CRISPR-Cas9基因敲除小鼠(行业精制)
《2024年利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》范文

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言基因编辑技术近年来在生物学和医学领域取得了巨大的突破,其中CRISPR-Cas9系统因其高效、精确的特性,已成为基因编辑的主要工具之一。
本文旨在探讨利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系的方法和过程,为基因编辑技术的进一步应用提供参考。
二、DUSP9基因与小鼠胚胎干细胞DUSP9基因是一种重要的蛋白磷酸酶基因,其编码的蛋白在细胞信号传导过程中具有重要作用。
小鼠胚胎干细胞(Mouse Embryonic Stem Cells, mESCs)是研究发育生物学和基因编辑的重要工具。
通过构建DUSP9基因敲除的小鼠胚胎干细胞系,可以研究DUSP9基因在细胞发育、分化以及疾病发生过程中的作用。
三、CRISPR-Cas9系统简介CRISPR-Cas9系统是一种基于细菌免疫系统的基因编辑技术,其原理是通过将特定的RNA与Cas9蛋白结合,形成复合物,然后识别并切割DNA序列,从而实现对基因的敲除或修改。
CRISPR-Cas9系统具有精确度高、效率高、成本低等优点,是现代基因编辑的主要手段。
四、构建DUSP9基因敲除小鼠胚胎干细胞系的步骤1. 载体构建:设计并合成针对DUSP9基因的特异性gRNA 序列,并将其与Cas9蛋白的表达载体一起构建成CRISPR-Cas9表达系统。
2. 细胞培养与转染:将小鼠胚胎干细胞培养至适当状态,然后利用转染技术将CRISPR-Cas9表达系统导入细胞中。
3. 基因编辑:通过CRISPR-Cas9系统识别并切割DUSP9基因的DNA序列,实现DUSP9基因的敲除。
4. 克隆筛选与鉴定:筛选并培养获得成功的DUSP9基因敲除小鼠胚胎干细胞克隆,通过PCR、测序等方法鉴定敲除效果。
5. 细胞系建立与保存:将成功构建的DUSP9基因敲除小鼠胚胎干细胞系进行保存与扩大培养,以备后续研究使用。
《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》范文

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言随着基因编辑技术的发展,CRISPR-Cas9系统已成为一种强大的工具,用于在生物医学研究中精确地编辑基因组。
DUSP9基因作为一种重要的基因,其功能在多种生物学过程中起着关键作用。
因此,构建DUSP9基因敲除小鼠胚胎干细胞系,对于研究DUSP9基因的功能及其在疾病发生发展中的作用具有重要意义。
本文旨在详细介绍利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系的过程。
二、材料与方法1. 材料小鼠胚胎干细胞(mESCs)、CRISPR-Cas9系统、相关基因编辑工具、培养基、生长因子等。
2. 方法(1)设计CRISPR-Cas9系统:根据DUSP9基因的序列信息,设计合适的CRISPR-Cas9系统,包括sgRNA和Cas9蛋白。
(2)制备mESCs细胞:培养mESCs细胞至合适的状态,以便进行基因编辑。
(3)转染与编辑:将CRISPR-Cas9系统转染至mESCs细胞中,利用Cas9蛋白对DUSP9基因进行切割。
(4)筛选与鉴定:通过PCR、Western blot、qRT-PCR等方法,筛选出成功敲除DUSP9基因的mESCs细胞,并进行鉴定。
三、实验过程1. 设计并构建CRISPR-Cas9系统,选择合适的sgRNA序列和Cas9蛋白表达载体。
2. 培养mESCs细胞至合适的状态,进行转染。
3. 观察转染后的细胞生长情况,确保Cas9蛋白的表达。
4. 利用PCR、Western blot、qRT-PCR等方法筛选出成功敲除DUSP9基因的mESCs细胞。
5. 对筛选出的细胞进行扩增培养,并保存于液氮中备用。
四、结果与讨论1. 结果(1)成功构建了CRISPR-Cas9系统,并将其转染至mESCs 细胞中。
(2)成功筛选出敲除DUSP9基因的mESCs细胞,并通过PCR、Western blot、qRT-PCR等方法进行了鉴定。
《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》范文

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言随着基因编辑技术的发展,CRISPR-Cas9系统已成为现代生物医学研究中常用的基因编辑工具之一。
它为科研人员提供了强大的基因敲除、插入或突变的能力,在多种模型动物制备及疾病研究领域具有广泛的应用前景。
本文旨在介绍利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系的过程,为相关研究提供技术参考。
二、材料与方法1. 材料(1) CRISPR-Cas9系统相关组件(包括Cas9蛋白、sgRNA 等);(2) 小鼠胚胎干细胞系;(3) DUSP9基因特异性敲除载体;(4) 培养基、试剂及其他实验耗材。
2. 方法(1) 设计并构建DUSP9基因敲除载体;(2) 准备小鼠胚胎干细胞系并进行细胞培养;(3) 将DUSP9基因敲除载体与胚胎干细胞共培养,实现基因编辑;(4) 筛选并扩增成功敲除DUSP9基因的胚胎干细胞;(5) 对敲除细胞进行鉴定及保存。
三、实验过程1. DUSP9基因敲除载体的构建根据DUSP9基因序列,设计并合成sgRNA序列,构建DUSP9基因敲除载体。
通过PCR扩增获得目的片段,将其克隆至载体中,构建成功后的载体通过测序验证其准确性。
2. 小鼠胚胎干细胞的培养与准备将小鼠胚胎干细胞置于适宜的培养条件下进行培养,待细胞生长至适宜状态时进行后续实验。
3. 基因编辑及筛选将DUSP9基因敲除载体与小鼠胚胎干细胞共培养,通过CRISPR-Cas9系统实现DUSP9基因的敲除。
随后,通过PCR、测序等方法筛选出成功敲除DUSP9基因的胚胎干细胞。
4. 鉴定与保存对筛选出的成功敲除DUSP9基因的胚胎干细胞进行鉴定,包括细胞形态观察、生长曲线绘制、基因型鉴定等。
将鉴定合格的细胞进行保存,以备后续实验使用。
四、结果与讨论1. 结果通过CRISPR-Cas9系统成功构建了DUSP9基因敲除小鼠胚胎干细胞系,并筛选出成功敲除DUSP9基因的细胞。
基因敲除型小鼠缩写

基因敲除型小鼠缩写基因敲除型小鼠(Knockout Mouse)是一种在实验室中经过基因编辑技术制造的小鼠模型。
通过敲除特定基因,科学家可以研究该基因在生物体发育、生理功能和疾病发展中的作用。
基因敲除型小鼠的出现为生命科学研究提供了重要工具,有助于我们更深入地理解基因对生命活动的影响。
在基因敲除型小鼠的制备过程中,科学家会选择目标基因,并使用基因编辑技术(如CRISPR/Cas9技术)将其敲除。
通过这种方式,小鼠的基因组中将不再包含目标基因的正常拷贝,从而使得该基因无法正常表达。
这样一来,科学家便可以观察到在缺失该基因的小鼠体内所产生的生理和行为变化。
基因敲除型小鼠的制备需要经过一系列复杂的实验操作。
首先,科学家需要设计合适的引物和合成寡核苷酸,用于引导CRISPR/Cas9系统精确剪切目标基因。
然后,利用载体将CRISPR/Cas9系统导入小鼠的受精卵中,使其在早期胚胎发育阶段就发挥作用。
经过一段时间的培养和发育,科学家便可以获得具有基因敲除突变的小鼠。
基因敲除型小鼠的制备不仅需要技术娴熟的实验操作,还需要对基因功能和细胞生物学等领域的深入理解。
科学家需要充分了解目标基因在生物体中的作用机制,以及其与其他基因的相互作用关系。
只有在对基因功能有清晰认识的基础上,才能准确选择目标基因,并制备出具有特定基因敲除突变的小鼠模型。
基因敲除型小鼠的应用范围非常广泛。
它们可以用于研究基因在发育过程中的作用,探究基因在特定组织和器官中的功能,以及揭示基因与疾病之间的关联。
通过对基因敲除型小鼠进行行为学、生理学和病理学等多方面的分析,科学家可以获取大量关于基因功能的宝贵信息。
基因敲除型小鼠是生命科学研究中的重要工具,为我们研究基因功能和疾病发展提供了有力支持。
通过制备基因敲除型小鼠模型,科学家可以更深入地了解基因的作用机制,从而为疾病的诊断和治疗提供新的思路和方法。
基因敲除型小鼠的研究将继续推动生命科学领域的发展,为人类健康做出更大的贡献。
crispr-cas9基因敲除小鼠原理
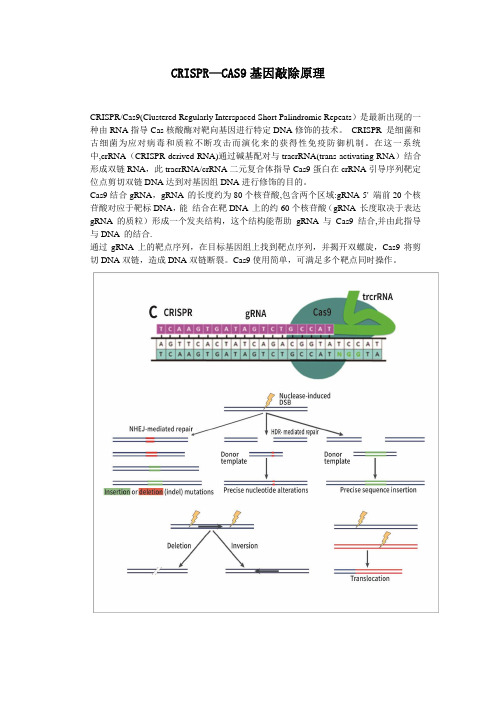
CRISPR—CAS9基因敲除原理
CRISPR/Cas9(Clustered Regularly Interspaced Short Palindromic Repeats)是最新出现的一种由RNA指导Cas核酸酶对靶向基因进行特定DNA修饰的技术。
CRISPR 是细菌和古细菌为应对病毒和质粒不断攻击而演化来的获得性免疫防御机制。
在这一系统中,crRNA(CRISPR-derived RNA)通过碱基配对与tracrRNA(trans-activating RNA)结合形成双链RNA,此tracrRNA/crRNA二元复合体指导Cas9蛋白在crRNA引导序列靶定位点剪切双链DNA达到对基因组DNA进行修饰的目的。
Cas9结合gRNA,gRNA 的长度约为80个核苷酸,包含两个区域:gRNA 5' 端前20个核苷酸对应于靶标DNA,能结合在靶DNA 上的约60个核苷酸(gRNA 长度取决于表达gRNA 的质粒)形成一个发夹结构,这个结构能帮助gRNA 与Cas9结合,并由此指导与DNA 的结合.
通过gRNA上的靶点序列,在目标基因组上找到靶点序列,并揭开双螺旋,Cas9将剪切DNA双链,造成DNA双链断裂。
Cas9使用简单,可满足多个靶点同时操作。
Insertion /deletion NHEJ HDR
gRNA
Cas9
Donor vector
基因敲除小鼠流程:。
基于CRISPR Cas9技术基因敲除小鼠(Cas9-KO)的制作方法-2018-2-28

基于CRISPR Cas9技术基因敲除小鼠(Cas9-KO)的制作方法一、CRISPR/Cas9靶向基因敲除小鼠制作的基本技术原理:通过CRISPR/Cas9基因敲除技术,crRNA通过碱基配对与tracrRNA(trans-activating RNA)结合,形成双链RNA。
这一tracrRNA:crRNA二元复合体指导Cas9蛋白在crRNA引导序列靶标的特定位点剪切双链DNA。
在与crRNA引导序列互补的位点,Cas9蛋白的HNH核酸酶结构域剪切互补链而Cas9 RuvC-like 结构域剪切非互补链,实现敲除目的基因的功能,制备基因敲除小鼠模型。
二、具体步骤如下:一)模型制作策略制作:利用生物信息学手段(NCBI&IMPC&MGI),分别仔细分析目的基因敲除后小鼠的生存能力及繁育能力,并结合邻近基因的影响,最终选择合适的敲除区域进行敲除方案的设计,出具相应的制作策略。
二)载体的设计和构建:使用麻省理工学院的CRISPR Design工具(/),依据中靶Score的高低及脱靶Score的高低设计一对长度为20bp的针对靶标DNA的寡聚核苷酸链序列用于制备sgRNA,并在该靶区域设计引物用于后续阳性小鼠的基因鉴定。
1、制备sgRNA的实验方法步骤:1)线性化pUC57-GDNA-T7载体中提pUC57-GDNA-T7载体,用BsaI线性化过夜。
胶回收保存备用。
2)引物退火及加磷酸将上下游引物(干粉)稀释,再进行引物退火及加磷酸。
3)连接&阳性菌落筛选取步骤二中的加磷酸产物与线性化载体pUC57-GDNA-T7进行连接,该连接反应在干式恒温器中进行。
对连接产物进行转化,涂板,37°C培养箱过夜培养。
再用PCR&测序的方法筛选阳性克隆,再将测序正确的克隆进行甘油菌保种,-80°C保存备用4)制备转录模板以构建好的sgRNA载体为模板进行PCR扩增,将PCR产物切胶回收,回收产物离心后倒掉上清留DNA沉淀,再溶解DNA。
《2024年利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》范文

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言在生物学研究领域,基因编辑技术日益显示出其巨大的潜力和应用前景。
CRISPR-Cas9系统作为一种强大的基因编辑工具,已在多个物种中成功用于构建基因敲除模型。
本文旨在介绍如何利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系,为相关研究提供参考。
二、材料与方法1. 材料(1)小鼠胚胎干细胞(mESCs);(2)CRISPR-Cas9系统相关试剂;(3)DUSP9基因敲除载体;(4)显微操作设备及试剂;(5)实验动物(小鼠)。
2. 方法(1)设计并构建DUSP9基因敲除载体;(2)将载体与mESCs共转染,使DUSP9基因发生双链断裂;(3)利用CRISPR-Cas9系统对断裂的DUSP9基因进行修复,形成基因敲除突变;(4)筛选并扩增基因敲除的mESCs;(5)将基因敲除的mESCs注射到小鼠囊胚中,生成转基因小鼠。
三、实验过程1. 载体构建及转染首先,设计并构建DUSP9基因敲除载体。
该载体应包含靶向DUSP9基因的识别序列、切割位点及修复模板。
随后,将载体与mESCs共转染,使DUSP9基因发生双链断裂。
此过程需在显微操作下进行,确保转染效率和准确性。
2. CRISPR-Cas9系统修复及筛选利用CRISPR-Cas9系统对断裂的DUSP9基因进行修复。
通过非同源末端连接或同源重组等方式,使基因发生突变,形成DUSP9基因敲除的突变体。
随后,通过PCR、测序等方法筛选并扩增基因敲除的mESCs。
3. 转基因小鼠生成及鉴定将扩增得到的基因敲除的mESCs注射到小鼠囊胚中,通过胚胎移植技术生成转基因小鼠。
随后,通过PCR、免疫组化等方法对转基因小鼠进行鉴定,确认DUSP9基因是否成功敲除。
四、结果与讨论1. 结果利用CRISPR-Cas9系统成功构建了DUSP9基因敲除的小鼠胚胎干细胞系。
通过PCR、测序等方法验证了DUSP9基因的敲除效率及准确性。
CRISPR-Cas9基因敲除小鼠

2021/3/7
CHENLI
26
Thanks!
2021/3/7
CHENLI
27
CHENLI
10
设计了两个剪切位点
2021/3/7
CHENLI
11
2021/3/7
有转入目的基因的
小鼠大多细胞目的基 因发生移码突变,目 的基因转录和翻译都 明显下调。
CHENLI
12
二、腺病毒转入大脑皮层额叶
2021/3/7
目标基因是NeuN
CHENLI
13
目的基因发生的改变
✓ 缺失一位 ✓ 缺失多位 ✓ 插入一位 ✓ 插入多位
2021/3/7
• 多为移码突变
CHENLI
21
Kras的基因变化
2021/3/7
CHENLI
22
P53和Lkb1不断被损坏, Kras则被修补变得越发 优势
2021/3/7
CHENLI
23
最终肺中出现肿瘤
2021/3/7
CHENLI
24
2021/3/7
CHENLI
25
总结
• 事实证明将Cre-dependent引入Cas9工具中, 可以大大扩展Cas9的用途。
2021/3/7
CHENLI
2
CRISPR-Cas9系统的原理
sgRNA
通过设计sgRNA来确定剪切位点 设计简单快速,无需重复构建核酸内切酶
2021/3/7
CHENLI
target
3
Cas9的局限性
• 受限于转基因范围,Cas9往往只用与细胞或胚 胎层面的实验。
CRISPRCas基因敲除小鼠

结语
谢谢大家!
• 使得CRISPR-Cas9应用更广泛。
2020/12/5
4
Cre-dependent Cas9 Rosa26 targeting 矢量图
Rosa26,使转 录可被诱导
荧光蛋白
2020/12/5
5
转入Cas9后与野生小鼠对比
2020/12/5
6
神经系统荧光对比
2020/12/5
7
三种实验测试效果:
荧光显示含有 Cre/Cas9的组织中NeuN表 达明显减少,而非转入非 目标的sgRNA则目标蛋白无 影响
15
NeuN被破坏的比例十分大
2020/12/5
Indel=insertions(插入) and deletions(删除) 16
多数为两个等位基因都被破坏,且其 中多为移码突变。
2020/12/5
2020/12/5
23
最终肺中出现肿瘤
2020/12/5
24
2020/12/5
25
总结
• 事实证明将Cre-dependent引入Cas9工具中, 可以大大扩展Cas9的用途。
• 以后用此学作 出贡献。
2020/12/5
26
Thanks!
2020/12/5
2
CRISPR-Cas9系统的原理
sgRNA
通过设计sgRNA来确定剪切位点 设计简单快速,无需重复构建核酸内切酶
2020/12/5
target
3
Cas9的局限性
• 受限于转基因范围,Cas9往往只用与细胞或胚 胎层面的实验。
• 本实验采用Cre-dependent Rosa26 Cas9 knockin mouse克服了这个局限性。
用CRISPR Cas9方案构建双基因敲除鼠, 获得双基因敲除纯合子小鼠的交配方案

双基因敲除小鼠繁殖工作:CRISPR/Cas9方案构建双基因敲除鼠,得到F0杂合子之后,如何建系才能获得双基因敲除纯合子小鼠?这是经常被问到的问题,下面就简单回答一下。
假设我们的目的基因为A和B,通常用CRISPR/Cas9方法得到的基因敲除鼠为杂合子,双杂合子小鼠基因型为AaBb,大写字母代表野生型(dominant),小写字母代表突变型(recessive)。
得到F0杂合子(AaBb)之后,可以用以下方案之一来获得双基因敲除纯合子小鼠:方案一:1.将双杂合子小鼠(AaBb)与野生鼠(AABB)交配,理论上将得到25%的野生型(AABB),25%基因A单杂合子(AaBB),25%基因B单杂合子(AABb)及25%双杂合子小鼠(AaBb)。
2.将所得到的双杂合子小鼠(AaBb)互交(inter-cross),理论上6.25%的后代将会是双基因敲除纯合子小鼠(aabb),见下图。
3.由于双基因敲除实验中一般都需要单基因敲除动物作为对照,所以在进行上面小鼠breeding的同时可以将基因A单杂合子(AaBB)互交,在后代中鉴定出基因A纯合子(aaBB),同样将基因B单杂合子(AABb)互交,在后代中鉴定出基因B纯合子(AAbb)。
方案二:将双杂合子小鼠(AaBb)与单基因纯合子(如aaBB)交配,所生小鼠中约25%为基因A纯合子而基因B杂合子(aaBb,见下图左)。
然后将aaBb小鼠互交,理论上后代小鼠中25%为双基因敲除纯合子小鼠(aabb),见下图右。
需要特别注意的几个问题:1)上面所讲的方法适用于位于不同的染色体两个基因的基因敲除,如果两个基因位于同一条染色体上,要通过上述方法得到双基因敲除纯合子小鼠很难;2)上述方法有赖于基因特异性的Genotyping PCR assays。
在开始setup breeding之前必须将两个目的基因特异性的Genotyping PCRassays 准备好;3)要事先研究一下目的基因敲除后有无胚胎致死性,是否影响其生长发育等。
小鼠大脑基因实验报告(3篇)

第1篇一、实验背景随着神经科学研究的深入,理解大脑的基因调控机制对于揭示神经疾病的发生机理和开发新的治疗方法具有重要意义。
本研究旨在通过基因编辑技术,探究特定基因在小鼠大脑发育和功能中的作用,为相关疾病的预防和治疗提供新的思路。
二、实验目的1. 利用CRISPR/Cas9基因编辑技术敲除小鼠大脑中特定基因。
2. 观察基因敲除对小鼠大脑发育、行为和认知功能的影响。
3. 分析敲除基因对小鼠大脑中相关通路和基因表达的影响。
三、实验材料与方法1. 实验材料- 小鼠胚胎干细胞(ES细胞)- CRISPR/Cas9系统- 实验小鼠(C57BL/6小鼠)- 实验试剂:DNA聚合酶、限制性内切酶、DNA连接酶、PCR引物等2. 实验方法(1)基因编辑1. 设计靶向特定基因的CRISPR/Cas9系统,包括sgRNA和Cas9蛋白。
2. 将sgRNA和Cas9蛋白导入小鼠ES细胞,进行基因编辑。
3. 对编辑后的ES细胞进行筛选,获得基因敲除的细胞系。
4. 将基因敲除的细胞系注射到C57BL/6小鼠的受精卵中,获得基因敲除的小鼠。
(2)小鼠行为和认知功能测试1. 观察基因敲除小鼠的生长发育、行为和运动能力。
2. 对小鼠进行认知功能测试,包括Morris水迷宫实验、Y迷宫实验等。
(3)基因表达分析1. 提取小鼠大脑样本,进行RNA提取和cDNA合成。
2. 利用PCR、RT-qPCR等方法检测敲除基因的表达水平。
3. 对小鼠大脑样本进行蛋白质组学分析,检测相关蛋白的表达水平。
四、实验结果1. 基因敲除成功敲除了小鼠大脑中特定基因,并通过PCR、RT-qPCR等方法验证了基因敲除的效果。
2. 小鼠行为和认知功能与野生型小鼠相比,基因敲除小鼠在Morris水迷宫实验中表现出明显的空间学习障碍,提示该基因可能参与小鼠的认知功能。
3. 基因表达分析敲除基因后,小鼠大脑中相关通路和基因表达发生了显著变化。
具体表现为:1. 神经递质合成酶的表达水平降低。
基因敲除小鼠的方法

基因敲除小鼠的方法
1. CRISPR/Cas9基因编辑技术,CRISPR/Cas9技术是一种高效的基因编辑工具,可以用来精确地敲除小鼠基因。
首先,科学家设计合成一段RNA序列,使其与目标基因序列相匹配,然后将这段RNA和Cas9蛋白复合体导入小鼠胚胎内。
复合体会通过识别并切割目标基因,导致基因敲除。
2. 胚胎干细胞技术,另一种常见的基因敲除小鼠方法是利用胚胎干细胞。
科学家可以将设计好的基因敲除载体导入小鼠胚胎干细胞中,使其发生基因敲除。
然后,这些修改过的干细胞可以被植入小鼠胚胎内,从而产生基因敲除小鼠。
3. 遗传改造小鼠技术,除了CRISPR/Cas9和胚胎干细胞技术,科学家还可以利用遗传改造技术来实现基因敲除。
这种方法涉及到选择性育种和杂交,通过选择性地交配和繁殖,最终得到具有特定基因敲除的小鼠品系。
总的来说,基因敲除小鼠的方法主要包括CRISPR/Cas9基因编辑技术、胚胎干细胞技术和遗传改造小鼠技术。
这些方法都是在实验室条件下进行的,需要经过严格的实验设计和操作流程,以确保
基因敲除的准确性和有效性。
同时,这些方法也为科学家提供了强大的工具,用于研究基因在生物体内的功能和作用机制。
SpCas9完全性敲除小鼠模型介绍

SpCas9完全性敲除小鼠模型介绍
CRISPR Cas9基因编辑技术( Clustered Regularly Interspaced Short Palindromic Repeats) 是最新出现的一种由gRNA介导Cas9蛋白核酸酶特异靶向到靶位点,从而对目的基因进行编辑的技术。
CRISPR是细菌和古细菌为应对病毒和质粒不断攻击演化而来的获得性免疫防御机制。
CRISPR基因编辑技术利用RNA与DNA杂交的原理,通过合成一个与特定DNA互补的gRNA,将CAS9蛋白靶向到靶位点,并进行切割产生DNA双链损伤。
这种DNA双链断裂可以提高这个特定位点的重组效率,从而可以利用细胞的同源重组和非同源末端结合机制来进行基因组的编辑,如基因的敲入、敲除和定点突变。
细胞质注射法获取F0转基因小鼠
制备流程 Cas9的结构
SpCas9完全性敲除小鼠应用
·简化了流程,缩短了制作周期,降低了成本
·可以制作多个基因同时敲除的动物
·可以制作不同种属的基因敲除动物.如猪、牛、斑马鱼。
基因敲除小鼠的实验流程

基因敲除小鼠的实验流程1.设计基因敲除小鼠实验方案在开始实验之前,需要明确研究目的,确定需要敲除的基因,并设计相应的实验方案。
一般可以使用 CRISPR-Cas9 系统来实现基因敲除,在设计基因敲除实验方案时,需要选择合适的 sgRNA 序列,以及设计恰当的引物用于检测突变。
2.获得基因敲除小鼠的胚胎干细胞为了实现基因敲除,需要获得基因敲除小鼠的胚胎干细胞。
一种常用的方法是利用胚胎干细胞对外源DNA的高度易感性,将敲除基因的质粒DNA转染到小鼠胚胎干细胞中。
3.筛选敲除基因的胚胎干细胞株系将转染了敲除基因的胚胎干细胞以悬浮培养的方式进行培养,培养一段时间后,利用一定的筛选条件来筛选出含有敲除基因的胚胎干细胞株系。
筛选条件可包括对抗生素的使用或筛选标记基因的表达。
4.制备敲除基因小鼠的固定胚胎干细胞系通过体外培养,将敲除基因的胚胎干细胞系定植在培养皿上,培养数代以后,将其冻存,以备后续的实验使用。
5.实施敲除基因小鼠的胚胎干细胞基因改造将固定的胚胎干细胞系重新激活,转染 Cas9 和 sgRNA,利用CRISPR-Cas9 系统使这些细胞具有敲除基因的突变。
6.识别敲除基因的胚胎干细胞阳性克隆株对转染了 Cas9 和 sgRNA 的胚胎干细胞进行筛选,通过 PCR、Western blot、Southern blot等技术方法识别出敲除了目标基因的阳性克隆株。
7.将敲除基因的胚胎干细胞注入小鼠的早期胚胎取出已受精的小鼠卵母细胞,利用显微操作将敲除基因的胚胎干细胞注入到小鼠的早期胚胎中。
利用体外受精或者通过体内或体外的胚胎移植方式将基因敲除干细胞注入受体小鼠。
8.制备基因敲除小鼠的嵌合小鼠将已注入敲除基因的胚胎干细胞的受体小鼠进行嵌合以产生基因敲除小鼠。
嵌合可以通过体内或体外的胚胎移植方式进行。
9.筛选识别基因敲除小鼠对产生的嵌合小鼠进行筛选,确认敲除基因是否成功。
可以通过 PCR、Western blot、Southern blot等技术方法对小鼠体细胞或组织进行分析。
《2024年利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》范文

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言基因编辑技术为现代生物学研究带来了巨大的突破。
在众多基因编辑技术中,CRISPR-Cas9系统以其高精度、高效率的特性备受关注。
本篇论文旨在探讨利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系的过程、结果以及相关讨论。
二、材料与方法1. 材料小鼠胚胎干细胞系、DUSP9基因序列信息、CRISPR-Cas9系统相关试剂等。
2. 方法(1)设计并合成针对DUSP9基因的CRISPR-Cas9系统指导RNA(gRNA)。
(2)将gRNA与Cas9蛋白共同转入小鼠胚胎干细胞中。
(3)通过PCR、Western Blot等方法检测DUSP9基因的敲除情况。
(4)对敲除后的胚胎干细胞进行扩增、培养,并分析其生物学特性。
三、实验结果1. DUSP9基因敲除效率分析通过PCR和Western Blot等方法,我们成功检测到DUSP9基因的敲除情况。
在转导了CRISPR-Cas9系统的胚胎干细胞中,DUSP9基因的敲除效率达到了XX%。
2. 胚胎干细胞的生物学特性分析通过对敲除后的胚胎干细胞进行扩增、培养,我们发现其增殖能力、分化能力等生物学特性均与野生型胚胎干细胞无显著差异。
3. 基因敲除小鼠模型的构建与验证将敲除DUSP9基因的胚胎干细胞注射到小鼠囊胚中,成功构建了DUSP9基因敲除小鼠模型。
通过对小鼠进行基因检测,验证了DUSP9基因的敲除情况。
四、讨论本实验利用CRISPR-Cas9系统成功构建了DUSP9基因敲除小鼠胚胎干细胞系,为进一步研究DUSP9基因的功能及其在相关疾病中的作用提供了有力工具。
同时,本实验也证明了CRISPR-Cas9系统在基因编辑领域的广泛应用和可靠性。
在实验过程中,我们注意到以下几点:首先,gRNA的设计与合成是影响基因敲除效率的关键因素之一,需要针对目标基因的序列信息进行精确设计。
一文教你如何利用CRISPRCas9技术建立小鼠敲除模型

一文教你如何利用CRISPRCas9技术建立小鼠敲除模型作者:Ryan来源:科研小助手公众号Manipulation of mouse genome with minimal off-target by microinjection of one-cell embryos with paired sgRNAs and nickaseWhile CRISPR/Cas9 technique has been widely used in genome editing regarding multiple organisms, the off-target effect can’t be neglected. Subsequently, to conquer the off-target effect, many strategies have been applied including longer sgRNA (about 26bp) and nickase combined with paired sgRNAs. Here we described a good menthod for generating the mutant mice/conditional knockout mice with minimal off-target by microinjection of one-cell embryos with paired sgRNAs and Cas9 nickase. Moreover, paired sgRNAs and nickase can also mutate multiple genes simultaneously, or to generate large deletions up to at least 10kb or more.Comparison of Cas9 and nickase (Cas9D10A)Figure 1. Cas9 nickase strategy. Cas9 nickase induces a double strand break adjacent CRISPR sites (TS1 and TS2) onopposite DNA strands. Constrastly, single-stand nicks at off-target sites (OTS) for either sgRNA will be corrected by the base-excision repair pathway, thus minimizing off-target mutations. P, PAM site.Plasmidsused in the protocolT7-Nickase(Cas9-D10A):T7-sgRNA:ReagentsPlasmids: T7-Nickase(Cas9-D10A) and T7-sgRNAmMESSAGE mMACHINE®T7 Ultra kit (Ambion, AM1345)MEGAshortscript TM Kit(Ambion, AM1354)RNeasy Mini Kit (QIAGEN,74104)MEGAclear TM Kit(Ambion, AM1908)RNAsecure TM Reagent(Ambion, AM7005)QIAprep Spin Miniprep Kit(QIAGEN, 27104)MiniElute PCRPurification Kit (QIAGEN, 28004)BsaI (NEB, R0535S)AgeI (NEB, R0552S)DraI (TAKARA, D1037A)T4 DNA Ripid ligation Kit(NEB,M2200S)PMSG (Sansheng, China,50IU/ml in normal saline, Aliquot and store at -80℃)HCG (Sansheng, China,50IU/ml in normal saline, Aliquot and store at -80℃)EmbryoMax® Injection Biffer (Millipore, MR-095-10F)ProteinaseK(Merck,1245680100, 20 mg/ml in water, Aliquot and store at -20℃)Lysis buffer (10 μMTris-H Cl, 0.4 M NaCl, 2 μM EDTA, 1% SDS) Phenol (Tris-saturated),Chloroform and alcoholPCR clearing Kit (Axygen,AP-PCR-50)T7EN1 (NEB, M0302L)PrimerSTAR HS DNA Polymerase (TAKARA, DR010A)pMD19T-vector kit(TAKARA, 3271)EquipmentCentrifuge(RT and 4℃)VortexOneDrop OD-1000+ SpectrophotometerThermocyclerThermomixerThermo-controlledwater bath(37℃,42℃ and 58℃)ProcedureConstructionof sgRNA expression vectors1. Design of paired sgRNA oligos.Select paired sgRNAs in a tail-to-tail orientation and separated by 10-30 bp, which have the sequence 5’-CCN(52-72)GG.All possible paired sites for mouse and human exons are available on website(/htgt/wge/). For each sgRNA, the 5’-GGN(19)GGmotif is preferred, however, 5’-GN(20)GG or 5’-N(21)GG are also satisfactory. BLAT or BLAST the sgRNA target sites in UCSC or ENSEMBL genome browsers to find those with few or no highly related sites in the genome.Order oligos as below:For 5’-GGN(19)GGmotifTop strand oligo:Bottom strand Oligo:For 5’-GGN(20)GG motifFor 5’-GGN(21)GG motif2. Annealing oligos prior to cloning.4.5μl Top Oligo (100 μM)4.5μl Bottom Oligo (100 μM)1μl NEB buffer 2Annealing oligos using a thermocycler with the following program:95℃,5 min; 95-85℃ at -2℃/s; 85-25℃ at -0.1℃/s; hold at 4℃.3. Preparation of T7-sgRNA plasmid.2 μg T7-sgRNA plasmid.1 μl CutSmart Buffer1 μl BsaIAdd H2O up to 50 μl and incubate at 37℃ for 2 h with occasional shake.Purify the digeston product using MinElutePCR Purification Kit.4. Ligation of annealed oligos with BsaI-digested T7-sgRNA4 μl annealed oligos2 μl (25 ng/μl) digested T7-sgRNA10×NEB ligation buffer 1 μlddH2O 2 μlNEB T4 DNA ligase 1 μlUp to 10 μlncubate at 22℃for 30 min5. Transformation and plate on Kan+plate (50 μg/ml).6. Confirm correct Insertion of sgRNA oligosby sequencing using M13-47 primer.7. Mini-prep T7-sgRNA plasmid using QIAprepSpin Miniprep Kit.Transcriptionof sgRNAs in vitro1. Ensurethat reagents, tubes and tips are RNase-free and that the work is done in aribonuclease-free enviroment.2. Digestpaired sgRNA plasmids with DraI and purify the digestion fragment.10 μg paired sgRNA plasmids (5 μg each)10 μl 10×Mbiffer5 μl DraI (15 U/μl)Add H2O up to 100 μl and incubate at 37℃ for 3 h with occasional shake.Check plasmids were digested completely bygel electrophoresis, loading 2 μl in 1% agarose gel.Two bands (1621 and 1152 bp) will beobserved. It is not necessary tio gel-purify the band harboring the sgRNAsequence.Add 4 μl RNAsecure and incubate at 60 ℃ for 10 min in a thermomixer.Purify and elute the digestion product with 10 μl RNase-free water usingMinElute PCR Purification Kit, 5-8 μg of DNA will be recovered.For mutiplexing experiments, two or more paired sgRNAs may be digested simultaneously in one tube.Alternatively, the transcription template containing the T7 promoter sgRNAsequence may be prepared by PCR amplification from a bacterial colony using thefollowing primers and PCR program:sgRNA-For:5’-TCTCGCGCGTTTCGGTGATGACGGsgRNA-Rev:5’-AAAAAAAGCACCGACTCGGTGCCACTTTTTCProgram:94℃,5min; ((98 ℃ ,10s; 72-62℃, -1℃/cycle, 15s; 72 ℃, 30s) 10 cycles, (98℃, 10s;62 ℃, 15s; 72 ℃, 30s) 25 cycles); 72 ℃, 5 min; hold at 4℃.Inactivate RNases byadding RNA secure and purify the PCR product using the MinElute PCR PurificationKit.3. Invitro transcription of sgRNAs using MEGAshortscriptTMKit.1 μl T710× Reaction Buffer1 μl T7 ATP Solution (75 mM)1 μl T7 CTP Solution (75 mM)1 μl T7 GTP Solution (75 mM)1 μl T7 UTP Solution (75 mM)4 μl purified template (more than 2 μg for plasmids, 700 ng-1000 ng for PCR products)1 μl T7 Enzyme Mix10 μl of transcription volume is OK.Incubate the reaction at 37 ℃ for 4-6 h in water bath or Thermocycler (Set thehot lid to 50 ℃).Add 1 μl TURBODNase and incubate at 37 ℃ for 15 min to remove the DNA template.4. Purify the sgRNAs by MEGAclearTM Kitaccording to the manufacturer’s instructions.RNA elutionoption 2 in the manual is preferred.Precipitatewith 5 M Ammonium Acetate and ethanol.Resuspendthe pellet using the 30 μl RNase free water.20-50 μgRNA will be obtained depending on the quality of DNA template.5.Assess sgRNA yield using the One Drop OD-1000+Spectrophotometer (or equivalent) and sgRNA quality by gel electrophoresis. RNAis loaded in DNA loading buffer and run on 1% agarose gel (180 V for 10 min).6.Aliquot and store at -80 ℃. The sgRNAs are stablefor one year without freeze-thaw cycles.Transcription of Nickase (Cas9-D10A) in vitro1. Ensure that reagents, tubes and tips are RNase-freeand that the work is done in a ribonuclease-free enviroment.2. Digest T7-Nickase (Cas9-D10A) plasmid with AgeIandpurify the digestion product.10 μgT7-Nickase (Cas9-D10A)10 μl NEBbuffer I4 μl AgeIAdd H2O upto 100 μl and incubate at 37 ℃ for 3 h with occasional shake.Add 4 μlRNAsecure and incubate at 60 ℃ for 10 min in a thermomixer.Check for complete digestion of the plasmid byelectrophoresis, loading 2 μl in 1% agarose gel.Purify and elute the digestion product with 10 μlRNas e-free water using MinElute PCR Purification Kit, 5-8 μg DNA will berecovered.3.In vitro transcribe Cas9-D10A using mMESSAGE mMACHINE® T7 Ultra Kit according to the manufacturer’s instruction.4. Purify the Nickase (Cas9-D10A) mRNA by RNeasy MiniKit ac cording to the manufacturer’s instructions.5. Assess sgRNA yield using the One Drop OD-1000+Spectrophotometer (or equivalent) and sgRNA quality by gel electrophoresis. RNAis loaded in DNA loading buffer and run on a 1% agarose gel (180V for 10 min).A yield of 30-60 μg mRNA is expected.Note: Due to the size of the Nickase (Cas9-D10A) mRNA, no visible size shift is seenafter poly-A tailing. The mRNA quality is good if a smear is not observed.6. Aliquot and store at -80 ℃. Nickase (Cas9-D10A)mRNA is stable for one year without freeze-thaw cycles.Collection of zygotes1. Superovulate 4-week-old female C57BL/6J (about12-14g) mice by intraperitoneal injection with PMSG (5 IU/100 μl) at 14:00 ofday 1 and with HCG (5 IU/100 μl) at 13:00 of day 3.2. Cross superovulated females with males (C57BL/6J orCBA).3. Identify plugged females at 9:00 of day4. Collectone-cell embryos as decribed in Reyon, D. et al, 2012.Preparation of microinjection mixture1. Thaw aliquot of the Cas9-D10A mRNA and sgRNAs onice. Dilute the Cas9-D10A mRNA with Embryo Max® Injection Buffer to a concentration of 20 ng/μl and sgRNAs (5 ng/μl each) in a final volume of 50 μl. Pipette the mixture upand down several times2. Centrifuge at 4 ℃ for 1 min at top sped, andcarefully transfer 45 μl supernatant to a new tube. Always keep the tube onice.Microinjection and embryo transferMicroinjection and embryo transfer are performed using standard methods for generation of transgenic mice as described in Andras,N. et al., 2003, Cold Spring Harb Protoc. We prefer to inject the RNA mixture into both the cytoplasm and larger (male) pronucelus.Genotyping founders1. Tail tips from founders (5-day-old) are collected and digested overnight at 55 ℃ with lysis buffer containing 100 μg/ml Proteinase K. Genomic DNA is extracted by phenol-chloroform and purified by ethanol precipitation.2. Target region(300-700 bp) are PCR amplified from genomic DNA and the products are purified with the PCR Cleanup Kit. Purified PCR products are denatured and reannealed in NEB buffer 2 in a thermocycler using the following programme;95℃,5 min; 95-85 ℃ at -2℃/s; 85-25 ℃ at -0.1℃/s; hold at 4℃.3. Hybridized PCR products are digested with 0.5 μlT7EN1 at 37℃ for 30 min and separated by 2% agarose gel. Mutant founders will yield lower molecular weight cleavage bands.4. Cloning and sequencing of PCR amplicons from genimic DNA of mutant founders is used to characterize the mutations. T-A cloning of PCR products us performed using the pMD19T kit (TAKARA) according to manufacturer’s instructions.TroubleshootingProblemSolution SgRNA expression plasmid does not contain insertpUC57-sgRNA vector is not digested completely.Extend the incubation time and shake thedigestion product occasinally. Colony PCR canbe used to identify the positive coloniesusing 5’-TTGTACTGAGAGTGCACCATATG-3’ and the bottom strand sgRNA oligoLow yield of sgRNAsa. Use the recommended kits to improve the quality of plasmids and templateb. Increase the amount of template or use the PCR product as template. Electrophoresis of sgRNAs shows more than one band a. sgRNAs can form dimers. Always keep sgRNAson ice. A low amount of dimer will not affectthe function of sgRNA.b. DNA template is incompletely digested.Circular template can produce longertranscripts. Extend the incubation time and shake the digestion product occasionally. c. DNA template contamination. Add more TURBO DNase and extend incubation time.Cas9-D10A mRNA produces a smear on an agrose gel a. Use RNAsecure to inactivate RNase contaminationb. Use the recommended kits to improve thequality of the DNA template.(QIAGEN Mini-prepand PCR clean-up kits are recommended)Time Taken4 days for the construction of sgRNA expression vectors.1 day for the in vitro transcription and preparation of sgRNAs. 1 day for the in vitro transcription and preparation of Cas9-D10A mRNA.4 days for the superovulationb of females, collection of 1-cell embryos and microinjections1 week for the genotyping of founder animals.。
基因敲除小鼠制备的流程

基因敲除小鼠的制备流程基因敲除小鼠已经成为现代生命科学基础研究和药物研发领域不可或缺的实验动物模型,在生命科学、人类医药和健康研究领域中发挥着重要的作用。
基于胚胎干细胞的基因打靶技术、EGE技术(基于Crispr cas9技术)是当下比较火热的基因敲除小鼠制备技术。
利用这两种技术制备基因敲除小鼠的流程是什么样的?一、基于胚胎干细胞的基因打靶技术制备基因敲除小鼠的流程:1.课题设计,订购课题BAC菌;2.按照课题设计,完成打靶载体设计和构建;3.将重组载体电转到胚胎干细胞中,用G418筛选转染后的胚胎干细胞,得到阳性克隆;4.进一步通过PCR和southern blot杂交技术(基因敲除小鼠检测金标准)对上一步得到的阳性克隆进行筛选,得到稳定整合外源基因的胚胎干细胞阳性克隆;5.将胚胎干细胞阳性克隆注射到小鼠囊胚中,并植入到假孕小鼠的子宫内;6.得到嵌合鼠,并获得F1阳性杂合子小鼠。
基于胚胎干细胞的基因打靶技术制备基因敲除小鼠是目前为止唯一一个可以满足几乎所有基因组修饰要求的打靶技术,但目前只应用在小鼠的基因敲除上,而且其周期长工作量大。
二、利用EGE技术(基于Crispr cas9技术)制备基因敲除小鼠的流程1.设计构建识别靶序列的sgRNA;2.设计构建致靶基因切割的EGE系统载体质粒;3.利用百奥赛图自主开发的UCA试剂盒对sgRNA/Cas9进行活性检测;4.设计构建打靶载体;5.体外转录sgRNA/Cas9 mRNA;6.小鼠受精卵原核注射sgRNA/Cas9 mRNA和打靶载体;7.获得Fo代小鼠,利用PCR对Fo代小鼠进行基因型鉴定;8.获得F1代小鼠,利用PCR和southern blot杂交技术(基因敲除小鼠检测金标准)对F1代小鼠进行基因型鉴定。
虽然EGE技术(基于Crispr cas9技术)制备基因敲除小鼠看似比基于胚胎干细胞的基因打靶技术制备基因敲除小鼠流程繁琐,其实不然,EGE技术(基于Crispr cas9技术)系统构建简单,基因敲除/敲入效率高,速度快,可实现多基因、多物种基因敲除/敲入,最快2个月即可得到F0代阳性鼠,5个月得到F1F1代杂合子小鼠。
CRISPRCas9基因敲除小鼠模型

CRISPRCas9基因敲除小鼠模型
根据基因序列设计合成sgRNA,针对Knockin插入点或点突变位置构建含Knockin片段或点突变的同源序列,与Cas9 mRNA共同注射到小鼠受精卵胞质,Cas9核酸酶、sgRNA、基因组靶序列结合并切割双链DNA,以含Knockin的同源序列为模版修复基因组DNA,最终获得在目的DNA序列插入Knockin片段或点突变的Knockin小鼠。
根据基因序列设计合成两个sgRNA,分别针对两个Loxp的插入点,构建含Loxp的同源序列,与Cas9 mRNA共同注射到小鼠受精卵胞质,Cas9核糖酶、sgRNA、基因组靶序列结合并切割双链DNA,以含Loxp同源序列为模板修复基因组DNA,最终获得在待敲除目的DNA序列两端各有一个Loxp序列的Flox小鼠。
基于CRISPRCas9构建小鼠UOX基因敲除模型

高尿酸血症(hyperuricemia)是由嘌呤代谢紊乱,尿酸排泄障碍引起的血尿酸异常为临床表现的异质性疾病。
尿酸病理性升高有5%~12%的风险导致尿酸结晶累积并损伤关节[1],引起痛风、急性及慢性关节炎等。
并且,高尿酸血症易引发并发症,如高血压、高血脂、2型糖尿病、冠心病。
临床上常规的高尿酸血症及痛风治疗手段为口服降尿酸药物[2],目前尚未有根治痛风的药物,新型降尿酸药物开发需要利用高尿酸动物模型进行降尿酸药物药效及药理筛选。
故建立稳定性好,更接近患者发病特征的高尿酸动物模型,能提高新型降尿酸药物开发效率及成药性,优化痛风治疗格局。
近年来, CRISPR/Cas9基因编辑技术凭借其简便性、特异性和高效性,在疾病动物模型构建领域应用日趋广泛和深入。
该研究利用CRISPR/Cas9,敲除小鼠基因组内尿酸氧化DOI:10.16662/ki.1674-0742.2020.07.032基于CRISPR/Cas9构建小鼠UOX基因敲除模型张茹君1,夏海林1,朱赟2,黄晶3,孟雨菡21.常州卡文斯实验动物有限公司,江苏常州213104;2.江苏科标医学检测有限公司,江苏常州213161;3.百格基因科技(江苏)有限公司,江苏常州213000[摘要]目的通过CRISPR/Cas9获得UOX基因敲除的小鼠纯合品系,为建立高尿酸血小鼠动物模型奠定基础。
方法根据小鼠UOX基因第三外显子前后两个位点设计双sgRNA,通过PCR、体外转录和纯化获得sgRNA和Cas9mRNA。
将sgRNA和Cas9mRNA显微注射进小鼠原核胚后,体外培养至二细胞胚阶段,进行胚胎移植至代孕母鼠。
F0代小鼠出生后,提取其DNA进行电泳分析和测序分析。
F0代交配繁殖至F2代获得UOX缺失的小鼠纯合品系。
结果显微注射sgRNA和Cas mRNA至原核胚,成功获得50枚囊胚。
移植后生育17只幼鼠。
其中,有8只小鼠UOX基因缺失突变,突变率为47.06%。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
专业分享
1
CRISPR-Cas9系统的原理
crRNA( CRISPR-derived RNA )通过碱基 配对与 tracrRNA (trans-activating RNA ) 结合形成 tracrRNA/crRNA 复合物,此复合物引 导核酸酶 Cas9 蛋白在与 crRNA 配对的序列靶 位点剪切双链 DNA。而通过人工设计这两种 RNA, 可以改造形成具有引导作用的sgRNA (short guide RNA ),足以引导 Cas9 对 DNA 的定点 切割。
• 使得CRISPR-Cas9应用更广泛。
专业分享
4
Cre-dependent Cas9 Rosa26 targeting 矢量图
Rosa26,使转 录可被诱导
荧光蛋白
专业分享
5
转入Cas9后与野生小鼠对比
专业分享
6
神经系统荧光对比
专业分享
7
三种实验测试效果:
三种转接方法: 纳米粒子、腺病毒转导、 慢病毒转导。 三个系统(器官): 免疫、神经、肺(癌)。
专业分享
2
CRISPR-Cas9系统的原理
sgRNA
通过设计sgRNA来确定剪切位点 设计简单快速,无需重复构建核酸内切酶
专业分享
target
3
Cas9的局限性
• 受限于转基因范围,Cas9往往只用与细胞或胚 胎层面的实验。
• 本实验采用Cre-dependent Rosa26 Cas9 knockin mouse克服了这个局限性。
专业分享
8
一、慢病毒转入树突状细胞
——转入树突状细胞实验过程
专业分享
9
通过荧光蛋白EGFP可以鉴别 Cas9是否转导成功
专业分享
10
设计了两个剪切位点
专业分享
11
有转入目的基因 的小鼠大多细胞目的 基因发生移码突变, 目的基因转录和翻译 都明显下调。
专业分享
12
二、腺病毒转入大脑皮层额叶
目标基因是NeuN
专业分享
16
多数为两个等位基因都被破坏,且其 中多为移码突变。
专业分享
17
三、腺病毒转入肺
专业分享
18
sgKras
sgp53
sgLkb1
Kras同源 修补
专业分享
19
P53的基因变化
专业分享
• 多为移码突变
20
Lkb1的基因变化
专业分享
• 多为移码突变
21
Kras的基因变化
专业分享
22
P53和Lkb1不断被损坏, Kras则被修补变得越发优势
专业分享
23
最终肺中出现肿瘤
ቤተ መጻሕፍቲ ባይዱ专业分享
24
专业分享
25
总结
• 事实证明将Cre-dependent引入Cas9工具中, 可以大大扩展Cas9的用途。
• 以后用此方法可以更好的对基因进行修整, 已达成更多的实验,为人类医学和生物学作 出贡献。
专业分享
26
Thanks!
专业分享
27
专业分享
13
目的基因发生的改变
✓ 缺失一位 ✓ 缺失多位 ✓ 插入一位 ✓ 插入多位
专业分享
14
荧光显示含有Cre/Cas9 的组织中NeuN表达明显减
少,而非转入非目标的 sgRNA则目标蛋白无影响
专业分享
15
NeuN被破坏的比例十分大
Indel=insertions(插入) and deletions(删除)