illumina_mapper_comparison_slides
Illumina测序介绍

Adapter sequence 接头序列
3' extension 3' 延伸
9 Part # 15045845_Rev.C
FOR RESEARCH USE ONLY
Original template 原始模板链
Discard 洗去丢弃
10 Part # 15045845_Rev.C
FOR RESEARCH USE ONLY
Newly synthesized
strand 新合成链
Single-Stranded DNA 单链DNA
NOTE: Single molecules bind to flow cell in a random pattern NOTE: 单个DNA 分子以随机 的方式与流动槽表面 结合
FOR RESEARCH USE ONLY
Illumina Sequencing Workflow Illumina 测序流程
tion 簇生成
cBot MiSeq NextSeq HiSeq 2500-Rapid
Sequencing 测序
Data Analysis 数据分析
3 Part # 15045845_Rev.C
FOR RESEARCH USE ONLY
HiSeq HiScan SQ
GA IIx MiSeq NextSeq
ICS/RTA CASAVA
MSR BaseSpace
Illumina Sequencing Workfl上能够与 测序仪配合的接头序列(Multiplexed, SR, PE)
illumina双端测序原理

illumina双端测序原理
Illumina双端测序原理是一种高通量测序技术,通过使用Illumina测序仪,分别从DNA序列的两端进行测序。
具体步骤如下:
1. DNA片段制备:将DNA样本进行随机剪切,然后在片段的末端加入适配器序列。
2. PCR扩增:将DNA片段进行PCR扩增,使其在适配器的
两端连续存在。
3. 测序芯片制备:将PCR扩增产物固定在质量好的玻璃芯片上,形成DNA片段阵列。
4. 簇生成:在测序芯片上进行扩增,使每个DNA片段形成簇。
5. DNA测序:利用碱基特异性的可逆终止,加入荧光标记的
碱基和DNA聚合酶,自由核苷酸流失后发出荧光信号,记录
每个碱基的测序信号。
6. 反向互补链合成:将测序芯片上的DNA链反转,形成反向
互补链。
7. 二次测序:与第一次测序相同的方式进行第二次测序。
8. 数据分析:对测序数据进行质量控制、过滤和比对分析。
双端测序的优点是能够提高测序的准确性和覆盖度,增强对重复序列的解读能力,并提供了更多的生物信息学分析的可能性。
illumina测序方法

illumina测序方法
Illumina测序方法是一种基于边合成边测序(Sequencing-By-Synthesis,SBS)的测序技术。
该方法包括以下步骤:
1. 向反应体系中同时添加DNA聚合酶、接头引物和带有碱基特异荧光标记的4种dNTP。
这些dNTP的3’-OH被化学方法所保护,因而每次只能添加一个dNTP。
2. 在dNTP被添加到合成链上后,所有未使用的游离dNTP和DNA聚合酶会被洗脱掉。
3. 加入激发荧光所需的缓冲液,用激光激发荧光信号,并有光学设备完成荧光信号的记录。
4. 利用计算机分析将光学信号转化为测序碱基。
这样荧光信号记录完成后,再加入化学试剂淬灭荧光信号并去除dNTP 3’-OH保护基团,以便能进行下一轮的测序反应。
Illumina测序方法的特点是每次只添加一个dNTP,这可以很好地解决同聚物长度的准确测量问题(Homopolymer错误)。
其主要的测序错误来源是碱基的替换,目前其测序错误率在1%-%之间。
以上信息仅供参考,如需了解更多信息,建议查阅生物信息学领域的学术文献或咨询相关专家。
illumina测序

illumina测序Illumina测序Illumina测序是一种高通量测序技术,广泛应用于基因组学和转录组学研究。
该技术基于DNA聚合酶链反应和桥式PCR技术,通过对DNA样本进行多次放大和测序,可以获得高分辨率的DNA测序结果。
Illumina测序的原理是在玻片上固定DNA片段,然后使用DNA聚合酶链反应将其复制成成千上万个复制品。
接下来,将DNA片段裂解成单链,并通过固定在玻片上的引物进行二次扩增。
这种二次扩增会在表面形成DNA聚集群,每个聚集群都包含数百万个相同的DNA片段。
在扩增过程中,每个DNA片段的末端都会加上一段DNA适配体,这些适配体可以与固定在玻片上的引物配对。
然后,在放大的DNA 群集上进行合成,使用荧光标记的二聚体核苷酸来读取每个DNA 片段的碱基。
通过逐个读取每个DNA片段的碱基,可以获得完整的DNA序列。
Illumina测序的优势在于其高通量和高精度。
由于使用了桥式PCR 技术,可以同时测序多个DNA片段,从而显著提高了测序效率。
此外,Illumina测序使用的荧光标记和高分辨率成像技术,可以准确读取每个碱基的荧光信号,进一步提高了测序的准确性。
利用Illumina测序技术,研究人员可以进行多种基因组学和转录组学研究。
通过对整个基因组的测序,可以识别出DNA序列的突变、插入和缺失等变异类型,从而帮助研究人员研究疾病的发生机制。
此外,Illumina测序还可以用于测定RNA浓度、检测转录本的表达水平以及鉴定RNA剪接变异。
虽然Illumina测序技术具有许多优势,但也存在一些限制。
首先,Illumina测序仍然需要较高的成本,特别是在进行大规模测序时。
其次,由于需要多次放大和拼接,Illumina测序在处理GC含量高的DNA序列时可能存在失真。
此外,Illumina测序对于DNA片段的长度也有一定的限制,通常在几百个碱基对的范围内。
尽管Illumina测序存在一些限制,但其高通量、高精度和广泛的应用范围使其成为目前最常用的测序技术之一。
Illumina各测序系统介绍与应用比较

On-Board ExAmp Mixing
On-Board Cluster Generation
Full 2x150bp Dual-Index
0.0 5O00B.0CG 1000.0 1500.0Re2a0d010.0 2500.0
i7 Prep
Index 1
I5 Prep
Index 2
P.E Turn
HiSeq 3000
750 Gb | 2.5B 2x150
Decreasing price per GB
2
Understand the Equipment Options
测序平台需要考虑的因素
我最常用的 应用?
哪些测序平台 最适合我的应用?
仪器之外的cost?
所选NGS平台 将如何影响
工作流及进程?
SERIOUS PRODUCTION SCALE!
PRODUCTION SCALE MANY SAMPLES | RUN
MiniSeq
MiSeq
NextSeq 500
LOW THROUGHPUT TGRS | WGS (MICROBES)
4
HiSeq X
Cost Per Human Genome
$3,000,000,000Human Genome Pro2je0c0t 3
Nanofabrication
Optics
Surface chemistry
Read length
Throughput
Informatics
10
Dye chemistry Enzymology
Fluidics Accuracy
Camera/sensors
Cycle time
illumina芯片介绍

Whole Genome
• WG Resequencing • Candidate • WG Gene Expr. Resequencing • miRNA Discovery • Chromosome & Profiling sequencing • WG ChIP-Seq • HT Biomarker • Gene Expr. Validation • WG GT • CNV Screening• Custom GT • Focused Expr. • Biomarker • FFPE Discovery
Detected duplication
B allele frequency data (AAB genotype) A shift in the LogR value is detectible, but the integration of B allele data improves the signal to noise ratio
From discovery to single target Validation
Sequencing & Arrays Platform Synergy
Discovery
>109 108 107
Focused Research/Validation
106 105 104 103
Screening
660kmarkerspersample4samplesperchipcomprehensivegenomiccoverage5000commoncnvregionsinfiniumhdassaylowdnainput200ng410persample700kmarkerspersample12samplesperchiphapmapallphases1000genomesprojectinfiniumhdassaylowdnainput200ng260persample?1millionmarkerspersample4samplesperchiphapmapallphases1000genomesprojectcnvprobesinfiniumhdassaylowdnainput200ng468persample??????????????????software???developedwithandforthecytogeneticistautomatedcrossmatchingandreportingfunctionsadjustabledatabaseofknownregions???numerousanalysisoptionsforcopynumberanalysisgraphicaltableanddatadisplayslinksto3rdpartysoftwareofferingsforresearchuseonlyhumancytosnp12beadchipoptimizedforefficientcytogeneticanalysishigherdensityincytohighvalueregions40ofgenomeallpericentromeresandsubtelomeressexchromosomescommonregionsofinteresteg
illumina测序原理

illumina测序原理
illumina测序技术是近几年来新出现的革命性的DNA测序技术,它可以非常快速、高效地检测出DNA序列。
其原理是将DNA分子片段分解成小片段,通过特殊的技术放大要测序的DNA片段,并利用激光扫描仪识别出每个放大的DNA片段的序列,从而完成DNA测序。
illumina测序的原理首先是在激光技术的基础上改进了DNA分
子的处理步骤。
它允许在DNA分子上更多种的分子标记,通过特殊的标记模式,使得每个DNA分子被不同的标记分子锁定在磁链上,从而实现分子的自动排序。
其次,illumina测序技术通过特殊的放大技术,利用水平分型
技术,放大DNA分子,从而形成一整块磁盘,其上分布着大量的DNA 分子,可以千分之一地检测DNA序列。
最后,illumina测序的最重要的技术是激光扫描技术。
激光扫
描仪将放大的DNA分子板发射的特定波长的激光做扫描,并从中提取出DNA分子的序列信息。
由此,以上三个技术组合在一起,就完成了illumina测序技术。
illumina测序技最大的优势在于可以快速、高效地检测出DNA
序列,同时可以非常大规模地阅读。
在分子遗传学研究、基因组学等方面有着巨大的应用前景,比如可以通过它进行大规模的基因组测序,可以分析DNA序列差异性以及突变等,可以帮助科学家更深入地了解生物的进化变化以及遗传病的发生机制。
总之,illumina测序技术是一种极大改进的DNA测序技术,它
比以前的技术更加快速、高效,更易于操作,对于基因组学研究有着极大的作用。
Illumina测序仪器故障问题处理(Troubleshooting)总结

3.
3.1.
现象描述:
在开始Read 1的照相前需进行校准对焦,第一步找平面有时会出错,如下图所示,提示“Exceeded maximum number of alignment”。
解决方法:
选择Normal Pause,先把网络连接问题解决,恢复网络后等待RTA运行将磁盘空间释放,再Resume Run。Hiseq2500的RTA可能会自行关闭了,找到RTA logs文件夹,运行里面的StartRTA.bat可以打开RTA。恢复网络后如果RTA不上传数据,查看大型机文件是否缺失并补齐。
(2)液路问题,重新安放FC和Mainfold,把FC和平台擦拭干净,FC往左下方靠,Mainfold垂直扣下压紧,如果仍然不通过,尝试更换Mainfold;更换Mainfold还是不通过的话,可能是注射器问题。
3.
cBot运行一个Run中可能发生错误而停止,原因可分为机械碰撞和通讯卡顿两类。
3.1.
解决方法:
(1)关闭HCS,重启电脑,等待电脑识别完成,出现DONOTEJECT盘再打开HCS软件,如果同样报错,尝试下一个方法。
(2)打开port manager软件或右键电脑——“Manager”——“Device Manager”,把所有COM删除,点击重启电脑,然后关闭仪器电源按钮,电脑重启后打开仪器电源按钮,等待识别完成,再打开HCS软件。若还不行,尝试下一个方法。
(3)把所有COM删除,点击重启电脑,然后关闭仪器电源按钮,电脑重启后打开仪器电源按钮,等待识别完成,再打开HCS软件。若还不行,尝试下一个方法。
Illumina测序技术在母血检测胎儿非整倍体中应用

W e , UANG Yo g h a,t 1 ( p rme to Obttis Jin me iH n — u e . De a t n f a serc , a g nCe ta s ia , i n n r lHo p t l J a gme 2 0 0 C ia n5 90 , hn )
I u n 测 序 技 术 在 母 血 检 测 胎儿 非 整倍 体 中应 用 l mia l
冯穗 华 王 . , , 威 黄泳 华 陈 芳 蒋馥 蔓 李 卫凯 麦巧娇 瞿京辉 张燕玲 张红云 陈 建勇 , , , , , , , ,
(. 门 市 中 心 医 院 产科 , 东 江 门 5 90 ;. 圳 华 大基 因研 究 院 ) I江 广 2 00 2深 摘要 : 的 目 景 。方 法 研 究 I u n 测 序 技 术 在 母 血 中 检 测 胎 儿 非 整 倍 体 的 可 行 性 , 其 在 无 创 性 产 前 诊 断 中 的应 用 前 l mia l 及
De e tn e a hr m o o a ne l i y Ilm i a s lx e ue i tc i g ft lc o s m la up ody b lu n - oe a s q ncngDN A n m a e na l s a FE N G - ua . A N G i tr lp a m Suih W
megn eh oo yfr h ses n f unrbepau [] rigtc nlg o eassmeto lea l lq eJ . t v
EurHe r 2 0 2 ( 1 1 8 . atJ, 0 7, 8 1 ): 2 3
[ ] rwnB Z a 6 B o G, hoXQ.sita aclrutao n h odsa d I rvsua l su dteg l tn — n r
Illumina NextSeq 500系统指南说明书

NextSeq500系统指南文档号15046563v07CHSILLUMINA所有2021年10月本文档及其内容归Illumina,Inc.及其附属公司(以下简称“Illumina”)所有,并且仅供其客户用于与本文档内所述产品用途相关的合同用途,不得用于其他任何目的。
若事先未获得Illumina的书面许可,不得出于任何其他目的使用或分发本文档及其内容,以及/或者以其他任何方式对其进行传播、披露或复制。
Illumina不通过本文档向第三方授权其任何专利、商标、所有权或普通法权利或类似权利。
必须由具备资质且受过相关培训的人员严格明确遵照本文档中的说明操作,以确保本文档中所述产品的使用适当且安全。
在使用此类产品之前,相关人员必须通读并理解本文档中的所有内容。
未能完整阅读并明确遵守本文档中包含的所有说明可能会导致产品损坏、对用户或其他人员造成人身伤害以及对其他财产造成损害,并且将导致产品适用的保证失效。
对于由不当使用本文档中描述的产品(包括其部件或软件)引起的任何后果,ILLUMINA概不承担任何责任。
©2021Illumina,Inc.保留所有权利。
所有商标均为Illumina,Inc.或其各自所有者的财产。
有关特定的商标信息,请参见/company/legal.html。
修订历史记录目录第1章概述1简介1更多资源1仪器组件2测序耗材概述5第2章入门9启动仪器9自定义系统设置9自定义运行设置11用户自备的耗材和设备12第3章测序13简介13使用Local Run Manager软件创建运行14使用NCS创建运行14准备试剂夹盒14准备流动槽14制备测序文库15设置测序运行16监控运行进度22运行后自动清洗23第4章维护24简介24执行手动清洗24更换空气过滤器26软件更新28关闭仪器29附录A故障诊断30简介30故障诊断文件30解决自动检查错误31废试剂容器已满31再次杂化工作流程32自定义配方和配方文件夹34系统检查34 RAID错误消息36网络存储错误36配置系统设置36附录B Real-Time Analysis39NextSeq500系统指南Real-Time Analysis概述39 Real-Time Analysis工作流程40测序输出文件43流动槽小区43输出文件夹结构46索引47技术协助50第1章概述简介1更多资源1仪器组件2测序耗材概述5简介Illumina®NextSeq™500系统完美融合了高通量测序的强大能力与台式测序仪器的简便特性。
illumina 测序试剂盒上的300cycles的意思

illumina 测序试剂盒上的300cycles的意思【原创实用版】目录1.Illumina 测序试剂盒的概述2.300cycles 的含义3.300cycles 在 Illumina 测序试剂盒中的应用4.300cycles 的优缺点正文一、Illumina 测序试剂盒的概述Illumina 是一家美国的生物科技公司,致力于开发基因组学、基因表达和基因变异等领域的高精度分析技术。
其中,Illumina 测序试剂盒是基因测序领域的重要产品,被广泛应用于科研和临床实验室。
二、300cycles 的含义在 Illumina 测序试剂盒中,300cycles 指的是在测序过程中,每个样本的 DNA 链经过 300 个循环的扩增。
这里的“循环”是指 DNA 聚合酶链反应(PCR)的扩增过程。
在每次循环中,DNA 的数量会翻倍,经过 300 个循环后,DNA 的数量将增加到原来的 2^300 倍。
三、300cycles 在 Illumina 测序试剂盒中的应用在 Illumina 测序试剂盒中,300cycles 主要用于实现高深度的 DNA 测序。
通过多次扩增,可以获取足够多的 DNA 分子,从而在测序过程中产生更多的数据,提高测序的准确性和分辨率。
此外,300cycles 也可以用于检测 DNA 样本中的微量模板,使其在测序过程中能够被有效检测到。
四、300cycles 的优缺点优点:1.提高测序深度:300cycles 可以增加 DNA 的数量,从而提高测序深度,有助于检测低频突变和罕见变异。
2.提高测序准确性:通过多次扩增,可以降低测序误差,提高测序结果的可靠性。
3.检测微量模板:300cycles 可以有效地扩增微量 DNA 样本,使其在测序过程中能够被检测到。
缺点:1.扩增偏倚:多次扩增过程中可能出现扩增偏倚,导致某些 DNA 片段的数量被过度扩增,影响测序结果的准确性。
2.扩增产物不均一:随着扩增次数的增加,扩增产物的均匀性可能会降低,导致部分扩增产物无法参与到后续的测序过程中。
illumina技术参数

Illumina技术是一种基于高通量测序平台的技术,具有以下主要参数:首先,Illumina测序平台具有极高的读长分辨率,可以用于组装出高质量的全基因组组装序列。
其次,该技术具有极低的实验偏差,可以更准确地反映基因组变异信息。
此外,Illumina 技术具有极高的灵敏度,可以检测到非常微小的基因型,这对于临床遗传病研究、罕见病研究等具有重要意义。
同时,Illumina技术具有通量高、样本通量多的优势,可以同时对多个样本进行测序分析。
最后,新一代测序技术具有较高的数据质量,数据质量高且稳定性好,这为后续实验数据的分析提供了良好的基础。
在具体应用方面,Illumina技术可以用于基因组学、转录组学、微生物组学、表组学等研究领域。
例如,在基因组学研究中,Illumina技术可以用于基因组组装、基因表达分析、单核苷酸多态性检测(SNP检测)等。
在转录组学研究中,Illumina技术可以对转录组进行高通量测序,分析基因表达谱、差异表达基因分析、基因调控网络分析等。
在微生物组学研究中,Illumina技术可以对微生物群落进行高通量测序,分析微生物多样性、种群结构分析、功能基因分析等。
总的来说,Illumina技术以其高精度、高通量、高灵敏度等特点,在生命科学领域中得到了广泛应用。
这些参数优势使得Illumina技术成为生命科学研究领域中的重要工具之一,为科研人员提供了更全面、更准确的数据支持。
需要注意的是,虽然Illumina技术具有许多优点,但也存在一些挑战和局限性。
例如,测序深度和覆盖度问题、质量控制问题、数据解读和注释难度等。
因此,在使用Illumina技术进行生命科学研究中,需要结合实际情况进行合理选择和应用。
同时,随着技术的不断发展和进步,相信未来Illumina技术将会在更多领域中发挥更大的作用。
illumina lrm 软件补丁说明指南说明书

简介Illumina ®注意到Local Run Manager 软件存在安全漏洞,并提供了软件补丁来防止他人远程利用此漏洞。
Local Run Manager 是一款独立的软件应用程序,隶属于以下系统的默认配置:•MiSeq •MiSeqDx *•NextSeq 500•NextSeq 550•NextSeq 550Dx *•MiniSeq •iSeq*供体外诊断使用。
本指南适用于上面列出的Illumina 仪器,以及安装了独立版Local Run Manager 的非仪器计算机。
该漏洞属于未经身份验证的远程命令执行(RCE ),未经缓解的CVSS 评分为10.0高风险(CVSS:3.1/AV:N/AC:L/PR:N/UI:N/S:C/C:H/I:H/A:H )。
上面列出的仪器需要执行以下缓解步骤,以防止未经授权的用户访问一台或多台仪器并执行远程访问攻击。
如果安装程序因为某些原因无法运行,请参见本文档结尾的其他缓解措施部分,或联系寻求更多协助。
请参见获取Local Run Manager 更新,了解可以通过哪几种方式下载或请求此补丁的副本。
•v1.0.0补丁—将更新Local Run Manager Web 配置并禁用远程Internet Information Services (IIS )访问。
获取Local Run Manager 安全补丁获取Local Run Manager 安全补丁的方式有四(4)种。
选项1—直接下载到您的仪器获取Local Run Manager 安全更新的最快方式是直接从托管网站将其下载到仪器。
1.通过安全电子邮件提供的链接,将补丁安装程序下载到您的仪器。
2.将文件传输到仪器上的C:\Illumina 文件夹。
3.按照应用Local Run Manager 安全补丁(第3页)中的说明操作。
LRM 软件补丁1.0说明指南选项2—将补丁安装程序下载到计算机,并通过U盘/共享文件夹传输到仪器如果无法将补丁安装程序下载到仪器,建议将其下载到单独的计算机,然后再传输到仪器。
illumina 测序试剂盒上的300cycles的意思

illumina 测序试剂盒上的300cycles的意思摘要:1.什么是illumina测序试剂盒?2.300cycles在illumina测序中的含义3.300cycles对测序结果的影响4.如何充分利用300cycles进行高效测序?正文:illumina测序试剂盒是一款广泛应用于基因组学、转录组学和表观遗传学等研究领域的测序工具。
它提供了灵活多样的文库制备方案、全系列多通量测序平台以及终的数据解读。
其中,300cycles是illumina测序中一个重要的参数,它表示测序反应中进行的循环次数。
300cycles在illumina测序中的含义是指在测序过程中,合成链增长的循环次数。
在测序过程中,通过循环次数的控制,可以实现对目标DNA序列的快速扩增和测序。
300cycles通常意味着在测序反应中,合成链有机会生长到约300个碱基对的长度。
在这个长度范围内,可以有效地区分不同的碱基序列,从而实现对目标序列的准确测序。
300cycles对测序结果的影响主要体现在测序深度和数据质量上。
较高的测序循环次数可以帮助获得更深的测序深度,提高序列覆盖度,从而提高测序数据的准确性。
此外,300cycles还能影响测序数据的通量,即在单位时间内可以获得的测序数据量。
合理的循环次数设置有助于提高测序效率,缩短项目周期。
为了充分利用300cycles进行高效测序,研究者需要根据项目需求和实验条件进行优化。
以下几点建议可以帮助实现高效测序:1.充分了解测序平台和试剂盒的特点,选择合适的测序策略。
2.根据实验目的和样本特性,合理设置测序深度。
对于复杂样本或需要高精度测序的项目,可以适当增加测序循环次数。
3.关注测序过程中的碱基转换效率和质量控制。
碱基转换效率受到测序试剂盒、仪器性能和实验操作等多方面因素的影响,研究者需要密切关注这些因素,确保测序数据的质量。
4.针对不同的项目需求,灵活调整测序策略。
例如,在某些情况下,可以采用多批次、小规模的测序策略,以提高测序效率和数据质量。
illumina平台个体重测序结题报告模板

1项目概况 (1)1.1合同关键指标 (1)1.2项目基本信息 (1)1.3项目执行情况 (2)1.4分析结果概述 (2)2项目流程 (3)2.1实验流程 (3)2.2信息分析流程 (4)3生物信息学分析方法和结果 (5)3.1测序数据质控 (5)3.1.1原始数据介绍 (5)3.1.2碱基测序质量分布 (6)3.1.3碱基类型分布 (7)3.1.4低质量数据过滤 (8)3.1.5数据质量统计 (9)3.2与参考基因组比对统计 (10)3.2.1比对结果统计 (10)3.2.2插入片段分布统计 (11)3.2.3深度分布统计 (11)3.3SNP检测与注释 (13)3.3.1样品与参考基因组间SNP的检测 (13)3.3.2样品之间SNP的检测(多个样品) (16)3.3.3SNP结果注释 (17)3.4Small InDel检测与注释 (19)3.4.1样品与参考基因组间Small InDel的检测 (19)3.4.2样品之间Small InDel的检测(多个样品) (20)3.4.3Small InDel的注释 (21)3.5SV检测与注释 (22)3.5.1SV的检测 (22)3.5.2SV的注释 (23)3.6CNV的检测与分析 (24)3.7各变异在基因组上的分布 (26)3.8DNA水平变异基因分析 (26)3.8.1DNA水平的变异基因检测 (26)3.8.2DNA水平的变异基因功能注释 (27)4数据下载 (32)4.1数据下载方法 (32)4.2结果文件查看说明 (32)摘要合同关键指标完成XX个样品的重测序,保证Q30达到85%。
数据评估:测序数据量,测序数据质量和GC含量的统计。
与基因组比对:比对率,基因组覆盖度,基因组覆盖深度统计。
变异检测和注释:SNP、InDel、SV、CNV的检测和注释。
DNA水平变异基因分析:检测编码区发生SNP非同义突变、InDel突变、SV突变的基因。
illumina 转录组测序简明实验流程(PE-oligodT NEB)
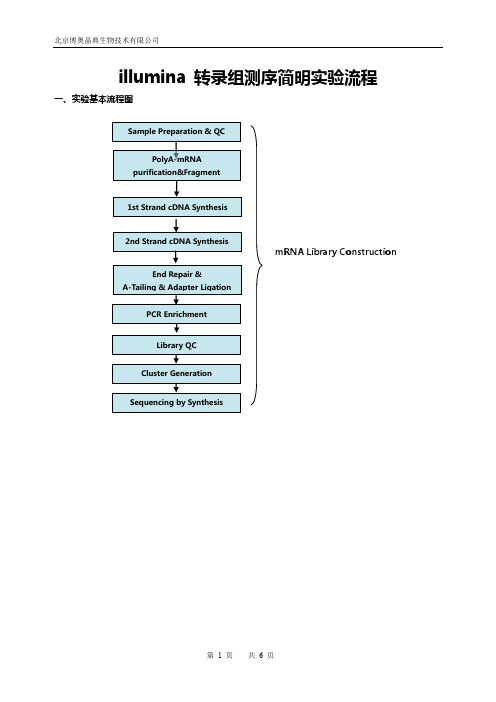
illumina 转录组测序简明实验流程一、实验基本流程图mRNA Library Construction二、mRNA建库流程1.材料准备1.2.1.3.2.样品准备和QC选择质量合格的Total RNA作为mRNA测序的建库起始样品,其质量要求通过Agilent 2100 BioAnalyzer检测结果RIN≥7,28S和18S的RNA 的比值大于或等于1.5:1,起始量的要求范围是0.1∽1ug。
用QUBIT RNA ASSAY KIT对起始的Total RNA进行准确定量。
3.建库实验步骤3.1.mRNA纯化和片段化3.1.1.mRNA纯化纯化原理是用带有Oligod(T)的Beads对Total RNA中mRNA进行纯化。
3.1.2.mRNA片段化3.2.1st Strand cDNA 合成3.3.2nd Strand cDNA 合成根据下表制备反应体系,然后在PCR仪上运行Program3,然后将第2链cDNA合成产物用144uL AMPure XP Beads进行纯化,最后用60µL的Nuclease free water进行重悬,取出55.5µL以备下一步使用;3.4.Perform End Repair/dA-tail3.5.Adaptor Ligation根据下表制备反应体系,然后在PCR仪上运行Program5、Program6,然后100uL AMPure XP Beads进行纯化后用52.5µL的Resuspension Buffer进行重悬,再用50uL AMPure XP Beads3.6.PCR扩增根据下表制备反应体系,然后在PCR仪上运行Program7,然后再45µL用AMPure XP Beads 进行纯化,最后用23µL的Resuspension Buffer进行重悬,取出20µL以备下一步使用;3.7. PCR 产物质控用QUBIT DNA HS ASSAY KIT 对PCR 产物进行准确定量。
用于Illumina测序平台数据分析的图像界面软件需求

用于Illumina测序平台数据分析的图像界面软件需求一、名称:专用于Illumina测序平台数据分析的图像界面软件二、用途:是一款生物信息学综合分析工具,其界面友好直观,操作简便,易于掌握。
除了基本的分子生物学工具外,如序列比对、进化树构建等,还可对所有主流的二代测序平台产生的测序结果进行数据分析,如重测序分析、转录组学分析、表观基因组学分析和从头拼接,并且该软件还可自定义标准化的工作流程(Workflow)用于Illumina数据拼接、分析,以及分析16S,18S,ITS及其他扩增区域的二代测序数据分析。
三、购置数量:1四、主要技术参数1、该软件是一个基因组工作台的综合分析包,它可以分析可视化数据,可以分析所有主要的NGS(二代测序)平台数据,比SOLiD、Roche454、Illumina、Ion Torrent 的数据。
2、适用的测序数据一代测序数据,Sanger平台;几乎所有的二代测序平台产生的数据,如 SOLiD、LifeIon Torrent、Complete Genomics、Roche454、Illumina Genome Analyzer;三代测序数据,PacBio平台。
3、提供De novo或公开数据库的OTU丛集比对。
4、使用自动分析流程,可快速获得病原种类及分型病毒实验室数据用于二代测序信息分析。
5、数据库支持,可以自动连接相关基因数据库5.1、可下载及分类NCBI全部微生物基因组数据库5.2、可下载整合 GO 数据库5.3、可下载整合 Protein Family-Pfam 数据库5.4、可下载整合病原物种序列数据库5.5、可下载整合抗药性基因数据库6、分类及组态分析6.1、分析16S, 18S, ITS及其他扩增区域的二代测序数据分析6.2、提供 De novo或公开数据库的OTU 丛集比对6.3、OTU 数据库: Greengenes, SILVA, and UNITE6.4、可针对不同分类层级的结果搭配样本注解一同呈现6.5、对微生物全基因组序列进行分类6.6、可针对特别功能性区域进行数据汇整分类7、统计分析7.1、计算群内群外的样本差异7.2、搭配样本注解提供PCA plot 结果呈现7.3、PERMANOVA 统计分析7.4、统计不同样本的微生物族群丰度7.5、测量均质化OTU丰度7.6、评估统计差异7.6、探索样本之间的关系系数8、使用自动分析流程, 可快速获得病原种类及分型:8.1、Multi locus sequence type (MLST)8.2、分类8.3、最接近的物种及参考序列8.4、微生物抗药性基因8.5、建立基因亲缘树8.6、根据分型及亲缘分析, 可以了解病源爆发演化的关联性8.7、提供数据运算管理及样本注解的整合介面.9、利用二代测序对微生物做全基因组测序并分型10、 Read mapping序列比对功能支持short reads、long reads 及paired reads;支持空位比对和非空位比对;支持Illumia、Sanger、Roche454 和SOLiD 的测序数据。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
bfast
bowtie
bwa
eland2
karma
maq
# incorrecly mapped reads
mosaik
novoalign
smalt
soap
2
Method 1 Read Pairs
• 2 Haplotypes (4 fastqs per read length) with SNPs and INDELs placed into reference • Classes: • REFERENCE (no variation) • SMALL_INS/DEL (INDEL 1-30 bp) • SNPS_1-5 (number of SNPs present)
0.85
0.80 37 54 76 108 read length
bfast
bowtie
bwa
eland2
karma
maq
mosaik
novoalign
smalt
soap
srprism
stampy
17
Sn (method 2)
1.00
0.75
Sn
0.50
0.25
0 37 54 76 108 read length
• • • •
7
CPU time (all)
soap
bowtie
bwa
smalt
srprism
stampy
eland2
maq
bfast
novoalign
0
75
150 average CPU mins/chunk
225
300
8
CPU time (method 1)
10000
1000
average CPU mins/chunk
bfast
bowtie
bwa
eland2
karma
maq
mosaik
novoalign
smalt
soap
srprism
stampy
18
Sn (DEL)
1.0
0.8
0.6
Sn 0.4 0.2 0
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 1 2 3 4 5 6 7 8 9 20 0
4
Splitting Into Chunks
• LSF farm @ Sanger: • Ideal job > 10 min, < 1 hr, < 8GB • Jobs > 8 hrs or > 16GB can pend for over a week • Tiny run time and memory also makes it suitable for the cloud • Must split input fastqs into chunks to speed up run time and reduce memory • Each input pair of fastqs split into chunks containing at most 50000000 bp total in the 2 files • Longer read length inputs have fewer total reads per
87.50 % of reads
93.75
100.00
% correct
% incorrect
% unmapped
14
PPV (all)
mosaik
eland2
karma
bfast
maq
novoalign
smalt
bwa
stampy
soap
srprism
bowtie
0.92
0.93
0.94
0.95
100
10
1 37 54 76 108 read length
bfast
bowtie
bwa
eland2
karma
maq
mosaik
ห้องสมุดไป่ตู้
novoalign
smalt
soap
srprism
stampy
9
CPU time (method 2)
10000
1000
average CPU mins/chunk
100
bfast
bowtie
bwa
eland2
karma
maq
mosaik
novoalign
smalt
soap
srprism
stampy
13
Sn (all)
novoalign
stampy
bwa
maq
mosaik
smalt
bfast
eland2
soap
srprism
karma
bowtie
75.00
81.25
Problems: • • • • karma & mosaik require > 16GB regardless of number of input reads - only ran on one split to get example CPU & memory usage mosaik (method 1 108 bp runs failed; excludes reads; performance statistics are overestimated) srprism (does not include all unmapped reads; includes multiple hits per read - stats wrong) razers2 (never worked at all)
stampy
srprism
bfast
0
2000
4000 maximum memory (MB)/chunk
6000
8000
11
Memory usage (method 1)
8000
6000
maximum memory usage (MB)
4000
2000
0 37 54 76 108 read length
bfast
bowtie
bwa
eland2
karma
maq
mosaik
novoalign
smalt
soap
srprism
stampy
12
Memory usage (method 2)
8000
6000
Maximum memory usage (MB)
4000
2000
0 37 54 76 108 read length
5
Mappers
• Completed running & analyzed successfully: • • • • • • • • • • bfast bowtie bwa eland2 karma maq (2 chunks initially failed due to > 8 hrs, but easy to repeat and complete since multi-step) novoalign (6 chunks failed due to > 8hrs, single step process requires running in long queue) soap stampy
bfast
bowtie
bwa
eland2
karma
maq
deletion length (bp)
mosaik
novoalign
smalt
soap
srprism
stampy
19
Sn (INS)
1.0
0.8
0.6
Sn 0.4 0.2 0 1 4 7 10 13 16 19 22 25 28 31 34 37 40 43 46 49 52 55 58 61 64 67 70 73 76 79 82 85 88 91 94 97 100 103 106 insert length (bp)
0.96 PPV
0.97
0.98
0.99
1.00
15
AP (all)
novoalign
stampy
bwa
maq
mosaik
smalt
bfast
eland2
soap
karma
srprism
bowtie
0.80
0.85
0.90 AP
0.95
1.00
16
Sn (method 1)
1.00
0.95
Sn
0.90
Illumina Mapper Comparison
(Draft V3, Sendu Bala, WTSI)
1
Simulated Read Pairs
• Generated by David Craig et al. @ Tgen • Error rates trained on 75th percentile Illumina reads from 1000 genomes pilot sequencing @ Sanger • Two methods, with read pairs of sequencing length 37, 54, 76 & 108 bp generated for each • Problem with ends of chromosomes; excluded from analysis. Problem resolved?