2015年版药典原子吸收分光光度法SOP
2015版中国药典修订内容

➢ 含量测定
✓ 主成分与辅料 ✓ 主成分与杂质
新技术与新方法的应用与实践
❖HPLC:系统适用性要求(正文)
灵敏度 ✓ 供试品溶液 ✓ 对照溶液或对照品溶液 ✓ 色谱系统 ✓ 检测器与参数设置 抛弃线
新技术与新方法的应用与实践
❖关于色谱柱
小粒径色谱柱,提速趋势
很多方法,都没有规定具体的色谱柱规格,便于企业根 据自己的实际情况,选择合适的规格色谱柱。
✓ 进行完善溶出度和释放度检查法,加强对现有常释口服 固体制剂(如降糖药等)和缓控释制剂有效性的控制; 加强肠溶制剂释放度和耐酸力、治疗胃酸药品的制酸力 的控制。
✓ 增加对难溶性晶型原料药的粒度、注射剂的复溶时间等 指标的研究与控制,提高产品的稳定性。
✓ 充分吸收现代分析技术用于药品的质量控制,同时强化 理化测定方法和生物测定方法的关联性研究。
色谱柱对流动相的要求 ✓纯度的要求 --超纯水,或用KMnO4处理过的双蒸水 --有机溶剂:色谱纯,并与填料相匹配 ✓缓冲液的pH值,在填料的允许范围(一般2-8)内 ✓缓冲液(盐)的浓度不要太高(≤100mM) ✓流动相对样品的溶解度 --调整有机溶剂和水的比例 --最好用流动相溶样
新技术与新方法的应用与实践
《中国药典》2015版四部通则
编码系列
0100系列 0200系列 0300系列 0400系列 0500系列 0600系列 0700系列 0800系列 0900系列 1000系列 1100系列 1200系列 2000系列 3000系列 8000系列 9000系列
类别
制剂通则 其他通则 一般鉴别实验 光谱法 色谱法 物理常数测定法 其他测定法 限量检查法 物理特性检查法 分子生物学技术 生物检查法 生物活性测定法 中药相关检测方法 生物制品相关检测方法 试剂与标准物质 指导原则
《中国药典》主要增修订内容

编号 0500
0502 0512 0531 0532 0600 0631 0632 0633 0700 0701 0702
通则名称 色谱法
薄层色谱法 高效液相色谱法 超临界流体色谱法(新增) 临界点色谱法(新增) 物理常数测定法
pH值测定法 渗透压摩尔浓度测定法
黏度测定法 其他测定法 电位滴定法与永停滴定法 非水溶液滴定法
0101片剂
口腔贴片应进行溶出度(通则0931)或释放度(通则9031)检查 分散片应进行溶出度(通则0931)和分散均匀性检查 缓释片应符合缓释制剂的有关要求(通则9013)并进行释放度(通则0931)
检查 肠溶片除另有规定外,应进行释放度(通则0931)检查 除冷冻干燥法制备的口崩片外,口崩片应进行崩解时限检查(通则0921)。
二部原附录名称 ⅠⅡⅢⅣⅤⅥⅧ ⅤB薄层色谱法 ⅤD高效液相色谱法
ⅥH pH值测定法 ⅨG渗透压摩尔浓度测定法
ⅥG黏度测定法
ⅦA电位滴定法与永停滴定法 ⅦB非水溶液滴定法
0800 0900
编号
0801 0821 0831 0832 0861
0901 0902 0903 0904 0921 0923 0931 0941 0942 0982
除另有规定外,片剂应进行以下相应检查 【重量差异】 【崩解时限】
崩解时限检查法(通则0921) 凡规定检查溶出度、释放度的片剂,一般不再进行崩解时限检查 【分散均匀性】 照崩解时限检查法(通则0921)检查,不锈钢丝网的筛孔内径为710μ m,水温为 15~25℃;取供试品6片,应在3分钟内全部崩解并通过筛网。 【微生物限度】 照非无菌产品微生物限度检查:微生物计数法(通则1105)和控制菌检查法(通则 1106)及非无菌药品微生物限度标准(通则1107)检查,应符合规定。
2015年药典需大型检验设备的中药材目录-一般委托检验
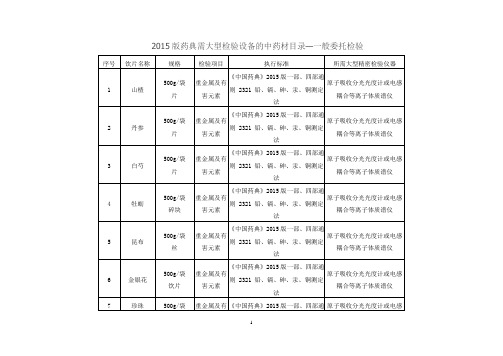
7
珍珠
500g/袋
饮片
重金属及有害元素
《中国药典》2015版一部、四部通则 2321 铅、镉、砷、汞、铜测定法
原子吸收分光光度计或电感耦合等离子体质谱仪
8
海螵蛸
500g/袋
饮片
重金属及有害元素
《中国药典》2015版一部、四部通则 2321 铅、镉、砷、汞、铜测定法
高效液相色谱-质谱仪
31
桃仁
500g/袋
饮片
黄曲霉毒素
《中国药典》 2015年版一部、四部通则 2351黄曲霉毒素测定法
高效液相色谱-质谱仪
32
蜈蚣
25/袋
饮片
黄曲霉毒素
《中国药典》 2015年版一部、四部通则 2351黄曲霉毒素测定法
高效液相色谱-质谱仪
33
槟榔
500g/袋
片
黄曲霉毒素
《中国药典》 2015年版一部、四部通则 2351黄曲霉毒素测定法
高效液相色谱-质谱仪
25
决明子
500g/袋
饮片
黄曲霉毒素
《中国药典》 2015年版一部、四部通则 2351黄曲霉毒素测定法
高效液相色谱-质谱仪
26
麦芽
500g/袋
饮片
黄曲霉毒素
《中国药典》 2015年版一部、四部通则 2351黄曲霉毒素测定法
高效液相色谱-质谱仪
27
陈皮
500g/袋
丝
黄曲霉毒素
《中国药典》 2015年版一部、四部通则 2351黄曲霉毒素测定法
原子吸收分光光度计或电感耦合等离子体质谱仪
9
海藻
500g/袋
段
原子吸收光谱法原理简述

原子吸收光谱法原理简述
原子吸收光谱法是一种用于分析物质中金属元素含量的方法。
它的原理简述如下:
当金属原子处于基态时,它们会吸收特定波长的光。
原子吸收光谱法利用这一特性来测量样品中金属元素的含量。
首先,样品被转化成气态原子或原子的气态化合物,然后通过光源发出的特定波长的光照射样品。
如果样品中含有被检测的金属元素,这些原子会吸收光,使得光源透过样品时的光强度减弱。
测量光源透过样品前后的光强度差异,就可以确定金属元素的含量。
原子吸收光谱法的原理基于不同金属元素吸收光的特性。
每种金属元素都有特定的吸收光谱线,这些谱线对应着特定波长的光。
因此,通过测量样品对不同波长光的吸收情况,可以确定样品中不同金属元素的含量。
此外,原子吸收光谱法还遵循比尔-朗伯定律,即吸收光强度与浓度成正比。
因此,可以通过测量吸收光强度的变化来确定金属元素的浓度。
总的来说,原子吸收光谱法利用金属原子对特定波长光的吸收特性,通过测量样品对光的吸收来确定其中金属元素的含量。
这一方法在分析化学和环境监测等领域有着广泛的应用。
2015年版药典标准变更项目
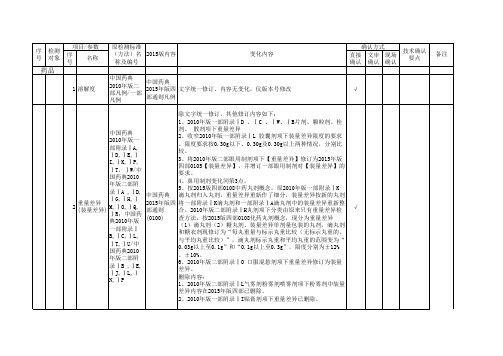
除文字统一修订,其他修订内容如下: 1、2010年版一部附录ⅠD 、ⅠC 、ⅠW、ⅠB片剂、颗粒剂、栓 剂、 散剂项下重量差异 中国药典 2、收窄2010年版一部附录ⅠL 胶囊剂项下装量差异限度的要求 2010年版一 。限度要求按0.30g以下、0.30g及0.30g以上两种情况,分别比 部附录ⅠA, 较。 ⅠD,ⅠE,Ⅰ 3、将2010年版二部眼用制剂项下【重量差异】修订为2015年版 I,ⅠK,ⅠP, 四部0105【装量差异】,并增订一部眼用制剂对【装量差异】的 ⅠT,ⅠW/中 要求。 国药典2010 4、鼻用制剂变化同第3点。 年版二部附 5、按2015版四部0108中药丸剂概念,原2010年版一部附录ⅠK 录ⅠA ,ⅠD, 中国药典 滴丸剂归入丸剂,重量差异重新作了细分,装量差异按新的丸剂 ⅠG,ⅠH,Ⅰ 重量差异 2015年版四 将一部附录ⅠK滴丸剂和一部附录ⅠA滴丸剂中的装量差异重新整 2 M,ⅠO,ⅠQ, (装量差异) 部通则 合。2010年版二部附录ⅠH丸剂项下分类由原来只有重量差异检 ⅠR;中国药 (0100) 查方法,按2015版四部0108化药丸剂概念,现分为重量差异 典2010年版 (1)滴丸剂(2)糖丸剂、装量差异单剂量包装的丸剂,滴丸剂 一部附录Ⅰ 和糖衣剂现修订为“每丸重量与标示丸重比较(无标示丸重的, B,ⅠC,ⅠL, 与平均丸重比较)”,滴丸剂标示丸重和平均丸重的范围变为“ ⅠT,ⅠU/中 0.03g以上至0.1g”和“0.1g以上至0.3g”,限度分别为±12% 国药典2010 、±10%。 年版二部附 6、2010年版二部附录ⅠO 口服混悬剂项下重量差异修订为装量 录ⅠB ,ⅠE, 差异。 ⅠJ,ⅠL,Ⅰ 删除内容: N,ⅠP 1、2010年版二部附录ⅠL气雾剂粉雾剂喷雾剂项下粉雾剂中装量 差异内容在2015年版四部已删除。 2、2010年版一部附录ⅠI贴膏剂项下重量差异已删除。
2015年版《中国药典》药用辅料标准有关问题的答复意见

附件:2015年版《中国药典》药用辅料标准有关问题的答复意见品种反馈问题答复意见聚山梨酯80(供注射用)【脂肪酸组成】项下对照品溶液浓度为每1ml中各含0.01mg的溶液(2015年版《中国药典》勘误第二批)认为该浓度下进样的对照品峰面积过小。
脂肪酸组成按面积归一化法计算,对照品仅仅作为定位用。
乳糖【性状】描述中白色的结晶性颗粒或粉末的理解问题。
【溶液的澄清度与颜色】溶液应澄清无色(不超过0.5号浊度标准液的浊度)。
认为标准过于严格,建议修订浊度限度为:与1号浊度液对比,不得深于对照。
【含量测定】项下以乙腈-水(70:30)为流动相基线不稳,建议以乙腈-水(61:39)为流动相。
加水溶解并稀释制成每1ml各含1mg的溶液响应值太低,导致积分不准确,重现性不好,系统适应性RSD不符合要求。
系统适用性试验理论板数以乳糖峰计算低于5000,达不到要求,建议修订为不低于4000。
1.对性状正确的理解应为:白色的结晶性颗粒,或者白色粉末。
2.乳糖作为注射剂的冻干保护剂使用,为保证药品的安全,药典标准应严格。
3.含量测定项下以乙腈-水(70:30)为流动相基线可以稳定。
每1ml各含1mg的溶液浓度合适,系统适应性RSD可以符合要求。
由于示差折光检测器需要较长时间稳定,建议增加稳定时间。
乳糖峰理论塔板数可以达到要求。
硬脂酸镁【镉盐】【镍盐】两个检测项目,照原子吸收分光光度法(通则0406第二法)测定,均未规定具体的原子化方法,建议两个检测项目标准中均加注“以石墨炉为原子化器,照原子吸收分光光度法(通则0406第二法)测定。
按照本版药典通则0406原子吸收分光光度法的规定,采用原子吸收分光光度法进行的检验,通常不指定原子化器。
检验人员应根据所测元素、仪器性能及工作经验选择相应的原子化器,即使选用了火焰原子化器,可根据被测元素及仪器性能改变燃气和助燃气的种类及比例控制火焰温度,以获得较好的火焰稳定性和测定灵敏度。
微生物限度检查操作规程(中国药典2015版四部通则)
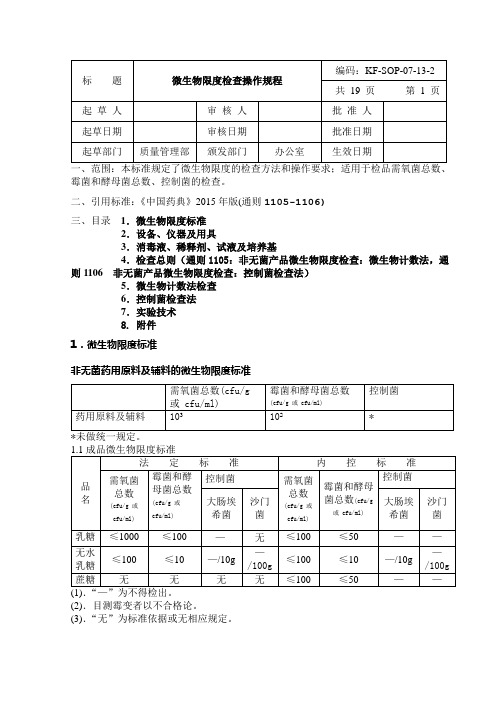
霉菌和酵母菌总数、控制菌的检查。
二、引用标准:《中国药典》2015年版(通则1105-1106)三、目录1.微生物限度标准2.设备、仪器及用具3.消毒液、稀释剂、试液及培养基4.检查总则(通则1105:非无菌产品微生物限度检查:微生物计数法,通则1106 非无菌产品微生物限度检查:控制菌检查法)5.微生物计数法检查6.控制菌检查法7.实验技术8. 附件1.微生物限度标准非无菌药用原料及辅料的微生物限度标准(2).目测霉变者以不合格论。
(3).“无”为标准依据或无相应规定。
准依据或无相应规定。
2.设施、仪器及用具2.1、设施:2.1.1.微生物限度检查室及相关设施:微生物计数试验环境应符合微生物限度检查的要求。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
单向流空气区域、工作台面及环境应定期进行监测。
2.1.2.其他设备:高压蒸汽灭菌器;细菌培养箱(30~35℃);霉菌培养箱(25~28℃);电炉(或其他适宜的加热装置);恒温水浴;电热干燥箱(250~300℃);电冰箱。
生化试剂储存箱。
2.2仪器及器皿2.2.1.菌落计数器;显微镜(1500X);电子天平或药物天平(感量0.1g);pH系列比色计。
2.2.2.玻璃器皿:锥形瓶(250~300ml,内装玻璃珠若干)、研钵(玻璃或陶瓷制,∮10~12cm)、培养皿(∮9 cm)、量筒(100 ml)、试管(18×180mm)及塞、吸管(1ml 分度0.01,10 ml分度0.1)、载玻片、盖玻片、玻璃消毒缸(带盖)。
2.2.3新购的玻璃器皿的清洁:先用流水冲洗,浸泡于1%~2%盐酸(工业用)液中约2~6小时,除去游离碱质,再用流水冲洗。
用于化学分析的玻璃仪器,需用重铬酸钾清洁液浸泡数分钟后,再用流水冲洗,最后以纯化水涮洗2~3次,晾干备用。
2.3用过的玻璃器皿:2.3.1未被病原微生物污染的器皿:可随时洗涤。
药企原子吸收URS

原子吸收分光光度计用户需求说明(URS)XXX制药公司2020年XX月文件审批:起草:审核:批准:目录1 简述和背景 (4)2 目的 (4)3 适用范围 (4)4 参考文献 (4)5 用户要求 (5)5.1 硬件要求 (5)5.2 软件要求 (5)5.3 附件要求 (6)5.4 外观及安全性要求 (7)5.5 性能要求 (7)5.6 法规要求 (9)5.7 培训要求 (9)5.8 验证、确认要求 (10)5.9 文件要求 (10)5.10供应商要求 (11)5.11 其他要求 (11)1 简述和背景原子吸收分光光度计是用于中药材、中药饮片重金属及有害元素测定的检测设备。
测量时依据要求可选择石墨炉法、火焰法、以及氢化物法。
设备由光源、原子化器、背景校正系统、自动进样系统和检测系统组成。
原子吸收分光光度计的测量对象是呈原子状态的金属元素和部分非金属元素,是基于测量蒸汽中原子对特征电磁辐射的吸收强度进行定量分析的一种仪器分析方法。
原子吸收分光光度法遵循朗伯比尔定律,一般通过比较对照品溶液和供试品溶液的吸光度,可计算供试品中待测元素的含量。
2 目的本文是中心实验室对需要购买的原子吸收分光光度计的用户需求说明。
主要阐述了我们对要购买原子吸收分光光度计的软硬件要求、安全要求、性能要求、法规要求、培训要求,文件要求、供应商要求、其他要求等。
使供应商能够提供满足我公司需求的原子吸收分光光度计。
3 适用范围供应商需达到本文的全部要求后,方可从其处购入设备。
仪器购入后安装在指定位置。
本URS 为仪器的基本要求,但实际过程中不仅限于此要求。
4 参考文献《药品生产质量管理规范》(2010版)《中华人民共和国药典》2015年版及附录《计算机化系统》《中华人民共和国药典》2020年版《药品GMP指南》(2011年版)《原子吸收光谱仪检定规程》JJG(教委) 023-1996《原子吸收分光光度计检定规程》JJG 694-2009《原子吸收分光光度计》GB/T 21187-20075 用户要求所购设备应满足以下要求:5.1 硬件要求5.2 软件要求5.3 附件要求5.4 外观及安全性要求5.5 性能要求5.6 法规要求5.7培训要求5.8验证、确认要求5.9文件要求5.10供应商要求5.11 其他要求。
紫外-可见分光光度法标准操作规程

1.目的:规范紫外-可见分光光度法检验操作,保证检验的质量。
2.范围:适于本公司紫外-可见分光光度法操作。
3.责任:质量管理科、中心化验室、检验员。
4.检验依据:《中国药典》2015年版四部紫外-可见分光光度法操作方法。
5.内容:5.1简述♦分光光度法是通过被测物质在特定波长处或一定波长范围内光的吸光度或发光强度,对该物质进行定性和定量分析的方法。
♦定量分析通常选择物质的最大吸收波长处测出吸光度,然后用对照品或百分吸收系数求算出被测物质的含量,多用于制剂的含量测定;对已知物质定性可用吸收峰波长或吸光度比值作为鉴别方法;若化合物本身在紫外光区无吸收,而杂质在紫外光区有相当强度的吸收,或杂质的吸收峰处化合物无吸收,则可用本法作杂质检查。
♦物质对紫外辐射的吸收是由于分子中原子的外层电子跃迁所产生的,因此,紫外吸收主要决定于分子的电子结构,故紫外光谱又称电子光谱。
有机化合物分子结构中如含有共轭体系、芳香环或发色基团,均可在紫外光区(200-400nm)或可见光区(400-760nm)产生吸收。
通常使用的紫外-可见分光光度计的工作波长范围为190-900nm,因此又称紫外一可见分光光度计。
♦紫外吸收光谱为物质对紫外区辐射的能量吸收图。
朗伯-比尔(Lambert 一Beer)定律为光的吸收定律,它是紫外-可见分光光度法定量分析的依据,其数学表达式为:1A = lg— = ECLT式中A为吸收度;T为透光率;E为吸收系数;C为溶液浓度;L为光路长度。
♦如溶液的浓度(C)为1% (g/ml),光路长度(L)为1cm,相应的吸收系数为百分吸收系数,以EfCM表示。
如溶液的浓度(C)为摩尔浓度(mol/L),光路长度为1cm时,则相应有吸收系数为摩尔吸收系数,以8表示。
5.2仪器♦紫外-可见分光光度计主要由光源、单色器、样品室、检测器、记录仪、显示系统和数据处理系统等部分组成。
♦为了满足紫外一可见光区全波长范围的测定,仪器备有二种光源,即氘灯和钨灯,前者用于紫外区,后者用于可见光区。
版《中国药典》药用辅料标准有关问题的答复意见

附件:2015年版《中国药典》药用辅料标准有关问题的答复意见品种反馈问题答复意见聚山梨酯80(供注射用)【脂肪酸组成】项下对照品溶液浓度为每1ml中各含0.01mg的溶液(2015年版《中国药典》勘误第二批)认为该浓度下进样的对照品峰面积过小。
脂肪酸组成按面积归一化法计算,对照品仅仅作为定位用。
乳糖【性状】描述中白色的结晶性颗粒或粉末的理解问题。
【溶液的澄清度与颜色】溶液应澄清无色(不超过0.5号浊度标准液的浊度)。
认为标准过于严格,建议修订浊度限度为:与1号浊度液对比,不得深于对照。
【含量测定】项下以乙腈-水(70:30)为流动相基线不稳,建议以乙腈-水(61:39)为流动相。
加水溶解并稀释制成每1ml各含1mg的溶液响应值太低,导致积分不准确,重现性不好,系统适应性RSD不符合要求。
系统适用性试验理论板数以乳糖峰计算低于5000,达不到要求,建议修订为不低于4000。
1.对性状正确的理解应为:白色的结晶性颗粒,或者白色粉末。
2.乳糖作为注射剂的冻干保护剂使用,为保证药品的安全,药典标准应严格。
3.含量测定项下以乙腈-水(70:30)为流动相基线可以稳定。
每1ml各含1mg的溶液浓度合适,系统适应性RSD可以符合要求。
由于示差折光检测器需要较长时间稳定,建议增加稳定时间。
乳糖峰理论塔板数可以达到要求。
硬脂酸镁【镉盐】【镍盐】两个检测项目,照原子吸收分光光度法(通则0406第二法)测定,均未规定具体的原子化方法,建议两个检测项目标准中均加注“以石墨炉为原子化器,照原子吸收分光光度法(通则0406第二法)测定。
按照本版药典通则0406原子吸收分光光度法的规定,采用原子吸收分光光度法进行的检验,通常不指定原子化器。
检验人员应根据所测元素、仪器性能及工作经验选择相应的原子化器,即使选用了火焰原子化器,可根据被测元素及仪器性能改变燃气和助燃气的种类及比例控制火焰温度,以获得较好的火焰稳定性和测定灵敏度。
原子吸收光谱仪 分光光度计

原子吸收光谱仪分光光度计
原子吸收光谱仪(Atomic Absorption Spectrophotometer,AAS)是一种用于分析金属元素含量的仪器。
它利用原子在特定波长
下吸收光线的原理,通过测量样品中金属元素吸收光线的强度来确
定其浓度。
分光光度计是AAS中的一个重要部分,它能够分解来自
样品中的光线,并测量吸收光线的强度。
AAS分光光度计的工作原理是基于原子在特定波长下吸收光线
的特性。
当样品被加热至高温时,其中的金属元素会被激发并跃迁
至高能级。
然后,通过向样品中传入特定波长的光线,可以使金属
原子吸收并跃迁至高能级。
分光光度计会测量样品吸收光线的强度,从而得出金属元素的浓度。
AAS分光光度计在许多领域都有广泛的应用,包括环境监测、
食品安全、药物分析等。
它具有高灵敏度、高选择性和高准确性的
特点,能够快速、准确地分析样品中金属元素的含量。
因此,AAS
分光光度计在科学研究和工业生产中发挥着重要作用。
总的来说,AAS分光光度计作为原子吸收光谱仪的核心部件,
是一种非常重要的分析仪器。
它的高灵敏度和准确性使其成为许多
行业中不可或缺的工具,为金属元素含量的分析提供了有力支持。
随着科学技术的不断发展,AAS分光光度计将会在更多领域展现其价值,为人类的发展和进步做出更大的贡献。
碳酸钙质量标准及检验操作规程

1 目的:本标准规定了碳酸钙的质量标准及检验操作规程,以规范操作。
2 范围:本标准适用于公司购进的辅料碳酸钙质量检验。
3 职责:质量部QA、QC人员对本标准实施负责。
4 内容:(质量标准)4.1 法定标准(中国药典2015年版二部)4.1.1 产品名称4.1.1.1 中文名:碳酸钙4.1.1.2 汉语拼音名: Tansuangai4.1.1.3 英文名:Calcium Carbonate不得少于98.5%。
本品按干燥品计算,含CaCO34.1.2 性状:本品为白色极细微的结晶性粉末;无臭,无味。
本品在水中几乎不溶,在乙醇中不溶;在含铵盐或二氧化碳的水中微溶;遇稀醋酸、稀盐酸或稀硝酸即发生泡沸并溶解。
4.1.3 鉴别4.1.3.1 取铂丝,用盐酸湿润后,蘸取本品在无色火焰中燃烧,火焰即显砖红色。
4.1.3.2 取本品约0.6g,加稀盐酸15ml,振摇,滤过,滤液加甲基红指示液2滴,用氨试液调至中性,再滴加稀盐酸至恰呈酸性,加草酸铵试液,即生成白色沉淀,分离,沉淀在醋酸中不溶,但在盐酸中溶解。
4.1.3.3 取本品适量,加稀盐酸即泡沸,产生二氧化碳气体,导入氢氧化钙试液中,即生成白色沉淀。
4.1.4 检查4.1.4.1 氯化物取本品0.10g,加稀硝酸10ml,加热煮沸2分钟,放冷,必要时滤过,依法检查(中国药典2015年版通则0801),与标准氯化钠溶液3.0ml制成的对照液比较,不得更浓(0.03%)。
4.1.4.2 硫酸盐取本品0.10g,加稀盐酸2ml,加热煮沸2分钟,放冷,必要时滤过,依法检查(中国药典2015年版通则0802),与标准硫酸钾溶液2.0ml制成的对照液比较,不得更浓(0.2%)。
4.1.4.3酸中不溶物取本品2.0g,加水10ml,混合后,滴加稀盐酸,随滴随振摇,待泡沸停止,加水90ml,滤过,滤渣用水洗涤,至洗液不再显氯化物的反应,干燥后炽灼至恒重,遗留残渣不得过0.2%。
2015新版药典4部

温度会影响分离效果,品种正文中未指明色谱柱温度时系指室温,应注意室温
变化的影响。为改善分离效果可适当提高色谱柱的温度,但不宜超过60℃。
品种正文项下规定的条件除填充剂类型、流动相组分、检测器类型不得改变外,
其余如色谱柱内径与长度、填充剂粒径、流动相速度、流动相组分比例、柱 温、进样量、检测器灵敏度等,均可适当改变,以达到系统适用性试验的要求。
1、微囊系指固态或液态药物被载体辅料包封成微小胶囊。通常粒径在1~250μ m之间的称 为微囊,而粒径在0.1~1μ m之间的称亚微囊,粒径在10~100nm之间的称纳米囊 2、微球系指药物溶解或分散在载体辅料中形成的微小球状体。通常粒径在1~250μ m之间 的称微球,而粒径在0.1~1μ m之间的称亚微球,粒径在10~100nm之间的称纳米球
有了大幅改变,主要目的是提出对生物等效性的设计、实施和评价的相关要 求,也讨论使用体外试验代替体内试验的可能性。
基于生物药剂学分类系统(BCS)的生物豁免仅限于人体吸收情况已知的高溶 解性药物,并且不应是窄治疗指数药物。这一概念适用于具有全身作用的普 通口服固体制剂的相同剂型。但是,它不适用于舌下制剂、颊制剂和调释制 剂。
置2分钟或适当时间脱气泡,小心开启容器,直接将供试品容器置于取样器上, 开启搅拌或以手缓缓转动,使溶液混匀(避免产生气泡),由仪器直接抽取
适量溶液(以不吸入气泡为限),测定并记录数据,弃第一次测定数据,取
后续测定数据的平均值作为测定结果。
增加第四法(桨碟法)、第五法(转筒法)
当品种项下规定需要使用沉降装置时,可将胶囊剂先装入规定的沉降装置内; 品种项下未规定使用沉降装置时,如胶囊剂浮于液面,可用一小段耐腐蚀的
药品检验员综合知识模拟练习题及参考答案

药品检验员综合知识模拟练习题及参考答案1、(单选题). 溶出度检查的温度应是( )A、37±1℃B、室温C、25℃D、20℃答案:A2、(单选题). 标准操作规程的英文简称为()A、SOPB、GSPC、GCPD、GMP答案:A3、(单选题). 不属于样品交接中需要协商的问题是( )A、测定项目B、检测方法C、采样方法D、测定误差答案:C4、(单选题). 根据《中国药典》(2015年版),甲芬那酸中2,3-二甲基苯胺的检查采用( )A、薄层色谱法B、原子吸收分光光度法C、气相色谱法D、高效液相色谱法答案:C5、(单选题). 在样品交接中应注意( )A、获取样品来源的详细信息,并记录B、确定测试时间和方法C、尽快对样品进行处理和测定D、对样品进行编号答案:A6、(单选题). 某甾体激素,加甲醇溶解后,加亚硝基铁氰化钠细粉碳酸钠与醋酸铵后,呈蓝紫色,该药物为( )A、雌二醇B、黄体酮C、醋酸地塞米松D、己烯雌酚答案:B7、(单选题). 红外压片时,盐酸盐药物宜采用的压片方法是( )A、NaBr压片法B、KCl压片法C、KBr压片法D、KI压片法答案:B8、(单选题). 使用万分之一分析天平减重法称量时,为使称量的相对误差在±0.1%以内,试样质量应( )。
A、在0.2g以上B、在0.2g以下C、在0.1G.以上D、在0.1g以下答案:A9、(单选题). 在高效液相色谱法中,使用的流动相应与仪器系统的原保存溶剂能互溶,如不互溶,则先取下上次的色谱柱,用()冲洗过渡,进样器和检测器的流通池也注入同样溶剂进行过渡,过渡完毕后,接上相应的右谱柱,换上本次使用的流动相。
A、乙猜B、异丙醇C、甲醇D、乙醇答案:B10、(单选题). 检验的准备包括熟悉规定的要求,选择检验方法制定( )规范。
A、实施B、规定C、评价D、检验答案:D11、(单选题). 影响药物溶解度的因素不包括( )。
A、药物的颜色B、药物的极性C、温度D、溶剂答案:A12、(单选题). 在没有其它规定的情况下,实验室样品管理记录应至少保留( )A、6个月B、3个月C、2个月D、1个月答案:B13、(单选题). 在分解试样时,硫酸的一个重要应用是除去( )A、挥发性酸B、不挥发性酸C、部分与硫酸根结合生成沉淀的阳离子D、水分答案:A14、(单选题). 鉴别诺氟沙星分子中的有机氟时,需加入的试剂是( )A、氢氧化钠B、靛蓝C、茜素氟蓝D、甲基红答案:C15、(单选题). 在色谱法中,载体在涂渍时,不正确的操作是()A、用水浴加热B、用红外灯照射C、用烘箱烘烤D、用玻璃棒轻轻搅拌答案:C16、(单选题). 旋光计的检定,可采用( )A、标准石英旋光管B、玻璃旋光管C、普通旋光管D、石英旋光管答案:A17、(单选题). 属于样品交接疑难问题的是( )A、标准文件的查找B、协议指标的确定C、实验前药品的准备D、实验室人员的培训答案:B18、(单选题). 称取氢氟酸试样时,一般采用 ( )进行称量。
中国药典2015年版甲硝唑检验原始记录

【性状】㈠.外观性状1.检验结果:本品为。
2.标准规定:本品为白色至微黄色的结晶或结晶性粉末;有微臭。
本品在乙醇中略溶,在水或三氯甲烷中微溶,在乙醚中极微溶解。
3.结论:□符合规定□不符合规定检验人/日期:复核人/日期:㈡.熔点1.仪器:熔点仪型号、编号鼓风干燥箱型号、编号2.操作:取供试品适量,研细,置105℃干燥,然后分取供试品粉末适量,置熔点测定用毛细管中,借助长短适宜的洁净玻璃管,垂直放在实验台面上,将毛细管自上口放入使自由落下,反复数次,使粉末紧密集结在毛细管的熔封端直至装入供试品的高度为3mm。
俟熔点仪温度上升至149℃时,将装有供试品的毛细管浸入传温液中部;继续加热,调节升温速率为每分钟上升1.0~1.5℃,记录供试品在初熔至全熔时的温度,重复测定3 次,取其平均值,即得。
3.检验结果:①℃~℃②℃~℃③℃~℃(极差≤0.5℃)平均值= ℃~℃4.标准规定:熔点为159℃~163℃。
5.结论:□符合规定□不符合规定检验人/日期:复核人/日期:㈢.吸收系数1仪器:电子天平型号、编号紫外-可见分光光度计型号、编号2.试剂:盐酸溶液(9→1000)配制批号3.操作:取本品,精密称取约16.3mg,置50ml量瓶中,加盐酸溶液(9→1000)溶解并稀释至刻度,摇匀,精密量取2ml,置50ml量瓶中,加盐酸溶液(9→1000)稀释至刻度,摇匀制成每1ml中约含13μg的溶液,照紫外-可见分光光度法,在277nm的波长处测定吸收度,吸收系数(E%11cm)为365~389。
AV4.计算公式及结果:E%11cm =----------100WW = g A = V(稀释倍数)=E%1= =1cm)为365~389。
5.标准规定:吸收系数(E%11cm6.结论:□符合规定□不符合规定检验人/日期:复核人/日期:【鉴别】㈠ .1.仪器:电子天平型号、编号2.试剂:氢氧化钠试液配制批号稀盐酸配制批号3.操作及结果:取本品 mg(约10mg),加氢氧化钠试液2ml,微温,即得溶液(应为紫红色);滴加稀盐酸使酸性后即变成(应为黄色),再滴加过量氢氧化钠试液则变成(应为橙红色)。
15 分光光度法检验操作规程
GMP质量管理(9)文件编号:ZL/SOP/TZ/01501分光光度法检验操作规程文件名:分光光度法检验操作规程文件编号:ZL/SOP/TZ/01501制定人:日期:年月日文件类型:工作标准审核人:日期:年月日版次:第一版批准人:日期:年月日印数:共 2份生效日期:年月日颁发部门:GMP办公室分发至:质量保证部、化验室1.目的:规范分光光度法检验操作,保证检验的质量。
2.适用范围:适于本公司原辅料、中间产品、成品的检验。
3.责任者:质量保证部经理、化验室主任、化验员。
4.检验依据:《中国兽药典》2010年版一部附录。
5. 内容:分光光度法是通过测定被测物质在特定波长处或一定波长范围内的分光光度或发光强度,对该物质进行定性和定量分析的方法。
1. 常用的波长范围为:200~400nm的紫外光区,400~760nm的可见光区,2.5~25μm(按波数计为4000~400cm-1)的红外光区。
2. 所用仪器为紫外分光光度计,可见光分光光度计(或比色计)、红外分光光度计或原子吸收分光光度计。
为保证测量的精密度和准确度,所用仪器应按照国家计量检定规程或本公司规定,定期进行校正检定。
3. 单色光辐射穿过被测物质溶液时,被该物质吸收的量与该物质的浓度和液层的厚度(光路长度)成正比,其关系如下式:GMP 质量管理(9) 文件编号:ZL/SOP/TZ/015011A=lg T =ECL 式中:A 为吸收度; T 为透光率; E 为吸收系数,采用的表示方法是(E ),即吸收度换算成溶液浓度为1% 1cm 1%(g /ml ),液层厚度为1cm 的数值; C 为100ml 溶液中所含被测物质的重量,(按干燥品或无水物计算),g ; L 为液层厚度,cm 。
物质对光的选择性吸收波长,以及相应的吸收系数是该物质的物理常数。
当已知某纯物质在一定条件下的吸收系数后,可用同样条件将该供试品配成溶液,测定其吸收度,即可由上式计算出供试品中该物质的含量。
《中国药典》2015年版:片剂

片剂片剂系指原料药物与适宜的辅料制成的圆形或异形的片状固体制剂。
中药还有浸膏片、半浸膏片和全粉片等。
片剂以口服普通片(也包括糖衣片、薄膜衣片)为主,另有含片、舌下片、口腔贴片、咀嚼片、分散片、可溶片、泡腾片、阴道片、阴道泡腾片、缓释片、控释片与肠溶片(包括肠溶衣片和结肠定位肠溶衣片)与口崩片等。
对片剂的质量要求除外观应完整光洁、色泽均匀,有适宜的硬度和耐磨性,以及药典品种项下规定的检验项目外,还应检查“重量差异”和“崩解时限”。
此外,阴道片应检查“融变时限”,阴道泡腾片应检查“发泡量”,分散片应检查“分散均匀性”,口腔贴片、阴道片、阴道泡腾片和1简述1.11.222.10.30g或0.30g2.22.333.13.244.1不得再放回供试品原包装容器内。
4.2遇有检出超出重量差异限度的药片,宜另器保存,供必要时的复核用。
4.3糖衣片应在包衣前检查片芯的重量差异,符合规定后方可包衣。
包衣后不再检查重量差异。
4.4薄膜衣片在包衣后也应检查重量差异。
5记录与计算5.1记录每次称量数据。
5.2求出平均片重(m),保留三位有效数字。
修约至两位有效数字,选择重量差异限度。
5.3按下表规定的重量差异限度,求出允许片重范围(m±m×重量差异限度)。
5.4再根据上表规定的重量差异限度作为判定的依据(避免在计算允许片重范围时受数值修约的影响)。
6结果与判定6.1每片重量均未超出允许片重范围(m±m×重量差异限度);或与平均片重相比较(凡无含量0.295g,应按0.30g的重量差异限度±5%计算。
(4)允许片重范围0.295±0.295×5%=0.280~0.310(g)(5)依法精密称定每片重量,保留三位有效数字,若均在上述允许片重范围内,则按6.1下判为符合规定;若上述供试品中有3片的片重分别为0.279g、0.311g、0.312g,超出允许片重范围(0.280~0.310g)但处于范围边缘,应按5.4项的要求与平均片重相比较,分别计算出该3片的重量差异百分率为-5.4%、5.4%与5.7%,因规定的限度为±5%,根据数值修约规定分别修约成-5%、5%与6%,超出重量差异限度的药片只有1片,按6.1项仍应判为符合规定;若超出重量差异限度的药片多于2片,按6.2项下判为不符合规定。
2015年版微生物限度检验操作规程

青岛**有限公司文件目的建立微生物限度检查操作规程,规范操作,保证结果的准确性。
范围成品、辅料、内包装袋及纯化水的检验。
责任品管部微生物限度检验人员内容概述:本检验操作规程依据中国药典2015年版四部《通则1105 非无菌产品微生物限度检查:微生物计数法》和《通则1106 非无菌产品微生物限度检查:控制菌检查法》进行检查。
微生物计数法一、计数方法1、微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。
2、计数方法本法包括平皿法、薄膜过滤法。
3、计数培养基适用性检查和供试品计数方法适用性试验供试品微生物计数中所使用的培养基应进行适用性检查。
供试品的微生物计数方法应进行方法适用性试验,以确定采用的方法适合于该产品的微生物计数。
4、菌种及菌液的制备4.1试验用菌株的传代次数不得超过5代(从菌种保藏中心获得的干燥菌种为第0袋),并采用适宜的菌种保藏技术进行保藏。
计数培养基适用性检查和计数方法适用性试验见表1。
4.2菌液制备按表1规定培养各试验菌株。
取金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、白色念珠菌的新鲜培养物,用PH7.0无菌氯化钠-蛋白胨缓冲液或0.9%无菌氯化钠溶液制成适宜浓度的菌悬液;取黑曲霉的新鲜培养物加入3-5ml含0.05%(ml/ml)聚山梨酯80的PH7.0无菌氯化钠-蛋白胨缓冲液或0.9%无菌氯化钠溶液,将孢子洗脱。
采用适宜的方法吸出孢子悬液至无菌试管中,用含0.05%(ml/ml)聚山梨酯80的PH7.0无菌氯化钠-蛋白胨缓冲液或0.9%无菌氯化钠溶液制成适宜浓度的黑曲霉孢子悬液。
菌液制备后若在室温下放置,应在2小时内使用;若保存在2-8℃,可在24小时内使用。
黑曲霉孢子悬液可保存在2-8℃,在验证过的贮存期内使用。
表1 试验菌液的制备和使用4.3阴性对照为确认试验条件是否符合要求,应进行对照试验,阴性对照试验应无菌生长。
4.4培养基适用性检查按照表1规定,接种不大于100cfu的菌液至胰酪大豆胨液体培养基或胰酪大豆胨琼脂培养基平板或沙氏葡萄糖琼脂培养基平板,置规定的条件下培养。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2015年版药典原子吸收分光光度法SOP页次:1/10 XXXXX公司检验方法标准操作规程编号:SOP-原子吸收分光光度法检查SOP批准人/日期:年月日审核人/日期:年月日制定人/日期:年月日生效日期:年月日颁发部门:分发部门:1. 目的明确原子吸收分光光度法检查的标准操作。
2. 范围适用于药品采用原子吸收分光光度法检查时的操作。
3. 职责3.1. 化验员负责本程序的实施,3.2. QC主管、质量保证部部长负责监督。
4. 定义无5. 规程5.1. 简述供试品在高温下经原子化产生原子蒸气时,如有一光辐射作用于原子,当辐射频率相应于原子中电子从基态跃迁到较髙能态所需要的能量时,即引起原子对特定波长的吸收。
吸收通常发生在真空紫外、紫外及可见光区。
原子吸收光谱为线光谱,通过测定该特征波长光谱线的吸光度可以计算出该待测元素的含量。
原子吸收一般遵守吸收分光光化阶段。
温度应升至能使样品转变成气态原子,该阶段的升温速度必须很快,加热时间应尽可能短,以延长石墨炉的寿命。
原子蒸气迅速从人射光束通道中扩散出去,形成一个瞬态吸收信号,用记录仪记录。
氢化物发生原子化器利用某些元素易形成低沸点氢化物的性质而设计的氢化物发生原子化器可以减少或避免因高温导致的背景干扰与化学干扰。
As、Sb、Bi、Ge、Sn、Pb、Se等元素在存在还原剂(除另有规定外,通常采用硼氢化钠)的酸性介质中易生成低沸点的易受热分解的氢化物,再依次由载气导人由石英管与加热器组成的原子吸收池中,在石英管中氢化物因受热而分解,并形成基态原子。
冷蒸气原子化器测汞时,在汞蒸气发生器中,汞离子被还原成汞,然后将汞蒸气直接导人原子吸收池中。
单色器通常用衍射光栅为色散元件。
仪器光路应能保证有良好的光谱分辨率和在相当窄的光谱带(0.2nm)下正常工作的能力。
单色器的结构与一般紫外-可见分光光度计相同。
检测器一般采用对紫外及可见光敏感的宽光谱工作范围的光电倍增管作为检测元件。
要求检测器的输出信号灵敏度高、噪声低、漂移小及稳定性好。
记录仪和数据处理系统原子吸收分光光度计常用绘图打印机记录测定结果。
数据处理系统需能测量信号积分值和制备标准曲线以及统计计算处理。
有的仪器将参数设定操作系统和数据处理系统放在一起工作。
背景干扰的消除背景吸收干扰是原子吸收测定中常见的现象。
造成背景干扰的原因多种多样,并往往随样品情况的变化而变化。
一般认为,背景来源于样品中共存组分及其在原子化过程中形成的次生分子或原子的热发射、光吸收和光散射。
其中有些干扰可以通过适当的样品前处理或优化原子化过程的条件得以消除或减少,但许多干扰仍难以避免。
必须另辟蹊径,通过改进仪器设计予以克服。
背景校正的基本原理是将分析谱线两侧的读数作为背景读数,然后从分析线的峰值读数中扣除之。
最常用的背景校正方法有三种:一是连续光源校正法,采用两个光源,主光源为线光源(即空心阴极灯),另一光源为连续光源,在紫外区通常用氘灯。
来自线光源的样品光束通过样品时,其吸光度读数为待测元素与背景吸收之和,来自连续光源的参比光束通过样品时测定背景读数,二者之差即为校正的待测元素的吸光度。
二是塞曼效应校正法,多电子原子的发射谱线通过强磁场时,由于空间量子化的缘故使谱线发生分裂,分裂后的中心线称Tt成分,两侧谱线称Z 成分。
7T成分作为样品光束测定样品和背景的总吸光度,Z 成分作为参比光束测定背景吸收,二者之差即为样品吸收。
作规程编号:SOP-原子吸收分光光度法检查SOP三是强脉冲自吸校正法,在空心阴极灯的工作周期内依次施加两个不同强度的脉冲,在弱脉冲作用下发射正常的谱线,在强脉冲作用下多谱勒效应和阴极溅射增强,从而使谱线变宽而且引起明显的自吸收,造成辐射能在中心波长处缺失而分布于中心波长的两侧。
将弱脉冲作用下的发射谱线作为样品光束,强脉冲作用下的自吸谱线作为参比光束,依次测定吸光度,以实施校正。
连续光源校正由于使用双光源,样品光束和参比光束的准直较为困难,导致在高背景时校正不足或补偿过度;另外,当共存元素的吸收线邻近分析线时,也往往造成补偿过度。
塞曼背景校正没有上述缺点,但当样品浓度较高时,工作曲线向浓度轴弯曲。
强脉冲自吸校正效果较好,但仍存在高浓度时工作曲线弯曲及灯寿命缩短等缺点。
5.2. 原子吸收分光光度计的检定5.2.1. 波长准确度与重复性根据中华人民共和国国家计量检定规程JJG694-90的规定,双光束原子吸收分光光度计的波长示值误差应不大于±0.5mn,波长重复性优于0.3nm。
5.2.2. 波长准确度与重复性检定方法按空心阴极灯上规定的工作电流,将汞灯点亮稳定后,在光谱带宽0. 2nm条件下,从汞、氖谱线253.7、365.0、435.8、546.1,640.2、724.5 和871.6nm中按均匀分布原则,选取3~5条逐一作3次单向(从短波长向长波长方向)测量最大能量波长示值,计算谱线波长测量值与标准值的平均误差。
波长重复性为3次测定中最大值与最小值之差。
5.2.3. 分辨率仪器光谱带宽为0.2nm时,应可分辨锰279.5nm和279.8nm的双线。
5.2.4. 分辨率检定方法将锰灯点亮,稳定后在光谱带宽为0.2nm时调节光电倍增管的高压,使 279.5nm谱线能量读数为100。
扫描测量锰双线,应能分辨出297.5nm和279.8nm两条谱线,且两线间峰谷能量应不超过40% 。
5.2.5. 基线稳定性火焰原子化法测定30min内静态基线和点火基线的稳定度,应不大于下表的指标。
火焰原子化法静态基线和点火基线的稳定度作规程编号:SOP-原子吸收分光光度法检查SOP项目使用中仪器(吸光度)静态基线最大零漂士 0.006最大瞬时噪声(峰-峰值) 0. 006点火基线最大零漂士 0. 008最大瞬时噪声(峰-峰值) 0. 0085.2.5.1. 基线稳定性检定法5.2.5.1.1. 静态基线稳定性的测定光谱带宽0.2nm、量程扩展10倍,点亮铜灯,原子化器未工作状态下测定。
单光束仪器与铜灯同时预热30min,用“瞬时”测量方法,或时间常数不大于0.5s,测定324. 7nm谱线的稳定性。
双光束仪器预热30min、铜灯预热3min后,按上述相同条件测定。
5.2.5.1.2. 点火基线稳定性的测定按测铜的最佳条件,用乙炔/空气火焰,吸喷去离子水lOmin后,在吸喷状况下重复(5.2.5.1.1)的测量。
5.2.5.2. 边缘波长能量带宽为0.2nm,响应时间不大于1.5s条件下,对砷193.7nm和铯852.lnm谱线进行测量,谱线的峰值应能调到100%,背景值/峰值应不大于2 %。
5min内谱线的最大瞬时噪声(峰-峰值)应不大于0.03A。
谱线能量为100%时,光电倍增管的高压应不超过最大高压值的85% 。
5.2.6. 火焰法测定铜的检出限[CL(n = 3)]和精密度(RSD)使用中的仪器应分别不大于0. 02μg/ml 和 1.5% 。
5.2.6.1. 检出限的检定仪器参数调至最佳工作状态,用空白溶液0.5mol/L HNO3调零,分别对3种铜标准溶液(0.50、1.00、3.00μg/ml)各进行3次重复测定,取3次测定平均值,按线性回归法求出工作曲线的斜率,即为仪器测定铜的灵敏度(S)。
S = dA/dc [A/(μg/ml)]在上述条件下,扩展标尺10倍,对空白溶液(或浓度3倍于检出限的溶液)进行11作规程编号:SOP-原子吸收分光光度法检查SOP次吸光度测量,并求出其标准偏差(SA),计算铜的检出限如下GL(n = 3)=3SA/S(μg /ml)5.2.6.2. 精密度的检定在(5.2.1)测定中选择标准溶液之一,其吸光度在0.1~0.3范围进行7次测定,求出相对标准偏差CRSD),即为仪器测铜的精密度。
5.2.7. 石墨炉法测定镉的检出限[QL(n = 3)],特征量(C.M.)和精密度(KSD),使用中的仪器应分别不大于4pg、2pg和7% 。
5.2.7.1. 检出限和特征量的检定仪器参数调至最佳工作状态,分别对空白溶液 (0.5mol/L HNO3)和3种镉标准溶液(0.50、1.00、3.OOng/ml)各进行3次重复测定,取3次测定平均值后,按线性回归法求出工作曲线的斜率,即为仪器测定镉的灵敏度(S)。
S = d A/dQ=dA/d(c×V) (A/pg)式中c为溶液浓度(ng/ml);V 为取样体积(pi)。
在上述条件下对空白溶液进行11次吸光度测定,并求出其标准偏差(SA)。
计算镉的检出限如下QL(w = 3) = 3SA/S(g)仪器测定镉的特征量计算如下 .C. M. = 0.0044(pg) S5.2.7.2. 精密度的检定在(5.2.6.1)测定中,对3.OOng/ml的镉标准溶液进行7次重复测定,即为仪器测镉的精密度。
5.2.8. 火焰法中样品溶液吸喷量(F)和表观雾化率(e) 应用本法可测定火焰原子化雾化的效率,样品的吸喷量应不小于3ml/min;雾化率应不小于8% 。
5.2.8.1. 吸喷量和表观雾化率的检定在与2. 5相同条件下,于10ml量筒内注人去离子水至10ml刻线,将毛细管插人筒底部,同时启动秒表,测量lmin时间内量筒作规程编号:SOP-原子吸收分光光度法检查SOP中水所减少的体积,即为吸喷量(F),取出进样毛细管,至废液管出口无废液排出后,将该管放入有一段水封的10ml量筒(量筒1)内。
另一量筒(量筒2)内注入10ml水,在上述条件下将毛细管插人水中,至10ml水全部吸喷完毕,废液管中无废液排出后,测量排出废液体积V(ml),并计算表观雾化率(ε)。
ε= 10-V×100% 105.2.9. 背景校正能力背景信号约为1A时,校正后的信号应不大于该值的1/30。
5.2.9.1. 火焰原子化器的仪器在镉228.8nm时先用无背景校正方式测量,调零后将吸光度约为1 A的屏网插入光路读得吸光度A1,再在背景校正方式调零,插人屏网读取吸光度A2,1/A2值应符合5.2.9的规定。
5.2.9.2. 石墨炉原子化器的仪器参数调至测镉的最佳状态,先用无背景校正方式,用移液管加人一定量的氯化钠(5.0mg/ml)溶液使产生1 A左右吸光度信号,读取吸光度(峰高法)A1,再用有背景校正方式全样测定,读取吸光度A2, A1/ A2的值应符合5.2.9的规定。
5.3. 样品测定操作方法5.3.1. 标准曲线法先配制一个被测元素的标准贮备液,通常可用该元素的基准化合物或纯金属按规定方法配制,亦可从有关单位中购得,用通常用作空白的溶液稀释成标准工作液。
再按测定方法的操作步骤配制一组合适的系列标准溶液。
在仪器推荐的浓度范围内,制备含待测元素的标准溶液至少3份,浓度依次递增,并分别加人供试品溶液配制中的相应试剂。