Bispecific Antibodies for Cancer Immunotherapy Current Perspectives
新型抗体临床前评价

血病-淋巴瘤和外周T细胞淋巴瘤(PTCL)的成年患者;
2013年11月Genentech的糖基化抗体obinutuzumab
(靶点为CD20)治疗难治性慢性淋巴细胞白血病 ;
Ecromeximab,Benralizumab等。
Amgen的双特异抗体blinatumomab(CD19和
CD3特异性)治疗急性B性(3个 月、 6个月)
种属
大鼠、猴 猴
ADC/游离细胞毒 分子
T-DM1 T-DM1
结果
食蟹猴30 mg/kg未见明显异常 大鼠60 mg/kg肝毒性、免疫毒性、动物死亡等 轻度、可逆的肝毒性(Kupffer细胞肥大、中性粒 细胞浸润、多核肝细胞);表皮细胞有丝分裂象 增多;坐骨神经、脊髓可见不可逆性轴突变性 HNSTD为 10 mg/kg 30 mg/kg无作用
Catumaxomab临床使用主要副作用为发热、疼痛、恶心和 呕吐,主要与细胞因子释放有关。 Blinatumomab说明书黑框警告两个副作用:细胞因子释放 综合征、神经毒性,可危及生命或致死。临床试验中还可 见可见发热、头痛、周围水肿、发热性中性粒细胞减少、 恶心、低血钾、震颤、皮疹和便秘等不良反应
3个月重复给药毒性
小鼠
鼠源性替 代分子 Blincyto
5周重复给药毒性
黑猩猩
组织交叉反应
猴、人
Blincyto
小鼠胚胎-胎仔发育
小鼠
鼠源性替 代分子
鼠源性替 代分子 N/A
0, 1,5 mg/kg/次,GD6和GD19各一次,未见胎仔毒性,母体毒 性与重复给药基本一致
0, 0.042,0.978 mg/kg,每天一次,共7天,未见明显异常 未进行
肿瘤细胞表达的免疫抑制蛋白

肿瘤细胞表达的免疫抑制蛋白
肿瘤细胞可以表达多种免疫抑制蛋白,以下是一些常见的免疫抑制蛋白:
1.PD-L1:程序性死亡受体-1(PD-1)与其配体PD-L1结合,可以抑制T细胞的免疫活性,从
而抑制免疫反应。
2.B7-H4:B7-H4是一种新的B7家族成员,它可以抑制T细胞的免疫应答,促进肿瘤细胞的
免疫逃逸。
3.FasL:FasL是一种细胞因子,它可以结合并激活Fas受体,从而诱导T细胞凋亡,进而抑制
免疫反应。
4.TGF-β:转化生长因子-β(TGF-β)可以抑制T细胞的增殖和免疫应答,同时还可以促进免疫
抑制性T细胞的分化。
5.IL-10:白细胞介素-10(IL-10)是一种抗炎细胞因子,它可以抑制抗原提呈细胞的功能,从
而抑制免疫应答。
这些免疫抑制蛋白的表达可以促进肿瘤细胞的免疫逃逸和进展,因此它们是肿瘤免疫治疗的重要靶点。
ppt-Bispecific antibodies

BiAb Trials
HRS-3/A9: anti-FCRIII x anti-CD20 for HD of B cell malignancy; MTD not reached at 64 mg/m2/dose MDX-H210: anti-CD64 x anti-Her2 for breast, ovarian, prostate CA; doses ranged from1-40 mg/m2 without DLT MDX-447: anti-CD64 x anti-EGFR for renal and head and neck cancer; Hypotension DLT, doses up to 40 mg/m2 H22x Ki-4: anti-CD64 x anti-CD30 for Hodgkin's Disease, doses up to 20 mg/m2 Common thread: deletion of Fc portions improved toxicity profiles
Combination of Cellular and Humoral Therapeutic Strategy
The specificity of monoclonal antibodies AND Non MHC restricted cytotoxicity mediated by T cells, NK cells, or other effector cells
Begin with the “End” In Mind
STRATEGY: Make T Cells better killers by redirecting or focusing their non-MHC restricted cytotoxicity on TAAs
细胞外基质金属蛋白酶诱导因子在肝癌细胞7721中对多药耐药的调节作用

PI RN的人肝癌多药耐药细胞 7 2/ d / N i 7 2/ d / N i 7 1 ml A 和 7 1 m2R A 细胞 。用反转录一 A R A 聚合酶链反应 ( T P R)流 R -C 、 式细胞技术检测 E MMP I 细胞表面多药耐药基 因( R 1mR A及其表达产物的表达水平 。噻唑蓝( r法 RN、 MD 一 ) N MT ) 检测 上述各细胞 的多药耐药性。 结果 2种多药耐药模型可用于肝癌多药耐药研究 。 多药耐药细胞 7 2 /d 和 7 1 ml A 72 / d 7 1 m2中 E A MMP I MD 一 R N, R 1的表达水平较 7 2 7 1细胞均升高 , 且增加了其对 多种化疗药物 的耐药性。 而利用
中国药物与临床 21 年 8 01 月第 1 卷第 8 C i s e ei &Ci c。 uut 0 1 o1,o 1 期 h e Rm de ne s li A gs21, 1 l . ns V. N8
・83 ・ 9
细胞 外基质 金属 蛋 白酶诱导 因子在 肝癌细胞 7 2 7 1中 对 多药耐药 的调节作用
s d h oe o MMP N i e eo me to ld u — eitn e f t y te rl fE u RI n d v lp n fmut rg rss c MDR i e ao aen ma c l . M eh d i a 1 n h p te io e s r2/d 1 72/d 2 Ni M Pd 71 m 和 7 1 m 细胞可致 M R 1 A A D 一 表达降低 ,增加 了其对化疗药物 的敏感性 。
结论 E P I MM R N是参与肝癌细胞 7 2 多药耐药且为调节多药耐药的重要分子。 71
成纤维细胞活化蛋白抑制剂在肿瘤诊疗中的研究进展
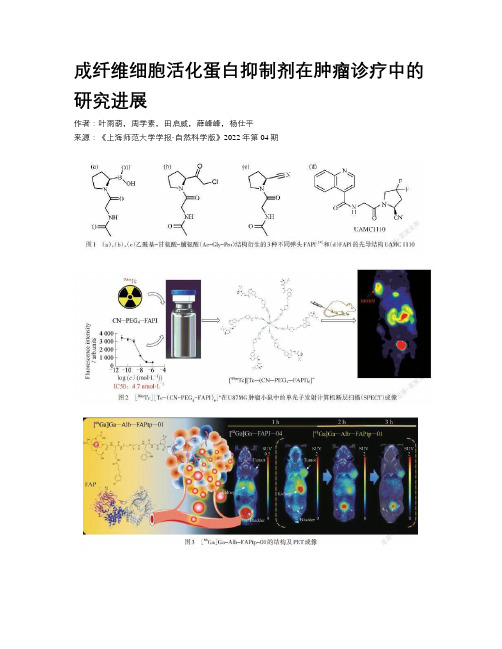
成纤维细胞活化蛋白抑制剂在肿瘤诊疗中的研究进展作者:叶雨萌,周学素,田启威,薛峰峰,杨仕平来源:《上海师范大学学报·自然科学版》2022年第04期摘要:成纤维细胞活化蛋白(FAP)在90%以上的上皮性癌间质中高表达,可以作为肿瘤成像和治疗的靶点.而一些已开发的成纤维细胞活化蛋白抑制剂(FAPI),由于对肿瘤的高亲和力和高肿瘤积聚,对肿瘤的诊断和治疗具有重大意义.文章综述了近年来FAPI在肿瘤诊疗方面的研究进展,重点阐述了新型FAPI在核医学上的诊疗应用,并且从构效关系上讨论了FAPI的靶向弹头结构,增强FAPI选择性及延长肿瘤保留时间的策略,进一步推动了FAPI向临床诊疗试剂转化.关键词:成纤维细胞活化蛋白抑制剂(FAPI); 核医学影像; 放射性治疗; 构效关系中图分类号: R 817.9 文献标志码: A 文章编号: 1000-5137(2022)04-0436-07Progress in fibroblast activation protein inhibitors for cancer diagnosis and treatmentYE Yumeng1, ZHOU Xuesu1, TIAN Qiwei1,2, XUE Fengfeng2, YANG Shiping1*(1. College of Chemistry and Materials Science, Shanghai Normal University, Shanghai 200234, China; 2. Shanghai Key Laboratory of Molecular Imaging, Shanghai University of Medicine and Health Sciences, Shanghai 201318, China)Abstract: Fibroblast activation protein(FAP) is highly expressed in more than 90% of epithelial carcinoma stroma and can be used as a target for tumor imaging and therapy. Some developed FAP inhibitors(FAPI) are of great significance in the diagnosis and treatment of tumors due to their high affinity for tumors and high tumor accumulation. Herein,the research progress of FAPI in tumor diagnosis and treatment in recent years was reviewed,with an emphasis on the clinical application of novel FAPI in nuclear medicine. In addition,FAPI targeting warhead structure and the strategies of enhancing FAPI selectivity and prolonging tumor retention time were discussed from the perspective of structure-activity relationship,which further promoted the transformation of FAPI into clinical diagnosis and treatment reagents.Key words: fibroblast activating protein inhibitors(FAPI); nuclear medical imaging; radiation therapy; structure-activity relationship0 引言癌相关成纤维细胞(CAFs)是一种异质性的成纤维细胞样细胞群,在肿瘤生长、迁移、转移、重构细胞外基质、治疗抵抗和免疫抑制中发挥关键作用,同时也是肿瘤微环境结构中最丰富的一类细胞[1-2].与癌细胞相比,CAFs的基因更稳定,更不易发生治疗耐药性[3-4],是癌症诊断和治疗的理想靶细胞.成纤维细胞活化蛋白(FAP)是一种II型膜结合的丝氨酸蛋白酶[5],在CAFs中过表达,而在健康成人组织中很少表达[6].有数据统计,FAP在90%以上的上皮性癌的间质中过表达[4].而且,在直肠癌、胰腺癌、卵巢癌等恶性肿瘤中,FAP的高表达与肿瘤局部浸润增加、淋巴结转移风险增加和患者生存期下降有关[7].从FAP与肿瘤组织的相关性、调节肿瘤行为的有效性可见,FAP是一个肿瘤靶向诊疗的理想靶点.因此,根据FAP在CAFs中的高表达及自身的蛋白酶特性,研究者们已经开发了一系列成纤维细胞活化蛋白抑制剂(FAPI).FAPI能够选择性地富集在肿瘤组织中,是一种有效的肿瘤靶向试剂,并且结合各种放射性同位素,展现出应用于癌症诊疗的巨大潜能.本文作者总结了近几年FAPI在肿瘤诊疗中的研究进展,重点介绍了新型FAPI在核医学领域肿瘤成像和治疗的应用,并从构效关系上讨论了增强FAPI选择性与延长保留时间的策略.1 FAPI的类型如图1所示,根据FAPI靶向弹头的伪肽结构,FAPI主要可分为下面几种类型:硼酸吡咯类、氯甲基酮类[8]和氰吡咯类.FAPI靶向弹头起抑制作用的机理是:FAPI中可分裂的肽键被不可分裂的亲电基团取代,引起FAP催化三联体中的丝氨酸羟基进行亲核攻击[9].硼酸吡咯类抑制剂由于对与FAP相关的多种脯氨酸肽酶有亲和力,对FAP的特异性受到了限制,并且还存在化学稳定性较低的缺点[10-11].而氰吡咯类抑制剂因为具有低纳摩尔FAP亲和性和高选择性等优异性质,已成为FAPI的主流构型.2014年,一种最有效的FAPI(简称:UAMC 1110)被开发出来,如图1(d)所示,它是一种典型的氰吡咯类抑制剂.目前,氰吡咯类抑制剂中具有代表性的是FAPI-02和FAPI-04,它们在临床实验中展现出高靶向性及高应用价值.另外,已有临床研究证明,相较于传统示踪剂氟代脱氧葡萄糖(FDG),FAPI-04在对各类肿瘤患者原發及转移灶的诊断上效果更优,尤其在肝转移瘤、腹膜癌、脑肿瘤的诊断上[12].FAPI-02和FAPI-04结构相似,两者的唯一区别在于FAPI-04的氰吡咯基团经二氟修饰,这增强了FAPI-04的疏水性,提高了抑制效力、配体效率和成纤维细胞活化蛋白与脯氨酰寡肽酶的比值(FAP/PREP)水平,提高了对FAP的选择性[13].2 增强FAPI选择性与延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 构建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设计了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫生组织(WHO)认定的分级为II级和III/IV级的异柠檬酸脱氢酶(IDH)突变星形细胞瘤的无创区分.虽然当前68Ga标记的FAPI在成像中得到了广泛关注,但由于68Ga的半衰期较短(半衰期(t1/2)为68 min),一次只能合成少量的放射性药物,并且不利于长距离的输送,这限制了68Ga-FAPI在实际医疗的应用.然而,放射性核素18F可以大量生产,并且它的发射器普遍可用,可以满足大量患者的需求.WANG等[19]开发了一种18F标记的铝与1,4,7-三氮杂环壬烷-N,N,N-三乙酸(NOTA)螯合的Al18F-NOTA-FAPI成像探针,可以在人工操作下制备,实现高放射性的批量生产.另外,如图4所示,HU等[20]通过临床研究证明一种18F标记的新型成纤维细胞活化蛋白抑制剂[18F]FAPI-42在各種癌症患者中表现出较高的病变检出率,可以成为68Ga-FAPI-04的替代品.在SPECT影像方面,由于SPECT具有低成本和广泛使用的特点,99 mTc标记的FAPI在实际患者的影像诊断中具有较大的应用潜能.LINDNER等[21]对一系列FAPI进行了99 mTc标记和临床研究,发现FAPI-34是一个优良的SPECT显像剂,可以通过快速肿瘤摄取和身体其他部位的快速清除获得高的对比度.TRUJILLO-BENITEZ等[22]还首次用99 mTc标记了硼酸吡咯类FAPI,结果显示:该示踪剂在人血清中的放射稳定性高,对FAP具有特异性识别,实现在肿瘤的高摄取和在肾脏中的快速清除.3.2 FAPI在肿瘤治疗中的应用目前,基于FAPI特异性靶向肿瘤的作用,许多对癌症具有靶向治疗效果的药物被开发出来.FAPI在肿瘤治疗上的应用大致可分为两类:一类是在FAPI上进行放射性核素标记用于放疗;另一类是通过化学合成在FAPI上偶联化疗药物进行化疗.在放疗上,WATABE等[23]使用半衰期较长的64Cu(t1/2为12.7 h)和225Ac(t1/2为10 d)标记FAPI,研究α-疗法治疗肿瘤的效果,结果表明:放射性铜和锕标记的FAPI-04(64Cu-FAPI-04和225Ac-FAPI-04)可用于治疗高表达FAP的胰腺癌.另一方面,如图5所示,为了研究短半衰期高能量同位素,如铼(188Re)、砹(21At)和铋(213Bi)等用于肿瘤放射治疗的效果,MA等[24]用与21At 化学性质相近的131I标记了FAPI-04,为后续21At放疗试剂的开发铺平道路.治疗结果表明:该种放射药物在胶质瘤近距离放疗中具有巨大的潜力.2 增强FAPI选择性与延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 構建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设计了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫生组织(WHO)认定的分级为II级和III/IV级的异柠檬酸脱氢酶(IDH)突变星形细胞瘤的无创区分.虽然当前68Ga标记的FAPI在成像中得到了广泛关注,但由于68Ga的半衰期较短(半衰期(t1/2)为68 min),一次只能合成少量的放射性药物,并且不利于长距离的输送,这限制了68Ga-FAPI在实际医疗的应用.然而,放射性核素18F可以大量生产,并且它的发射器普遍可用,可以满足大量患者的需求.WANG等[19]开发了一种18F标记的铝与1,4,7-三氮杂环壬烷-N,N,N-三乙酸(NOTA)螯合的Al18F-NOTA-FAPI成像探针,可以在人工操作下制备,实现高放射性的批量生产.另外,如图4所示,HU等[20]通过临床研究证明一种18F标记的新型成纤维细胞活化蛋白抑制剂[18F]FAPI-42在各种癌症患者中表现出较高的病变检出率,可以成为68Ga-FAPI-04的替代品.在SPECT影像方面,由于SPECT具有低成本和广泛使用的特点,99 mTc标记的FAPI在实际患者的影像诊断中具有较大的应用潜能.LINDNER等[21]对一系列FAPI进行了99 mTc标记和临床研究,发现FAPI-34是一个优良的SPECT显像剂,可以通过快速肿瘤摄取和身体其他部位的快速清除获得高的对比度.TRUJILLO-BENITEZ等[22]还首次用99 mTc标记了硼酸吡咯类FAPI,结果显示:该示踪剂在人血清中的放射稳定性高,对FAP具有特异性识别,实现在肿瘤的高摄取和在肾脏中的快速清除.3.2 FAPI在肿瘤治疗中的应用目前,基于FAPI特异性靶向肿瘤的作用,许多对癌症具有靶向治疗效果的药物被开发出来.FAPI在肿瘤治疗上的应用大致可分为两类:一类是在FAPI上进行放射性核素标记用于放疗;另一类是通过化学合成在FAPI上偶联化疗药物进行化疗.在放疗上,WATABE等[23]使用半衰期较长的64Cu(t1/2为12.7 h)和225Ac(t1/2为10 d)标记FAPI,研究α-疗法治疗肿瘤的效果,结果表明:放射性铜和锕标记的FAPI-04(64Cu-FAPI-04和225Ac-FAPI-04)可用于治疗高表达FAP的胰腺癌.另一方面,如图5所示,为了研究短半衰期高能量同位素,如铼(188Re)、砹(21At)和铋(213Bi)等用于肿瘤放射治疗的效果,MA等[24]用与21At 化学性质相近的131I标记了FAPI-04,为后续21At放疗试剂的开发铺平道路.治疗结果表明:该种放射药物在胶质瘤近距离放疗中具有巨大的潜力.2 增强FAPI选择性與延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 构建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设计了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫生组织(WHO)认定的分级为II级和III/IV级的异柠檬酸脱氢酶(IDH)突变星形细胞瘤的无创区分.虽然当前68Ga标记的FAPI在成像中得到了广泛关注,但由于68Ga的半衰期较短(半衰期(t1/2)为68 min),一次只能合成少量的放射性药物,并且不利于长距离的输送,这限制了68Ga-FAPI在实际医疗的应用.然而,放射性核素18F可以大量生产,并且它的发射器普遍可用,可以满足大量患者的需求.WANG等[19]开发了一种18F标记的铝与1,4,7-三氮杂环壬烷-N,N,N-三乙酸(NOTA)螯合的Al18F-NOTA-FAPI成像探针,可以在人工操作下制备,实现高放射性的批量生产.另外,如图4所示,HU等[20]通过临床研究证明一种18F标记的新型成纤维细胞活化蛋白抑制剂[18F]FAPI-42在各种癌症患者中表现出较高的病变检出率,可以成为68Ga-FAPI-04的替代品.在SPECT影像方面,由于SPECT具有低成本和广泛使用的特点,99 mTc标记的FAPI在实际患者的影像诊断中具有较大的应用潜能.LINDNER等[21]对一系列FAPI进行了99 mTc标记和临床研究,发现FAPI-34是一个优良的SPECT显像剂,可以通过快速肿瘤摄取和身体其他部位的快速清除获得高的对比度.TRUJILLO-BENITEZ等[22]还首次用99 mTc标记了硼酸吡咯类FAPI,结果显示:该示踪剂在人血清中的放射稳定性高,对FAP具有特异性识别,实现在肿瘤的高摄取和在肾脏中的快速清除.3.2 FAPI在肿瘤治疗中的应用目前,基于FAPI特异性靶向肿瘤的作用,许多对癌症具有靶向治疗效果的药物被开发出来.FAPI在肿瘤治疗上的应用大致可分为两类:一类是在FAPI上进行放射性核素标记用于放疗;另一类是通过化学合成在FAPI上偶联化疗药物进行化疗.在放疗上,WATABE等[23]使用半衰期较长的64Cu(t1/2为12.7 h)和225Ac(t1/2为10 d)标记FAPI,研究α-疗法治疗肿瘤的效果,结果表明:放射性铜和锕标记的FAPI-04(64Cu-FAPI-04和225Ac-FAPI-04)可用于治疗高表达FAP的胰腺癌.另一方面,如图5所示,为了研究短半衰期高能量同位素,如铼(188Re)、砹(21At)和铋(213Bi)等用于肿瘤放射治疗的效果,MA等[24]用与21At 化学性质相近的131I标记了FAPI-04,为后续21At放疗试剂的开发铺平道路.治疗结果表明:该种放射药物在胶质瘤近距离放疗中具有巨大的潜力.2 增强FAPI选择性与延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 构建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设計了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫。
比利时科学家发现遏制肿瘤生长新抗体

MI' , 察不 同化 疗药 物 ( r 法 观 T I 阿霉 素 、 春 新碱 、 叶 长 足
乙甙) 在不 同浓 度 下 以及 联 合应 用 异 搏 定 , 胶 质瘤 对
细胞 U 5 存 活度 的影 响 , 21 以推 测 化 疗药 物 的不 同剂
量 以及联 合用 药 与 胶 质瘤 化 疗 敏 感 性 的 关 系 。结 果 发 现 , 质瘤 细胞 U 5 胶 2 1的存 活 率 随 着 化疗 剂 量 增 加 而 减少 , 并且 异搏 定 对 化疗 药 有 一 定 的协 同作 用 , 证 明化疗 敏感 性与化 疗 药 物 的剂 量 及 联 合使 用 存 在 一
维普资讯
・
论
善 ・
J eRsa2 8 o3 N. d eJ 0, 17 o M ,n0 V. 1
不 明显 。足 叶 乙甙 药物 加 入异搏 定 后 , 细胞存 活 率无
明显 变化 。
加 入异搏 定 后 , 阿霉 素低 浓度 区域 细胞 存 活率 在 有 明显 下 降。见 表 6及 第 1 9页 彩 图 4 2 。长 春 新 碱 低 浓度 区域 , 加入 异搏 定后 , 细胞 存 活率 也有 下 降 , 但
活 度减 少 的程度 并不 成正 比 , 阿霉 素 的浓 度 在 0 3 如 .
~
3 祁 红 , 红 专 , 菊妹 , . 昔 帕 明 单用 和 合 用 替 尼 泊 苷 对 大 鼠脑 陈 冯 等 地 胶质瘤 c 6细胞 增 生 的 调 控 作 用 . 国 癌 症 杂 志 ,00,1 1 :2 中 20 0( ) 6
比利 时 勒 芬 大 学 医院 的研 究 人 员 最 近发 现 了 遏 制 肿 瘤 生 长 的 新 抗 体 , 种 抗 体 的优 点 是 不 良反应 小 , 可 适 用 于 孕 妇 、 童 这 将 儿 或 非 常 虚 弱 的癌 症 患 者 。研 究 人 员 在 《 细胞 》 志 上 报 告 说 , 杂 目前 临 床 使 用 的抗 癌 药 物 大 多 针 对 血 管 内皮 细 胞 生 长 因 子 , 类 药 这
蓖麻毒蛋白免疫毒素的制备及其体外靶向抗肿瘤效应

蓖麻毒蛋白免疫毒素的制备及其体外靶向抗肿瘤效应陈海霞,陈祥胜△,黄婧,祝香芝(湖北中医药大学,湖北武汉 430065)[摘要] 目的制备蓖麻毒蛋白(Ricin)A链(RTA)与转铁蛋白受体(TFR/CD71)mAb偶联的免疫毒素(IT),并考察其体外靶向抗肿瘤活性。
方法以蓖麻子为原料,提纯RTA,在偶联剂N-琥珀酰胺-3-(2-吡啶二硫)丙酸酯(SPDP)的作用下,构建IT;并以间接ELISA法检测IT的免疫反应性,以CCK-8法测定并比较IT在体外对人肝癌SMMC-7721细胞和正常肝细胞LO2的细胞毒效应。
结果 SPDP偶联法制备的IT对肿瘤细胞的免疫反应性无明显变化,体外实验结果显示,各浓度IT 对SMMC-7721细胞的杀伤作用较LO2细胞差异具有统计学意义(P<0.05)。
结论 CD71 mAb-RTA免疫反应性稳定,对肿瘤和正常体细胞的体外细胞毒效应差异明显,提示其对肿瘤细胞有较好的靶向杀伤作用,有望开发成为肿瘤的靶向治疗药物。
[关键词]免疫毒素;蓖麻毒蛋白;CD71;恶性肿瘤;靶向治疗[中图分类号] R735.7 [文献标识码] APreparation of Ricin immunotoxin and studying its targeted therapyanti-tumor effect in vitroCHEN Hai-xia, CHEN Xiang-sheng△, HUANG Jing, ZHU Xiang-zhi(Hubei University of Traditional Chinese Medicine, Wuhan 430065, China)[Abstract]Objective To prepare the immune toxin (IT) of Ricin A chain (RTA) and transferrin receptor (TFR/CD71) mAb coupling, and to investigate the anti-tumor activity in vitro. Methods IT was constructed by using castor beans as raw materials, purified RTA, under the action of the coupling agent N - succinamide-3-(2-pyridine disulfide) propionate (SPDP). In order to detect the immune reactivity of IT in the indirect ELISA method, the cck-8 method was used to determine and compare the cytotoxic effects of IT in vitro for human liver cancer cell SMMC -7721 and normal hepatocellular LO2. Results SPDP coupling method of IT there was no significant alteration of the immune response to tumor cells, in vitro experimental results showed that the concentration IT on LO2 cells SMMC-7721 cell killing effect significantly enhanced (P<0.05). Conclusion CD71 mAb - RTA immune reactivity is stable, and there was obvious difference betweem the tumor and normal cells in vitro cytotoxic effect. It has better targeted killing effect on tumor cells,and is expected to develop into tumor targeted therapy drugs.[Keywords] immunotoxin; Ricin; CD71; malignant tumor; targeted therapy资助项目:湖北省卫计委资助项目(WJ2015MB132);湖北省教育厅科研项目(B2013129)作者简介:陈海霞,女,硕士,主管中药師,研究方向:药物,E-mail:598121112@;陈祥胜,通信作者,男,硕士,副教授,研究方向:生物药物,E-mail:515887025@。
治疗晚期癌症的新免疫合剂

任意 的作 为 神 经化肥 应 用 途径
“
可 能 不是 最 佳 的
正 在 几 个 医 学 中 心 进行 的 I 期 试 验 发 现 睫 状 体 的 神 经 营 养 因 子 可 以减 慢 肌 萎 缩 性 脊 髓 侧
.
韩 秀厦 译 白 M W N 3 4 ( 1 1 )
.
13
No
v
19 93
敏江校
索硬 化 的 进展
马 萨诸 塞 大 学 医 学 中 心 神 经 病
一
5
一
新 研 制 的 癌 疫 苗
.
美 国消 息 次给 晚期 结 肠 制 的癌疫 苗 隔
3
、
匹 兹 堡 癌症 研 究所
首
所 具有 的免 疫 反 应
胰腺 给
、
乳腺 癌症 患者 服用 新研
例 无 望 治愈 的患 者
,
粘 蛋 白疫苗存 在 于 浓缩的 粘蛋 白肤 中 添加 吸 引免 疫细 胞 的佐剂 后制 成 的 免 疫 细 胞 与 粘 蛋 白 相 遇 的机 0 会 究.ຫໍສະໝຸດ 宿 主 体 内后 再 分裂
尽 管在美 国
s
t
e v e n
特别是 在 国 立 癌症 研究 所 的
g 的方 案 中
,
疫反 应 来 抵 抗 无 法 控 制 的 晚 期 癌 症
匹 兹堡 医学 院 外 科 医师
R
o s e n
b
,
e
r
正在 应 用其 他 的
Jo s h
u a
R
u
b i
n
J L种 肿 瘤 疫 苗
`
4
病 人 背部下 方一 侧的 5 个 部位
7
最 大剂 量 达到
重组抗体药物

重组抗体药物抗体药因其靶向性强、副作用小等显著特点已经成为药物研发的热点,由于传统单抗容易诱发人抗鼠抗体反应(Human anti mouse antibody, HAMA),近年来逐渐兴起了通过基因克隆以及DNA重组技术将鼠源单克隆抗体进行人源化的重组抗体技术1。
目前已重组抗体药物已经在肿瘤的治疗中取得了显著的疗效,迅速占据市场。
重组抗体药物研发的关键是要保持和提高抗体的特异性与亲和性,同时降低抗体的免疫原性,从而提高抗体的作用效果。
目前这类治疗性抗体主要包括以下几种类型2:1) 抗体依赖的细胞介导细胞毒性作用(antibody-dependent cell-mediated cytotoxicity, ADCC)增强效应的抗体药物; 2)抗体-药物偶联物(Antibody-drug conjugate, ADC);3)双靶点特异性抗体(Dual targeting bispecific antibody, BsAb)。
ADCC是抗体类药物杀伤癌细胞的主要机制之一,近年来研究者们通过重组抗体技术对抗体进行修饰以提高ADCC活性已经取得了长足的进展。
EGFR是肿瘤治疗中一个常见的靶点,研究者已经通过对EGFR单抗进行糖基化修饰来增强其ADCC活性,并证明在针对EGFR阳性的实体瘤的治疗过程中,这种修饰的抗体比已上市的西妥昔单抗(cetuximab)有更强的激活ADCC的能力3。
EphA3是实体瘤与白血病中都表达的一个非常有潜力的治疗靶点,研究者从Fab抗体库中筛选出与EphA3亲和力极高的片段,并将其与人源的IgG1k保守区链接形成重组抗体,检测发现这种重组抗体针对EphA3阳性白血病细胞有很强的ADCC活性4。
系统性肥大细胞增生症(Systemic mastocytosis, SM)是一种极为罕见的骨髓增生性肿瘤,在成熟的肥大细胞表面有Siglec-8的表达,研究者利用重组抗体技术构建了一种IgG1类型的Siglec-8抗体,并发现这种抗体对SM病人来源的肥大细胞有ADCC活性5。
BIRC5在肝癌中的表达及其表达与临床病理特征的关系

BIRC5在肝癌中的表达及其表达与临床病理特征的关系背景BIRC5(baculoviral inhibitor of apoptosis repeat-containing 5)是一种抑制凋亡的蛋白。
它有着广泛的表达和功能分化,能够在多种细胞类型中抑制细胞凋亡,包括癌细胞。
已有研究表明,在肝癌中BIRC5的高表达与肝癌的发生、发展、转移和恶化密切相关。
此外,BIRC5还与肝癌的临床表现有关。
BIRC5在肝癌中的表达BIRC5在肝癌中的表达呈现多样性。
一些研究发现,BIRC5在肝癌组织中的表达显著高于正常肝组织。
例如,Cui et al.(2013)对160例肝癌患者进行了BIRC5的免疫组化检测,发现肝癌组织中BIRC5阳性率为68.1%,而正常肝组织中BIRC5阳性率仅为5%。
此外,Wang et al.(2019)的研究表明,BIRC5在肝癌组织中的表达水平与肝癌的组织学分级、血管浸润和肿瘤大小呈正相关。
然而,也有研究发现,BIRC5在肝癌中的表达并不一定高于正常肝组织。
例如,张云等人对22例肝癌患者进行了BIRC5的实时荧光定量PCR检测,发现BIRC5在19例(86.4%)肝癌组织中表达下降,BIRC5表达与肝癌组织分级、肿瘤径和临床病理分期没有相关性。
这一研究结果提示了BIRC5在肝癌发生、发展和转移中可能起更为复杂的作用。
BIRC5表达与肝癌的临床病理特征关系BIRC5的表达与肝癌的临床病理特征是有关联的。
一些研究表明,BIRC5在肝癌中的高表达与肝癌患者的预后较差有关。
例如,Villanueva等(2006)的研究表明,BIRC5的高表达与肝癌患者的肿瘤组织学分级和预后密切相关。
他们对198例肝癌患者进行分析,结果表明,BIRC5在肝癌组织中的阳性率与肿瘤组织学分级呈正相关,BIRC5在肝癌组织中的高表达与肝癌患者生存期缩短密切相关。
此外,BIRC5的表达还与肝癌的侵袭、转移、血管浸润等病理参数有关。
以牛血清白蛋白为中间载体的血卟啉衍生物与抗胃癌单克隆抗体交联物的抗肿瘤作用

以牛血清白蛋白为中间载体的血卟啉衍生物与抗胃癌单克隆抗
体交联物的抗肿瘤作用
张永健;王耐勤;刘彤;董志伟
【期刊名称】《药学学报》
【年(卷),期】1990(25)12
【摘要】以牛血清白蛋白为中间载体,将血卟啉衍生物(HPD)与抗胃癌单克隆抗体3H11交联。
交联物3H11-BSA-HPD的克分子比为1:1:200。
本文对3H11-BAS-HPD的体内外抗肿瘤作用进行了研究,并与直接交联物3H11-HPD进行比较。
3H11-BSA-HPD和3H11-HPD对胃癌靶细胞BGC-823的细胞毒效应相似,并均明显比游离HPD强。
在接种靶细胞(2×10~5细胞/只)的裸鼠中,对照组和HPD组均于接种后13天内形成瘤块,而间接和直接交联物处理组在34天实验期内仅有
1/6动物形成肿瘤。
结果表明3H11-BSA-HPD和3H11-HPD在HPD相等剂量下,具有相似的导向杀伤肿瘤细胞的作用。
【总页数】5页(P886-890)
【关键词】单克隆抗体;血卟啉衍生物;抗肿瘤药
【作者】张永健;王耐勤;刘彤;董志伟
【作者单位】北京市肿瘤防治研究所
【正文语种】中文
【中图分类】R979.12
【相关文献】
1.单克隆重抗体与血卟啉衍生物交联物抗胃癌作用的实验研究 [J], ;
2.抗胃癌单克隆抗体与血卟啉衍生物的交联物... [J], 杨丽;王耐勒
3.单克隆抗体与血卟啉衍生物交联物对胃癌杀伤效应的实验研究 [J], 林克
4.单克隆抗体与血卟啉衍生物交联物的制备及其对胃癌靶细胞的杀伤… [J], 张长弓;陈瑞川
因版权原因,仅展示原文概要,查看原文内容请购买。
《2024年新型砷代谢产物联合隐丹参酮对人乳腺癌MCF-7细胞凋亡的作用研究》范文

《新型砷代谢产物联合隐丹参酮对人乳腺癌MCF-7细胞凋亡的作用研究》篇一一、引言乳腺癌是全球女性最常见的恶性肿瘤之一,其发病率逐年上升,严重威胁着女性的生命健康。
目前,尽管有手术、化疗和放疗等多种治疗方法,但乳腺癌的复发率和死亡率仍然较高。
因此,寻找新的治疗方法和药物,以提高治疗效果和降低复发率,成为当前乳腺癌研究的重点。
近年来,新型砷代谢产物和隐丹参酮在抗肿瘤方面的研究逐渐受到关注。
本研究旨在探讨新型砷代谢产物联合隐丹参酮对人乳腺癌MCF-7细胞凋亡的作用及机制。
二、材料与方法1. 材料新型砷代谢产物、隐丹参酮、人乳腺癌MCF-7细胞株、DMEM培养基、胎牛血清、胰酶等。
2. 方法(1)细胞培养:将人乳腺癌MCF-7细胞株置于DMEM培养基中,加入10%胎牛血清,在37℃、5%CO2的条件下培养。
(2)药物处理:将新型砷代谢产物和隐丹参酮按照不同浓度梯度处理MCF-7细胞,观察细胞形态变化。
(3)MTT法检测细胞增殖:采用MTT法检测不同浓度药物处理后MCF-7细胞的增殖情况。
(4)流式细胞术检测细胞凋亡:采用流式细胞术检测细胞凋亡情况,分析新型砷代谢产物联合隐丹参酮对MCF-7细胞凋亡的影响。
(5)Western blot检测相关蛋白表达:采用Western blot检测相关凋亡蛋白的表达情况,探讨其作用机制。
三、结果1. 细胞形态变化新型砷代谢产物和隐丹参酮单独或联合处理MCF-7细胞后,细胞形态发生明显变化,表现为细胞体积缩小、胞质浓缩、核碎裂等凋亡特征。
2. 细胞增殖情况MTT法检测结果显示,新型砷代谢产物和隐丹参酮单独或联合处理MCF-7细胞后,细胞增殖受到抑制,且呈浓度依赖性。
3. 细胞凋亡情况流式细胞术检测结果显示,新型砷代谢产物联合隐丹参酮处理MCF-7细胞后,细胞凋亡率明显升高,且呈时间依赖性。
4. 相关蛋白表达情况Western blot检测结果显示,新型砷代谢产物联合隐丹参酮处理MCF-7细胞后,凋亡相关蛋白Bax、Caspase-3等表达上调,而抗凋亡蛋白Bcl-2表达下调。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
90
Müller & Kontermann
antigen [CEA]) were selected.f'-' Although the principle and the targets on effector and tumor cells have remained fundamentally the same over the time, the format of the bispecific antibody has varied considerably, influenced by the technology progress and guided by the clinical requirements. Thus, the first generation of bispecific antibodies was created by hybridization of two antibody-producing cells, or by chemical cross-linking of two different monoclonal antibodies or fragments thereof. The development of recombinant antibody technology then allowed the design of the second generation, which consists of several smaller formats where the antibody is essentially reduced to the domains involved directly in the antigen binding. The main problems of the first generation, consisting of low efficacy and severe adverse effects such as cytokine storm syndrome, could be ascribed to immunogenieity and Fc-mediated effects, whieh were addressed and improved by the next generation.'^-*'
Current Perspectives
Dafne Müller and Roland E. Kontermann
Institut für Zellbiologie und Immunologie, Universität Stuttgart, Stuttgart, Germany
Contents
Abstract 1. Recombinant Bispecific Antibody Formats 2. Trifunctionai Antibodies for Cancer immunotherapy 3. Bispecific T-Ceii Enganger for Concer immunotherapy 4. Other Formats for T-Celi fîetargeting 5. Dual Targeting Strategies 6. Bispecific Antibodies for Pre-Targeting Radiotherapy 7. Targeting of Tumor Necrosis Factor for improved Radiotherapy 8. Haif-Life Extension Strategies 9. Conciusions 89 90 91 93 93 94 94 95 95 96
the most potent effector cells of the immune system, which cannot be activated by the effector mechanism of conventional therapeutic monoclonal antibodies. In this context, CD3 has become widely accepted as the appropriate trigger molecule. Nevertheless, retargeting of other effector cells such as natural killer cells, macrophages, and neutrophils have also been on trial, where the Fc receptors FcyRIII (CD16), FcyRI (CD64). and FcaR (CD89) were chosen accordingly as trigger molecules. On the side ofthe tumor cell, established tumor-associated antigens of hématologie malignancies (e.g. CD19, CD20, CD30) and solid tumors (e.g. epidermal growth factor receptor [EGFR], human epidermal growth factor receptor 2 [HER2/ERBB2/neu], epithelial cell adhesion molecule [EPCAM], cardnoembryonic
REVIEW ARTICLE
Bloörußs 2010. 24 (2)\ 8 5 ^ 6 1173-88tM/10/0002-0389/549 <?b/0 ® 2010 Adh Data Intofmotlon BV. A< rtghtt reaarved.
Bispecific Antibodies for Cancer Immunotherapy
assemble into different immunoglobulins, including nonfunctional, monospecific, but also bispecific antibodies. These bispeciilc antibodies resemble more or less normal IgGs, depending on the heavy chain subclass. For example, fusion of two hybridomas, one mouse and the other rat, as utilized by trifunctional triomabs (see section 2), results in hybrid bispeeific IgGs possessing one mouse y2a heavy chain and one rat 72b heavy chainj^^l Alternatively, bispecific antibodies can be produced from purified antibodies by chemical conjugation of whole immunoglobulins or their antigen-binding (Fab') fragments (figure 1). A plethora of different recombinant bispecific antibody formats have been developed during the last two decades.''-' These formats include IgG-Hke molecules utilizing the Fc region for dimerization. but also small recombinant formats such as tandem single-chain variable fragment (scFv) molecules (taFv), diabodies (Db), single-chain Db (scDb), and various derivatives thereof^^' (figure 1). Fc-mediated dimerization is often used to produce bispecific tetravalent molecules, i.e. molecules possessing two binding sites for each antigen. This can be achieved, for example, by fusing a single-chain Fv fragment to the C-terminus of an antibody heavy chain or substituting the Fab arm with a bispecific single-chain antibody
The application of bispecific antibodies in cancer immunotherapy is based mainly on the concept of retargeting effector cells to tumor cells, but new strategies have emerged recently, for example, the recruitment of effector molecules.t'l Effector cell retargeting is achieved by simultaneous binding of the bispecific molecule to a tumor antigen on the target cell and a trigger molecule on the effector cell, leading to effective effector cell activation and ultimately to tumor cell destruction. Thus, the redirection of an immune response to the tumor site, bypassing Fc-mediated or major histocompatibility complex (MHC)-restricted activation of effector cells, is achieved. Among the effector cells, cytotoxic T cells have been mostly employed for this approach, considering that they constitute