射安全非医疗应用的X射线和密封Y射线源的安装能量达10MevGeneral radiation safety - Installations using non-medical X-ray andsealed gamma-ray sources, energies up to 10 MeVANSI N43.6-1997 密封放射性源的分类Classification of SealedRadioactive SourcesANSI N43.9-1991Y射线照相仪器的设计和试验规范Gamma Radiography - Specifications for Design and Testing of ApparatusANSI N322-1996直接和间接读取石英纤维袖珍剂量计的检验和试验规范Inspection and Test Specifications for Direct and Indirect Reading QuartzFiber Pocket Dosimeters ASME Boiler & Pressure VesselCode(ASME锅炉压力容器规范)第V卷《无损检测》2004版,第2篇“射线检测”,强制性附录-包含动态射线照相、实时射线成像检测内容ASME SE-1647确定射线照相对比灵敏度的推荐实施方法ASME Code Case 2476使用荧光成像板的射线照相Radiography using phosphor imaging platesMIL-HDBK-55-66射线照相无损检测手册(已由MIL-HDBK-7285取代)MIL-STD-139A-65射线检测铝镁合金铸件的完好性要求MIL-STD-453C-88射线照相检测MIL-STD-746A-63铸造爆破器材的射线照相检测要求MIL-STD-779-68钢焊缝参考X射线照片(由ASTM E390取代)MIL-STD-1257A-87钻铭合金枪管射线照相及目视检验MIL-R-11470A-71对射线检验设备,操作方法和操作人员的合格审查(由MIL-STD-453取代) MIL-I-36013B-72折迭式X射线观片灯MIL-R-45226-62石墨的射线照相检测(已停用)MIL-R-45774A(92)铝,镁导弹零件熔焊完好性要求-射线照相检测MIL-STD-1948(91)中子射线照相检验的术语和定义汇编MIL-HDBK-7285(92)射线照相检验MIL-HDBK-733(92)复合材料无损检验方法-射线照相法MIL-STD-1166A(91)固体火箭推进剂射线照相检验要求MIL-STD-1264B(93)钢焊缝完好性射线照相检验-与ASTM E390各级参考底片比较MIL-STD-1265A(92)钢铸件射线照相检验分类和完好性要求MIL-STD-1894A(86)不完全焊透钢焊缝的射线照相参考标准及射线照相程序MIL-STD-1895A(86)不完全焊透铝焊缝的射线照相参考标准及射线照相程序BAC 5915(美国波音公司)射线检验DPS 4.736(美国麦道公司)射线检验API 1104(美国石油协会)管道及有关设备的焊接AWS B 5.15射线照相评片资格技术条件。
外文翻译(英文)利用IR,SEM和维尔卡技术检测硅酸盐水泥的早起水化及其制备过程

Early hydration and setting of Portland cement monitored by IR,SEM and Vicat techniquesRikard Ylmén,Ulf Jäglid,Britt-Marie Steenari,Itai Panas ⁎Department of Chemistry and Biotechnology,Environmental Inorganic Chemistry,Chalmers University of Technology,S-41296Gothenburg,Swedena b s t r a c ta r t i c l e i n f o Article history:Received 26November 2007Accepted 30January 2009Keywords:HydrationCalcium-silicate-hydrate (C-S-H)Spectroscopy Cement paste Portland cementDiffuse Re flectance Infrared DR-FTIR spectroscopy is employed to monitor chemical transformations in pastes of Portland limestone cement.To obtain a suf ficient time resolution a freeze-dry procedure is used to instantaneously ceasing the hydration process.Rapid re-crystallization of sulphates is observed during the first 15s,and appears to be complete after ~30min.After ~60min,spectroscopic signatures of polymerizing silica start to emerge.A hump at 970–1100cm −1in conjunction with increasing intensity in the water bending mode region at 1500–1700cm −1is indicative of the formation of Calcium Silicate Hydrate,C-S-H.Simultaneously with the development of the C-S-H signatures,a dip feature develops at 800–970cm −1,re flecting the dissolution of Alite,C 3S.Setting times,180(initial)and 240(final)minutes,are determined by the Vicat bining DR-FTIR,SEM and Vicat measurements it is concluded that the setting is caused by inter-particle coalescence of C-S-H.©2009Elsevier Ltd.All rights reserved.1.IntroductionToday,Portland cement is a widely used binder in concrete construction.C 3S (alite)and C 2S (belite)is essential to the build-up of strength in Portland cement.These two calcium-silicate phases are formed above 800°C,where C 3S is preferentially formed upon elevating the temperature and increasing amount of added burned lime,CaO.C 3S is responsible for short term strength development (days to months)while C 2S displays the better long term strength development performances (~years).The quest for increasingly shorter setting time and early strength has seen the C 3S/C 2S ratio increase in commercial Portland cement.In recent years,the increased attention on environmental aspects of material conversion has in fluenced research towards possible modi fications of Portland cement to better meet the increasing demands for sustainability in the construction sector.This is done by using additives and changing the composition of the cement.Many different experimental techniques have been employed to investigate the effects on material conversion as Portland cement is dissolved and transformed into calcium-silicate-hydrate,C-S-H.For determination of setting times,Vicat measurements are often employed.At later stages in the hydration process,an ultrasonic cement analyser may be used to determine changes in the elastic modulus of the mortar [1,2].Calorimetry is employed to monitor the heat released upon hydration [3–7],whereas X-ray diffraction [8–13],nuclear magnetic resonance [14–16]and Fourier transform infrared spectroscopy,FTIR,are used toobtain chemical information.Morphological information may be obtained by means of scanning electron microscopy and transmission electron microscopy [11,12,15,17].Spectroscopic methods are commonly used to study the chemistry of cement hydration.In the present work the hydration of Portland cement has been monitored mainly by means of infrared spectroscopy.In infrared spectroscopy one utilizes that molecules or groups of atoms on large molecules absorbs different wavelengths of infrared light depend-ing on which atoms that constitute the molecule or group,its geometry and its immediate surroundings.It can therefore be used to study both crystalline and amorphous samples.The sample is irradiated with infrared light with a span of different wavelengths.The sample will absorb some of the light at wavelengths that are characteristic to its chemical composition.To see at which wavelengths the sample has absorbed light the intensity at each wavelength is measured with and without sample.IR radiation only penetrates about 1wavelength into the sample (~10µm for 1000cm −1),making it ideal in the study of surface processes.In previous studies where FTIR was used to study the hydration of cement and its components,the sample was prepared by mixing the cement with KBr and pressing the mixture into pellets [18–21].The usefulness of Diffuse Re flectance Fourier Transform Infrared Spectro-scopy,DR-FTIR,as a tool for studying the hydration of cement has also been demonstrated in previous work [22,23].A comparison between DR-FTIR and the KBr pellet technique has been done by Delgado et al.[24],who showed that the methods produce similar spectra.The advantage of the KBr technique is that it provides better de fined bands than DR-FTIR,but the sample preparation is more labour intensive.The results of the present study suggest that the DR-FTIR technique employed is indeedCement and Concrete Research 39(2009)433–439⁎Corresponding author.Tel.:+46317722860;fax:+46317722853.E-mail address:itai@chalmers.se (I.Panas).0008-8846/$–see front matter ©2009Elsevier Ltd.All rights reserved.doi:10.1016/j.cemconres.2009.01.017Contents lists available at ScienceDirectCement and Concrete Researchj ou r n a l h o m e pa g e :ht t p ://e e s.e l s e v i e r.c o m /C E MC ON /d e f a ul t.a s ppreferred in that external physico-chemical interference is minimized,i.e.the hydration products are studied in the proper cement matrix with a minimum of sample tampering,and avoiding contact with foreign chemicals.Differential IR light absorption of samples which have been allowed to hydrate for different times is reported here.Water displays strong absorption in the mid-IR range,which makes it virtually impossible to perform in situ studies of cement hydration.A second draw back of in situ DR-FTIR for the study of cement hydration is that the surface of the cement paste,while hydrating,may become too flat for the diffuse re flectance technique to be ef ficiently used.These considerations validate selection of an ex situ DR-FTIR approach.To study very early hydration using an ex situ technique,it is imperative that the hydration is stopped instantaneously at a predetermined time.To satisfy this requirement,a freeze-dry technique is adopted in this research.The freezing of the sample with liquid nitrogen ensures that all chemical processes are very much retarded,while the subsequent water evaporation step at low temperature minimizes any thermally induced chemical transforma-tions other than water removal while drying.Indeed,earlier microscopy work [25–27]has shown that freezing is a relatively mild method to stop hydration.The drying will of course affect the structures of some phases.Bound water,like in ettringite,could be partially removed,and morphological properties may change upon removal of water.The purpose of the present study is to demonstrate the ef ficiency of the freeze-dry procedure in conjunction with DR-FTIR spectroscopy for studying the complex hydration chemistry of Portland cement.An attempt to correlate relevant spectroscopic signatures to the devel-opment of strength in the system is also made.Strength development is monitored here by means of Vicat measurements.2.ExperimentalThe Portland cement used was a Portland limestone cement,“byggcement Std PK Skövde CEM II/A-LL 42,5R ”,from Cementa AB.An automatic/manual mortar mixer 39-0031from ELE International was used.The cement was mixed with distilled deionized water that was poured into the mixing bowl before adding the cement.The ratio of water to as received dry cement was 0.4by weight in both DR-FTIR and Vicat measurements.The cement was carefully added and the paste was mixed at 140rpm on the mixing blade and 62rpm on the mixing head.The hydration time was measured from the instant when the cement was added to the water.2.1.DR-FTIRThe spectrometer used was a Nicolet Magna-IR 560with an insert cell for diffuse re flectance spectroscopy.The measurement range liesbetween 400and 4000cm −1.The diffuse re flectance technique is utilized,in which the incident beam is allowed to be re flected off the ground sample towards an overhead mirror upon which the diffusely scattered rays are collected and measured in the detector.A more detailed description is given by Fuller and Grif fiths [28].The sample is scanned 64times with a resolution of 2.0cm −1and the presented data is an average value.Each sample was prepared and analyzed 3times and the final spectrum was an average of these 3measurements to minimize differences due to sample preparation.The batch size was 200g of as received dry cement.As the cement hydration was studied from 15s the cement paste was only mixed for 15s.However,the chemical development of the cement paste was found to be insensitive of mixing time as long as the cement was completely wetted [29].Samples were prepared in plastic dishes of 35mm in diameter.The thickness of the paste in the dishes was ~2–3mm.Lids were placed over the dishes while they hydrated to prevent water from evaporating.The samples were hydrated between 15s and 360min in normal laboratory environment,then frozen by immersion in liquid nitrogen and subsequently placed in the freeze drier overnight.Measurements were made the following day.Before measurement the sample was ground and placed in the sample holder of the DR-FTIR spectrometer.To obtain good reproducibility,great care was taken when grinding the samples and placing them in the sample cup to make the samples as similar as possible.2.2.VicatThe batch size was 300g of as received dry cement and the cement paste was mixed for 2⁎90s with a stop in between for 15s to scrape the paste from the inside walls.The Vicat apparatus used was a Vicatronic automatic recording apparatus E040and measurements were performed in a 40mm mould with a calibrated weight of 300g and a cylindrical needle with flat tip area of 1mm 2.2.3.Scanning electron microscopyThe microscope used was a FEI Quanta 200FEG ESEM operated in secondary electron detection mode with high-vacuum and an acceleration voltage of 2kV.Some of the freeze-dried samples were pulverized.Since the freeze-dried samples were barely holding together this was easily done with a metal spoon.Some of the powder was placed on carbon tape attached to the sampleholder.Fig.1.Vicat measurement showing the depth of penetration of the Vicat needle into the cement as function of time.The height of the mould was 40mm.Table 1Possible assignment to some of the peaks observed in Figs.2–5.Wave number [cm −1]Possible assignment Reference656–658υ4of SiO 4[21,40]714υ4of CO 3[22,32,35,37]847–848Al –O,Al –OH [21,35]877–878υ2of CO 3[21,22,35,37]1011–1080Polymerized silica [19]~1100–1200υ3of SO 4[19,22,31,32]1200–1202Syngenite,thenardite [32–34]1400–1500CO 3[19,21,22,35,37]1620–1624υ2of water in sulphates [22,31,33]1640–1650υ2H 2O[21,35,36]1682–1684υ2of water in sulphates [22,31,33]1795–1796CaCO 3Own measurement,[22]2513–2514CaCO 3Own measurement,[22]2875–2879CaCO 3Own measurement,[22]2983–2984CaCO 3Own measurement,[22]3319–3327Syngenite,thenardite [32–34]3398–3408υ3of H 2O,capillary water [36]3457υ1+υ3of H 2O[21,36]3554υ3of H 2O in gypsum [22,31]3611Bassanite [22]3641–3644Ca(OH)2Own measurement,[20,23,24,37]434R.Ylmén et al./Cement and Concrete Research 39(2009)433–439Several regions were examined to make sure that the observed structures were representative of the sample.3.ResultsThe present study attempts to correlate setting with the evolution of spectral features in DR-FTIR spectra during early hydration of cement.The Vicat setting time measurement for the used Portland cement is displayed in Fig.1.Initial andfinal set are seen to occur at 180min and240min respectively.In Section3.1,the overall time evolution of DR-FTIR absorption intensities is presented.Possible assignments of the different bands are shown in Table1,and interpreted in Sections3.1.2–3.1.4.3.1.Time resolved spectra of hydrating cementThe hydration process was monitored for thefirst six hours by applying the freeze dry method,grinding of sample and subsequently acquiring the DR-FTIR spectra.The recorded absolute spectra of dry and hydrated cement are displayed in Fig.2.It shows the spectra of theas received dry cement together with the cement just after it has been mixed(15s),after180min and360min of hydration.Weak signatures of hydration can be seen in the900–1200cm−1region.To enhance these effects,various difference spectra were constructed.In Fig.3,the difference spectra employ as received dry cement as reference.Now, the spectroscopic features can be seen significantly clearer and we observe the development and saturation band at1100–1200cm−1 already after15s.This is complemented by a more slowly growing feature at900–1100cm−1.Because the bands that developed after 15s cannot be associated with the actual hardening of cement paste, the15s spectrum was taken as reference in Figs.4and5.Fig.3 supports the overall procedure in that a smooth background is observed in the relevant spectral regions.Having found this,Fig.5 focuses on the500–2000cm−1interval and the spectra for twelve different hydration times are displayed.3.1.1.Sulphate bandsThe sulphates originally present in Portland cement are gypsum (CaSO4·2H2O),hemihydrate(bassanite,CaSO4·0.5H2O)and anhy-drite(CaSO4).The latter ones are formed when the gypsum is ground with the cement clinker.The heat makes some of the crystal water in the gypsum to dissociate.When water is added to the cement the sulphates react with the aluminate and ferrite phases of the cement to produce AFt phase.This phase in turn reacts further with the aluminate and ferrite phases to form the AFm phase[30].Characteristic sulphate absorption bands are generally found in the range1100–1200cm−1due to theυ3vibration of the SO42−-group in sulphates[19,22,31,32].It is very difficult to interpret this area by studying FTIR-spectra only,since the many forms of sulphates give rise to several peaks here and cause lots of overlaps,but also because the υ3vibration of the SiO42−-group can absorb in this region,especially when it has polymerized[21].Therefore no in-depth analysis of it will be done in this work.In the DR-FTIR spectrum of as received dry cement(Fig.2,bottom spectrum),a broad feature is seen in1100–1200cm−1region reflecting mainly amorphous sulphates.Immedi-ately after mixing with water,some sharp absorption bands develop at 1100cm−1,1200cm−1and3320cm−1,indicative of very rapid dissolution of sulphates followed by crystallization(Fig.2,15s spectrum).This can also be inferred by considering the15s difference spectrum in Fig.3.This spectrum corresponds to the difference between that acquired after15s of hydration,and the spectrum of dry cement.Spectral signatures of sulphate chemistry after15s of hydration,corresponding to re-crystallization are obtained.Appar-ently,crystalline sulphate phases form very early in the hydration process,after which they become inactive spectator phases.The extent to which this holds true can be assessed by replacing the as received dry cement reference spectrum for that of15s hydrated cement(Figs.4and5).From Fig.5we observe significant changes in the sulphate absorption bands up to30min of hydration.Apparently, intermediate phases are formed consistent with theabsorptionFig.2.Absorbance of as received dry cement and cement that has been allowed tohydrate for15s,180min and360min after the cement was added to the water.Thespectra are shown offset forclarity.Fig.3.Difference spectra where the absorbance spectrum of as received dry cement hasbeen subtracted from the absorbance spectra of cement hydrated for15s,180min and360min.The spectra are shown offset forclarity.Fig.4.Difference spectra in the range400–4000cm−1where the absorbance spectrumof the freshly mixed cement(15s)has been subtracted from the absorbance spectra ofcement hydrated for30s,5min,120min and360min.The spectra are shown offset forclarity.435R.Ylmén et al./Cement and Concrete Research39(2009)433–439spectra of syngenite (K 2Ca(SO 4)2·H 2O)and thenardite (Na 2SO 4)or closely related compounds [32–34].At any rate,after 60min,little changes can be seen in the sulphate absorption region of the spectra.3.1.2.Water associated bandsIn the spectrum for as received dry cement there is a peak at 1623cm −1and a smaller one at 1684cm −1.These are caused by the bending vibration υ2of water in sulphates,mainly gypsum [22,31,33].The peak at 3554cm −1is caused by the υ3vibration of water in gypsum [22,31]and the peak at 3611cm −1could be caused by bassanite (CaSO 4·0.5H 2O).As hydration progresses there is a broad feature forming with its centre at ~1650cm −1,caused by the bending vibration υ2of irregularly bound water [21,35,36].The consumption of gypsum can be seen as dips in this feature at 1623cm −1and 1680cm −1(Figs.4and 5).A small increase in gypsum during the first 10min is implied,and may be due to the transformations of anhydrite and bassanite.The “background ”level for wave numbers N 1600cm −1is steadily increasing with increasing hydration times.Since there seems to be no corresponding decrease in any other area,this is probably caused by the incorporation of water.The absorption intensities due to the υ2vibration mode of water at ~1650cm −1and the υ1+υ3modes at ~3450cm −1and results from Mollah et al.and Yu et al.support this observation [21,36].3.1.3.Silica associated bandsAfter about 2h of hydration new spectral intensity shifts are observed from ~900cm −1towards ~1000–1100cm −1(see Figs.3–5),neither associated with sulphates nor water,suggestive of rearrange-ments in the silica subsystem.These dip-hump features are taken to re flect dissolution of alite and simultaneously the polymerization ofsilica [21,23,37,38]to form calcium silicate hydrate C-S-H (vide infra ).In order to focus on the silica chemistry,the 15s reference spectrum is replaced by that acquired after 30min (see Fig.6),i.e.after the sulphate chemistry has stopped.Monotonous growth of the C-S-H associated absorption intensities (970–1100cm −1)is observed.The dip in the absorption spectrum at 800–970cm −1,which deepens with time,is due to the dissolution of the C 3S clinker phase [39].The intensities in the dip (800–970cm −1)and hump (970–1100cm −1)regions in Fig.6were integrated in an attempt to correlate the clinker dissolution with the silica polymerization.A horizontal line at the intensity at 970cm −1was used as baseline.The result is plotted in Fig.7.3.1.4.Hydroxides and carbonatesThe peak at 3643cm −1(see Table 1and Figs.2and 3)corresponds to Ca(OH)2,which is formed as silicate phases in the cement dissolve.The peaks at 1796cm −1,2513cm −1,2875cm −1,2983cm −1and the shoulder at 1350–1550cm −1are due to that portion of calcium carbonate,which is added to the cement by the manufacturer after clinker calcination.The amount of calcium carbonate is seen to decrease as the hydration progresses,i.e.negative absorption bands in the difference spectra of Figs.3and 4.This may partly be due to the reaction of calcite with the aluminate to form less crystalline phases such as carboxyaluminates [40,41]or the carbonate ion can substitute for sulphate ions in Aft and AFm phases [13,30].The peak growing at ~1070cm −1could be the υ1vibration of CO 3-group in the formed carbonates [33,35],but this observation would contradict theoverallFig.6.Difference spectra in the range 500–2000cm −1where the absorbance spectrum of cement hydrated for 30min has been subtracted from the absorbance spectra of cement with hydration times from 60–360min.The spectra are shown offset forclarity.Fig.7.Integrated value of the absorbance in the intervals 800–970cm −1(upper dots)and 970–1100cm −1(lower dots)in Fig.6as function of hydration time of the cement.The lines are drawn on free hand to guide the eye and does not represent a mathematicalmodel.Fig.5.Difference spectra in the range 500–2000cm −1where the absorbance spectrum of the freshly mixed cement (15s)has been subtracted from the absorbance spectra of cement with hydration times from 30s to 360min.The spectra are shown offset for clarity.436R.Ylmén et al./Cement and Concrete Research 39(2009)433–439Fig.8.SEM pictures of cement at different stages of hydration.a)Surface of unhydrated particle.b)Surface of particle hydrated for 15s.c)Surface of particle hydrated for 120min.d)Surface of particle hydrated for 240min.e)Surface of particle hydrated for 480min.f)Surface of particle hydrated for 480min at larger magni fication.437R.Ylmén et al./Cement and Concrete Research 39(2009)433–439reduction of carbonate absorption intensities with time.A more plausible candidate for this absorption band is the stretching vibration of Si–O,which is also found in jennite(Ca8(Si6O18H2)(OH)8Ca·6H2O) [37,38].3.2.SEMSEM pictures of cement grains at different stages of hydration are displayed in Fig.8.The surfaces of the unhydrated particles are bare, with debris lying on top(Fig.8a).After15s and120min of hydration (Fig.8b,c)the surfaces of the cement particles are still found to be bare,but lumps and platelets have formed in addition to the debris present already on the unhydrated particles.Fig.8d shows cement after240min of hydration.Now a carpet is covering the cement particles.The carpet has grown even more after480min of hydration and is seen to consist of needle-like protruding structures(Fig.8e,f).4.DiscussionA longstanding issue concerns the roles of various phases during early hardening of Portland cement.In particular the roles of sulphates,added to the Portland cement as anhydrous(CaSO4), hemihydrate(CaSO4·0.5H2O),and gypsum(CaSO4·2H2O)have been much discussed in this context.Indeed,the general consensus is that the dissolution and re-crystallization of the various sulphate contain-ing phases is completed well before the setting occurs[42,43].Yet,due to the complexity and instability of the early cement chemistry,the sulphates,besides their well known function as water absorbents, have been empirically found to affect the morphology of the hydrating paste both by providing a background ionic strength and by forming intermediate phases,which suppress“flash setting”.In the present study,results show that the sulphate related DR-FTIR absorption bands display large changes in the1100–1200cm−1interval but that this occurs mainly during thefirst10min of hydration,during which the development of sharp bands imply the formation of crystalline phases.The appearing platelets and hexagonal crystals seen with SEM are possibly associated with these phases.After30min,the inter-conversion of sulphate phases has apparently stopped.The sulphates formed are most probably ettringite or monosulphate,as earlier studies on cement hydration have shown that these sulphates are formed during thefirst minutes of hydration[11,43,44].In this study of the evolution of the C-S-H absorption bands,the30min spectrum was chosen as reference.The degree to which the sulphate chemistry is completed at this time can be appreciated by studying1100–1200cm−1region in Fig.6,keeping in mind that C-S-H also displays absorption bands in this interval.By DR-FTIR spectroscopy,detectable amounts of polymerized silica are formed after approximately1h of hydration,as seen in Fig.6in the 900–1100cm−1interval.It is gratifying to note how well the integrated intensities at800–970cm−1as function of time(Fig.7) correlate with the quantitative X-ray diffraction study on C3S hydration by Taylor et al.[45],who interpreted their results to imply C-S-H formation.The fact that the growth of the hump feature at970–1100cm−1follows the C3S dissolution process implies that the signature of polymeric silica indeed corresponds to C-S-H formation.It can be noted how the formation of polymerized silica(970–1100cm−1) is correlated in time with an increased incorporation of water in the structure as seen in the absorption interval at1500–1700cm−1.This supports further that calcium silica hydrate C-S-H is a major product formed upon early Portland cement hydration,as C-S-H consists of polymerized silica and calcium ions with water incorporated.It becomes interesting to attempt to correlate the materials conversion observed with DR-FTIR with morphological changes as seen with SEM.The acceleration phase of C-S-H formation starts somewhere between120and180min(Figs.6and7).Simulta-neously a growth of a needle-like phase is developed on the cement particles(Fig.8).This phase has been attributed to C-S-H in previous studies of alite,C3S,where no other phase than C-S-H and portlandite(Ca(OH)2)is formed[25,46].It is seen in Fig.1that the setting starts after180min,and that it is completed after240min. Since the conversion of the sulphates occurs during thefirst30min, the possibility that the needle-like phase is due to sulphates is ruled out.However,the acceleration phase of C-S-H formation(vide supra) occurs on the same time scale as the formation of the needle-like phase seen by SEM as well as that of the setting process.An identification of C-S-H as the phase responsible for the setting of the Portland cement is thus arrived at.Support is produced to the claim that C-S-H is responsible for the initial development of strength in Portland cement pastes.Also,it is suggested that C-S-H is formed continuously during hydration and in particular so prior to the setting.This implies that the actual setting is due to coalescence of clinker grains and that it is associated with the formation of sufficient amounts of C-S-H,to increase friction and bridge the inter-grain distances.The presentfindings are consistent with those of Chen and Odler [43],who reach the conclusion that setting in ordinary Portland cement is mainly due to the formation of C-S-H as long as the ratio between sulphates and C3A+C4AF is balanced,else“false setting”results due to the formation of ettringite or monosulphate.5.ConclusionsCement is a complex material,and its hydration possibly provides additional complexity.Indeed,as yet no single method exists which completely determines all chemical reactions taking place in a cement structure from the mixing and onward.Therefore several comple-mentary techniques must be used.In the present study,signatures of early setting of an untampered limestone Portland cement were extracted by correlating DR-FTIR,SEM, and Vicat measurements.The objective of this paper was to demonstrate how diffuse reflectance Fourier transform infrared spectroscopy in combination with freeze-drying may add a piece of the puzzle regarding material conversion during the very early stages of cement hydration, down to fractions of a minute.Whereas setting of each unique cement must be addressed separately,a method to monitor the material conversions during early hydration has been presented.Summarizing:•the time evolution of the sulphate chemistry displays very rapid crystallization followed by a slow recrystallization phase,which is completed within approximately30min;•the appearance of a broad absorption hump at970–1100cm−1after 60min of hydration is due to polymeric silica.It is correlated with the development of water bending vibration bands(1500–1700cm−1). This implies the formation of calcium silicate hydrate,C-S-H;•time dependent changes in morphology due to the hydration process,as monitored with SEM,were found to correlate with the DR-FTIR signatures of C-S-H formation,•the growth of a dip feature in the spectra at800–970cm−1,identified as the dissolution of C3S Alite,correlates with the formation of C-S-H.Vicat setting begins after180min and is completed after240min. This occurs well after the sulphate reactions have stopped.However, the C-S-H formation in the acceleration phase of C3S dissolution, displays the same time dependence as that of the setting process.The observations support the understanding of setting in terms of coalescing C-S-H coated Portland cement particles.AcknowledgementsThe support from the Knowledge foundation(KK stiftelsen),the Swedish Research Council,and Eka Chemicals Inc.,Bohus is gratefully acknowledged,as well as valuable discussions with Inger Jansson.438R.Ylmén et al./Cement and Concrete Research39(2009)433–439。
近红外光谱仪 国际标准

近红外光谱仪国际标准英文回答:The International Organization for Standardization (ISO) has developed a number of standards for near-infrared (NIR) spectroscopy. These standards cover a variety of topics, including:The following are some of the key ISO standards for NIR spectroscopy:ISO 12099:2010: Plastics Determination of the composition of plastics using near infrared spectroscopy.ISO 18593:2017: Foodstuffs Determination of protein content using near infrared spectroscopy.ISO 21445:2018: Cosmetics Determination of alcohol content using near infrared spectroscopy.ISO 21516:2018: Pharmaceuticals Determination ofactive ingredient content using near infrared spectroscopy.These standards provide guidance on the use of NIR spectroscopy for a variety of applications. They can help to ensure that NIR spectroscopy is used in a consistent and reliable manner.中文回答:国际标准化组织(ISO)制定了许多关于近红外(NIR)光谱的标准。
国外环境影响评价概述及国内外工作程序的比较

国外环境影响评价概述及国内外工作程序的比较作者:董文婉来源:《科技视界》 2013年第31期董文婉(中国石油安全环保技术研究院HSE评价中心,中国北京 102206)【摘要】伴随经济发展,环境也越来越受到人们的关注,而环境影响评价,作为一个新兴的行业,也逐渐走进人们的视野。
本文简述了国外环境影响评价工作的概况及工作程序,并与国内的环评程序相比较,讨论其中的差异和发展。
【关键词】环境影响评价;工作程序;国内外比较随着全球经济的发展,人类对环境的影响越来越大,而人类活动所造成的空气污染,土地沙漠化,植被破坏,物种灭绝等环境问题也成为人类自身发展的隐患。
人们开始关心环境,治理环境,并不断寻求“防患于未然”的更好的保护环境的方法。
环境影响评价(Environmental Impact Assessment,简称EIA)因此孕育而生。
1 国外环境影响评价的发展1969年,美国通过《国家环境政策法》,成为第一个把环境影响评价列入法律并确定其相应评价制度的国家。
1974年,新南威尔士州成为澳大利亚第一个建立环评导则的州政府。
1985年,欧洲共同体颁布了欧洲第一部环境影响评价相关的导则,随后1988年,英国正式为本国的环境影响评价立法[1]。
上世纪90年代到本世纪初,外国的环境影响评价迅速发展起来。
在英国环评立法的最初两年,每一年进行环评的项目只有约20个,而从1990年至1999年,平均每年进行环境影响评价的项目数量增长到约300个。
到了2003年,进行环评的项目已经超过600个。
同样的发展速度也出现在欧洲其他国家。
如丹麦、奥地利、西班牙等国,上世纪90年代到本世纪初,环评项目的年平均数均成倍的增长[2]。
这种发展趋势,不仅说明环评作为一个专业学科的兴起,也反应世界人民在近20年对于环境的关心程度日益增强。
2 国外环境影响评价的主要内容根据各国的国情不同,对于环境影响评价的要求也不尽相同。
笼统来说,环境影响评价的目的在于从环境(主要自然环境,也包括社会和经济环境)的角度,来评价、判断一个项目或者工程是否应该行进,怎样进行。
淀粉及其衍生物二氧化硫含量的测定

淀粉及其衍生物二氧化硫含量的测定GB/T 16886.5-2003Biological evaluation of medical devices--Part 5:Tests for in vitro cytotoxicity医疗器械生物学评价第5部分:体外细胞毒性试验GB/T 3048.2-2007Test methods of AC electrical motors - Part 2: Temperature rise test交流电动机试验方法第2部分:温升试验GB/T 16656.48-2010Industrial automation systems and integration - Product data representation and exchange - Part 48: Integrated generic resource: Core工业自动化系统与集成产品数据表达与交换第48部分:集成通用资源:核心GB/T 35370-2017Technical specification for designing and constructing of low voltage switchgear and controlgear assemblies低压成套开关设备和控制设备设计施工技术规范GB/T 2820.1-2009Reciprocating internal combustion engines - Exhaust emission - Part 1: Non-road mobile machinery往复式内燃机排气污染物排放第1部分:非公路移动机械GB/T 5590.2-1985Method for the measurement of the apparent sound power level of noise sources using sound pressure--Precision method for anechoic rooms用声压法测量噪声源表观声功率级绝对室精密法GB/T 30490-2014Metallic and other inorganic coatings―Electroplated coatings of zinc with supplementary treatments on iron or steel金属和其他无机覆盖层铁或钢上电镀锌及补充处理GB/T 12164-2008Test methods for properties of paper and board for food packaging 食品包装用纸及纸板性能试验方法GB/T 17625.1-1998Electromagnetic compatibility--Limits--Limits for harmonic current emissions(equipment input current≦16A per phase)电磁兼容限值谐波电流发射限值(设备输入电流≦每相16A)GB/T 2910.13-2009Textiles - Quantitative chemical analysis - Part 13: Mixtures of polyamide 6,6 and polyamide 6,polyamide 4,6 and polyamide 4,6,6,6纺织品定量化学分析第13部分:聚酰胺6,6与聚酰胺6,聚酰胺4,6及聚酰胺4,6,6,6的混合物GB/T 20255.2-2006Cranes - Design principles for loads and load combinations - Part 2: Mobile and locomotive cranes起重机载荷和载荷组合的设计原则第2部分:。
超高效液相色谱-串联质谱法测定化妆品中15种N-亚硝胺化合物

第42 卷第 11 期2023 年11 月Vol.42 No.111469~1478分析测试学报FENXI CESHI XUEBAO(Journal of Instrumental Analysis)超高效液相色谱-串联质谱法测定化妆品中15种N-亚硝胺化合物汪毅1,梁文耀1,何国山1,陈张好2,周智明2,吴谦1,席绍峰1,谭建华1*(1.广州质量监督检测研究院,国家化妆品质量检验检测中心(广州),广东广州511447;2.广东省药品检验所,广东广州510663)摘要:采用超高效液相色谱-串联质谱(UPLC-MS/MS)建立了化妆品中15种痕量N-亚硝胺化合物的分析方法。
水剂样品以水或乙腈分组超声提取,膏霜乳液样品采用亚铁氰化钾-乙酸锌溶液沉淀大分子或者饱和氯化钠-乙腈盐析分组处理后,以Agilent Poroshell 120 SB-Aq(100 mm×3.0 mm,2.7 μm)色谱柱分离,经大气压化学电离源(APCI)电离,多反应监测模式检测,以同位素内标法定量。
结果表明,15种N-亚硝胺化合物在相应质量浓度范围内线性关系良好(r2>0.995),检出限和定量下限分别为5~15 ng/g和15~45 ng/g。
水、乳、膏霜3种化妆品基质在25、50、100 ng/g加标水平下的平均回收率为88.0%~111%,相对标准偏差(RSD,n=6)为1.4%~9.8%。
该方法用于市售化妆品检测,发现13批次样品检出N-亚硝基二乙醇胺(NDELA),其中1批次超限量值。
方法的专属性强,灵敏度高,精密度好,解决了N-亚硝胺化合物稳定性差、易被干扰等问题,适用于化妆品中15种N-亚硝胺化合物的痕量测定。
关键词:N-亚硝胺化合物;化妆品;超高效液相色谱-串联质谱法(UPLC-MS/MS);大气压化学电离源中图分类号:O657.63;O623.732文献标识码:A 文章编号:1004-4957(2023)11-1469-10 Determination of Fifteen N-nitrosamine Compounds in Cosmetics by Ultra Performance Liquid Chromatography-TandemMass SpectrometryWANG Yi1,LIANG Wen-yao1,HE Guo-shan1,CHEN Zhang-hao2,ZHOU Zhi-ming2,WU Qian1,XI Shao-feng1,TAN Jian-hua1*(1.Guangzhou Quality Supervision and Testing Institute,National Quality Supervision and Testing Center for Cosmetics(Guangzhou),Guangzhou 511447,China;2.Guangdong Institute for Drug Control,Guangzhou 510663)Abstract:An ultra performance liquid chromatography-tandem mass spectrometric(UPLC-MS/MS)method was established for detecting 15 trace N-nitrosamine compounds in cosmetics. The final estab⁃lished method involved ultrasonic extraction of cosmetics using water or acetonitrile for different com⁃pounds. The samples were treated with potassium ferrocyanide-zinc acetate solution for precipitating macromolecules or saturated sodium chloride-acetonitrile for salting out.An Agilent Poroshell 120 SB-Aq(100 mm × 3.0 mm,2.7 μm) chromatography column was used for separation,followed by atmospheric pressure chemical ionization(APCI) source and multiple reaction monitoring mode detec⁃tion in the isotope internal standard method for quantification. The result showed good linearity(r2> 0.995) for the 15 N-nitrosamine compounds in their respective concentration ranges,with detection and quantitation limits of 5-15 ng/g and 15-45 ng/g,respectively.The average recoveries for the three cosmetic matrices(aqueous,emulsion,cream) at spiked levels of 25,50,100 ng/g were be⁃tween 88.0% and 111%,with relative standard deviations(RSD,n=6) of 1.4%-9.8%. The method was applied to the detection of commercial cosmetics and N-nitrosodiethanolamine(NDELA) was de⁃tected in 13 batches,with one batch exceeding the limit. The strong specificity,high sensitivity,and good precision made the method could solve the problems of poor stability and easy interference ofdoi:10.19969/j.fxcsxb.23051602收稿日期:2023-05-16;修回日期:2023-06-10基金项目:广东省药品监督管理局化妆品风险评估重点实验室专项(2021ZDZ03);广东省市场监督管理局科技项目(2022CZ06)∗通讯作者:谭建华,博士,正高级工程师,研究方向:色谱-质谱检测技术研究,E-mail:tanjianhua0734@第 42 卷分析测试学报N-nitrosamine compounds,and was suitable for the trace determination of 15 N-nitrosamine com⁃pounds in cosmetics.Key words:N-nitrosamine compounds;cosmetics;ultra performance liquid chromatography-tan⁃dem mass spectrometry(UPLC-MS/MS);atmospheric pressure chemical ionization(APCI) sourceN-亚硝胺化合物是一类具有N-亚硝基结构的化合物,因取代基的不同,形成了种类繁多的同系物,目前已发现超过300种[1]。
利用近红外光谱技术快速测定航空煤油物理性质

航空煤油具有低温流动性好、热值高、燃烧性能好、腐蚀小等优点,评价航空煤油的指标有多种,如密度、馏程、冰点、闪点等。
传统的评价需要多种分析方法来完成,其不足之处有:检测周期长、操作繁琐、维护复杂、成本高等。
因此有必要采用一种简单可靠的分析方法,快速测定航空煤油的性能参数,及时指导工艺生产,只是在需要时对其进行验证,进而减少实测次数,降低分析成本。
80年代以来,随着计算机技术和化学计量技术的迅速发展,现代近红外光谱快速分析技术迅速崛起。
因其具有分析速度快、重复性好、分析成本低、不破坏样品等特点,近年来已成功应用于许多分析领域。
例如,近红外光谱已成功用于原油实沸点收率、密度等,汽油辛烷值和族组成,柴油的十六烷值等参数的测定[1-4],在线近红外光谱技术在汽油调合、重整、乙烯裂解中也均取得了良好的应用效果[5-6]。
近红外光谱主要是有机物分子中X-H(X为C、O、N、S等)基团的红外基频的倍频及合频吸收。
从理论上讲,样品的物理性质和组成有关。
只要组成的变化在光谱上有反映,这些物理性质即可通过近红外技术来测定。
本文采用傅里叶变换近红外光谱仪,成功测定了航空煤油的冰点、密度、闪点、初馏点、终馏点等性质,分析结果均能达到标准方法的误差要求,满足化验和生产需求。
1 实验部分1.1 实验仪器傅里叶变换近红外光谱仪,InAs检测器,分辨率1~64cm-1可调(步长为2),0.5mm 光程CaF2样品池。
1.2 样品光谱采集在近红外光谱仪上,以空样品池为参比,采用8cm-1分辨率,在3800~10000cm-1范围内采集光谱,扫描32次取平均光谱。
1.3 样品来源及基础数据测定为减少工作量,样品取自中石化股份有限公司天津分公司炼油部航煤加氢装置的馏出口航煤。
航煤冰点、密度、闪点和馏程等数据采用馏出口控制班的分析数据。
收集数据期间,尽量由经验丰富、分析数据比较准确的化验工分析这些项目,出现问题时,必须进行复测,确保样品和数据准确。
简版SpectRx近红外光谱系统课件

Incoming Inspection and Verifications 输入检验和确认
In-process Inspection
在线检测
Maintains Consistent Quality
保持品质一致性
Accuracy ( Concentration ): 0.01% 含量精确度:万分之一
Active Ingredients 活性成分 Impurities 杂质 Concentration 浓度 Homogeneity 同质性
简版SpectRx近红外光谱系统课件
6
SpectRx™ NIR
Tablets/Capsules
Diffused Reflectance 漫反射系数 Tablet Hardness 硬度 Tablet Disintegration 崩解 Tablet/Capsule Moisture Contents 湿度含量 Tablet Friability 脆性 Tablet Capsule Active Ingredients 活性组分 Tablet/Capsule Impurities 杂质 Tablet Cracks 裂纹 Capsule Deterioration 变质 Tablet/Capsule Homogeneity 药片-胶囊同质性
库), 95%的机会可以发现一个未知的成分,除非组成的信号信噪比太小。
简版SpectRx近红外光谱系统课件
10
On Line Production 在线生产 (Including raw materials release 包括原材料放行)
简版SpectRx近红外光谱系统课件
11
Rotary Inspection in Lab (实验室旋转式检测)
有机波谱分析课件一.绪论

波谱分析法被广泛地应用于各科研生产中,本 校化学化工学院实验室具有:
HP6890/5973 GC/MS
产于美国PE公司
GC-14B
产于日本岛津公司
HPLC 产于美国Waters公司
FT-IR 产于美国PE, 伯乐公司
UN-Vis 产于日本岛津公司
诺贝尔化学奖享有世界最高荣誉,设有物理、
化学、生物或医学、文学和和平五项奖。1901年, 瑞典皇家科学学院成立,根据诺贝尔遗愿,诺贝尔 奖金委员会首次颁奖。截止到2003年,诺贝尔奖的 颁发已有一百多年的历史了。期间共颁发化学奖94 次,8年因第一、二次世界大战末颁奖。
从最近几十年来诺贝尔化学奖颁奖的情况来看,化学科学逐 渐出现了和生命科学相融合的趋势。在20世纪的最后25年里, 诺贝尔化学奖有三分之一左右给予化学与分子生物学领域研究 的成果。其中大部分获得项目往往直接或间接地在波谱分析方 面获得了突破.如:2002年诺贝尔化学奖由在质谱和核磁共振这 两个重要领域的科学家们分享。获奖者,质谱领域的 John.B.Fenn 和KoIchi Tanaka(田中耕一),核磁共振领域 的Kort Wuthrich,对生物化学领域的进一步发展作出了贡献。
质谱技术早在十九世纪末就出现了,最早用于小分 子分析是由 Joseph J. Thompson 在1912年报道的。二十 世纪有许多诺贝尔奖直接依赖于质谱分析。例如, Harold Urey 发现氘(获得1934年诺贝尔化学奖),富勒烯, “足球碳”的发现使Robert Carl, Sir Harold Kroto和 Richard Smalley 获得1996年诺贝尔化学奖。
核磁共振在生物大分子中的应用
质谱能够回答诸如“是什么”和“有多少种”蛋白质这 样的问题,而核磁共振则回答这种蛋白质“是什么样子”的问 题。
ASTM欢迎相关方参与制定拉曼光谱学标准

1
的 工 作 。 目前 正 在 进 行 F 2 6 17 的 多 实 验 室 研 究 工 作 。
摘 自: ASTM n e t
AS T M欢迎 相关方参与制定拉曼光谱学标准
A S T M 技 术 委 员会 E 1 3 分 “ 子 光 谱 学 和 分 离 科 学 ”
分 委 员会 E 13 0 8 .
试 验 方 法 》。
该 新 标 准 由分 委 员会 F40 0 1 .
试 “ 验 方
法” 制定。
A S TM F 2 6 17 描述 了 筛选 和 量 化用 于 生 产 消 费 者
产 品 的聚合体 中元 素含量 方法 。 技 术委 员 会 副主 席
Ta e o v a n
称 d e r M a t e n
‘‘拉 曼 光 谱 学 ”
正 在制定 一
项 新标
准 W K l 8 8 1 4 《拉 曼 光 谱 学 相 对 强 度 校 正 导 则 》。 该 委
员会 欢 迎 所 有 相 关 方 , 特 别 是 来 自鉴 定 科 学 的 终 端
鼠 户的 参 与 i
拉曼光谱学是一 种应用 广泛 的发射技术 , 用 于 测 定样本的化学 成分 。 美 国国家标准技术研 究所
除了海军 , 美 国交通 部 、 联 邦公路委员会 、 大
型船所有者和建筑者 、 涂料公 司和符合ASTM E250 1
《检 验 荧 光 粉 涂 层 用 光 源 产 品 的 规 范 》的 检 验 色 料 生
产商所都可采纳ASTM E 2630 。 其他潜在用户还 包括
如海上 石油钻塔等海上钢结构所有者和经 营者 、 管 道 和 石 油贮 油罐行业 以及 市政水箱所有 者 。
那 么理 论上 说最终 产品也 是安全 的。 F 26 17 在制定 中
突破传统测量技术,福伊特和PTS Heidenau使用近红外测量技术测量造纸黏性和非黏性污染物

突破传统测量技术,福伊特和P T S本刊讯(Voith 消息) 近期,福伊特与德国PTS Heidenau的研究和服务机构合作,推出了一种新的测量方法来检测黏性污染物。
该测量方法使用近红外(NIR)相机,可以在造纸过程中可靠地检测污染物,与传统方法相比,测量精度和应用范围表现更好。
测量精度更高,应用范围更广与造纸行业的传统方法相比,该测量技术利用近红外相机确定污染物的数据信息,测量精度高,将获得的光谱测量结果与已知污染物的数值进行比较和分配,这样就可以根据污染物的化学成分对其进一步分类。
此外,使用近红外测量技术可以对黏性和非黏性污染物分别进行识别和分类。
鉴定更可靠,减少实验室工作量新的测量方法可以获得更可靠的信息,同时减少了实验室的工作量。
“使用近红外技术测量造纸污染物的黏性,使造纸企业能够根据单次测量结果,以可靠和足够的精度确定黏性平衡,”福伊特造纸研发纤维系统高级经理Linus Friedrich博士说:“而使用传统方法,由于结果差异较大,测量过程会有一定局限,不利于测量精度的提高。
”新的测量方法可以使用实验室片材成形器生产加工样品,使样本包含纤维、黏性和非黏性污染物,与传统方法相比,省去了样品制备的众多中间步骤。
例如,库存样品中的黏性污染物不再需要经过浓缩才能进行分析。
与应用传统方法相比,实验室成片的准备时间明显缩短,测量不易出现误差。
此外,新的近红外测量方法除了可用于评估最终纸样外,还可用于评估库存样品。
造纸行业的新标准由于具有技术优势,近红外测量技术有可能逐渐取代现有的测量黏性的方法。
关于样品制备和测量性能技术规则的发布,为制定造纸行业的新标准创造了基础条件。
新的测量技术可在位于德国海德海姆(Heidenheim)的福伊特造纸技术中心进行进一步研发和客户试验活动。
这项创新技术还可用于服务操作和客户审核等。
INFORMATION77Sep., 2020 Vol.41, No.18China Pulp & Paper Industry。
SPECTRUM MEASURING DEVICE AND SPECTRUM MEASURING M
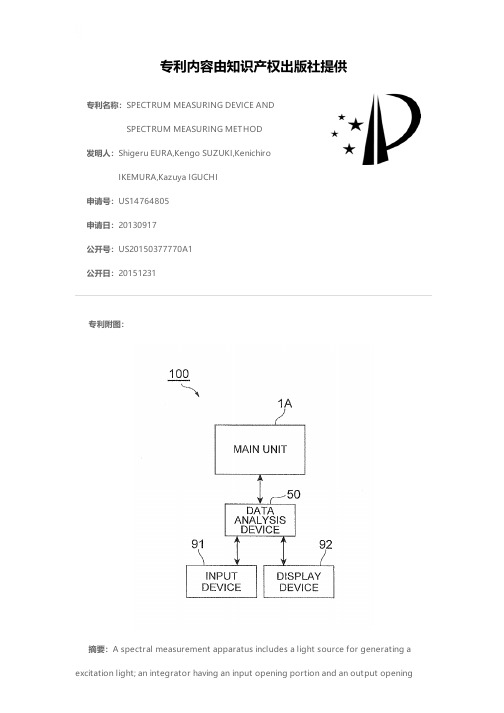
专利名称:SPECTRUM MEASURING DEVICE ANDSPECTRUM MEASURING METHOD发明人:Shigeru EURA,Kengo SUZUKI,KenichiroIKEMURA,Kazuya IGUCHI申请号:US14764805申请日:20130917公开号:US20150377770A1公开日:20151231专利内容由知识产权出版社提供专利附图:摘要:A spectral measurement apparatus includes a light source for generating a excitation light; an integrator having an input opening portion and an output openingportion; a housing portion arranged in the integrator and for housing a sample; an incidence optical system for making the excitation light incident to the sample; a photodetector for detecting a light to be measured output from the output opening portion; and an analysis means for calculating a light absorptance of the sample, based on a detection value detected by the photodetector, and an irradiation area with the excitation light at a position of incidence to the sample is set larger than an irradiated area of the sample, and the analysis means performs an area ratio correction regarding the irradiation area with the excitation light and the irradiated area of the sample, with respect to the light absorptance calculated.申请人:HAMAMATSU PHOTONICS K.K.地址:Hamamatsu-shi, Shizuoka JP国籍:JP更多信息请下载全文后查看。
CMS50 CMS52 无线电测试仪说明书

~ Applications
• Radiocom Tests • Portable and Handhr.ld Mobile • Phone Test
ForyouJ'local n,!;lrollix J'I'j}),(!I'oJlllllil'o 80(! II", /i"
ill th" hac!; of this r:(Jlok'M or oliisido till" U.S. (',,/I: '.s. 1~S03-(i27-Jfl33. iI1sirl" the 1 mil: 1-1J()()~42(i-2:!O().
Modulation frequency range (CMS52): f <8 MHz, DC to 10kHz; f ~8 MHz, DC to 20 kHz. (CMS50): 15 Hz to 10kHz. kB MHz.
翻译:可见-近红外光谱分析技术,鱼肉鲜度评价的新工具

可见/近红外光谱分析技术:鱼肉鲜度评价的新工具?H. NILSEN, M. ESAIASSEN, K. HEIA, AND F. SIGERNES摘要:利用可见/近红外光谱分析技术(VIS/NIR)来分析鳕鱼(Gadus morhua)和鲑鱼(大西洋鲑)冰冻储藏的时间,进而评鉴其新鲜度。
依托多元统计分析对光谱数据和储藏时间的相关性进行建模。
对于鳕鱼而言, 最佳拟合模型应该根据可见波长的范围来建立, 这样,评估的相关性可以达到0.97,误差可以精确到1.04 天。
对于鲑鱼, 最佳拟合模型应根据来自于近红外光谱数的范围来建立,评估的相关性可以达到0.98 ,误差可以精确到1.20天。
由此证明, 可见/近红外光谱分析技术对于鱼肉鲜度的评价是非常有益的。
关键字:可见/近红外光谱分析技术, 鳕鱼, 鲑鱼, 储藏时间概论“鱼肉新鲜度”这个术语通常用在科研和工业领域。
其实,根据对其内容的不同理解和翻译,新鲜度这个词是一个歧义词, 因而如果没有交代明确,不应使用 (Bremner and Sakaguchi 2000)。
在本文中, “鲜度”这个术语是指代一个冰藏时间的函数; 冰藏时间越短, 鱼就会越新鲜。
如果采后的时间短暂,鱼就可视为是保持其原始特征的.在储藏过程中, 这些特征将会由于一些生物化学过程、化学过程、物理过程、微生物过程而发生变化,直至腐坏,这些因素是由时间和温度这两个因素共同影响的(Ashie and others 1996)。
因此, 将新鲜度以冰藏时间的函数来定义,对于其作为估算剩余货架时间的手段来说,是十分有益的。
.由于基于互联网和其他媒体的电子商务的增加,计算机信息技术在鱼肉鲜度领域的应用将比不可少。
现有的确定鱼肉鲜度方法曾经被布莱姆和Sakaguchi (2000)以及Olafsdóttir等人(1997)很好的审查过. 据后来的作者总结,在渔业中,感官评价是目前评价鲜度的最重要的方法。
新购红外光谱仪样品测试相关说明

新购红外光谱仪样品测试相关说明
红外光谱仪用途:
主要用于有机、无机、高分子及生物等材料结构研究和化学组成分析。
主要技术参数及指标:
红外光谱仪(中红外/近红外波段,Spectrum 10)
测试方式:透过,反射,ATR
厂家:美国PE公司
主要性能指标:
分辨率:优于0.4 cm-1(中红外区)
0.1-6.4 nm,1000 nm处(近红外区)
光谱范围:15000-450 cm-1
信噪比(RMS):优于250000:1(4 cm-1, 1 min,FR-DTGS检测器,KBr分束器)OPD 速度:0.10 to 4.00 cm/sec 可调
纵坐标精度:优于0.05 %T
波数精度:优于0.008 cm-1
收费标准:
校内测试20元/个
校外测试100元/个
样品要求:
粉体,液体,薄膜均可。
分析测试中心
2014-3-20。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
a rXiv:as tr o-ph/411477v116Nov24Site Evaluation and RFI spectrum measurements in Portugal at the frequency range 0.408-10GHz for a GEM polarized galactic radio emission experiment Rui Fonseca a Domingos Barbosa a ,d ,1Luis Cupido h Dinis M.dos Santos b ,c George F.Smoot e ,f Camilo Tello g a CENTRA,Instituto Superior T´e cnico,Av.Rovisco Pais,1049-001Lisbon,Portugal b Dep.Electr´o nica e Telecomunica¸c ˜o es,University of Aveiro,Campus Universit´a rio de Santiago,3810-193Aveiro,Portugal c Instituto de Telecomunica¸c ˜o es,Campus Universit´a rio de Santiago,3810-193Aveiro,Portugal d Centro de Astrof´ısica da Universidade do Porto,Rua das Estrelas,4150-762Porto,Portugal e Astrophysics Group,MS 50-205,Lawrence Berkeley National Laboratory,1Cyclotron Rd.,Berkeley CA 94720,USA f Physics Dpt.,University of California,366Le Conte Hall,Berkeley CA 94720,USA g Instituto Nacional de Pesquisas Espaciais,Divis˜a o de Astrof´ısica,Caixa Postal 515,12210-070,S˜a o Jos´e dos Campos SP,Brasil h Centro de Fus˜a o Nuclear,Instituto Superior T´e cnico,Av.Rovisco Pais,1049-001Lisboa,Portugalbackground;polarization;1email:barbosa@astro.up.ptFig.1.Topfigure represents a schematic view of the scanning strategy:azimuthal dish rotation for a given elevation.Bottomfigure is a scheme of scanning instrument and mounting.(seefigure1).The resultant maps obtained at several locations would then be merged to produce templates covering large areas of the sky with constant angular resolution from408MHz up to10GHz and good absolute calibration of the zero-level of the maps.With foreground cartography as one of the main task currently being pursued by CMB teams,GEM project is evolving towards the measurements of galac-tic synchrotron polarization characterization at higher frequencies5-10GHz (Tello et al.,in preparation).Originally,the Berkeley team has developed a compact and portable5.5-m diameter radio antenna,which has been used for thefirst-stage observations.Observations were made from different locations, like near Bishop,California(fall1993through fall1994),Leyva,Colombia (close to the equator1995)and is currently working at Cachoeira Paulista, Brazil(12;13)having now coverd most of the southern hemisphere in1.4and 2.3GHz.To also cover the northern hemisphere,and thus produce a tem-plate of most of the sky,one needs a good site in the northern hemisphere with suitable conditions for clima,RFI and infrastructure capabilities.Thus, Portugal was surveyed tofind a suitable location to host a second antenna to complement the original GEM southern hemisphere maps.The GEM workingfrequencies were chosen because below20GHz,atmosphere contribution is negligible(3).Also,for polarization measurements,besides water lines around 22GHz,oxygen only contaminates polarization measurements for frequencies higher than60GHz,where oxygen molecular rotational modes may be an important concern for ground experiments(7;8).1.2RFI sourcesRadio observations need above all sites with no radio frequency interference (RFI).Perhaps,the biggest threat lies on Global System for Mobile Communi-cations(GSM)networks,meaning frequency dispute with radio astronomers. GSM mobile phone networks use several bands around900MHz,1.8GHz and the near future Universal Mobile Telecommunications System(UMTS)to be shortly implemented will use several2.1GHz bands.Other telecommuni-cations concerns are radio broadcast emissions,analog and digital television broadcast Broadcast emissions,aeronautical communications-mainly along air corridors and airports,satellite communications(communications,meteo-rological and GPS)and amateur radio services,these using theoretically a wide range of bands from3.5GHz to250GHz.Besides telecommunications,recent wireless computer networks use heavily the2.37GHz band with prospects for a near future5GHz upgrade.Also,microwave ovens will present leaks at2.4GHz despite the fact that they are in accordance with industrial stan-dards.These leaks are insignificant to human health,but readily detected as bursts of microwave signal in nearby radiometers working at these frequen-cies.Besides these main concerns,secondary concerns as sources of RFI can be high-voltage power lines and old motorcycle engines.To conclude,one must avoid contact with human settlements.Of course,not all attributed frequen-cies by the telecommunications regulation authorities are susceptible of being contaminated.Either because they are used sporadically-easily eliminated in data processing-or because they are used heavily only in urban areas and do not appear in rural,isolated areas.2Sites evaluationTofind the best locations to host the experiment,several sites were selected after analyzing long term data of important weather variables-temperature, relative humidity,insolation rate(giving an idea of good weather days)from their geographic areas.The data statistics is publicly available from the Por-tuguese Instituto de Metereologia e Geof´ısica(http://www.meteo.pt).Por-tugal shows a temperate Atlantic climate with large variations despite its size. Main differences rest on altitude(mountainous and medium-high north/centerTable1Typical Sources of RFI contaminations.Actually,while Radio Amateur service bands can go up to250GHz,they rarely exceed1300MHz.RF sourceGSM networks1900-1980;2110-2170Radio broadcast475-870Satellite commu.2450Computer Wireless Net.0.177-1300Location latitudeCalif´o rnia37o18’W07o52’N839mTable2Best sites coordinates.andflat-low south)and humidity,depending on sea distance.Although At-lantic winds induce high levels of rain in the winter season and mean high levels of humidity nationwide except for some parts in the interior center and south,spring and summer mean relative humidities around20-30%or less in the country’s interior.These regions are also those with lower human den-sity with some villages living still outside the GSM world.After correlating with GSM coverage maps,two sites in southern Portugal and one in central Portugal were therefore chosen for RFI measurements.The sites main characteristics show a similar pattern for annual RH varia-tion of20-30%during most of the year.Temperatures tend to be high in summer(∼30−35o),with high thermal amplitudes between night and day specially for site1(average night temperature of∼15o C or below).These values indicate good conditions for night observations throughout the dry sea-son,implying low water absorption at the GEM considered frequencies(4) Precise full characterization of sites weather conditions,specially of Castan-heira da Serra,will be available locally after installing instrumentation for weather measurements in the exact places and should enable a robust check on variations of temperature and relative humidity.We plan to proceed with campaigns to check explicitly for the sites for annual,monthly,diurnal direct variations of water vapor content and temperature.Fig.2.The three disconic antennas used in our setup(right).They correspond to the bands:1-[350-1050]GHz;2-[1.1-3.6]GHz;3-[3.6-10.8]GHz.2.1RFI measurementsAfter correlating both climatic and GSM service coverage,we selected sev-eral sites for survey.A primary search on GSM residual coverage reduced our target sample to three sites.Since GEM scanning strategy involves azimuthal rotation of360degrees circles,the presence of even a single localized source of RFI signals can produce a substantial cut in the sky area surveyed.For this reason,we chose disconic antennas for its omnidirectionality and large band receiver capabilities(seefigure2).While disconic antennas may theoret-ically work for bands with initial frequency10times larger than the central frequency(10:1),in practice we opted to optimally cover a frequency band range like3:1.For the wide frequency range[100MHz-10GHz]we built three disconic antennas covering optimally the bands[350-1050]MHz,[1.1-3.6]GHz, [3.6-10.8]GHz.Thefirst antenna is,by above,still capable of detecting strong signals below100MHz.The antennas were then put on a2.5-m high rod and connected to a spec-trum analyzer(an HP8563A).Measurements were taken at different time to check for source variability.The results are shown infigures1,2.It is quite clear that RFI appears to be concentrated,as expected,in three wide bands (radio∼88−100MHz,tv∼500MHz and GSM0.9-1.1GHz.).Simultane-ously,the relative humidity measured was23%.The site of Castanheira da Serra,herafter site1,shows promising conditions with only one RFI source appearing intermittently(tv).While not showing any other expected source, it showed some very low frequency signals(shortwave signals),most likely due to ionospheric reflections.One may ask however,that weaker signals could be present and masked under our setup noise that could become important whena sensible receiver is setup on an antenna.Ideally,the best situation would beto test for RFI with a very sensitive amplifier circuit as close as possible to our desired sensitivity.For5-10GHz,we expect the galactic emission to be on the order of1-100mK.The total power emitted at these frequencies,considering a300MHz bandwidth(12)is about-134dBm2,quite below our noisefloor around-90dBm.This shows that we should be aware of weak,distant sig-nals that may be lurking below this preliminary survey noise sensitivity and carefully shield the instrument.We note that the expected dish will survey at elevations higher than45o to avoid horizon and ground problems,so sidelobes pick up would be much smaller.Typically a cassegrain antenna,optimized for low sidelobes,at an elevation of45o to60o has an horizontal pick up lower than-40dB.A problem could be the very weak signals coming from an almost isotropic distribution of distant sources,leaking in our bands.However,human density(and village density)and as a consequence transmission antennas den-sity in the areas we surveyed is very sparse.For site1,the nearest important settlement is at a distance of15Km,and there are several mountain rings in between.We note since we are going to operate near a protected radioastron-omy band where no interference other than harmonics are expected,the main concern would be the intermodulation generated products intrinsic to our re-ceiver front-end.Harmonics and spurious of the GSM base stations are obliged by law to be at least60dB below carrier,therefore any unwanted signals that may fall inside our band would be at least60dB below the actuall received GSM signals.Also,RFI due to telecommunications may show strong variabil-ity with time and there could be the case where measurements were taken in a quiet period.We did check for this and registered several intermittent signals,most likely due to the occasional use of GSM(figure2).In site1,the intermittent signal seems to be a distant TV broadcast leaking through.We did,however,check GSM and radio coverage from portuguese operators with the official portuguese frequency and radio communications board(ANACOM -http:www.anacom.pt)and found the area of site1to be a blank area(free of coverage).This was also checked directly at the nearest village,where GSM coverage is totally absent.Site1also shows a very convenient orography with the presence of several higher mountains rings(∼1000m)screening signals that could pass from nearby villages.Finally,to test our setup,we checked the antennas response,with the settings we used,at the laboratory(Instituto de Telecomunica¸c˜o es),to several gener-ated signals and tested for GSM and weak wireless computer network signals down to the noisefloor of-90dBm.Fig.3.RFI spectrum for the two sites.Signal amplitude are given in dBm.Solid lines are permanent signals and dashed lines represent intermittent signals detected during measurements.RFI lines appear,as expected by bunches:Radio services appears mainly at100-200MHz;tv broadcast bunch around the500MHz band and GSM services clearly peak around the900MHz and the1.1GHz bands.3ConclusionsWithin the context of the Galactic Emission project,we surveyed several sites for climatic and RFI measurements.Two of the sites show good conditions -low humidity,high number of good weather days,stable geograpy and low RFI on the survey bands to host a GEM antenna,with one-Castanheira da Serra clearly showing a clean radio spectrum,free of RFI in the importantfrequency range of2-10GHz.4AcknowledgmentsWe would like to thank Juan Pardo for helpful discussions on atmosphere polarization.We acknowledge Miguel Lacerda and M´a rio Rui Santos at In-stituto de Telecomunica¸c˜o es for all the help.We thank Ana Mour˜a o for the encouragement and Prof.Armando Rocha for invaluable tips on antennas.DB acknowledges support from FCT through grant contract SFRH/11640/2002. This research was supported by FCT Project POCTI/FNU/42263/2001. References[1] C.L.Bennet et al.,ApJS,148,1[2] A.Kogut et al.,ApJS,148,161[3]W.N.Brandt,ApJ,424,1,1994[4] B.Butler,VLA Scientific Memo,176,1998[5]L.Danese,R.B.Partridge,ApJ,342,604-615,1989[6]H.J.Liebe,l.Waves,10,631-650,1989[7]S.Hanany,P.Rosenkranz,New Astronomy Reviews,47,1159,2003[8]J.Pardo,private communication[9]G.F.Smoot et al.,ApJ,21,1,1992[10]W.Reich,A&AS,48,219-297,1982[11]W.Reich,A&AS,63,205-292,1986[12]C.Tello,PhD Thesis,INPE,1999[13]C.Tello et al.,A&A,12,1,1992[14]S.Torres et al.,Ap&SS240:225-234,1996[15]S.Torres,G.Smoot,G.De Amici,G.,Becerra,M.,Chaux,E.Gomez,J.,and Umana,A.,Rev.Col.Fis.,25,23,1993[16]E.M.Leitch et al,Nature,420,763,2002[17]J.Kovac et al.,Nature,420,772,2002[18]A.da Costa et al.,PRD,68,083003,2003。