神经病学综合征:下运动神经元综合征(Lowermotorneuronsyndrome)
神经病综合征

syndromes
of nervous diseases
无法入睡 抑郁症困扰
神奇的多囊肾新疗法!聋哑耳鸣、头痛新疗法!
疤痕疙瘩、瘩痤疮找中医
神经系统疾病主要产生感觉与运动两大障碍,其表现形式多种多样。一种疾病可以出现各种不同的症状和体征,反之,一种症状或体征可由不同疾病(或病因)引起,而同一疾病因损害部位不同,其临床表现亦不尽相同。临床上把不同致病因素引起的各种神经征候群称为神经病综合征。本节列举临床上常见的各种神经病综合征,就其病因、症候及其病因的鉴别诊断做了较为详细的描述,便于查找与校对,为临床诊断及鉴别诊断提供线索。
【鉴别诊断】
(一)椎—基底动脉供血不足(vertebrobasilar ischemia)
椎—基底动脉系统的短暂脑缺血发作时有时仅表现头昏、眼花、走路不稳等含糊症状而难以诊断。若有局灶性症状如眩晕、复视、构音障碍、吞咽困难、交叉性或双侧肢体瘫痪和感觉障碍、共济失调等则诊断较为明确,其发作特点为起病突然,历时短暂,一般无意识障碍,可反复发作。伴有Avellis综合征的短暂脑缺血发作提示延髓受累。
(三)患者有各种程度的精神发育迟滞,而脑部缺乏特异性病理改变。视乳头水肿较少见,视神经萎缩较多见。
【鉴别诊断】
(一)Crouzon氏综合征 又称遗传性头颜面骨发育不全,为一种特殊类型的颅骨缝早闭。多有家族遗传史。头面部畸形的特点为多数颅骨缝过早闭合,上颌骨发育差和脑积水,颅骨的前后径短,两眼分离并向外斜视,鼻底退缩、鼻弓增宽,眼眶下缘缩小,眼球向前突出,上、下齿呈反咬合。除头面部畸形外,常有头痛和脑挤压征象。
(二)小头畸形 常因胎儿期的有害环境因素所致。偶然见于常染色体隐性遗传。本症中脑部和颅骨的发育均有障碍,发育完全后脑重不超过1000克,最大颅周径一般不超过47厘米。头颅形状亦有特异性变化,额部和枕部平坦、狭小、顶部略呈尖形,和发育完整的面骨形成强烈对照。头皮增厚,头发粗密。身材矮小,智能发展停留于白痴阶段。
神经肌病---下运动神经元综合征的诊断,怎么破?

神经肌病---下运动神经元综合征的诊断,怎么破?在神经科门诊当中,经常会碰到一些患者表现为对称或不对称的肌无力伴肌肉萎缩,没有明显的感觉障碍和上运动神经元损害证据,根据临床定位诊断可考虑脊髓前角、前根、运动神经轴索或运动神经髓鞘的病变,从临床描述性诊断而言,在未完善进一步检查之前,可考虑诊断为下运动神经元综合征(Lower motor neuron syndromes)。
随着对疾病认识的深入和检查手段的发展,我们肯定不能囿于LMN综合征的诊断,必须进行细分。
在此之前,有必要对LMN综合征的概念进行一番梳理。
LMN综合征是一类以肌肉萎缩无力、腱反射减弱而无感觉障碍为特点的临床综合征,其责任病变部位可以为脊髓前角、前根或运动神经,其病因较多,包括遗传性和获得性两大类,遗传性包括脊肌萎缩症、肯尼迪病、远端遗传性运动神经病等,获得性包括散发性连枷臂/连枷腿型运动神经元病、慢性格林巴利综合征纯运动变异型、多灶性运动神经病等。
电生理检查对于进一步鉴别诊断至关重要,通过电生理定位与前角/前根、运动轴索或运动髓鞘所对应的疾病有云泥之分,鉴于部分获得性疾病(譬如MMN或CIDP)具可治性,非常有必要对LMN综合征进行精确诊断。
与其他很多临床综合征类似,LMN综合征的诊断思路主要包括了解疾病分类----认识疾病特点----建立诊断流程几个步骤。
只有了解了疾病的分类,才能为后续的“对号入座”打下基础,而掌握了疾病的特点,才能厘清不同“座位”的特征,形成鉴别诊断图谱,碰到具体病人才能按图索骥,对号入座。
而建立诊断流程则是在上述基础之上,形成诊断策略。
LMN综合征的临床特点总结见表1:LMN综合征的诊断流程图见图1:在上述的LMN综合征诊断体系中,特别要注意远端遗传性运动神经病(dHMN)和单肢肌萎缩(MMA)两类疾病,前者是目前我们容易忽略的品种,鉴于基因型和临床表型的关联不足,大家对此类疾病的认识非常欠缺;而后者可以解释临床上一些有良性病程的定位于前角细胞(颈段或腰段)的中老年患者,年轻人我们有平山病这个标签可以使用,但对于中老年人,应该用什么诊断一直是个问题,个人建议现阶段就可以用单肢肌萎缩(MMA)做“垃圾桶”暂时存放一下吧!缩写列表:AMAN急性运动轴索神经病CHMP2B染色质修饰蛋白2BCIDP慢性炎性脱髓鞘性神经病dHMN远端遗传性运动神经病FUS融合肉瘤GBS吉兰-巴雷综合征LMN下运动神经元MMA单肢肌萎缩MMN多灶性运动神经病MND运动神经元病SBMA脊髓延髓肌萎缩症SMA脊肌萎缩症SOD1超氧化物歧化酶1TMS经颅磁刺激VAPB囊泡相关膜蛋白B参考文献:Differentiating lower motor neuron syndromes. J Neurol Neurosurg Psychiatry. 2016 Dec 21. pii: jnnp-2016-313526. doi: 10.1136/jnnp-2016-313526.。
神经系统疾病常见综合征

神经系统疾病常见综合征神经系统疾病常见综合征Bell sigh(贝尔征):面神经炎患者,闭眼时双眼球向外上方转动,露出白色巩膜,称为贝尔征。
Fisher综合征:表现为眼外肌麻痹、共济失调及腱反射消失三联征,伴脑脊液蛋白-细胞分离。
CTS(腕管综合征):各种原因致正中神经在腕管内受压,出现桡侧三个手指感觉障碍、麻木、疼痛及大鱼际肌萎缩称腕管综合征。
Froin征:椎管严重梗阻时脑脊液蛋白-细胞分离,细胞数正常,蛋白含量超过10g/L时,黄色的脑脊液流出后自动凝固,称为Froin征。
Meige综合征:主要表现为眼睑痉挛和口-下颌肌张力障碍。
帕金森综合症:静止性震颤、运动迟缓、肌强直、姿势步态障碍。
Charcot三主征:眼震、意向震颤和吟诗样语言。
Lhermitte sigh(莱尔米特征):被动屈颈会诱导出现刺激感或闪电样感觉,自颈部沿脊柱放散至大腿或足部,称为莱尔米特征。
手足口综合征:EV71颅高压三主征:头痛、恶心呕吐、视乳头水肿。
脑膜刺激征:颈强直、Kernig征、Brudzinski征。
巴宾斯基等位征:1 Chaddock征2 Oppenheim征3Schaeffer征4 Gordon征5 Gonda征6 Pussep征无动性缄默征:又称睁眼昏迷。
病变在脑干上部和网状激活系统,病人无目的的注视,似觉醒状态但缄默不语,肢体不能活动。
脊髓前动脉综合征:脊髓梗死正常发生在脊髓前动脉供血区,以中胸段和下胸段多见,病损水平的相应部位出现根痛,短时间内即发生截瘫,痛温觉丧失,大小便障碍,深感觉保留,称为~parinnaud 综合征:上丘的破坏性病变可引起两眼向上同向运动不能。
Millard-Gubler 综合征:一侧脑桥病变时可出现同侧面神经和展神经麻痹,对侧偏瘫。
Brow-Sequard 综合征:又称为脊髓半切综合征,损伤平面以下同侧上运动神经元瘫痪和深感觉缺失,对侧痛温觉缺失。
Weber综合征:一侧中脑大脑脚受损,同侧动眼神经麻痹及对侧中枢性偏瘫。
神经病学名词解释整理(3)

神经病学名词解释整理(3)神经病学名词解释整理37. Foville syndrome综合征:①脑桥内侧部肿瘤破坏同侧展神经、内侧纵束和锥体束②引起两眼向病灶对侧凝视、病侧面瘫及对侧肢体偏瘫35. Weber综合征:又称中脑腹侧部综合征。
①中脑腹侧及大脑脚底肿瘤破坏同侧动眼N和锥体束②引起同侧动眼神经麻痹和对侧肢体偏瘫③多小脑幕裂孔疝。
38. Parinaud综合征:又称中脑顶盖综合征。
见于松果体瘤,两侧中脑顶盖受累。
双眼垂直运动麻痹,不能向上仰视。
若侵及中脑被盖则瞳孔对光反射消失。
39. 脊髓半切综合征(Brown-Séquard syndrome):半侧脊髓损害(如髓外肿瘤早期、外伤),损害平面以下同侧中枢性瘫痪、深感觉缺失,对侧痛温觉缺失。
40. 三偏综合征:①内囊病变可引起锥体束全部受损而致对侧偏瘫②如病损波及内囊后肢的后部,阻断传导对侧半身感觉的丘脑皮质束及传导两眼对侧视野的视放射,则可伴有对侧偏身感觉缺失和对侧同向偏盲③内囊病变最常见于脑血管意外。
41. Horner综合征:①一侧眼交感神经麻痹,出现同侧瞳孔缩小、上睑下垂、眼裂凹陷,眼结膜充血及面部无汗②病变可在患侧脑干,颈段脊髓或病侧颈内动脉壁。
42. 去大脑皮质综合征(decorticate syndrome):①双侧大脑皮质广泛损害,功能丧失而皮质下功能仍保存②常见于严重脑外伤、缺氧或感染后③患者能无意识的睁眼、闭眼或转动眼球但眼球不能随光线或物品而转动,貌似清醒但对外界刺激无反应④有抓握、吸吮、咳嗽等反射,有无意识的吞咽活动,四肢肌张力增高,双侧锥体束征阳性,上肢屈曲,下肢伸直者称为去皮质强直(decorticate rigidity)。
(去大脑强直四肢均为伸性强直)43. 威廉斯环(Willis circle):①脑底动脉环,是脑底沟通颈内动脉和椎-基底动脉系的动脉环,使两侧大脑半球及一侧大脑半球的前后部有充分供血②由双侧大脑前动脉、颈内动脉、大脑后动脉、前交通动脉和后交通动脉组成③当环的某处血供障碍时,可对脑血液供血发挥调节和代偿作用。
(完整word版)神经病学必考大题与名词解释

一、名词解释(1) Brown-Sequard综合征:又称为脊髓半切损害,表现为病侧损害节段的根性疼痛和感觉过敏带,病侧损害平面以下的上运动神经元瘫痪和深感觉消失,对侧痛、温觉缺失。
常见于髓外肿瘤、外伤、脊髓血肿等。
(2) 闭锁综合征:又称去传出状态,是双侧脑桥基底部病变,典型临床症状为意识清醒,咽部、四肢不能活动,仅以眼部的某些动作与外界交流(3) Horner征:由于颈上及脑干网状结构等交感纤维损害,临床表现为病侧眼球凹陷、瞳孔缩小、眼裂变小,可伴有同侧面部少汗或无汗。
(4) Broca失语:损伤部位在额下回后部,又称运动性失语。
临床特点以口语表达障碍最为突出,呈典型非流利型口语,口语理解相对较好。
(5) Wernicke失语:即感觉性失语,受损部位位于优势半球的颞上回后部。
表现为对口语无法理解,但运动性语言正常(6) 低颅压头痛:是脑脊液压力降低(<60mmHg)所致的头痛,多为体位性,患者通常在直立15分钟内出现头痛或头痛明显加重,卧位后头痛缓解或消失。
(7) 痛性抽搐:原发性三叉神经痛患者发作时疼痛可引起反射性面肌抽搐,口角牵向患侧,并有面红、流泪和流涎,称痛性抽搐。
(8) Hunt综合征:病变在膝状神经节时,除了有周围性面瘫、舌前2/3味觉丧失、听觉过敏,还有患侧乳突疼痛、耳廓和外耳道感觉减退或异常,外耳道或鼓膜出现疱疹。
(9) Bell麻痹:是指茎乳孔内面神经非特异性炎症导致的周围性面瘫(10) Bell征:面神经炎时出现周围性面瘫,闭眼时瘫痪侧眼球向外上方转动显露出白色巩膜。
(11) 脑脊液蛋白细胞分离:脑脊液中蛋白含量增高明显而细胞数目正常,在格兰-巴雷综合征中此现象是该病的特征之一(12) 多发性神经病:是四肢远端对称性感觉障碍、下运动神经元瘫痪和自主神经功能障碍的临床综合征。
(13) 短暂性脑缺血发作(TIA):是指因脑血管病变引起的短暂性、局限性脑功能缺失或视网膜功能障碍,临床症状多在10~20分钟,多在1小时内缓解,最长不超过24消失,不遗留神经功能缺损症状,结构性影像学检查无责任病灶。
30种精神病名称、特征及症状

1、第欧根尼综合症:Diogenes又名肮脏混乱综合症或众议院综合征症状特点:1.生活脏乱,2.极度自卑感3.又强迫性的囤积行为,无法舍弃财物,过度的购买欲4.有强烈的隐居欲望,拒绝他人帮助5.主要出现在老年人上,有时伴随老年痴呆症2、外地口音综合症:Foreignaccentsyndrome:外国口音综合症(Foreignaccentsyndrome)是一种临床上很罕见的病症通常伴随着严重的脑损伤,此病导致患者说母语时如同带有外国口音,例如:一个美国人可能说话带着法国口音3、科塔尔综合症科塔尔综合征(Cotardsyndrome):以虚无妄想(nihilisticdelusion)和否定妄想(delusionofnegation)为核心症状患者主要是认为自身躯体和内部器官发生了变化.部分或全部已经不存在了如某患者称自己的肺烂了肠子也烂了甚至整个身体都没了.患者认为自己已经死了不复于人世或者五脏六腑已经被掏空即使正和外人说话也不认为自己是活着的4、卡普格拉妄想综合症:卡普格拉妄想症(Capgrasdelusion)命名自第一个介绍这个心理疾病的法国心理医师,患有这种病的人会认为,自己的爱人被一个具有同样外貌特征的人取代了5、Fregoli妄想综合症又名人身变换症和卡普格拉妄想症(Capgrasdelusion)相反,这类患者认为身边许许多多的人其实都是同一个人的伪装,以上都属于错觉认知综合症的一种6、被爱妄想症(Erotomania)是一种少见的心理疾病,患者会陷入另一个人(通常有较高的社会地位)和他谈恋爱的妄想之中。
被爱妄想症又被称为克雷宏波综合症,“oldmaid’spsycho si s”、“eroticparanoia”、“eroticself-referentdelusions以纪念法国精神病学家克雷宏波(1872-1934)于1921年发表了题目为“LesPsychosesPassionelles”的一篇论文。
《神经病学》常考名词解释

Hunt综合征:表现为耳后、乳突剧烈疼痛,外耳道疱疹及周围性面神经麻痹,伴有泪腺、唾液腺分泌障碍,见于带状疱疹病毒引起的膝状神经节炎。
Brown-sequard综合征:脊髓半切征,病变侧损伤平面以下深感觉障碍和上级运动神经元瘫痪,对侧损伤平面以下痛、温度觉缺失。
见于脊髓占位性病变、脊髓外伤。
Gerstman综合征:四主症,即计算不能(失算)、手指失认,左右侧认识不能,书写不能(失写),见于优势半球顶叶角回皮质损伤。
三偏综合征:内囊完全损伤导致病变对侧偏瘫、偏盲和偏身感觉障碍,常见于脑出血和脑梗塞。
wallenberg综合征:①眩晕、恶心、呕吐、眼震(前庭神经核损害),②真性球麻痹:吞咽困难、构音障碍、同侧软腭麻痹及咽反射消失(舌咽、迷走神经及疑核损伤),③病灶侧肢体共济失调(绳状体、部分小脑损害)④Horner综合征(颈部交感神经受损)⑤同侧面部痛、温度觉丧失,触觉存在,呈核性分布(三叉神经脊束核受损)⑥对侧痛温度觉消失(脊髓丘脑侧束损伤)⑦呃逆(网状结构中呼吸中枢损伤)Millard-Gubler综合征:脑桥腹下部综合征,表现为①病变侧展神经麻痹及周围性面神经麻痹,②对侧中枢性偏瘫,③对侧偏身感觉障碍。
见于脑桥基底部的展神经、面神经、锥体束、脊髓丘脑侧束及内侧丘系损伤。
Weber综合征:①病变侧动眼神经麻痹,②对侧肢体中枢性偏瘫,主要见于损害大脑脚底,影响了动眼神经和锥体束。
Benedict综合征:红核综合征,表现为①病变侧动眼神经麻痹,②对侧肢体不自主运动或共济失调。
动眼神经、黒质、红核受损而锥体束未受损。
吉兰-巴雷综合征:又称为急性炎症性脱髓鞘性多发性神经病,是一组急性或亚急性发病,病理改变为周围性神经炎性脱髓鞘,临床表现为四肢对称性弛缓性瘫痪,伴有手套或袜套样感觉障碍及脑神经损害的自身免疫病,实验室检查有脑脊液蛋白-细胞分离现象及神经电生理异常。
foster-kennedy综合征:表现为病变侧因肿瘤压迫而出现视神经萎缩,对侧颅内压增高导致视神经乳头水肿。
神经病学名词解释与简答

神经病学:是研究神经系统疾病和肌肉疾病病因、发病机制、临床表现、诊断和鉴别诊断、预防和治疗及康复等内容的一门临床学科。
感觉性失语:患者能听见对方和自己说话的声音,但不能理解说话的含义。
运动性失语:又称Broca失语,由优势侧额下回后部病变引起,以口语表达障碍为突出特点,听理解相对较好,呈非流利型口语。
脑疝:是颅内压增高的严重后果,是部分脑组织因颅内压力差而造成移位,当位移超过一定的解剖界限时则称之为脑疝。
三偏综合征:指内囊包含大量上、下行纤维,一侧内囊小范围损伤时,可引起对侧肢体偏瘫(皮质脊髓束、皮质核束损伤)和偏身感觉障碍(丘脑中央辐射受损),大范围损伤还可以有对侧同向性偏盲(视辐射受损),即出现“三偏综合征”Horner综合征:颈8~胸1节段侧角细胞受损,瞳孔缩小(病损同侧),眼球内陷(眼眶肌麻痹),眼裂变小(眼睑肌麻痹),同侧面部出汗减少脊髓休克:当脊髓与高位中枢断离时,脊髓暂时丧失反射活动的能力而进入无反应状态的现象。
颈膨大:颈部上部神经出入处形成膨大,相当于C5~T2,两上肢呈下运动神经元性瘫痪,两下肢呈上神经元性瘫痪。
腰膨大:腰部下肢神经出入处形成膨大,相当于L1~S2,受损时出现双下肢下运动神经元性瘫痪,双下肢及会阴部各种感觉缺失,括约肌障碍。
延髓麻痹:舌咽、迷走神经彼此邻近,有共同的起始核,常同时受损,表现为声音嘶哑、吞咽困难、饮水呛咳及咽反射消失,也称真性延髓麻痹。
假性延髓麻痹:舌咽、迷走神经的运动核受双侧皮质脑干束支配,当一侧损害时不出现延髓麻痹症状,当双侧皮质延髓束损伤时才出现构音障碍和吞咽因难,而咽反射存在,称假性延髓麻痹。
昏迷:是一种严重的意识障碍。
患者意识完全丧失,各种强刺激不能使其觉醒,无有目的的自主活动,不能自发睁眼。
脑膜刺激征:为脑膜受激惹的表现,脑膜病变导致脊髓膜受到刺激并影响到脊神经根,当牵拉刺激时引起相应肌群反射性痉挛的一种病理反射。
见于脑膜炎,蛛网膜下腔出血和颅内压增高等。
神经病学名词解释

神经病学名词解释之杨若古兰创作1、Horner综合征:表示为一侧瞳孔缩小.眼裂变小、眼球内陷,可伴随同正面部少汗.2、感觉过度:因为刺激阈增高与反应时间耽误,刺激必须达到很强的程度方有感觉,在刺激后需经一潜伏期才干感到强烈的定位不明确的不适感,且持续一段时间才消逝.3、感觉过敏:表示为轻微的刺激即惹起强烈的感觉,系因对触痛觉的敏感性加强或感觉阈值降低所致.4、假性球麻痹:又称假性延髓麻痹,两侧皮质延髓受累时出现舌咽,迷走神经麻痹症状,表示为发音嘶哑,吞咽困难,咽反射消逝.5、活动性失语:Broca失语,口语表达妨碍突出,理解绝对好,次要累及上风半球额下回后部.6、感觉性失语口语理解严重妨碍为其突出特点,故以往称为感觉性失语.患者对他人和本人讲的话均不睬解,或仅理解个别词或短语;口语表达有适当的语法结构但缺乏实质词,表示为语量多,讲话不费力,发音清晰,腔调正常,短语是非正常,即所谓流利型口语,病变位于上风半球Wernicke区(颞上回后部).7、缺血半暗带:急性脑梗死病灶是由中间坏死区及其四周的缺血半暗带(ischemic penumbra)构成.而缺血半暗带内因仍有侧支轮回存在,可获得部分血液供给,尚有大量可存活的神经元,如果血流敏捷恢复,损伤仍为可逆的,脑代谢妨碍可得以恢复,神经细胞仍可存活并恢复功能.8、TIA:指反复发作的局灶性脑缺血导致的突发、短暂、可逆性神经功能妨碍的临床综合征.9、闭锁综合症(Locked-in syndrome):见于双侧脑桥基底部损害.患者认识清楚,但四肢及面部瘫痪不克不及张口说话和吞咽,仅保管闭眼和眼球垂直活动功能,并以此表达本人的志愿.10、Weber综合征(中脑腹侧综合征):中脑病变导致同侧动眼神经瘫痪,对侧中枢性偏瘫.11、Wallenberg综合征(延髓外侧综合征):系椎动脉梗阻,狭隘或小脑后下动脉梗阻惹起的临床症状.表示为眩晕/恶心/呕吐/眼震,病灶同侧软腭及声带麻痹/吞咽困难/声音嘶哑,病灶同侧共济失调,病灶同侧Horner(,病灶同正面部浅感觉妨碍,对侧半身浅感觉妨碍.12、Millard-Gubler综合征:脑桥外侧梗死,表示为病变侧外展神经和面神经四周性麻痹,病变对侧中枢性舌下神经和肢体瘫痪.13、腔隙形态:多发性腔隙性梗死累及双侧锥体束,出现严重精神妨碍,痴呆,假性延髓麻痹,双侧锥体束征,类帕金森病和大小便失常. 14、分水岭脑梗死:相邻血管供应区之间的分水岭区域或边沿带的局部缺血.此种梗死约占全部脑梗死的10%15、脊髓半切综合征(脊髓半侧损害):次要特征是病变同侧损害节段以下上活动神经元性瘫痪,同侧深感觉妨碍及病变对侧损害节段以下痛温觉减退或丧失,而触觉坚持良好,病变侧损害节段以下血管舒缩功能妨碍.16、脊髓休克:脊髓病变初期表示为肌肉松弛,肌张力低,腱反射消逝、病理征阴性和尿潴留等,普通持续1~6周.17、Beevor征:T10-11病变时下半部腹直肌有力,当患者仰卧位用力抬头时,可见脐孔被腹直肌上半部牵拉向上挪动.18、鞍区回避:髓内压榨感觉妨碍自病叛变段向下发展,鞍区(S3-S5)感觉保存至最初才受累.19、三偏综合征:内囊损害累及视纤维时,表示为对侧肢体偏瘫,对侧偏生感觉妨碍,对侧同向偏盲.20、分离性感觉妨碍:脊髓丘脑束损伤时,对侧损害平面以下痛温觉缺失,而触觉和深感觉仍保管.21、颈膨大:颈膨大相当于C5~T2节段,是安排上肢神经的起源,受损可出现双上肢下活动神经元性瘫,双下肢上活动神经元性瘫,病变平面以下各种感觉缺失,肩部及上肢可有放射性根痛,括约肌妨碍.22、腰膨大:腰膨大相当于L1~S2节段,是安排下肢神经的起源,受损出现双下肢下活动神经元瘫,双下肢及会阴部各自感觉缺失,尿便妨碍.23、脊髓压榨症:是一组椎管内占位性病变惹起脊椎受压综合征,可有脊髓半切或横贯性损害和椎管梗阻,脊神经根和血管受累的临床表示.24、上升性脊髓炎:脊髓损害节段呈上升性,在起病1-2天甚至数小时内慢慢上升至延髓,或瘫痪由下肢敏捷涉及上肢甚至延髓安排的肌群,并出现吞咽困难、构音不良、呼吸肌瘫痪而死亡.25、华勒变性:指内伤使轴突断裂后,因为不再有轴浆运输提供轴突所需的养分成分,断端远侧的轴突很快字近端向远端发生变性、解体,解体的轴突和髓鞘可有同样的变更.但普通只到比来的1-2个郎飞结而不再继续,接近细胞体的轴突断伤可使细胞体坏死.26、轴突变性:因为中毒或养分妨碍等缘由,使细胞体合成蛋白质等物资发生妨碍或者轴浆运输阻滞,导致最远端的轴突不克不及得到须要的养分,是以其变性通常从轴突的最远端开始向近端发展.27、MS:即多发性硬化,是以中枢神经零碎脱髓鞘病变成特点的本身免疫性疾病,临床上表示为病变部位的多发性及发病时间的多发性. 28、三叉神经痛:面部三叉神经分布区域内反复发作的、短暂的、阵发性的剧痛称为三叉神经痛.29、Bell征:表示为患正面部脸色肌瘫痪、额纹消逝,不克不及皱眉,眼裂扩大、闭目不全,闭目时瘫痪侧眼球转向上方,露出白色巩膜. 30、特发性面神经麻痹:是指茎乳孔内面神经非特异性炎症所致的单侧四周性面瘫.31、扳机点(触发点):“扳机点”亦称“触发点”,常位于上下唇、鼻翼、齿龈、口角、舌、眉等处,即为三叉神经痛患者口角,鼻翼,颊部或舌部敏感区,轻触或刺激扳机点可激发疼痛发作.32、痛性抽搐:三叉神经痛惹起发射性的面肌抽搐,患者口角牵向患侧,可伴面部皮肤发红、流泪和流涎.33、Hunt综合征:为带状疱疹病毒感染所致,病变累及膝状神经节,出现同正面部脸色肌瘫痪,舌前2/3味觉丧失与听觉过敏,尚有瘫痪侧的乳突部疼痛,耳廓与外耳道感觉减退,外耳道或鼓膜出现疱疹.34、脑脊液蛋白细胞分离:指脑脊液蛋白增高而细胞数正常或轻度增高.是格林- 巴利综合征特征性改变之一.病程3~6周蛋白增高最明显.35、癫痫:是一组脑神经元异常过度放电惹起的慢性反复发作性短暂脑功能失调综合征,以突然发生反复发作为特征,是发作性认识丧失的罕见缘由.36、杰克逊(Jackson)发作:部分活动性发作如放电沿大脑皮质活动区分布逐步扩展,临床常表示为抽搐自对侧拇指沿腕部、肘部和肩部扩展.37、癫痫持续形态:是癫痫持续发作之间认识尚未恢复又频繁再发,或癫痫发作持续30分钟以上不自行停止.38、痫性发作:每次神经元的阵发性放电或短暂的脑功能异常称为痫性发作.39、Todd瘫:癫痫部分活动性发作后如遗留临时性(数分至数日)局部体瘫痪或有力,称Todd瘫痪.40、全身性强直性发作:又称大发作,次要表示为全身肌肉强直和阵挛,伴认识丧失及自立神经妨碍.41、失神发作:失神经发作是癫痫痫性发作的一种表示,以认识妨碍为次要表示.又分典型和不典型失神二种.典型失神经发作:病人活动突然停止,发呆,呼之不该,手中物体落地,部分可机械反复原本的简单动作,每次发作持续数秒钟,每天可发作数十、上百次,醒后不克不及回忆.42、主动症:复杂部分性发作的活动表示以调和的不自立活动为特征,癫痫发作过程中或发作后认识形态,出现必定程度上调和的,有适应性的有认识活动,发作后不克不及回忆.43、帕金森病:是发生于中年以上的黒质和黒质纹状体通路变性的疾病,临床四大主征是肌强直、震颤、活动迟缓、姿式步态异常.44、急性横贯性脊髓炎:系指一种非特异性炎症,急性脊髓白质的脱髓鞘性病变或坏死性病变,临床表示为急性完整性或不完整性截瘫.可能与炎性脱髓鞘性疾病有关.好发部位:胸段(T3~5).45、面具脸:面部脸色肌活动减少,经常双眼凝视,瞬目减少,呈现面具脸.46、慌张步态:患者自卧位、座位起立困难,迈步后即以极小的程序向前冲,越走越快,不克不及及时停步或转弯.47、静止性震颤:震颤出此刻肢体处于静止形态时,在自立活动时减轻或消逝.其频率普通为每秒4~8次,见于帕金森综合症.48、折刀样强直:视部位分歧,只累及部分肌群,主动活动关节市,阻力在开始时较明显,随后敏捷减弱,呈现所谓折刀景象,为锥体束损害时的体征,常伴随腱反射亢进和病理征.49、铅管样强直:屈肌和伸肌同时受累,主动关节时始终坚持增高的肌力,类似曲折软铅管的感觉.50、齿轮样强直:部分患者伴随震颤,检查时可感到在均匀的阻力中出现规律而断续停顿,如同动弹齿轮感.51、写字过小征:帕金森病患者因壁肌及手指肌的强直,上肢不克不及做精细动作,表示为书写困难,所写的字曲折不正,越写越小.52、疲劳试验(Jolly test):嘱患者持续上视出现上睑下垂或两臂持续平举后出现上臂下垂,歇息后恢复则为阳性.53、危象:因为本身病情加重或医治不当惹起呼吸肌有力所致的严重呼吸困难形态.54、肌有力危象:当中正肌有力患者本人病情加重或者医治不当惹起呼吸肌有力所致的严重呼吸困难形态.55、胆碱能危象:因为抗胆碱药物过量所惹起肌有力加重.56、反拗危象:重症肌有力患者对抗胆碱酯酶药物不敏感,出现肌有力加重、呼吸困难等.57、Miller-Fisher综合征(书P105):次要表示为眼外肌麻痹、共济失调和腱反射消逝三联征.58、偏头痛:是一种多种病因惹起的,颅内外血管神经功能妨碍导致的,以发作性单或双侧头痛为特征的疾病.。
2021年神经外科的20个综合征(全文)

2021年神经外科的20个综合征(全文)一、Foster-kennedy syndrome:额叶底面肿瘤(常见于嗅沟脑膜瘤)可出现同侧嗅觉缺失和视神经萎缩,对侧视乳头水肿,称为福斯特-肯尼迪综合征。
二、Gerstmann syndrome:古茨曼综合征为优势半球角回损害所致,主要表现有:计算不能(失算症)、手指失认、左右辨别不能(左右失认症)、书写不能(失写症)及有时伴失读。
三、Parinaud syndrome:帕里诺综合征又称上丘脑综合征、中脑顶盖综合征或者上仰视性麻痹综合征。
由上丘脑病变(多为松果体肿瘤压迫中脑四叠体)而导致的眼球垂直同向运动障碍,向上凝视麻痹、瞳孔对光反射消失(上丘受损);神经性聋(下丘受损);小脑性共济失调(结合臂受损)。
症状多为双侧。
四、Wallenberg syndrome:瓦伦贝格综合征,又称为延髓背外侧综合征常见的原因为小脑后下动脉、椎基底动脉或外侧延髓动脉缺血性损害。
表现为:1.眩晕、恶心、呕吐及眼震(前庭神经核损害);2.病灶侧软腭、咽喉肌瘫痪,表现为吞咽困难、构音障碍、同侧软腭低垂及咽反射消失(疑核及舌咽、迷走神经损害);3.病灶侧共济失调(绳状体损害);4.霍纳综合征(交感神经下行纤维损害);5.交叉性偏身感觉障碍,即同侧面部痛、温觉缺失(三叉神经脊束及脊束核损害),对侧偏身痛、温觉减退或丧失(脊髓丘脑侧束损害)。
五、Horner syndrome:霍纳综合征,又称颈交感神经麻痹综合征是由于交感神经中枢至眼部的通路上受到任何压迫和破坏,引起患侧瞳孔缩小、眼球内陷、上睑下垂及患侧面部无汗的综合征。
据受损部位可分为中枢性障碍、节前障碍及节后障碍的损害。
六、CushingⅠsyndrome:桥小脑角综合征(1)病变部位:脑桥小脑角。
(2)常见病因:听神经瘤、胆脂瘤、胶质瘤、桥小脑角脑膜瘤或蛛网膜炎、蛛网膜囊肿、结核性脑膜炎、血管畸形和动脉瘤。
(3)受累颅神经:Ⅴ、Ⅶ、Ⅷ,有时伴Ⅵ,Ⅸ,Ⅹ。
神经病学名词解释资料汇总

神经病学名词解释资料汇总黄斑回避:一侧枕叶视中枢病变引起两眼对侧视野同向偏盲,视野中心视力常保存。
一个半综合征:一侧脑桥的外展旁核和双侧内侧纵束受损。
表现为两眼向病侧注视时,同侧眼球不能外展,对侧眼球不能内收。
向对侧凝视时,对侧眼球能外展,病侧眼球不能内收。
但双眼内聚运动仍正常。
帕里诺(Parinaud)综合征:上丘的破坏性病变(如松果体瘤)引起两眼向上同向运动不能,称帕里诺(Parinaud)综合征。
动眼危象:上丘的刺激性病变表现为眼球发作性转向上方,称动眼危象。
见于脑炎后帕金森综合征。
Horner氏征:颈上交感神经节及其纤维损害所致,表现为同侧瞳孔缩小、眼球内陷(眼眶肌瘫痪)、眼裂变小(睑板肌瘫痪)、同侧面部出汗减少。
Hunt Syndrome:一侧周围性面N麻痹伴有外耳道疼痛和疱疹时,提示膝状N节的带状疱疹病毒感染。
眩晕vertigo:是机体对空间位置关系的定向障碍,表现为视物旋转感或自身旋转感,常伴恶心、呕吐、面色苍白、出汗和眼球震颤。
假性球麻痹:由于双侧皮质脑干束损害引起舌咽和迷失N支配区肌肉的麻痹,表现为发音嘶哑,饮水呛咳,吞咽困难等球麻痹症状。
可见双侧软腭弓下垂和活动受限。
多伴长束体征及额叶释放征(强哭强笑,出现抓握反射),咽反射存在。
真性球麻痹:由于周围性舌咽、迷走神经损害引起舌咽和迷失N支配区肌肉的麻痹,表现为发音嘶哑,饮水呛咳,吞咽困难等球麻痹症状。
可见单侧或双侧软腭弓下垂和活动受限。
无长束体征(延髓核性损害可伴)无额叶释放征咽反射消失。
颈静脉孔综合征:舌咽、迷走、副神经均自颈静脉孔出颅,后颅凹病变常使三者同时受累断连休克现象(脊休克):脊髓与高位中枢离断后反射活动能力暂时丧失而进入无反应状态的现象。
表现为损伤平面以下肌张力降低,深反射和浅反射消失,病理反射引不出。
是脊髓突然失去高位中枢的易化性调节所致。
一段时间后,脊髓功能恢复,变现为肌张力增高,深反射亢进,出现病理反射。
三偏征:内囊损害所致的对侧偏瘫、对侧偏身感觉障碍,对侧同向偏盲。
神经病学名词解释-3

神经病学名词解释-3神经病学Neurology名词解释●缺损症状:是指神经结构受损时,正常功能的减弱或消失。
●刺激症状:是指神经结构受刺激后引起的过度兴奋的表现。
●释放症状:是指高级中枢受损后,原来受其抑制的低级中枢因抑制解除而出现功能亢进。
●断联休克症状:是指中枢神经系统局部发生急性严重损害时,引起功能上与受损部位有密切联系的远隔部位神经功能短暂丧失。
●Wallenberg综合征:即延髓背外侧综合征。
表现为:①眩晕、恶心、眼震;②病灶侧软腭、咽喉肌瘫痪(吞咽困难、构音障碍);③病灶侧共济失调;④Horner综合征(单侧瞳孔缩小、眼裂变窄、眼球内陷、眼睑下垂,同侧额部无汗);⑤交叉性感觉障碍(同侧面部痛温觉缺失,对侧偏身痛温觉缺失),常见于椎-基底动脉缺血。
●闭锁综合征:即去传出状态,双侧脑桥基底部病变。
表现为双侧中枢性瘫痪,只能以眼球上●脊髓束和皮质脑干束。
功能是发放和传递随意运动冲动至下运动神经元,并控制和支配其活动,损伤后可产生中枢性瘫痪(痉挛性瘫痪)。
●下运动神经元:包括脊髓前角细胞、脑神经运动核及其发出的轴突,是接受锥体系统、锥体外系和小脑系统冲动的最后通路,是冲动到达骨骼肌的唯一通路,损伤可产生周围性瘫痪(迟缓性瘫痪)。
●去皮质综合征:因双侧皮质广泛受损导致皮质功能减退或丧失,皮质下功能仍保留。
表现为意识丧失,对外界刺激无反应,反射存在,四肢肌张力增高,双侧锥体束征阳性,姿势呈去皮质状态(上肢屈曲内收,下肢伸直、足屈曲)。
●真性球麻痹:即延髓麻痹,是一侧或双侧舌咽、迷走神经下运动神经元受损引起唇、腭、舌和声带麻痹或肌肉无力导致声音嘶哑、吞咽困难、饮水呛咳及咽反射消失。
●假性球麻痹:是指当双侧皮质脊髓束损伤时出现的构音障碍和吞咽困难,而咽反射存在的现象。
●Broca失语:即运动性失语。
由优势半球额下回后部病变引起,表现为口语表达障碍,非流利,口语理解能力相对保留,有不同程度的复述、命名、阅读和书写障碍。
神经病学 名词解释及简答

1.上运动神经元锥体系:包括上、下两个运动神经元。
上运动神经元胞体主要位于大脑皮质躯体运动中枢的锥体细胞,这些细胞的轴突组成下行的锥体束,下行至脊髓的称皮质脊髓束,至脑干运动神经核的纤维为皮质核束。
上运动神经元损伤可引起痉挛性瘫痪,肌张力增高,深反射亢进,可出现病理反射,早期不出现肌萎缩。
2.下运动神经元:下运动神经元胞体位于脑干脑神经运动核和脊髓前角运动细胞,它们的轴突分别组成脑神经和脊神经,支配全身骨骼肌的随意运动。
下运动神经元受损时,出现弛缓性瘫痪,肌张力降低、肌萎缩,深反射消失。
3.视神经萎缩(optic atrophy):是指任何疾病引起视网膜神经节细胞和其轴突发生病变,致使视神经全部变细的一种形成学改变,为病理学通用的名词,一般发生于视网膜至外侧膝状体之间的神经节细胞轴突变性。
4.一个半综合征(one-and-a-half syndrome):又称脑桥麻痹性外斜视,若一侧桥脑侧视中枢(外展旁核)及双侧内侧纵束同时受到破坏,则出现同侧凝视麻痹(一个),对侧核间性眼肌麻痹(半个),即两眼向病灶侧注视时,同侧眼球不能外展,对侧眼球不能内收,向病灶对侧注视时,对侧眼球能外展,病灶侧眼球不能内收,两眼内聚运动仍正常。
5.帕里诺综合征(Parinaud综合征):又称上丘脑综合征、中脑顶盖综合征、上仰视性麻痹综合征。
由中脑上丘的眼球垂直同向运动皮质下中枢病变而导致的眼球垂直同向运动障碍,累及上丘的破坏性病灶可导致两眼向上同向运动不能。
6.阿-罗瞳孔(Argyell-Robertson瞳孔):发生在神经梅毒病人中,在半数左右的脊髓痨或者麻痹性痴呆病人中出现,主要是瞳孔缩小、光反射消失、而调节反射存在(调节反射是在看东西有远向近时,两眼向中间辐辏、瞳孔缩小)。
7.艾迪瞳孔:又称强直性瞳孔,其临床意义不明。
表现一侧瞳孔散大,只在暗处用强光持续照射时瞳孔缓慢收缩,停止光照后瞳孔缓慢散大。
调节反射也缓慢出现喝缓慢恢复。
运动神经元疾病分类

运动神经元疾病分类运动神经元疾病是一类累及运动神经元的神经系统疾病,其特点是运动障碍和肌无力。
这些疾病可以分为两大类:上行性运动神经元疾病和下行性运动神经元疾病。
一、上行性运动神经元疾病上行性运动神经元(UMN)位于中枢神经系统,包括大脑皮质的锥体束和脑干的皮质脊髓束。
当这些上行性运动神经元受损时,会导致肌张力增高、反射亢进和肌肉抽搐等临床表现。
1. 脊髓性肌萎缩(spinal muscular atrophy,SMA)脊髓性肌萎缩是一种常见的遗传性上行性运动神经元疾病,主要特征是进行性的肌无力和肌萎缩。
这种疾病通常在婴儿期或儿童早期发生,并且会导致呼吸困难和吞咽困难等严重并发症。
2. 运动神经元病(motor neuron disease,MND)运动神经元病是一组罕见的上行性运动神经元疾病,包括肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)和原发性进行性肌萎缩等。
ALS是最常见的运动神经元病,其特征是进行性的上行性和下行性运动障碍。
二、下行性运动神经元疾病下行性运动神经元(LMN)位于脊髓前角和脑干的脑神经核。
当这些下行性运动神经元受损时,会导致肌张力降低、反射减弱或消失以及肌无力等临床表现。
1. 肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)ALS在上述上行性运动神经元疾病中已有所提及,在此再次强调其同时累及了上行性和下行性运动神经元。
这种疾病通常在成年期发生,并且会导致进行性的肌无力、肌萎缩和呼吸困难等严重并发症。
2. 帕金森氏综合征(Parkinson's disease,PD)帕金森氏综合征是一种常见的下行性运动神经元疾病,其特征是进行性的肌张力降低、震颤和运动缓慢。
这种疾病通常在中老年人发生,并且会导致平衡障碍和语言障碍等临床表现。
3. 肌萎缩性侧索硬化(progressive muscular atrophy,PMA)肌萎缩性侧索硬化是一种罕见的下行性运动神经元疾病,其特征是进行性的肌无力和肌萎缩。
神经皮肤综合征

版权所有:解放军总医院磁共振成像中心
神经纤维瘤病I型
女,28岁,腰骶部不适1年,发现盆腔占位22余天,盆腔肿瘤穿刺活检病理为神经源 性肿瘤;其母亲、哥哥均有神经纤维瘤病。
版权所有:解放军总医院磁共振成像中心
神经纤维瘤病I型(续)
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
Patient’s ID: 126480 F 8Y Limping for 1 year
术后病理诊断为:神经纤维瘤病
版权所有:解放军总医院磁共振成像中心
Patient’s ID: 126480 F 8Y Limping for 1 year
术后病理诊断为:神经纤维瘤病
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
Patient’s ID: 126480 F 8Y Limping for 1 year 术后病理诊断为:神经纤维瘤病
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
男性, 32岁。 1996年诊为NF-2,行双侧听神经瘤及颈部椎管内肿瘤切除术。 影像号:20636 门诊号: 住院号: 病理号:
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
版权所有:解放军总医院磁共振成像中心
下运动神经元综合征?
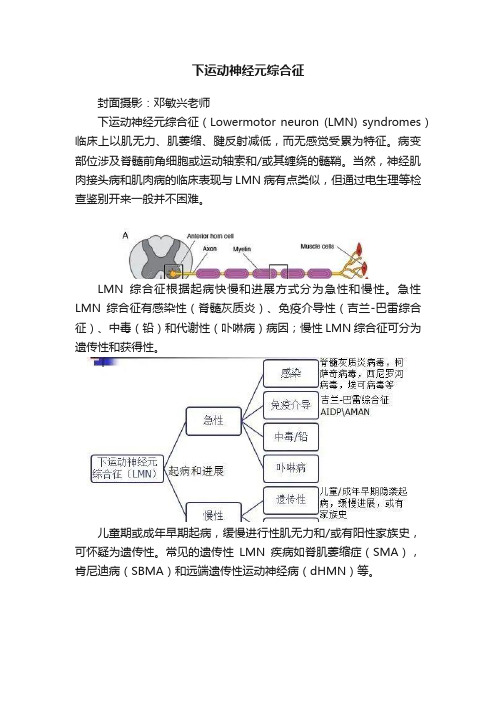
下运动神经元综合征封面摄影:邓敏兴老师下运动神经元综合征(Lowermotor neuron (LMN) syndromes)临床上以肌无力、肌萎缩、腱反射减低,而无感觉受累为特征。
病变部位涉及脊髓前角细胞或运动轴索和/或其缠绕的髓鞘。
当然,神经肌肉接头病和肌肉病的临床表现与LMN病有点类似,但通过电生理等检查鉴别开来一般并不困难。
LMN综合征根据起病快慢和进展方式分为急性和慢性。
急性LMN综合征有感染性(脊髓灰质炎)、免疫介导性(吉兰-巴雷综合征)、中毒(铅)和代谢性(卟啉病)病因;慢性LMN综合征可分为遗传性和获得性。
儿童期或成年早期起病,缓慢进行性肌无力和/或有阳性家族史,可怀疑为遗传性。
常见的遗传性LMN疾病如脊肌萎缩症(SMA),肯尼迪病(SBMA)和远端遗传性运动神经病(dHMN)等。
获得性LMN综合征主要包括平山病和运动神经元病的变异型,如进行性肌萎缩(PMA),连枷臂/腿综合征,节段性MND等。
当然通过电生理检查找出另外两个免疫介导性疾病,如多灶性运动神经病(MMN)和慢性炎性脱髓鞘性多发神经病(CIDP)变异型至关重要,因为他们对免疫治疗是有效的。
因此,对于LMN综合征的诊断,重点是做以下三件事:第一是明确肌无力的特点:对称/不对称,近端/远端受累,上肢/下肢为著,是否存在球部受累。
第二是神经传导和肌电图检查,这对于明确病变是神经源性非常关键,应重点评价:肌无力是否对称,是否为长度依赖性,是否存在局部运动传导阻滞或脱髓鞘改变,是否存在临床下感觉异常等。
第三是完善辅助检查,包括影像学(神经核磁/超声),血/脑脊液抗体检测,分子遗传学分析,高级神经生理技术(经颅磁刺激)等,这些都是临床评价的延伸。
下面对各种LMN综合征做简要介绍:晚发型脊肌萎缩症(Late-onset SMA) SMA是一组遗传性疾病,因脊髓前角细胞和脑干运动神经核变性,导致肌无力进行性加重,且以近端为著,伴腱反射减低或消失。
神经系统疾病常见综合征

神经系统疾病常见综合征神经系统疾病常见综合征Bell sigh(贝尔征):面神经炎患者,闭眼时双眼球向外上方转动,露出白色巩膜,称为贝尔征。
Fisher综合征:表现为眼外肌麻痹、共济失调及腱反射消失三联征,伴脑脊液蛋白-细胞分离。
CTS(腕管综合征):各种原因致正中神经在腕管内受压,出现桡侧三个手指感觉障碍、麻木、疼痛及大鱼际肌萎缩称腕管综合征。
Froin征:椎管严重梗阻时脑脊液蛋白-细胞分离,细胞数正常,蛋白含量超过10g/L时,黄色的脑脊液流出后自动凝固,称为Froin征。
Meige综合征:主要表现为眼睑痉挛和口-下颌肌张力障碍。
帕金森综合症:静止性震颤、运动迟缓、肌强直、姿势步态障碍。
Charcot三主征:眼震、意向震颤和吟诗样语言。
Lhermitte sigh(莱尔米特征):被动屈颈会诱导出现刺激感或闪电样感觉,自颈部沿脊柱放散至大腿或足部,称为莱尔米特征。
手足口综合征:EV71颅高压三主征:头痛、恶心呕吐、视乳头水肿。
脑膜刺激征:颈强直、Kernig征、Brudzinski征。
巴宾斯基等位征:1 Chaddock征 2 Oppenheim征 3 Schaeffer征4 Gordon征5 Gonda征6 Pussep征无动性缄默征:又称睁眼昏迷。
病变在脑干上部和网状激活系统,病人无目的的注视,似觉醒状态但缄默不语,肢体不能活动。
脊髓前动脉综合征:脊髓梗死正常发生在脊髓前动脉供血区,以中胸段和下胸段多见,病损水平的相应部位出现根痛,短时间内即发生截瘫,痛温觉丧失,大小便障碍,深感觉保留,称为~parinnaud 综合征:上丘的破坏性病变可引起两眼向上同向运动不能。
Millard-Gubler 综合征:一侧脑桥病变时可出现同侧面神经和展神经麻痹,对侧偏瘫。
Brow-Sequard 综合征:又称为脊髓半切综合征,损伤平面以下同侧上运动神经元瘫痪和深感觉缺失,对侧痛温觉缺失。
Weber综合征:一侧中脑大脑脚受损,同侧动眼神经麻痹及对侧中枢性偏瘫。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
神经病学综合征:下运动神经元综合征 ( Lower motor neuron
syndrome)
运动神经元( Lower motor neuron syndrome ,LMN )
综合征临床表现为肌肉萎缩,肌无力,腱反射减退,但不伴有感觉受累。
从远端运动神经至前角细胞通路上的任何损伤
均可导致LMN 综合征。
神经肌肉接头疾病和肌肉疾病有时
可与LMN 综合征混淆,需注意鉴别。
LMN 综合征可粗分
为遗传性,散发性和免疫介导。
免疫介导的神经病,例如多灶性运动神经病,慢性炎症性脱髓鞘性多发性神经病,与散发性以及遗传性LMN 综合征鉴别非常重要,因其是可治的疾病。
LMN 表现的运动神经元病通常为散发性,但也已经
发现少数遗传突变。
其他遗传性LMN 综合征主要包括脊髓的临床诊断首先要明确起病和进展方式,例如,运动神经元病进展较快,而脊髓性肌萎缩症和免疫性神经病相对较慢。
性肌肉萎缩症,远端型遗传性运动神经病等。
LMN 综合征肌无力的分布也很重要,如对称或非对称,近端或远端,上肢为主或下肢明显,伴或不伴球麻痹等。
神经传导速度检测和肌电图是证实神经源性损害的必要手段,检查重点在于受累类型,包括是否对称或长度依赖,有无局灶性运动传导阻滞或脱髓鞘特征,是否合并亚临床的感觉受累。
1脊髓性肌萎缩症
脊髓性肌萎缩症(Spinal muscular atrophy ,SMA ),又称脊肌萎缩症,是儿童期较常见的常染色体隐性遗传病,主要表现为进行性、对称性四肢和躯干肌肉无力、萎缩,重症患儿常
死于呼吸衰竭。
该病在活产婴儿中的发病率约为体5q13 上运动神经元存活基因1(survival motor neuron ,
1/6000-1/10000 ,中国人群中SMA 的携带率约为1/42 。
染色SMN1 )纯合缺失或复合杂合突变(即1 个SMN1 基因缺失,另1 个SMN1 基因存在点突变)引起SMN 蛋白表达下降是
SMA的主要致病机制。
(1)SMA I型:又称Werding-Hoffman
病,是严重的亚型(重型),约占确诊SMA 患者的一半。
此型发病急、进展快,一般在出生6 个月之内发病;患者表现为广泛的肌张力减退,肌肉严重无力,因此无法抬头,不能坐或走,多因喂养和呼吸困难于2 岁之前死亡。
(图1:SMA
I 型患者)
2)SMA II 型:又称Dubowitz 病,为慢性婴儿型(中间型),通常在7-18 个月内发病,患者能坐但不能站立行走,大多可以生存至10-20 岁。
(图2:SMA II 型患者)
3)SMA III 型:又称Kugelberg-Welander 病,为青少年型
轻型)。
其症状表现具有很大的异质性,根据发病时间和
行走能力再分型,例如在出生后3年内发病为a型,有44%
的患者20岁之前可以行走;出生3年后发病为b 型,90%的患者能够在20 岁前站立和行走。
此型病情发展缓慢,患
者肌肉无力,但寿命不受影响。
(图3:SMA III 型患者)
4) SMA IV 型:为成年型(极轻型) 般于20-30 岁以
后发病,主要表现为缓慢发生的上下肢近端无力和肌肉萎缩,成年期都能够行走,寿命正常。
2脊髓延髓肌萎缩脊髓延髓肌萎缩 ( Spinobulbar muscular atrophy ,SBMA ),又称为Kennedy 病,是一种少见的成人发病的性连锁隐性遗传
运动神经元变性疾病。
其特征是缓慢进展的延髓、面部及四肢肌肉无力和萎缩。
1968 年由Kennedy 等首先报道,致病基因定位于性染色体长臂近侧端( Xq11-12 ),主要是由于雄激素受体( androgen receptor,AR )基因1 号外显子编码多聚谷氨酰胺的CAG 异常扩增所致。
一般为30-60 岁发病,疾病初期多为非特异性症状,如姿势性震颤和肌肉痉挛。
姿势性震颤多出现在患者肢体无力前数年甚至数十年前,值得引起临床医生重视,有研究发现其原因可能与临床下的感觉障碍和运动单位减少有关。
早期临床症状包括面部收缩震颤或言语不清,多有肌束震颤,尤其是口周和舌头。
肢体无力是患者最常见问题,其次为延髓功能障碍,延髓肌受累可导致构音障碍和吞咽困难。
虽然目前认为肯尼迪病为下运动神经元病,但也有研究表明,肯尼迪病患者存在轻度认知功能障碍,表现为言语流畅性障碍、概念形成异常以及记忆力下降。
部分肯尼迪病患者表现出雄激素不敏感的迹象,如乳房
发育、睾丸萎缩、勃起障碍、生育能力降低。
女性携带者通常无症状。
体格检查发现以下运动神经元体征为主,可见轻度的肌肉萎缩、束颤,肌力轻度减退,肢体近端更明显,腱反射减弱或消失和感觉缺失。
大多数患者血清中的肌酸激酶水平升高,部分合并高血压、高血脂症、轻度肝功能异常、葡萄糖耐受不良等。
肯尼迪病患者电生理检查以广泛的慢性神经源性损害为最主要改变,多伴感觉和运动神经传导异常,且感觉异常比运动异常更多见。
肯尼迪病的进展通常非常缓慢,晚期可因吸入性肺炎死亡。
(图4:Kennedy 病患者面部不对称无力和舌肌萎缩)
视频1:Kennedy 病患者口周肌束颤动)
视频2:Kennedy 病患者男性乳房发育,两侧面部肌肉萎缩,舌
肌颤动,双手姿势性和动作性震颤,步态正常)
3远端型遗传性运动神经病远端型遗传性运动神经病(Distal hereditary motor neuropathy,dHMN )进展缓慢,肌无力呈长度依赖性(即主要累及远端),其包含了一组遗传异质性的疾病,其临床表型多变,且可有重叠。
多数为常染色体显性遗传,但也有常染色体隐性遗传和X 连锁遗传的报道。
通常在儿童或青年期起病,但成人患者也不少见。
一些患者可表现为上肢明显
dHMN V ),声带麻痹(dHMN VII ),呼吸窘迫(dHMN VI )和锥体束征。
本病无明显的感觉受累,可与轴索型
Charcot-Marie-Tooth 病鉴别,尽管两者可有共同的基因突变。
图5:dHMN 患者下肢远端肌肉萎缩)表2 远端型遗传性运动神经病分型亚型遗传方式临床表型基因染色体位点
dHMN I 型AD 青少年起病伴远端萎缩和无力
HSPB1HSPB8GARSDYNC1H1 ------- dHMN II 型AD 成人起病伴远端萎缩和无力HSPB1HSPB8BSCL2HSPB3 (很可能)
- dHMN III 型AR 缓慢进展的肌萎缩和无力未知
11q13dHMN IV 型AR 缓慢进展的肌萎缩和无力伴膈肌麻痹未知
11q13dHMN V 型AD 上肢为主GARSBSCL2--dHMN VI
型AR 脊髓性肌萎缩症合并呼吸窘迫1 型IGHMBP2-dHMN
VII 型AD 成人起病伴声带麻痹DCTN1TRPV4 未知--2q14X
连锁dHMNX 连锁远端起病的肌萎缩和无力ATP7A-dHMN
合并锥体束征ADdHMN 合并锥体束征SETX*BSCL2 未知未知--
4q34-q357q34-q36 约旦杰拉什地区dHMNAR 起源于约旦杰拉什地区的dHMN 合并锥体束征未知9p21.1-p12 先天性远端脊髓性肌萎缩症AD 出生时远端无力合并关节挛缩
TRPV4-。