Tivantinib_DataSheet_MedChemExpress
齐多夫定结构式

齐多夫定结构式一、齐多夫定基本信息齐多夫定(Zidovudine,AZT)是一种核苷类逆转录酶抑制剂,用于治疗艾滋病和某些癌症。
它是由美国制药公司开发,于1987年获美国食品药品监督管理局批准上市。
齐多夫定的化学名称为3'-叠氮-3'-脱氧胸腺嘧啶核苷,是一种白色结晶粉末,易溶于水。
二、化学结构式化学结构式为:化学式: C10H13N5O4分子量: 267.24结构式: 1-(3-叠氮-2,3-二脱氧-β-D-呋喃核糖基)-5-甲基嘧啶-2,4(1H,3H)-二酮三、合成路线齐多夫定的合成路线主要有两条:一条是以胸腺嘧啶为原料,经过还原、溴代、消除、氧化等步骤得到;另一条是以2',3'-二脱氧胸腺嘧啶核苷为原料,经过还原、溴代、消除、氧化等步骤得到。
具体合成过程较为复杂,涉及多步化学反应。
四、药理作用齐多夫定通过抑制逆转录酶活性,从而阻断病毒DNA的合成。
具体来说,它能够与逆转录酶结合,抑制酶的活性,从而阻止病毒DNA链的延长。
此外,齐多夫定还能掺入病毒DNA,阻止病毒DNA的复制。
五、药物代谢齐多夫定口服后易吸收,血药浓度达峰时间为0.5-1.5小时。
主要分布在细胞外液,易透过胎盘屏障。
在体内代谢为5'-单磷酸酯和3'-叠氮-5'-腺苷酸。
主要通过肾脏排泄,约40%-50%以原形排出,其余为代谢产物。
消除半衰期为1.1-1.9小时。
六、临床应用齐多夫定主要用于治疗艾滋病和某些癌症,如急性淋巴细胞性白血病等。
在艾滋病的治疗中,齐多夫定常与其他抗逆转录病毒药物联合使用,以抑制HIV 病毒的复制。
在某些癌症治疗中,齐多夫定可作为辅助治疗手段,与其他化疗药物联合使用。
七、不良反应齐多夫定的不良反应主要包括骨髓抑制、胃肠道反应、过敏反应等。
骨髓抑制表现为贫血、白细胞减少和血小板减少等。
胃肠道反应包括恶心、呕吐、腹泻等。
过敏反应表现为皮疹、荨麻疹等。
黄芩素对人乳腺癌细胞系MDA-MB-231_侵袭、迁移、上皮间充质转化的调控作用及其机制

黄芩素对人乳腺癌细胞系MDA -MB -231侵袭、迁移、上皮间充质转化的调控作用及其机制陈林1,2,梁秋果1,2,吉杨丹1,2,王恒1,21 黔南民族医学高等专科学校基础医学部,贵州都匀558013;2 黔南州天然产物与组分功效重点实验室摘要:目的 观察黄岑素对人乳腺癌细胞系MDA -MB -231的侵袭、迁移及上皮间充质转化(EMT )的调控作用,探讨其可能作用机制。
方法 取对数生长期MDA -MB -231细胞分为一组、二组、三组及对照组,一组、二组、三组分别加入2.5、5、10 μmol /L 的黄岑素,对照组不做任何处理。
培养48 h 时采用划痕修复实验观察四组细胞迁移能力、采用Transwell 侵袭实验观察四组细胞侵袭能力,采用Western Blotting 法检测细胞EMT 标志物波形蛋白(vimentin )及E -钙黏蛋白(E -cadherin )、整合素αv 、β3、磷酸化黏着斑激酶(p -FAK )、磷酸化磷脂酰肌醇3激酶(整合素p -PI3K )。
结果 与对照组相比,黄岑素组细胞迁移率降低、侵袭细胞数少,细胞E -cadherin 相对表达量高,vimentin 、整合素αv 、整合素β3、p -FAK 、p -PI3K 蛋白相对表达量低,且呈剂量依赖性(P 均<0.05)。
结论 黄芩素抑制MDA -MB -231细胞的侵袭、迁移及EMT 。
黄岑素可能通过抑制整合素αv 、整合素β3表达,进一步抑制p -FAK 、p -PI3K 蛋白表达,抑制MDA -MB -231的侵袭、迁移及EMT 。
关键词:黄芩素;乳腺癌;细胞侵袭;细胞迁移;上皮间质转化;波形蛋白;E -钙黏蛋白;整合素αv 、整合素β3;黏着斑激酶;磷脂酰肌醇3激酶doi :10.3969/j.issn.1002-266X.2024.06.003中图分类号:R965.1 文献标志码:A 文章编号:1002-266X (2024)06-0010-04Regulatory effects of baicalein on invasion , migration , and EMT in human breast cancer cell line MDA -MB -231CHEN Lin 1, LIANG Qiuguo , JI Yangdan , WANG Heng1 Department of Basic Medicine , Qiannan Medical College for Nationalities , Duyun 558013, ChinaAbstract : Objective To investigate the regulatory effects of baicalein on the invasion , migration , and epithelial -mesenchymal transition (EMT ) of human breast cancer cell line MDA -MB -231 and to explore its potential mechanism of action. Methods MDA -MB -231 cells in the logarithmic growth phase were divided into the Group 1, Group 2, Group 3, and Control group. Baicalein was added to cells in the Group 1, Group 2, and Group 3 at concentrations of 2.5, 5, and 10 μmol /L , respectively , while cells in the Control group were not treated. After 48 h of incubation , Scratch repair experi‐ment was used to observe cell migration capacity , Transwell invasion experiment was performed to assess cell invasion ca‐pacity , and Western blotting was utilized to detect the expression levels of EMT markers including vimentin and E -cad‐herin , as well as integrin αv , integrin β3, phosphorylated focal adhesion kinase (p -FAK ), and phosphorylated phosphati‐dylinositol -3 kinase (p -PI3K ) proteins. Results Compared with the control group , the cell migration rate decreased , the number of invading cells decreased , the relative expression level of E -cadherin was higher , and the relative expression levels of vimentin , integrin αv , integrin β3, p -FAK , and p -PI3K protein were lower in the baicalein group , with a dose -de‐pendant manner (all P <0.05). Conclusions Baicalein inhibits the invasion , migration , and EMT of MDA -MB -231cells. Baicalein may inhibit the expression of integrin αv , integrin β3, and further inhibit the expression of p -FAK and p -PI3K proteins , thereby inhibiting the invasion , migration , and EMT of MDA -MB -231 cells.Key words : baicalein; breast carcinoma; cell invasion; cell migration; epithelial -mesenchymal transition; vimentin;基金项目:黔南民族医学高等专科学校校基金(qnyz202013,qnyz202206);黔南民族医学高等专科学教育教学科研基金项目(qnyzjx202205);黔南民族医学高等专科学校大学生科技创新项目(qnyz202201)。
Tivozanib_SDS_MedChemExpress

Inhibitors, Agonists, Screening LibrariesSafety Data Sheet Revision Date:May-24-2017Print Date:May-24-20171. PRODUCT AND COMPANY IDENTIFICATION1.1 Product identifierProduct name :TivozanibCatalog No. :HY-10977CAS No. :475108-18-01.2 Relevant identified uses of the substance or mixture and uses advised againstIdentified uses :Laboratory chemicals, manufacture of substances.1.3 Details of the supplier of the safety data sheetCompany:MedChemExpress USATel:609-228-6898Fax:609-228-5909E-mail:sales@1.4 Emergency telephone numberEmergency Phone #:609-228-68982. HAZARDS IDENTIFICATION2.1 Classification of the substance or mixtureGHS Classification in accordance with 29 CFR 1910 (OSHA HCS)Acute toxicity, Oral (Category 4),H302Acute aquatic toxicity (Category 1),H400Chronic aquatic toxicity (Category 1),H4102.2 GHS Label elements, including precautionary statementsPictogramSignal word WarningHazard statement(s)H302 Harmful if swallowed.H410 Very toxic to aquatic life with long lasting effects.Precautionary statement(s)P264 Wash skin thoroughly after handling.P270 Do not eat, drink or smoke when using this product.P273 Avoid release to the environment.P301 + P312 IF SWALLOWED: Call a POISON CENTER or doctor/ physician if you feel unwell.P330 Rinse mouth.P391 Collect spillage.P501 Dispose of contents/ container to an approved waste disposal plant.2.3 Other hazardsNone.3. COMPOSITION/INFORMATION ON INGREDIENTS3.1 SubstancesSynonyms:AV⁻951; AV951; AV 951Formula:C22H19ClN4O5Molecular Weight:454.86CAS No. :475108-18-04. FIRST AID MEASURES4.1 Description of first aid measuresEye contactRemove any contact lenses, locate eye-wash station, and flush eyes immediately with large amounts of water. Separate eyelids with fingers to ensure adequate flushing. Promptly call a physician.Skin contactRinse skin thoroughly with large amounts of water. Remove contaminated clothing and shoes and call a physician.InhalationImmediately relocate self or casualty to fresh air. If breathing is difficult, give cardiopulmonary resuscitation (CPR). Avoid mouth-to-mouth resuscitation.IngestionWash out mouth with water; Do NOT induce vomiting; call a physician.4.2 Most important symptoms and effects, both acute and delayedThe most important known symptoms and effects are described in the labelling (see section 2.2).4.3 Indication of any immediate medical attention and special treatment neededTreat symptomatically.5. FIRE FIGHTING MEASURES5.1 Extinguishing mediaSuitable extinguishing mediaUse water spray, dry chemical, foam, and carbon dioxide fire extinguisher.5.2 Special hazards arising from the substance or mixtureDuring combustion, may emit irritant fumes.5.3 Advice for firefightersWear self-contained breathing apparatus and protective clothing.6. ACCIDENTAL RELEASE MEASURES6.1 Personal precautions, protective equipment and emergency proceduresUse full personal protective equipment. Avoid breathing vapors, mist, dust or gas. Ensure adequate ventilation. Evacuate personnel to safe areas.Refer to protective measures listed in sections 8.6.2 Environmental precautionsTry to prevent further leakage or spillage. Keep the product away from drains or water courses.6.3 Methods and materials for containment and cleaning upAbsorb solutions with finely-powdered liquid-binding material (diatomite, universal binders); Decontaminate surfaces and equipment by scrubbing with alcohol; Dispose of contaminated material according to Section 13.7. HANDLING AND STORAGE7.1 Precautions for safe handlingAvoid inhalation, contact with eyes and skin. Avoid dust and aerosol formation. Use only in areas with appropriate exhaust ventilation.7.2 Conditions for safe storage, including any incompatibilitiesKeep container tightly sealed in cool, well-ventilated area. Keep away from direct sunlight and sources of ignition.Recommended storage temperature:Powder-20°C 3 years4°C 2 yearsIn solvent-80°C 6 months-20°C 1 monthShipping at room temperature if less than 2 weeks.7.3 Specific end use(s)No data available.8. EXPOSURE CONTROLS/PERSONAL PROTECTION8.1 Control parametersComponents with workplace control parametersThis product contains no substances with occupational exposure limit values.8.2 Exposure controlsEngineering controlsEnsure adequate ventilation. Provide accessible safety shower and eye wash station.Personal protective equipmentEye protection Safety goggles with side-shields.Hand protection Protective gloves.Skin and body protection Impervious clothing.Respiratory protection Suitable respirator.Environmental exposure controls Keep the product away from drains, water courses or the soil. Cleanspillages in a safe way as soon as possible.9. PHYSICAL AND CHEMICAL PROPERTIES9.1 Information on basic physical and chemical propertiesAppearance Light brown to brown (Solid)Odor No data availableOdor threshold No data availablepH No data availableMelting/freezing point No data availableBoiling point/range No data availableFlash point No data availableEvaporation rate No data availableFlammability (solid, gas)No data availableUpper/lower flammability or explosive limits No data availableVapor pressure No data availableVapor density No data availableRelative density No data availableWater Solubility No data availablePartition coefficient No data availableAuto-ignition temperature No data availableDecomposition temperature No data availableViscosity No data availableExplosive properties No data availableOxidizing properties No data available9.2 Other safety informationNo data available.10. STABILITY AND REACTIVITY10.1 ReactivityNo data available.10.2 Chemical stabilityStable under recommended storage conditions.10.3 Possibility of hazardous reactionsNo data available.10.4 Conditions to avoidNo data available.10.5 Incompatible materialsStrong acids/alkalis, strong oxidising/reducing agents.10.6 Hazardous decomposition productsUnder fire conditions, may decompose and emit toxic fumes.Other decomposition products - no data available.11.TOXICOLOGICAL INFORMATION11.1 Information on toxicological effectsAcute toxicityClassified based on available data. For more details, see section 2Skin corrosion/irritationClassified based on available data. For more details, see section 2Serious eye damage/irritationClassified based on available data. For more details, see section 2Respiratory or skin sensitizationClassified based on available data. For more details, see section 2Germ cell mutagenicityClassified based on available data. For more details, see section 2CarcinogenicityIARC: No component of this product present at a level equal to or greater than 0.1% is identified as probable, possible or confirmed human carcinogen by IARC.ACGIH: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by ACGIH.NTP: No component of this product present at a level equal to or greater than 0.1% is identified as a anticipated or confirmed carcinogen by NTP.OSHA: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by OSHA.Reproductive toxicityClassified based on available data. For more details, see section 2Specific target organ toxicity - single exposureClassified based on available data. For more details, see section 2Specific target organ toxicity - repeated exposureClassified based on available data. For more details, see section 2Aspiration hazardClassified based on available data. For more details, see section 212. ECOLOGICAL INFORMATION12.1 ToxicityNo data available.12.2 Persistence and degradabilityNo data available.12.3 Bioaccumlative potentialNo data available.12.4 Mobility in soilNo data available.12.5 Results of PBT and vPvB assessmentPBT/vPvB assessment unavailable as chemical safety assessment not required or not conducted.12.6 Other adverse effectsNo data available.13. DISPOSAL CONSIDERATIONS13.1 Waste treatment methodsProductDispose substance in accordance with prevailing country, federal, state and local regulations.Contaminated packagingConduct recycling or disposal in accordance with prevailing country, federal, state and local regulations.14. TRANSPORT INFORMATIONDOT (US)This substance is considered to be non-hazardous for transport.IMDGUN number: 3077Class: 9Packing group: IIIEMS-No: F-A, S-FProper shipping name: ENVIRONMENTALLY HAZARDOUS SUBSTANCE, SOLID, N.O.S.Marine pollutant: Marine pollutant.IATAUN number: 3077Class: 9Packing group: IIIProper shipping name: Environmentally hazardous substance, solid, n.o.s.15. REGULATORY INFORMATIONSARA 302 Components:No chemicals in this material are subject to the reporting requirements of SARA Title III, Section 302.SARA 313 Components:This material does not contain any chemical components with known CAS numbers that exceed the threshold (De Minimis) reporting levels established by SARA Title III, Section 313.SARA 311/312 Hazards:No SARA Hazards.Massachusetts Right To Know Components:No components are subject to the Massachusetts Right to Know Act.Pennsylvania Right To Know Components:No components are subject to the Pennsylvania Right to Know Act.New Jersey Right To Know Components:No components are subject to the New Jersey Right to Know Act.California Prop. 65 Components:This product does not contain any chemicals known to State of California to cause cancer, birth defects, or anyother reproductive harm.16. OTHER INFORMATIONCopyright 2017 MedChemExpress. The above information is correct to the best of our present knowledge but does not purport to be all inclusive and should be used only as a guide. The product is for research use only and for experienced personnel. It must only be handled by suitably qualified experienced scientists in appropriately equipped and authorized facilities. The burden of safe use of this material rests entirely with the user. MedChemExpress disclaims all liability for any damage resulting from handling or from contact with this product.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
Afatinib_dimaleate_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Afatinib dimaleate is an irreversible, dual EGFR /HER2 inhibitor, shows potent activity against wild–type and mutant forms of EGFR and HER2, with IC 50 of 0.5 nM, 0.4 nM, 10 nM and 14 nM for EGFR wt , EGFR L858R , EGFR L858R/T790M and HER2, respectively.IC50 & Target: IC50: 0.5 nM (EGFR wt ), 0.4 nM (EGFR L858R ), 10 nM (EGFR L858R/T790M ), 14 nM (HER2)[1]In Vitro: In cell–free in vitro kinase assays, Afatinib (BIBW2992) dimaleate shows potent activity against wild–type and mutant forms of EGFR and HER2, similar to Gefitinib in potency for L858R EGFR, but about 100–fold more active against the Gefitinib–resistant L858R–T790M EGFR double mutant, with an IC 50 of 10 nM. BIBW2992 is furthermore comparable to Lapatinib and Canertinib for in vitro potency against HER2, with an IC 50 of 14 nM. The most sensitive kinase in this evaluation is lyn with an IC 50 of 736 nM [1].Afatinib is an irreversible inhibitor of these ErbB family receptors. Esophageal squamous cell carcinoma (ESCC) cell lines are sensitiveto Afatinib with IC 50 concentrations at lower micro–molar range (at 48 hour incubation: HKESC–1=78 nM, HKESC–2=115 nM,KYSE510=3.182 μM, SLMT–1=4.625 μM and EC–1=1.489 μM; and at 72 hour incubation: HKESC–1=2 nM, HKESC–2=2 nM,KYSE510=1.090 μM, SLMT–1=1.161 μM and EC–1=109 nM) with a maximum growth inhibition over 95%. Afatinib can strongly induce G 0/G 1 cell cycle arrest in HKESC–2 and EC–1 in a dose– and time–dependent manner [2].In Vivo: Afatinib (15 mg/kg) strongly inhibits the growth of HKESC–2 tumor once the treatment began. Average tumor sizes of vehicle and treatment at end point are 348±24 mm 3 and 108±36 mm 3 respectively, showing significantly difference between them. And apparently tumor size does not bounce back in a short period of time after the end of Afatinib administration. Without rapid change of body weight throughout the treatment shows that the toxicity of Afatinib is minimal and this drug is well tolerated [2].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]The EGFR kinase domain–GST fusion proteins are extracted from Sf9 biomasses, 72 hours post infection, with HEPEX (20 mM HEPES pH 7.4, 100 mM NaCl, 10 mM β–glycerophosphate, 10 mM para–nitro–phenylphosphate, 30 mM NaF, 5mM EDTA, 5% glycerol, 1% Triton X–100, 1 mM Na 3VO 4, 0.1% SDS, 0.5 μg/mL pepstatin A, aprotinin 20 KIU/mL, Leupeptin 2 μg/mL,Benzamidine 1 mM, 2.5 μg/mL 3,4–dichloroisocoumarin, 2.5 μg/mL trans–epoxysuccinyl–L–leucyl–L–amido butane and 0.002%PMSF) and used for the determination of the IC 50 values. Each 100 μL enzyme reaction contains 10 μL of Afatinib (BIBW2992) in 50% Me 2SO, 20 μL of substrate solution (200 mM HEPES pH 7.4, 50 mM Mg–acetate, 2.5 mg/mL poly (EY), 5 μg/mL bio–pEY) and 20 μL enzyme preparation. The enzymatic reaction is started by addition of 50 μL of a 100 μM ATP solution made in 10 mM MgCl 2.Assays are carried out at room temperature for 30 minutes and terminated by the addition of 50 μL of stop solution (250 mM EDTA in 20 mM HEPES pH 7.4). 100 μL are transferred to a streptavidin coated microtiterplate, after an incubation time of 60 min at room temperature the plate is washed with 200 μL of wash solution (50 mM Tris, 0.05% Tween20). A 100 μL aliquot of a HRPO– labeled anti–PY antibody (PY20H Anti–Ptyr:HRP supplied by Transduction Laboratories) 250 ng/mL are added to the wells. After 60 min of incubation, the plate is washed three times with a 200 μL wash solution. The samples are then developed with a 100 μL TMB Peroxidase Solution (A:B=1:1). The reaction is stopped after 10 min. The plate is transferred to an ELISA readerProduct Name:Afatinib (dimaleate)Cat. No.:HY-10261A CAS No.:850140-73-7Molecular Formula:C 32H 33ClFN 5O 11Molecular Weight:718.08Target:EGFR; EGFR; Autophagy Pathway:JAK/STAT Signaling; Protein Tyrosine Kinase/RTK; Autophagy Solubility:DMSO: ≥ 35 mg/mLand extinction is measured at OD450nm. All data points are performed in triplicates[1].Cell Assay: Afatinib is dissolved in DMSO (100 mM) and stored (–80°C), and then diluted in corresponding medium justbefore addition to cell cultures[2].[2]Human ESCC cell lines, EC–1, HKESC1 and HKESC2, SLMT1, and KYSE510 are cultured in RPMI with 10% fetal bovine serum (FBS). Cytotoxicity is assessed by a colorimetric assay using MTT. Tumour cells are cultured in 48–well plates (3000–8000 cells per well) in respective culture medium. Afatinib in complete medium is added at 24 hr after cell plating and incubated at 37°C with 5% CO2 for 48 and 72 hr. Cell growth inhibition is expressed as the percentage of absorbance of control cultures measured at 570 nm with a microplate reader and 50% of the maximum growth inhibition (IC50) is calculated by GraphPad PRISM. In each experiment, triplicate wells are performed for each drug concentration (n=3), and assay is repeated in three independent experiments[2].Animal Administration: Afatinib is dissolved in 0.5% methylcellulose (Mice)[2].[2]Mice[2]Six weeks old female athymic nude mice (nu/nu) weighing about 16–20 gram are used. ESCC xenografts are established by inoculating HKESC–2 (6×104 cells re–suspended in 50 μL of HBSS–buffer) subcutaneously into both flanks of the nudemice. When tumor size reached to 4–6 mm diameter, they are randomized in either treatment (15 mg/kg) or vehiclecontrol group. Afatinib for treatment is prepared by dissolving in 0.5% methylcellulose before administration. Either drugor vehicle is administered to mouse by oral gavage in a schedule of 5 days on plus 2 days off for two weeks. Drug efficacyis evaluated by monitoring the change in tumor size with caliper. Tumor volume is calculated with the formula TumorVolume=(width2×length)/2.References:[1]. Li D, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008 Aug 7;27(34):4702–11.[2]. Wong CH, et al. Preclinical evaluation of afatinib (BIBW2992) in esophageal squamous cell carcinoma (ESCC). Am J Cancer Res. 2015 Nov 15;5(12):3588–99.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
西咪替丁的化学结构式

西咪替丁的化学结构式1. 西咪替丁的概述西咪替丁(Simvastatin)是一种用于降低胆固醇和脂蛋白水平的药物,属于他汀类药物。
它通过抑制胆固醇合成的关键酶HMG-CoA还原酶,从而减少胆固醇在体内的合成。
西咪替丁是一种处方药,常用于治疗高胆固醇和高脂蛋白血症,预防心血管疾病的发生。
2. 西咪替丁的化学结构式西咪替丁的化学名为(1S,3R,7S,8S,8aR)-8-{2-[(2R,4R)-4-羟基-6-氧代-3,5-二甲基-4-甲硫基-4-氧代-5-氮-6-甲基-1,2,3,4-四氢-2-吡啶基]乙基}-3,7-二甲基-1,2,3,7,8,8a-六氢-1-萘酮。
西咪替丁的化学式为C25H38O5S,分子量为418.57 g/mol。
西咪替丁的结构式如下所示:3. 西咪替丁的合成途径西咪替丁的合成途径相对复杂,主要包括以下几个步骤:3.1 邻氨基苯甲酸的合成首先,通过邻氨基苯甲酸的合成作为起始原料。
邻氨基苯甲酸是通过对硝基苯甲酸的氢化还原得到的。
3.2 吡咯的合成邻氨基苯甲酸与乙酰乙酸乙酯反应生成吡咯化合物。
该反应需要碱催化。
3.3 吡咯的环化吡咯化合物通过烷基化反应得到环化产物。
该反应需要环化试剂和酸催化。
3.4 吡咯的氧化环化产物经氧化反应生成相应的醛。
该反应需要氧化剂。
3.5 醛的还原醛经还原反应生成相应的醇。
该反应需要还原试剂。
3.6 醇的酯化醇经酯化反应生成相应的酯。
该反应需要酯化试剂和酸催化。
3.7 酯的水解酯经水解反应生成相应的酸。
该反应需要水解试剂。
最终,通过以上合成步骤,得到西咪替丁。
4. 西咪替丁的药理作用西咪替丁通过抑制HMG-CoA还原酶的活性,阻断胆固醇的合成途径,从而降低体内胆固醇水平。
此外,西咪替丁还可以增加低密度脂蛋白受体的表达,促进低密度脂蛋白的清除,进一步降低胆固醇水平。
西咪替丁的主要药理作用包括:4.1 降低胆固醇水平西咪替丁通过抑制胆固醇的合成,可以显著降低总胆固醇、低密度脂蛋白胆固醇和甘油三酯的水平。
Baricitinib_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Baricitinib is a selective orally bioavailable JAK1/JAK2 inhibitor with IC 50 of 5.9 nM and 5.7 nM, respectively.IC50 & Target: IC50: 5.9 nM (JAK1), 5.7 nM (JAK2), >400 (JAK3), 53 nM (Tyk2)[1]In Vitro: In cell–based assays, Baricitinib (INCB028050) proves to be a potent inhibitor of JAK signaling and function. In PBMCs,Baricitinib inhibits IL–6–stimulated phosphorylation of the canonical substrate STAT3 (pSTAT3) and subsequent production of the chemokine MCP–1 with IC 50 values of 44 nM and 40 nM, respectively. In isolated naive T–cells, INCB028050 also inhibits pSTAT3stimulated by IL–23 (IC 50=20 nM). Importantly, this inhibition prevented the production of two pathogenic cytokines (IL–17 and IL–22) produced by Th17 cells–a subtype of helper T cells with demonstrable inflammatory and pathogenic properties–with an IC 50value of 50 nM. In stark contrast, the structurally similar but ineffective JAK1/2 inhibitors INCB027753 and INCB029843 has no significant effect in any of these assays systems when tested at concentrations up to 10 μM [1].In Vivo: Baricitinib (INCB028050) treatment, compares with vehicle, inhibits the increase in hind paw volumes during the 2 wk of treatment by 50% at a dose of 1 mg/kg and >95% at doses of 3 or 10 mg/kg. Because baseline paw volume measurements are taken on treatment day 0–in animals with significant signs of disease–it is possible to have >100% inhibition in animals showing marked improvement in swelling [1]. Baricitinib (0.7 mg/day) treated mice exhibits substantially reduced inflammation asassessed by H&E staining, reduced CD8 infiltration, and reduced MHC class I and class II expression when compared withvehicle–control treated mice. CD8+NKG2D + cells, critical effectors of disease in murine and human alopecia areata (AA), are greatlydiminished in Baricitinib treated mice compare with vehicle control treated mice [2].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]Enzyme assays are performed using a homogeneous time–resolved fluorescence assay with recombinant epitope tagged kinase domains (JAK1, 837–1142; JAK2, 828–1132; JAK3, 718–1124; Tyk2, 873–1187) or full–length enzyme (cMET and Chk2)and peptide substrate. Each enzyme reaction is performed with or without test compound (11–point dilution), JAK, cMET, or Chk2enzyme, 500 nM (100 nM for Chk2) peptide, ATP (at the K m specific for each kinase or 1 mM), and 2.0% DMSO in assay buffer. The calculated IC 50 value is the compound concentration required for inhibition of 50% of the fluorescent signal. Additional kinase assays are performed at Cerep using standard conditions at 200 nM. Enzymes tested included: Abl, Akt1, AurA, AurB, CDC2, CDK2,CDK4, CHK2, c–kit, EGFR, EphB4, ERK1, ERK2, FLT–1, HER2, IGF1R, IKKα, IKKβ, JNK1, Lck, MEK1, p38α, p70S6K, PKA, PKCα, Src, and ZAP70[1].Cell Assay: Baricitinib(INCB 028050) is dissolved in stock solutions, and then diluted with appropriate media before use [1].[1]Human PBMCs are isolated by leukapheresis followed by Ficoll–Hypaque centrifugation. For the determination of IL–6–induced MCP–1production, PBMCs are plated at 3.3×105 cells per well in RPMI 1640+10% FCS in the presence or absence of various concentrations of INCB028050 (1 nM, 10 nM, 100 nM, 1 μM, and 10 μM). Following preincubation with compound for 10 min at room temperature,cells are stimulated by adding 10 ng/mL human recombinant IL–6 to each well. Cells are incubated for 48 h at 37°C, 5% CO 2.Product Name:Baricitinib Cat. No.:HY-15315CAS No.:1187594-09-7Molecular Formula:C 16H 17N 7O 2S Molecular Weight:371.42Target:JAK; JAK; JAK Pathway:Epigenetics; Stem Cell/Wnt; JAK/STAT Signaling Solubility:DMSO: 25 mg/mLSupernatants are harvested and analyzed by ELISA for levels of human MCP–1. The ability of INCB028050 to inhibit IL–6–induced secretion of MCP–1 is reported as the concentration required for 50% inhibition (IC50). Proliferation of Ba/F3–TEL–JAK3 cells is performed over 3 d using Cell–Titer Glo[1].Animal Administration: Baricitinib (INCB 028050) is suspended in 0.5% methylcellulose (Rat)[1].[1][2]Rat[1]Female rats (n=6 per gender per group) are given a dose of 10 mg/kg Baricitinib and given by oral gavage at 10 mL/kg. The first three rats are bled at 0 (predose), 2, 8, and 24 h, and the second three rats are bled 1, 4, and 12 h after dosing. EDTA is used as the anticoagulant, and samples are centrifuged to obtain plasma. An analytical method for the quantification of INCB028050 has been developed and used to analyze samples from toxicology studies. The method combines a protein precipitation extraction with 10% methanol in acetonitrile and LC/MS/MS analysis. The method has demonstrated a linear assay range 1–5000 nM using 0.1 mL of study samples. Data are processed using Analyst 1.3.1. A standard curve is determined from peak area ratio versus concentration using a weighted linear regression (1/x2).Mice[2]The C3H/HeJ graft–recipient mouse model of AA is used for these experiments. Briefly, alopecic skin from a C3H/HeJ mouse that spontaneously developed hair loss is grafted onto 8–10 week old C3H/HeJ mice free of disease. At the time of grafting, an osmotic pump that administered approximately 0.7 mg/day of Baricitinib or placebo is implanted. Osmotic pumps are changed monthly. A time–to–event survival analysis for interval censored data is performed. The survival and interval packages in R are used to perform log–rank tests. The hypothesis that the survival distributions are equal in the (n=10) Baricitinib–treated mice and (n=10)placebo–treated mice is rejected at the 5% level using Sun's score to perform an exact log–rank two–sample test with the p–value of 0.0035.References:[1]. Fridman JS, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010 May 1;184(9):5298–307.[2]. Jabbari A, et al. Reversal of Alopecia Areata Following Treatment With the JAK1/2 Inhibitor Baricitinib. EBioMedicine. 2015 Feb 26;2(4):351–5.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
Imatinib_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Imatinib is a known inhibitor of the c–Kit , Bcr–Abl , and PDGFR tyrosine kinases, inhibits the SLF–dependent activation of c–Kit wt kinase with IC 50 of ~100 nM, which is similar to the concentration requires for inhibition of Bcr–Abl and PDGFR.IC50 & Target: IC50: ~100 nM (c–Kit, Bcr–Abl, and PDGFR)[1]In Vitro: Imatinib (STI571) inhibits c–Kit autophosphorylation, activation of MAPK, and activation of Akt without altering totalprotein levels of c–kit, MAPK, or Akt. The concentration that produces 50% inhibition for these effects is approximately 100 nM [1].Imatinib (STI571) is very effective (in vitro IC 50 of 25 nM) against the chronic myeloid leukemia–causing kinase Bcr–Abl. Imatinib also efficiently inhibits Kit (in vitro IC 50, 410 nM) and PDGFR (in vitro IC 50, 380 nM)[2]. Imatinib (STI571) is a multi–target inhibitor of v–Abl,c–Kit and inhibits Bcr/Abl, v–Abl, Tel/Abl, the native PDGFβ receptor, and c–Kit, but it does not inhibit Src family kinases, c–Fms, Flt3,the EGFR or multiple other tyrosine kinases. Imatinib inhibits tyrosine phosphorylation and cell growth of Ba/F3 cells expressing Bcr/Abl, Tel/Abl, Tel/PDGFβR, and Tel/Arg with an IC 50 of approximately 0.5 μM in each case, but it has no effect on untransformed Ba/F3 cells growing in IL–3 or on Ba/F3 cells transformed by Tel/JAK2[3]. The IC 50s of Imatinib(STI571) is a multi–target inhibitor of v–Abl, c–Kit and on BON–1 and H727 cells after exposure for 48 h are 32.4 and 32.8 μM, respectively [4].In Vivo: In the phosphorothioate antisense oligodeoxynucleotides (PS–ASODN) group, tumor growth is inhibited by 59.437%, which is markedly higher than in the Imatinib (STI571) is a multi–target inhibitor of v–Abl, c–Kit and group (11.071%) and liposomenegative control group (2.759%). Telomerase activity is significantly lower (P<0.01) in the PS–ASODN group (0.689±0.158) compare with the Imatinib group (1.838±0.241), liposome negative control group (2.013±0.273), and saline group (2.004±0.163)[5]. Imatinib (25 mg/kg/day, p.o.) suppresses the growth of endometriotic tissue and reduces the number of ovarian follicles in a rat model.Imatinib effectively treats experimental endometriosis by its inhibitor effects on angiogenesis and cell proliferation [6].PROTOCOL (Extracted from published papers and Only for reference)Cell Assay: Imatinib (STI571) is dissolved in DMSO and diluted with appropriate media [4].[4]BON–1 cells (7,500 per well) andNCI–H727 cells (5,000 per well) are seeded into flat–bottomed 96–well plates in triplicate and allowed to adhere overnight in 10%fetal bovine serum–supplemented DMEM or RPMI 1640 complete medium, respectively; the medium is then exchanged forserum–free medium (negative control) or serum–free medium containing serial dilutions of Imatinib. After 48 h (control cultures do not reach confluence), the number of metabolically active cells is determined by the3–(4,5–dimethylthiazol–2–yl)–2,5–diphenyltetrazolium bromide assay, and absorbance is measured in a Packard Spectra microplate reader at 540 nm. Growth inhibition is calculated. Experiments are done in triplicates [4].Animal Administration: Imatinib (STI571) is prepare in saline (Mice and Rat)[5][6][5][6]Mice [5]The 40 tumor–bearing SCID mice are randomly divided into four groups (10 mice per group): the PS–ASODN group (5 μM, each mouse receives 0.2 mL by intratumor injection once daily); Imatinib group (0.1 mg/g body weight); liposome negative control group (0.01 mL/g); and saline group (0.01 mL/g). The mice in each group receive the relevant treatment by intra–tumor injection once dailyProduct Name:Imatinib Cat. No.:HY-15463CAS No.:152459-95-5Molecular Formula:C 29H 31N 7O Molecular Weight:493.60Target:c–Kit; Bcr–Abl; PDGFR; Autophagy Pathway:Protein Tyrosine Kinase/RTK; Protein Tyrosine Kinase/RTK; Protein Tyrosine Kinase/RTK; Autophagy Solubility:DMSO: 40 mg/mLfrom day 7 to day 28 after implantation. After 28 d, the mice are sacrificed, and tumor weight and longest and shortest diameters are measured by electronic scale and vernier caliper, respectively. Inhibition of tumor growth is calculated.Rat[6]Adult female Wistar–Albino rats (220–240 g) are used. Twenty–one days after the first surgical procedure, the rats undergo a second laparotomy to evaluate the occurrence of endometriosis. Twenty–four rats have visually confirmed endometriotic implants and are randomized into three groups to receive Imatinib (25 mg/kg/day, p.o.), Anastrozole (0.004 mg/day, p.o.), or normal saline (0.1 mL, i.p.) for 14 days.References:[1]. Heinrich MC, et al. Inhibition of c–kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000 Aug 1;96(3):925–32.[2]. Guida T, et al. Sorafenib inhibits imatinib–resistant KIT and platelet–derived growth factor receptor beta gatekeeper mutants. Clin Cancer Res. 2007 Jun 1; 13(11):3363–9.[3]. Okuda K, et al. ARG tyrosine kinase activity is inhibited by STI571.Blood. 2001 Apr 15;97(8):2440–8.[4]. Yao JC, et al. Clinical and in vitro studies of imatinib in advanced carcinoid tumors. Clin Cancer Res. 2007 Jan 1;13(1):234–40.[5]. Sun XC, et al. Depletion of telomerase RNA inhibits growth of gastrointestinal tumors transplanted in mice. World J Gastroenterol. 2013 Apr 21;19(15):2340–7.[6]. Yildiz C, et al. Effect of imatinib on growth of experimental endometriosis in rats. Eur J Obstet Gynecol Reprod Biol. 2016 Feb;197:159–63.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
Amuvatinib_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Amuvatinib (MP–470) is a potent and multi–targeted inhibitor of c–Kit, PDGFRα and Flt3 with IC50 of 10 nM, 40 nM and 81 nM,respectively.IC50 Value: 10 nM(c–KitD816H); 40 nM(PDGFRαV561D); 81 nM(Flt3D835Y) [1]Target: c–Kit; PDGFRα; FLT3in vitro: The hydrochloride salt of MP–470 also inhibits several mutants of c–Kit, including c–KitD816V, c–KitD816H, c–KitV560G, and c–KitV654A, as well as a Flt3 mutant (Flt3D835Y) and two PDGFRα mutants (PDGFRαV561D and PDGFRαD842V), with IC50 of 10 nM to8.4 μM. MP–470 potently inhibits the proliferation of OVCAR–3, A549, NCI–H647, DMS–153, and DMS–114 cells, with IC50 of 0.9μM–7.86 μM [1]. MP–470 also inhibits c–Kit and PDGFRα, with IC50 values of 31 μM and 27 μM, respectively. MP–470 demonstrates potent cytotoxicity against MiaPaCa–2, PANC–1, and GIST882 cells, with IC50 of 1.6 μM to 3.0 μM. MP–470 also binds to and inhibits several c–Kit mutants, including c–KitK642E, c–KitD816V, and c–KitK642E/D816V [2]. In MDA–MB–231 cells, MP–470 (1 μM) inhibits tyrosine phosphorylation of AXL [3]. In LNCaP and PC–3, but not DU145 cells, MP–470 exhibits cytotoxicity with IC50 of 4 μM and 8μM, respectively, and induces apoptosis at 10 μM. In LNCaP cells, MP–470 (10 μM) elicits G1 arrest and decreases phosphorylation of Akt and ERK1/2 [4].in vivo: In mice xenograft models of HT–29, A549, and SB–CL2 cells, MP–470 (10 mg/kg–75 mg/kg via i.p. or 50 mg/kg–200 mg/kg via p.o.) inhibits tumor growth [1]. In mice bearing LNCaP xenograft, MP–470 (20 mg/kg) combined with Erlotinib significantly induces tumor growth inhibition (TGI) [4].PROTOCOL (Extracted from published papers and Only for reference)Cell assay [4]For routine analysis of apoptosis, treated cells are examined for apoptotic morphology using a fluorescence staining technique. Briefly,cells are exposed to DMSO or differing doses of MP470, Erlotinib, or IM (1 to 10 μM) for 24 h and are harvested by trypsinization.After staining with a mixed dye solution containing 100 mg/ml each acridine orange and ethidium bromide the morphology of the cells is observed by fluorescence microscopy, and the number of apoptotic cells is quantified. In all cases a minimum of 200 cells are counted for each sample. Using Annexin V staining to detect apoptosis, treated cells are harvested by trypsinization and rinsed with cold PBS once. After centrifugation for 5 min, cells are resuspended in 500 μl of 1× Annexin V binding buffer and then added 1 μl of Annexin V–FITC and 1 μl of Propidium Iodide. After incubation for 5 min at room temperature in the dark, the samples were analyzed by flow cytometry.Animal administration [4]Forty eight 6–7 week–old SCID male mice are injected with 2× 107 LNCaP cells subcutaneously into the right hind flank. One month after inoculation, when tumors reach a volume of ~100 mm3, animals are divided randomly (pair–matched) into four test groups eachProduct Name:Amuvatinib Cat. No.:HY-10206CAS No.:850879-09-3Molecular Formula:C 23H 21N 5O 3S Molecular Weight:447.51Target:c–Kit; FLT3; PDGFR Pathway:Protein Tyrosine Kinase/RTK; Protein Tyrosine Kinase/RTK; Protein Tyrosine Kinase/RTK Solubility:10 mM in DMSOwith 12 mice: control group (DMSO), Erlotinib (80 mg/kg) group, MP470 (50 mg/kg) group and Erlotinib plus MP470 group. TKIs is administered IP daily from days 1 to 24. The control group is injected with 5% DMSO. A second study is also conducted with MP470 at 10 mg/kg and 20 mg/kg with 80 mg/kg Erlotinib to assess for biological efficacy (pharmacodynamics) and efficacy with 12 mice per group with the control arm of 5% DMSO. The length (L) and width (W) of the subcutaneous tumors are measured by calipers and the tumor volume (TV) was calculated as: TV = (L × W2)/2. Mice are sacrificed at the end of treatment (2–3/group), end of study or if they reached 2000 mm3 at any time during the study. Excised tumors are either fixed in paraffin or snap frozen for immunohistochemical analysis.References:[1]. Bearss DJ, et al. US Patent, US/2008/0226747.[2]. Hurley LH, et al. World Patent, WO/2005/037825.[3]. Mahadevan D, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene, 2007, 26(27), 3909–3919.[4]. Qi W, et al. MP470, a novel receptor tyrosine kinase inhibitor, in combination with Erlotinib inhibits the HER family/PI3K/Akt pathway and tumor growth in prostate cancer. BMC Cancer, 2009, 9, 142.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
伊利替康 结构式 -回复

伊利替康结构式-回复伊利替康(Elidel)是一种局部用药治疗湿疹(atopic dermatitis)的药物。
它的主要成分是酯类化合物pimecrolimus。
伊利替康是一种非激素药物,被广泛使用于针对湿疹患者的长期治疗中,有效减轻患者的症状,并且具有较少的副作用。
本文将详细解析伊利替康的结构式,为读者提供更深入的了解。
首先我们来看一下伊利替康的结构式:酯类化合物pimecrolimus的化学名为(1S, 9S, 12S, 13E, 21R, 23R)-9, 21-dihydroxy-12-isopropyl-12-(2-methyl-1-oxobutoxy)-2,5,11,13,1 9-pentamethyl-8,21-dioxo-1H,20H-[1,4]oxazino[3,4]quinolizino[2,1 -a]isoquinoline-13-yl-2-methylpropanoate。
分析伊利替康的结构式,我们可以看到它由几个部分组成。
首先是氧杂吡咯(oxazino)的环结构,它是伊利替康的骨架。
吡咯环上连接着一个氮杂喹啉环(quinolizino)和一个异戊酸酯侧链。
在氮杂喹啉环上有一个吡美沙酮(pimecrolimus)基团。
伊利替康的结构中还有一些关键的官能团。
首先是两个羟基基团(hydroxyl group),它们分别连接在吡美沙酮基团的2号和21号碳原子上。
这些羟基基团在药效中扮演着重要的角色。
接下来是一个异丙基基团(isopropyl group),连接在吡美沙酮基团的12号碳原子上。
最后是一个异丁基基团(2-methyl-1-oxobutoxy),连接在吡美沙酮基团的12号羟基上。
伊利替康属于一类被称为calcineurin抑制剂(calcineurin inhibitors)的药物。
它通过抑制T细胞的活化和炎性细胞因子的释放,从而减轻过敏反应引起的炎症和瘙痒症状。
与传统的皮质类固醇类药物(如氢化可的松)相比,伊利替康作用于皮肤外层而不影响全身免疫系统,因此具有较少的副作用,尤其是在儿童和长期使用患者中。
cas66981-73-5_Tianeptine_技术参数MedBio

1185241-83-1
10mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED16717
Azelastine HCl
Azelastine HCl
79307-93-0
200mg
≥98%
cas
1、产品物理参数:
常用名
噻奈普汀
英文名
Tianeptine
CAS号
66981-73-5
分子量
436.952
密度
1.4±0.1 g/cm3
沸点
609.2±65.0 °C at 760 mmHg
分子式
C21H25ClN2O4S
熔点
129-131°C
闪点
322.2±34.3 °C
2、同类产品列表:
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED16879
ML 289
ML 289
1382481-79-9
50mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED16787
BMY 7378
BMY 7378
21102-95-4
50mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
BQCA338747Fra bibliotek41-410mg
≥98%
品牌
货号
中文名称
英文名称
Etomoxir_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Etomoxir is a potent inhibitor of carnitine palmitoyltransferase–I (CPT–1).IC50 & Target: CPT–1[1]In Vitro: Etomoxir binds irreversibly to the catalytic site of CPT–1 inhibiting its activity, but also upregulates fatty acid oxidation enzymes. Etomoxir is developed as an inhibitor of the mitochondrial carnitine palmitoyltransferase–1 (CPT–1) located on the outer mitochondrial membrane. Etomoxir, in the liver can act as peroxisomal proliferator, increasing DNA synthesis and liver growth.Thus, etomoxir, in addition of being a CPT1 inhibitor could be considered as a PPARalpha agonist [1]. Etomoxir is a member of the oxirane carboxylic acid carnitine palmitoyl transferase I inhibitors and has been suggested as a therapeutic agent for the treatment of heart failure. Acute Etomoxir treatment irreversibly inhibits the activity of carnitine palmitoyltransferase I. As a result, fatty acid import into the mitochondria and β–oxidation is reduced, whereas cytosolic fatty acid accumulates and glucose oxidation is elevated. Prolonged incubation (24 h) with Etomoxir produces diverse effects on the expression of several metabolic enzyme [2].In Vivo: Etomoxir is an inhibitor of free fatty acid (FFA) oxidation–related key enzyme CPT1. P53 interacts directly with Bax, which is inhibited by Etomoxir, further confirming the direct interaction of P53 and Bax, and the involvement of FAO–mediatedmitochondrial ROS generation in db/db mice [3]. Rats are injected daily with Etomoxir, a specific CPT–I inhibitor, for 8 days at 20mg/kg of body mass. Etomoxir–treated rats display a 44% reduced cardiac CPT–I activity. The treatment of Lewis rats for 8 days with 20 mg/kg Etomoxir does not alter blood glucose, which is in line with comparable etomoxir–feeding studies. Similarly, Etomoxir feeding does not affect general growth characteristics such as gain in body mass, nor does it affect hindlimb muscle mass.However, heart mass and liver mass are both significantly increased by 11% in Etomoxir–treated rats [4].PROTOCOL (Extracted from published papers and Only for reference)Cell Assay: Etomoxir is dissolved in DMSO and stored, and then diluted with appropriate medium before use [2]. [2]Rat heart H9c2 myoblastic cells are incubated in DMEM containing 10% fetal bovine serum until near confluence. In some experiments,cells are preincubated for 2 h with DMEM (serum–free) in the absence or presence of 1–80 μM Etomoxir and then incubated for 2 h with 0.1 mM [1–14C]oleic acid (10 μCi/dish, binds to BSA in a 1:1 molar ratio). In other experiments, cells arepreincubated for 2 h plus or minus 40 μM Etomoxir and then incubated for 2 h with 0.1 μM or 0.1 mM [1,3–3H]glycerol (10μCi/dish), 0.1 mM [1–14C]oleic acid (2 μCi/dish, binds to BSA in a 1:1 molar ratio), 0.1 mM [1–14C]palmitic acid (2μCi/dish, binds to BSA in a 1:1 molar ratio), 28 μM [3H]ethanolamine (2 μCi/dish), 28 μM [methyl–3H]choline (2 μCi/dish), 0.4mM [3H]serine (20 μCi/dish), or 40 μM myo–[3H]inositol (10 μCi/dish). The medium is removed and the cells washed twice withice–cold saline and then harvested from the dish with 2 mL methanol–water (1:1, v/v) for lipid extraction. An aliquot of thehomogenate is taken for the determination of total uptake of radioactivity into cells. Phospholipids are then isolated andradioactivity in these determined [2].Animal Administration: Etomoxir is dissolved in 0.9% (w/v) NaCl (Rat)[4].[3][4]Mice [3]Product Name:Etomoxir Cat. No.:HY-50202CAS No.:124083-20-1Molecular Formula:C 17H 23ClO 4Molecular Weight:326.82Target:Others Pathway:Others Solubility:10 mM in DMSO80 male C57BLKS/J lar–Lepr db/db mice and 20 wild type littermates (8 week) are used. db/db mice are randomly divided into four groups: db/db group, Etomoxir group, MitoQ group, and PFT–α group. In the Etomoxir group, mice are intraperitoneally injected with 1 mg/kg Etomoxir twice every week. In the MitoQ group, 50 μM MitoQ is given to the mice in water. Water bottles, containing either MitoQ, are covered with aluminum foil, and all bottles are refilled every 3 days. In the PFT–α group, mice are intraperitoneally injected with 1 mg/kg PFT–α twice every week. WT mice are administrated with vehicle instead. The experimental period is 8 weeks. At the end, peripheral blood samples and bone marrow cells are harvested for the assays.Rat[4]Male Lewis rats, weighing 150–200 g, are used in the present study. Animals are kept on a 12 h:12 h light/dark cycle and fed a Purina Chow diet and water ad libitum. The rats are divided into two groups: (1) control and (2) Etomoxir. Etomoxir (20 mg/kg of body weight) is dissolved in 0.9% (w/v) NaCl and administered intraperitoneally for 8 days. Control rats receive saline. The last injection is given 24 h before the experiment. Animals are anaesthetized with an intraperitoneal injection of a nembutal and heparin (3:1) mixture. Subsequently, the heart is removed for LCFA uptake studies and for analyses of transporter protein contents.References:[1]. Rupp H, et al. The use of partial fatty acid oxidation inhibitors for metabolic therapy of angina pectoris and heart failure. Herz. 2002 Nov;27(7):621–36.[2]. Xu FY, et al. Etomoxir mediates differential metabolic channeling of fatty acid and glycerol precursors into cardiolipin in H9c2 cells. J Lipid Res. 2003 Feb; 44(2):415–23.[3]. Li J, et al. FFA–ROS–P53–mediated mitochondrial apoptosis contributes to reduction of osteoblastogenesis and bone mass in type 2 diabetes mellitus. Sci Rep. 2015 Jul 31;5:12724.[4]. Luiken JJ, et al. Etomoxir–induced partial carnitine palmitoyltransferase–I (CPT–I) inhibition in vivo does not alter cardiac long–chain fatty acid uptake and oxidation rates. Biochem J. 2009 Apr 15;419(2):447–55.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
Ibrutinib-biotin_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Ibrutinib–biotin is a probe that consists of Ibrutinib linked to biotin via a long chain linker, extracted from patent WO2014059368A1Compound 1–5, has an IC 50 of 0.755–1.02 nM for BTK .IC50 & Target: IC50: 0.755–1.02 nM (BTK), 1.71–2.29 nM (TEC), 3.66–4.27 nM (BLK), 4.42–5.57 nM (BMX), 5.99–6.92 nM (LCK),33.8–36.3 nM (Src), 0.187–0.198 μM (ITK),5.34–4.87 μM (JAK3)[1]In Vitro: Ibrutinib–biotin (Compound 1–5) is a probe that consists of Ibrutinib linked to biotin via a long chain linker. Ibrutinib is a TEC family kinase inhibitor. The TEC kinase family is composed of five members, TEC, BTK (Bruton's Tyrosine Kinase), ITK(interleukin–2–inducible T–cell kinase)/EMT/TSK, BMX and TXK/RLK. The TEC family kinases participate in phosphotyrosine–mediated and phospholipid–mediated signaling systems. Many TEC family proteins are abundantly expressed in hematopoietic tissues, and play important roles in the growth and differentiation processes of blood cells [1].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]In this study, assay formats are tested for sensitivity, specificity, range, and to determine most suitable anti BTK Antibody. An aliquot of DOHH2 cell lysate (1 mg/mL) is inhibited with 1 μM Ibrutinib then labeled with probe (1 μM). Negativecontrols: Untreated DOHH2 and Jurkat cell lysates (l mg/mL) are labeled with probe (1 μM) and Untreated DOHH2 cell lysate. Standard Streptavidin plate (5–pack); Read Buffer T (50 mL), SULFO TAG Goat Anti–mouse (50 μg); SULFO TAG Goat Anti–rabbit (50 μg); SULFO TAG streptavidin (50 ug); MSD Standard Plates; MSD Blocker A; Protease inhibitor cocktail; Positive Control lysates from BTK expressing cell line (DOHH2); Negative Control lysates; Ibrutinib (PCI); probe compound 1–5 (biotinylated probe).References:[1]. Betty Y. CHANG, et al. Companion diagnostics for tec family kinase inhibitor therapy. WO 2014059368 A1.Product Name:Ibrutinib–biotin Cat. No.:HY-100342CAS No.:1599432-18-4Molecular Formula:C 56H 80N 12O 9S Molecular Weight:1097.37Target:Btk Pathway:Protein Tyrosine Kinase/RTK Solubility:10 mM in DMSOCaution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@ Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
Alda-1_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Alda–1 is a potent ALDH2 agonist, which significantly improves ALDH2 activity.In Vivo: Alda–1 treatment results in a significant decrease of 4–HNE–protein content in the plasma of apoE -/- mice. Alda–1administration leads to a slight increase in gene expression related to neurogenesis (Nog ), mitochondrial biogenesis (CYTB , ND1),and apoptosis (Bax , Gsk3b ) in the Hp of apoE -/- mice. Alda–1 administration leads to 2 and 10 differentially expressed proteins in theFCx and Hp of apoE -/- mice, respectively [1]. Alda–1 (1.5 mg/kg, b.w., i.p.) administration significantly increases the climbing time,tends to reduce the immobility time and increases the swimming time of the prenatally stressed rats in the forced swim test.Moreover, treatment of prenatally stressed rats with Alda–1 significantly increases number of entries into the open arms of the maze and the time spent therein, as assessed by elevated plus–maze test [2]. Alda–1 (8.5 mg/kg, i.p.) with glucose significantly lowers 4–HNE and FJB–positive cells in the cerebral cortex of Alda–1–treated rats than in DMSO–treated rats 24 h after glucose administration [3]. Alda–1 (10 mg/kg per day) treatment prevents aldehydic overload, mitochondrial dysfunction and improves ventricular function in post–MI cardiomyopathy rats [4].PROTOCOL (Extracted from published papers and Only for reference)Cell Assay:[2]Spleen cells (4×106 cells/mL) are stimulated by optimal concentrations of concanavalin A (Con A; 2.5μg/mL and 0.6 μg/mL) and lipopolysaccharide (LPS, 5 μg/mL) and are incubated in 96–well plates at final volume of 0.2mL for 72 h. Cell proliferation is determined by adding 0.5 μCi of [3H]–thymidine per well at 16 h before the end of the incubation.The cultures are harvested with an automatic cell harvester, and [3H] thymidine incorporation is assessed using a liquid scintillationcounter.Animal Administration: Alda–1 is dissolved in 1 mL/kg b.w. DMSO/water 50/50.[2]After behavioral verification at three months of age,the animals are divided into the following four groups: control, control + Alda–1, prenatally stressed and prenatally stressed + Alda–1(6 animals per group). Alda–1 injections are given intraperitoneally (i.p.) once daily at a dose of 1.5 mg/kg b.w. (dissolved in 1 mL/kg b.w. DMSO/water 50/50) for 14 days. At the same time, the control and prenatally stressed rats receive 1 mL/kg b.w. DMSO/water 50/50. The injections of Alda–1 and vehicle are given between 10 a.m and 11 a.m. In the last five days of Alda–1 treatment the behavioral parameters in the elevated plus maze test and then in the forced swim test are measured.References:[1]. Stachowicz A, et al. Proteomic Analysis of Mitochondria–Enriched Fraction Isolated from the Frontal Cortex and Hippocampus of Apolipoprotein E Knockout Mice Treated with Alda–1, an Activator of Mitochondrial Aldehyde Dehydrogenase (ALDH2). Int J Mol Sci.[2]. Stachowicz A, et al. The impact of mitochondrial aldehyde dehydrogenase (ALDH2) activation by Alda–1 on the behavioral and biochemical disturbances in animal model of depression. Brain Behav Immun. 2016 Jan;51:144–53.Product Name:Alda–1Cat. No.:HY-18936CAS No.:349438-38-6Molecular Formula:C 15H 11Cl 2NO 3Molecular Weight:324.16Target:Aldehyde Dehydrogenase (ALDH)Pathway:Metabolic Enzyme/Protease Solubility:DMSO: ≥ 51 mg/mL[3]. Ikeda T, et al. Effects of Alda–1, an Aldehyde Dehydrogenase–2 Agonist, on Hypoglycemic Neuronal Death. PLoS One. 2015 Jun 17;10(6):e0128844.[4]. Gomes KM, et al. Aldehydic load and aldehyde dehydrogenase 2 profile during the progression of post–myocardial infarction cardiomyopathy: benefits of Alda–1. Int J Cardiol. 2015 Jan 20;179:129–138.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
Omarigliptin-Standard-DataSheet-MCE

[2]. Li X, et al. Omarigliptin alleviates cognitive dysfunction in Streptozotocin-induced diabetic mouse. Bioengineered. 2022 Apr;13(4):9387-
Caution: Product has not been fully validated for medical applications.
For research use only.
Tel: 400-820-3792*************Fax: ************E-mail: **********************
Omarigliptin (Standard)是 Omarigliptin 的分析标准品。本产品用于研究及分析应用。Omarigliptin (MK-
3102) 是一种有效的、选择性的、具有口服活性的并能穿过血脑屏障的二肽基肽酶 4 dipeptidyl peptidase 4
(DPP-4) 抑制剂。Omarigliptin 显示出抗帕金森病活性。Omarigliptin 具有改善糖尿病相关认知功能障碍的神
1/1Masles —您身边的抑制剂大师
[3]. Ayoub BM, et al. Repurposing of Omarigliptin as a Neuroprotective Agent Based on Docking with A2A Adenosine and AChE Receptors, Brain GLP-1 Response and Its Brain/Plasma Concentration Ratio after 28 Days Multiple Doses in Rats Using LC-MS/MS. Molecules. 2021
Tivantinib_LCMS_07439_MedChemExpress

=====================================================================Acq. Operator : Li Shan(LCMS-02) Seq. Line : 32Acq. Instrument : HY-LCMS-02 Location : Vial 30Injection Date : 12/31/2014 12:02:41 PM Inj : 1Inj Volume : 3.000 µlAcq. Method : D:\AGLIENT 1260\DATA\20141231\20141231 2014-12-31 09-27-51\100-1000MS+3MIN( 0.02%FA).MLast changed : 12/31/2014 9:27:51 AM by Li Shan(LCMS-02)Analysis Method : D:\AGLIENT 1260\DATA\20141231\20141231 2014-12-31 08-59-26\100-1000MS+3MIN( 0.02%FA).M (Sequence Method)Last changed : 12/31/2014 1:19:39 PM by Li Shan(LCMS-02) (modified after loading)Method Info : Postive,MS:100-1000,Column ID:A-RP-102,40℃Catalog No : HY-50686 Batch#07439 A-RP-32Additional Info : Peak(s) manually integratedmin0.511.522.53mAU 02505007501000125015001750 DAD1 B, Sig=214,4 Ref=off (D:\AGLIENT...0\DATA\20141231\20141231 2014-12-31 09-27-51\CPK2014-D31-07439.D)1.3111.6591.816===================================================================== Area Percent Report =====================================================================Sorted By : Signal Multiplier : 1.0000Dilution : 1.0000Do not use Multiplier & Dilution Factor with ISTDsSignal 1: DAD1 B, Sig=214,4 Ref=offPeak RetTime Type Width Area Height Area # [min] [min] [mAU*s] [mAU] %----|-------|----|-------|----------|----------|--------| 1 1.311 MM 0.0440 6.96597 2.63651 0.1281 2 1.659 MM 0.0583 9.69770 2.77457 0.1784 3 1.816 MM 0.0486 5420.18994 1859.45325 99.6935Totals : 5436.85360 1864.86432===================================================================== *** End of Report ***=====================================================================Acq. Operator : Li Shan(LCMS-02) Seq. Line : 32Acq. Instrument : HY-LCMS-02 Location : Vial 30Injection Date : 12/31/2014 12:02:41 PM Inj : 1Inj Volume : 3.000 µlAcq. Method : D:\AGLIENT 1260\DATA\20141231\20141231 2014-12-31 09-27-51\100-1000MS+3MIN( 0.02%FA).MLast changed : 12/31/2014 9:27:51 AM by Li Shan(LCMS-02)Analysis Method : D:\AGLIENT 1260\DATA\20141231\20141231 2014-12-31 08-59-26\100-1000MS+3MIN( 0.02%FA).M (Sequence Method)Last changed : 12/31/2014 1:20:51 PM by Li Shan(LCMS-02) (modified after loading)Method Info : Postive,MS:100-1000,Column ID:A-RP-102,40℃Catalog No : HY-50686 Batch#07439 A-RP-32Additional Info : Peak(s) manually integratedmin0.511.522.5380000100000120000140000160000180000200000220000240000 MSD1 TIC, MS File (D:\AGLIENT 1260\DATA\20141231\20141231 2014-12-31 09-27-51\CPK2014-D31-07439.D) ES-API, Pos, S1.813MS Signal: MSD1 TIC, MS File, ES-API, Pos, Scan, Frag: 50 Spectra averaged over upper half of peaks. Noise Cutoff: 1000 counts.Reportable Ion Abundance: > 10%.Retention Mol. Weight Time (MS) MS Area or Ion1.813 852571 371.10 I 370.10 I 213.15 I 130.15 I 124.15 I 100.15 Im/z10020030040050060070080020406080100*MSD1 SPC, time=1.781:1.853 of D:\AGLIENT 1260\DATA\20141231\20141231 2014-12-31 09-27-51\CPK2014-D31-07439.D ES-API Max: 54034105.1214.1124.2235.1762.3213.1392.1371.1370.1*** End of Report ***。
托伐普坦Tolvaptan杂质汇总列表
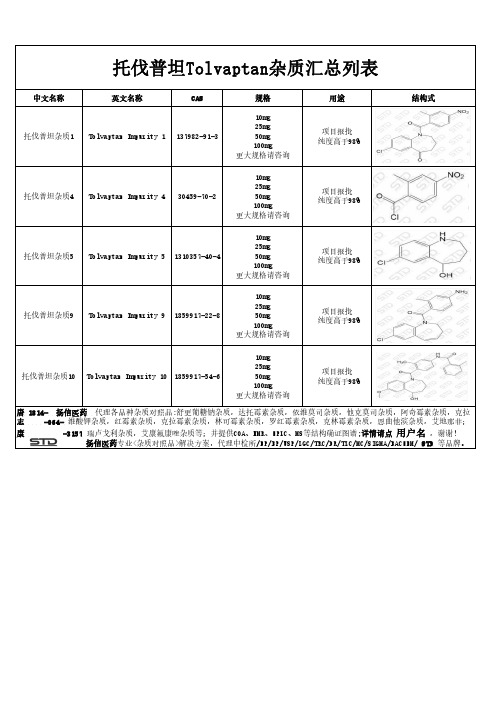
志 -064- 维酸钾杂质,红霉素杂质,克拉霉素杂质,林可霉素杂质,罗红霉素杂质,克林霉素杂质,恩曲他滨杂质,艾地那非;
康
-3157 瑞卢戈利杂质,艾康氟康唑杂质等;并提供COA、NMR、HPLC、MS等结构确证图谱;详情请点 用户名 ,谢谢!
扬信医药专业<杂质对照品>解决方案,代理中检所/EP/BP/USP/LGC/TRC/DR/TLC/MC/SIGMA/BACHEM/ STD 等品牌。
托伐普坦Tolvaptan杂质汇总列表
中文名称
英文名称
CAS
托伐普坦杂质1 Tolvaptan Impurity 1 137982-91-3
规格
10mg 25mg 50mg 100mg 更大规格请咨询
用途
项目报批 纯度高于98%
结构式
托伐普坦杂质4 Tolvaptan Impurity 4 30459-70-2
10mg 25mg 50mg 100mg 更大规格请咨询
项目报批 纯度高于98%
托伐普坦杂质10 Tolvaptan Impurity 10 1859917-54-6
10mg 25mg 50mg 100mg 更大规格请咨询
项目报批 纯度高于98%
唐 1814- 扬信医药 代理各品种杂质对照品:舒更葡糖钠杂质,达托霉素杂质,依维莫司杂质,他克莫司杂质,阿奇霉素杂质,克拉
10mg 25mg 50mg 100mg 更大规格请咨询
项目报批 纯度高于98%
托伐普坦杂质5
Tolvaptan Impurity 5 1310357-40-4
10mg 25mg 50mg 100mg 更大规格请咨询
项目报批 纯度高于98%
托伐普坦杂质9ຫໍສະໝຸດ Tolvaptan Impurity 9 1859917-22-8
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Tivantinib is a novel and highly selective c–Met tyrosine kinase inhibitor with K i of 355 nM.IC50 & Target: Ki: 355 nM (c–Met)[1]In Vitro: Tivantinib (ARQ 197) selectively inhibits c–Met activity in cell–free and cell–based assays. c–Met–expressing cancer cell lines treated with Tivantinib display either a dose–dependent loss of proliferative capacity or caspase–dependent apoptosis that positively correlates with either ligand–dependent c–Met activity or constitutively active c–Met. To examine the biochemical mode of inhibition of Tivantinib, kinetic analyses are done using recombinant human c–Met in a filtermat–based assay. The K m of ATP is 50.5±2.2 μM,which is similar to the K m value of ATP. In these kinetic studies, Tivantinib inhibits human recombinant c–Met with a calculated inhibitory constant (K i ) of ~355 nM. In vitro exposure to Tivantinib inhibits constitutive c–Met phosphorylation in HT29 and MKN–45cells, and HGF–induced c–Met phosphorylation in MDA–MB–231 and NCI–H441 cells with an IC 50 of 100 to 300 nM [1]. Tivantinib is a low–molecular–weight compound, and is the first in class orally available selective inhibitor of c–Met [2].In Vivo: Pharmacodynamically, the phosphorylation of c–Met in human colon xenograft tumors (HT29) is strongly inhibited by Tivantinib (ARQ 197), as assessed by a dramatic reduction of c–Met autophosphorylation 24 hours after a single oral dose of 200mg/kg of Tivantinib. This same dosage in mice shows that tumor xenografts are exposed to sustained plasma levels of Tivantinib,consistent with the observed pharmacodynamic inhibition of c–Met phosphorylation and inhibition of proliferation of c–Met harboring cancer cell lines. A C max of 5.73 μg/mL (13 μM), an area under the concentration–time curve of 12.1 μg/mL h, and a t 1/2 of2.4 hours are measured. Plasma levels of Tivantinib 10 hours after dosing are determined to be 1.3 μM, >3–fold above thebiochemical inhibitory constant of Tivantinib for c–Met [1].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]Recombinant c–Met protein (50 ng) comprising residues 974 to 1390 is incubated in a reaction buffer [50 mM Tris–HCl (pH 7.5), 2 mM DTT, 0.1 mM Na 3VO 4, 10 mM MgCl 2, 1 mM EGTA, 0.02 mg/mL bovine serum albumin, and 10%glycerol] with various concentrations of Tivantinib or DMSO in a total volume of 20 μL. After incubating for 20 minutes atroom temperature, 20 μL of 20 μM poly–Glu–Tyr substrate and 20 μL of increasing amounts of cold ATP containing 1.5 μCi of[γ–33P]ATP are added to initiate the reaction. The reaction is stopped after 5, 10, 20, 40, and 60 minutes by the addition of 10%phosphoric acid and 10 μL aliquots of the reaction mixture are spotted onto P30 filtermat in triplicate. The filters are washed thrice for 5 minutes with 0.75% phosphoric acid and once with methanol for 2 minutes. The level of radioactivity is determined by using a Wallac TriLux MicroBeta liquid scintillation counter. Nonspecific binding is determined by conducting the assay in the absence of enzyme and then subtracting the value from each of the experimental values. Reaction rates are determined in the linear range of each reaction and GraphPad Prism software is used to calculate the K m and V max values [1].Cell Assay: Tivantinib (ARQ 197) is prepared in DMSO and stored, and then diluted with appropriate medium before use [1].[1]HT29,MKN–45, MDA–MB–231, and NCI–H441 (lung cancer) human cancer cells are seeded in 96–well plates overnight in a medium withProduct Name:Tivantinib Cat. No.:HY-50686CAS No.:905854-02-6Molecular Formula:C 23H 19N 3O 2Molecular Weight:369.42Target:c–Met/HGFR Pathway:Protein Tyrosine Kinase/RTK Solubility:10 mM in DMSO10% FBS. Each cell line is optimized for seeding cell number to ensure a similar degree of confluence at the end of the experimentin nontreated (control) wells. The next day, cells are treated with different concentrations of Tivantinib for 24 hours at 37°C. After ARQ 197 treatment, the drug–containing medium is removed, and cells are washed twice with PBS and incubated in a drug–free medium for an additional 48 hours. Cells are then incubated and stained for 4 hours with the MTS reagent (final concentration of 0.5 mg/mL) per well and are lysed. The results are quantitated by spectrophotometry at λ=450 nm[1].Animal Administration: Tivantinib (ARQ 197) is formulated in polyethylene glycol 400/20% Vitamin E tocopheryl polyethylene glycol succinate (60:40) (Mice)[1].Tivantinib (ARQ 197) is prepared by dissolving 10 mg in 10 mL methanol (1 mg/mL). The stock solution is furtherdiluted with methanol to obtain working solutions at several concentration levels (Rat)[2].[1][2]Mice[1]Female athymic nude mice are acclimated to the animal housing facility for at least 1 week before the study. Efficacy studies are done in athymic mice bearing HT29, MKN–45, or MDA–MB–231 tumor xenografts to determine the effect of ARQ 197 on tumor growth. Tumor cells [5×106 (HT29) and 8×106 (MKN–45 and MDA–MB–231) cells/animal] are inoculated s.c. on day 0. Tumor dimensionsare measured by a digital caliper and tumor volumes are calculated as length×width2/2. When tumors reached a volume of ~100 mm3, mice are randomized into groups and treated daily with orally administered vehicle control or 200 mg/kg Tivantinib formulated in polyethylene glycol 400/20% Vitamin E tocopheryl polyethylene glycol succinate (60:40) at 30 mg/mL, for 5 consecutive days, followed by a 2–day dosing holiday for four cycles. Therefore, each animal received a total of 20 doses. Results are expressed as mean tumor volume±SEM. To assess differences in tumor size between groups, a Mann–Whitney nonparametric t test is performed and significance is defined as P<0.05.Rat[2]Six male Sprague–Dawley rats (180–220 g) are used to study the pharmacokinetics of Tivantinib. Diet is prohibited for 12 h before the experiment but water is freely available. Blood samples (0.3 mL)References:[1]. Munshi N, et al. ARQ 197, a novel and selective inhibitor of the human c–Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010 Jun; 9(6):1544–53.[2]. Bai YL, et al. Quantitative analysis of tivantinib in rat plasma using ultra performance liquid chromatography with tandem mass spectrometry. J Pharm Biomed Anal. 2016 Jul 15;126:98–102.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。