SNP芯片数据分析
snp基因芯片原理

snp基因芯片原理SNP基因芯片原理引言:随着基因组学和生物技术的快速发展,人类对于基因及其与疾病关联性的研究也越来越深入。
SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是一种常见的基因变异形式,它在人类遗传变异中占据重要地位。
为了研究SNP与疾病之间的关系,科学家们开发了SNP基因芯片,它是一种高通量、高灵敏度的分子生物学工具。
本文将详细介绍SNP基因芯片的原理以及应用。
一、SNP基因芯片的定义及分类SNP基因芯片是一种利用高通量平行杂交技术检测SNP位点的工具。
根据其设计原理和应用领域的不同,SNP基因芯片可以分为两类,即基于探针的SNP芯片和基于测序的SNP芯片。
1. 基于探针的SNP芯片基于探针的SNP芯片利用DNA探针与待测样品中的基因组DNA 序列特异性杂交的原理,通过检测杂交信号来识别不同基因型。
这种芯片设计简单、成本较低,适用于小规模的SNP检测。
2. 基于测序的SNP芯片基于测序的SNP芯片采用高通量测序技术,可以直接测定待测样品中的SNP位点。
这种芯片设计复杂、成本较高,但可以同时检测数百万个SNP位点,具有更高的灵敏度和准确性。
二、SNP基因芯片的工作原理SNP基因芯片的工作原理主要包括芯片设计、杂交反应、信号检测和数据分析四个步骤。
1. 芯片设计芯片设计是SNP基因芯片的关键步骤。
首先,需要确定待测SNP 位点的基因型信息,包括目标基因型和野生型等。
然后,根据基因型信息设计一组特异性的DNA探针,这些探针可以与待测样品中的目标SNP位点特异性杂交。
2. 杂交反应杂交反应是SNP基因芯片的核心步骤。
将待测样品中的DNA与芯片上的DNA探针进行杂交反应,使其结合形成DNA双链。
杂交反应的条件包括温度、时间和缓冲液成分等,需要根据具体实验要求进行优化。
3. 信号检测信号检测是SNP基因芯片的关键步骤。
通过荧光染料或放射性同位素等标记探针,使其与芯片上的杂交DNA结合,形成信号。
SNP芯片数据分析

Affymetrix SNP芯片数据分析方案项目一、基本分析包括:芯片原始数据的处理和基因分型,我们给出有统计意义的SNP列表。
描述性统计,如minor allele frequency,Hardy-Weinberg equilibrium等。
显著性检验,实验组与对照组的差异,假阳性率(FDR)的计算等。
SNP的关联分析,建立线性模型或logistic回归模型等。
(所有的统计可以选择由SAS,SPSS,或S-Plus/R给出)项目二、Copy Number Variation(CNV)的计算。
CNV是目前的一个热点研究内容。
SNP芯片数据可以用于精确地计算CNV。
我们提供针对SNP芯片的基于CNAG(Copy Number Analyser for GeneChip), dChip(DNA-Chip Analyzer)和CNAT(Chromosome Copy Number Analysis Tool)等算法的CNV计算结果。
项目三、SNP注释通过SNP在染色体上的位置,利用寻找SNP可能影响的基因( or EST)。
我们也可以对相应基因进行功能的注释(gene ontology ,pathway和转录因子分析等),进而解释SNP可能的作用机理。
该部分可以参考常规表达谱芯片的分析。
项目四:基于模式识别的SNP挖掘传统的SNP挖掘使用统计学的方法来进行,往往在敏感性与特异性上有一定的限制。
利用一些模式识别/机器学习的方法可以更好解决SNP筛选问题。
我们提供基于决策树等SNP挖掘算法。
Hsiang-Yu Yuan et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Research 2006 34(Web Server issue):W635-W641项目五:诊断模型建立利用筛选到的SNP建立人工神经网络(ANN)、SVM、PAML等诊断模型,在临床上具有重要意义。
#分析流程#SNP芯片分析流程简析
#分析流程#SNP芯片分析流程简析许多的这些单核苷酸多态性可引起不同的遗传性状,即遗传的多态性(polymorphism),如ABO血型位点标记,白细胞HLA位点标记和个体药物代谢差异等。
了解这些DNA序列的差异和单核苷酸多态性以及这些差异所表现的意义将疾病的预测、诊断、预后和预防带来革命性的变化。
现已发现的单核苷酸多态性在人类基因组上就已经达到了三千万以上。
SNP分析无论是对于疾病的诊治、药物的开发还是物种群体的进化都具有十分重要的意义。
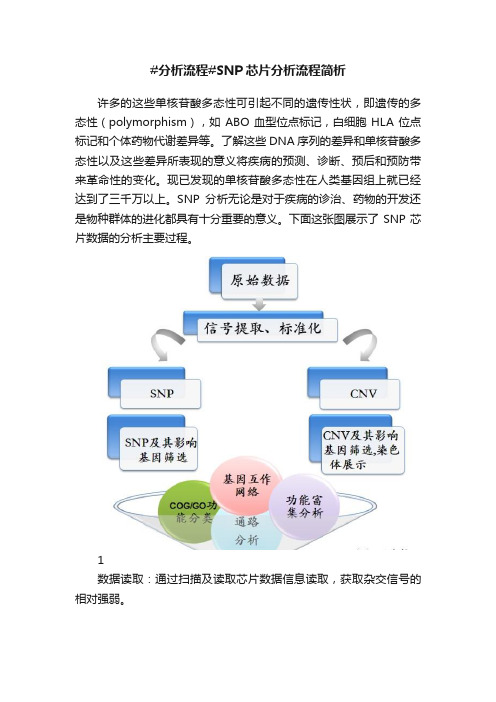
下面这张图展示了SNP芯片数据的分析主要过程。
1数据读取:通过扫描及读取芯片数据信息读取,获取杂交信号的相对强弱。
2数据标准化:数据标准化旨在除去数据中所包含的非生物学变化,这些变化可能来自实验的任何一步,包括芯片制作、RNA提纯、cDNA标记、DNA杂交、或者芯片扫描。
3SNP分析:通过信号强度,标准化的等位基因强度比,标记之前的距离和B等位基因的群体频率等参数推断SNP数目和特定染色体的区域,给出有统计意义的SNP列表。
4CNV/LOH分析:展示CNV在染色体上的分布情况及目标基因上所有CNV的分布及类型等位基因处于杂合时,会出现丢失或突变成另一个基因的趋势,称为杂合性缺失(LOH)。
杂合性缺失在肿瘤中是一种非常常见的DNA变异。
长期的细胞遗传学的研究证实,几乎所有的肿瘤细胞都存在染色体片断的非随机性丢失。
而基因拷贝数的变化,往往与神经功能、细胞生长的调节、新陈代谢以及某些疾病有关。
5COG/KOG功能注释及分类分析:对基因功能进行COG或KOG 分类,通过COG分类可以对变化基因所调节的功能有直观和感性的认识,从而了解待研究因子对于生物功能的影响,并对后续生物学实验的进行提供指导作用。
6GO Enrichment:对于得到的特定基因分类,采取DAVID、EasyGO等GO分析软件对所得基因进行功能富集分析,并得到可能的富集功能。
7KEGG Pathway分析:基于KEGG等数据库,采取超几何分布检验等统计手段,得到显著富集的生物信号通路或者代谢通路。
基因组学中的SNP分析

基因组学中的SNP分析SNP(Single Nucleotide Polymorphism)是指基因组中的单个核苷酸突变。
SNP分析是基因组学研究中的重要分析方法之一,为了更好地了解SNP分析在基因组学中的作用,我们需要从以下几个方面进行逐步的了解。
一、SNP的特征SNP是常见的继承性遗传变异,主要发生在基因组中7-10%的位置。
它具备许多有价值的特征,例如高度多态性、共有性基因性和容易鉴定性等。
SNP的多态性使其成为研究人类及其他物种遗传标记的优良素材。
SNP基于其出现的频率可以分为高频和低频。
高频SNP在人类人群中具有普遍性,低频SNP在某些群体中出现的频率很低。
SNP在基因组中的位置也非常有规律,即位于编码区、非编码区、隐形区,以及转录因子结合区等重要区域中。
二、SNP分析的方法SNP分析的方法根据分析的目的和数据场景不同,可以分为不同的方法。
常见的SNP分析技术包括测序分析、芯片分析和PCR分析等。
测序分析是快速发展的分析技术,包括全基因组测序和目标基因测序两种。
芯片分析是目前应用比较广泛的SNP分析技术,可快速、准确地进行大规模的SNP检测。
PCR分析适用于单个SNP的检测和测序后验证,具有快速、灵敏度高、操作简单等优点。
三、SNP分析的应用SNP分析在基因组学中的应用非常广泛,主要应用于以下几个方面:1、研究遗传多样性SNP在人群中的频率不同,可以用于描述人类、动植物的遗传多样性,推断人类或种群的出现时间及演化过程等。
2、研究遗传病理学SNP分析也可用于研究不同类型的疾病和病态的发生机制,便于快速准确地识别和分析疾病易感性基因。
3、研究药理学SNP分析也可以帮助研究药物代谢方面的基因,寻找药物作用机制、筛选新药等。
4、研究育种学SNP不仅可应用于人类、动植物的遗传多样性研究中,还可以帮助育种与遗传改良中研究重要基因资源。
四、SNP分析的未来SNP分析虽然已经在基因组学研究中得到了广泛的应用,但随着科技的不断进步,SNP分析的应用范围将会更广泛。
SNP分析命令范文

SNP分析命令范文SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是一种常见的基因变异形式,它在基因组中的单个核苷酸位置上出现了多个可能的碱基。
SNP分析是研究和鉴定SNP在个体或种群中的分布和相互关系的方法。
对于研究人类和其他生物种群的基因变异和相关性,SNP分析被广泛应用于基因组学、进化生物学、人类遗传学和相关疾病的研究。
1.样本准备:首先需要准备好所需样本,并提取其中的DNA。
样本可以是血液、组织、唾液等。
DNA提取可以使用各种商用DNA提取试剂盒或标准的有机/无机方法。
2. Genotyping(基因分型):SNP分析的第一步是进行基因型(基因组型)鉴定,确定样本中每个SNP位点上的碱基。
常见的基因分型方法包括PCR-RFLP(聚合酶链反应-限制性片段长度多态性)、TaqMan探针分型、SNP芯片分析和高通量测序等。
3.数据处理和分析:获得基因型数据后,需要进行数据处理和分析。
常见的数据处理包括质量控制筛选、错误纠正和填充缺失值等。
数据分析可以使用各种统计学和生物信息学方法来研究SNP在个体或种群中的频率、关联性和相关性等。
常用的分析方法包括关联分析、群体结构分析、遗传多态性评估等。
4.功能注释:SNP是可能会对基因功能产生影响的遗传变异。
因此,在SNP分析中,经常需要对鉴定的SNP进行功能注释。
这使得我们可以了解SNP是否位于编码区、非编码区、转录因子结合位点等,从而评估其对基因功能的影响。
5.生物特征和关联研究:SNP的分析还可以用于研究SNP与个体生理特征、疾病易感性、药物反应等之间的关联。
通过比较不同个体之间的SNP分布,我们可以发现与特定生理特征或疾病相关的SNP。
1.PLINK:一款常用的用于执行SNP数据管理和基因关联分析的软件。
可以用于数据质量控制、基因型质量控制、关联性分析、基因型-表型关联等。
2. GATK (Genome Analysis Toolkit):是一款用于基因组数据分析的强大软件,包括对SNP和INDEL的鉴定与拼接、变异注释等。
SNP芯片介绍之SNP
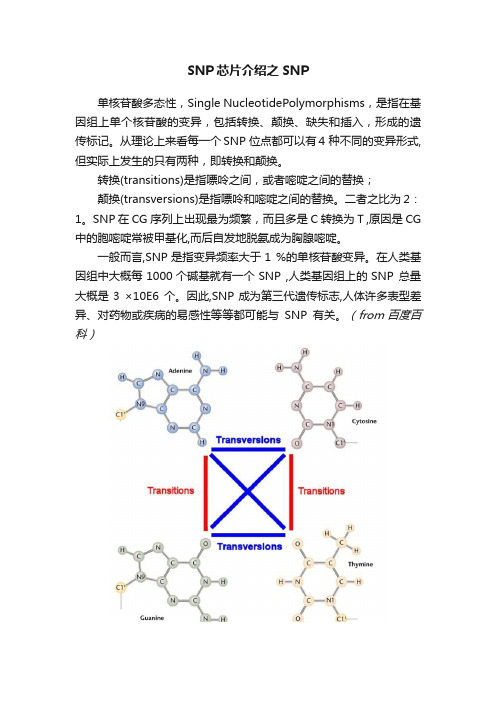
SNP芯片介绍之SNP单核苷酸多态性,Single NucleotidePolymorphisms,是指在基因组上单个核苷酸的变异,包括转换、颠换、缺失和插入,形成的遗传标记。
从理论上来看每一个SNP 位点都可以有4 种不同的变异形式,但实际上发生的只有两种,即转换和颠换。
转换(transitions)是指嘌呤之间,或者嘧啶之间的替换;颠换(transversions)是指嘌呤和嘧啶之间的替换。
二者之比为2:1。
SNP 在CG序列上出现最为频繁,而且多是C转换为T ,原因是CG 中的胞嘧啶常被甲基化,而后自发地脱氨成为胸腺嘧啶。
一般而言,SNP 是指变异频率大于1 %的单核苷酸变异。
在人类基因组中大概每1000 个碱基就有一个SNP ,人类基因组上的SNP 总量大概是3 ×10E6 个。
因此,SNP成为第三代遗传标志,人体许多表型差异、对药物或疾病的易感性等等都可能与SNP有关。
(from 百度百科)图 1. SNP 转换和颠换SNP研究意义1、SNP具有已知性、可遗传性、可检测性,可用于疾病基因的定位、克隆和鉴定,寻找疾病相关的突变位点。
2、SNP构成的个体差异的遗传基础,可用于疾病易感性研究、个性化医疗等。
3、器官移植中供体/受体配对分析。
预测启动子SNPs位点与转录因子结合的编码去SNPs对蛋白质的空间结构,生物功能的影响等。
SNP常见数据库HapMap (/):International project involving scientistsand fundingagencies from Japan, the United Kingdom, Canada, China, Nigeria,and the UnitedStates. It is the reference database in many SNP studiesdbSNP(/projects/SNP/) :The largest SNP database and ishosted at the National Center for Biotechnology Information。
基于SNP芯片数据分析不同奶牛场基因组近交系数及筛选功能性基因

畜牧兽医学报 2023,54(7):2848-2857A c t a V e t e r i n a r i a e t Z o o t e c h n i c a S i n i c ad o i :10.11843/j.i s s n .0366-6964.2023.07.017开放科学(资源服务)标识码(O S I D ):基于S N P 芯片数据分析不同奶牛场基因组近交系数及筛选功能性基因王振宇1,张赛博1,刘文慧1,梁 栋1,任小丽2,闫 磊2,闫跃飞2,高腾云1,张 震2,3*,黄河天1*(1.河南农业大学动物科技学院,郑州450046;2.河南省奶牛生产性能测定中心,郑州450045;3.河南省种业发展中心,郑州450046)摘 要:旨在利用基因组长纯合片段(r u n s o f h o m o z y g o s i t y,R OH )信息评估河南省不同中国荷斯坦牛群体的全基因组近交水平,并通过R OH 检测鉴定基因组R OH 富集区域,筛选与奶牛经济性状相关的候选基因㊂本研究基于G G P B o v i n e 150K 芯片对来自河南省7个牧场900头荷斯坦牛进行全基因组R OH 检测,统计R OH 在荷斯坦群体中的数目㊁长度及频率,根据R OH 计算基因组近交系数(F R O H ),并对高频R OH 区域进行基因注释㊂结果表明,在全部900个体中共检测出55908个R OH 片段,平均长度4.23M b ㊂7个牧场平均近交系数(F R O H )的变化范围从0.082(H 7)到0.123(H 2),平均F R O H 为0.106㊂在R OH 的高频区域内共鉴定到79个与奶牛经济性状相关的基因,如与牛体型㊁体高有关的基因A K A P 3㊁C 5H 12o r f 4㊁F G F 6,与胴体及繁殖性状相关的基因C A P N 3,与妊娠维持和胎儿生长直接相关的基因C H S T 14,影响牛奶蛋白质组成的基因I L 5R A ,参与调节胎儿卵泡生成的基因F G F 10㊂其中,在14号染色体上检测到一个高频率的R OH 区域(22.78~23.38M b ),超过80%的个体都在该区域内发生R OH 片段,并在此区域鉴定到与生长和饲料转化率相关的基因T G S 1㊁L Y N ㊁C H C HD 7㊂基于R OH 信息的奶牛近交评估可为奶牛场的选种选配提供指导,在高频R OH 区域鉴定到的候选基因可作为奶牛分子育种中进行标记辅助选择的基因㊂关键词:长纯合片段(R OH );基因组近交系数;候选基因;中国荷斯坦牛中图分类号:S 823.91 文献标志码:A 文章编号:0366-6964(2023)07-2848-10收稿日期:2022-11-22基金项目:国家现代农业产业技术体系(C A R S 36);河南省现代农业(奶牛)产业技术体系建设专项资金(H A R S -22-14-S );河南省重点研发专项(221111111100);河南省科技攻关项目(222102110342;222102110254)作者简介:王振宇(1996-),男,河南永城人,硕士生,主要从事动物遗传育种研究,E -m a i l :w z yh a n 2017@163.c o m *通信作者:黄河天,主要从事动物遗传育种研究,E -m a i l :h u a n gh t @h e n a u .e d u .c n ;张 震,主要从事动物遗传育种与繁育研究,E -m a i l :z z gx u @163.c o m G e n o m i c I n b r e e d i n g C o e f f i c i e n t A n a l y s i s a n d F u n c t i o n a l G e n e S c r e e n i n gi n D i f f e r e n t D a i r y F a r m s B a s e d o n S N P C h i p Da t a WA N G Z h e n y u 1,Z H A N G S a i b o 1,L I U W e n h u i 1,L I A N G D o n g 1,R E N X i a o l i 2,Y A N L e i 2,Y A N Y u e f e i 2,G A O T e n g yu n 1,Z H A N G Z h e n 2,3*,HU A N G H e t i a n 1*(1.C o l l e g e o f A n i m a l S c i e n c e a n d T e c h n o l o g y ,H e n a n A g r i c u l t u r a l U n i v e r s i t y ,Z h e n g z h o u 450046,C h i n a ;2.H e n a n D a i r y H e r d I m p r o v e m e n t C e n t e r ,Z h e n gz h o u 450045,C h i n a ;3.H e n a n S e e d I n d u s t r y D e v e l o p m e n t C e n t e r ,Z h e n gz h o u 450046,C h i n a )A b s t r a c t :T h i s s t u d y a i m e d t o e s t i m a t e w h o l e -g e n o m e i n b r e e d i n gl e v e l s o f C h i n e s e H o l s t e i n c a t t l e f r o m d i f f e r e n t h e r d s i n H e n a n p r o v i n c e b y u s i n g t h e r u n s o f h o m o z y g o s i t y (R O H ),a n d i d e n t i f yR O H e n r i c h e d r e gi o n s a n d s c r e e n c a n d i d a t e g e n e s a s s o c i a t e d w i t h t h e t r a i t s o f e c o n o m i c i n t e r e s t .7期王振宇等:基于S N P芯片数据分析不同奶牛场基因组近交系数及筛选功能性基因A t o t a l o f900C h i n e s e H o l s t e i n c a t t l e f o r m7d a i r y h e r d s i n H e n a n p r o v i n c e w e r e u s e d t o d e t e c t g e n o m e-w i d e R OH b y t h e G G PB o v i n e150K B e a d c h i p.T h e n u m b e r,l e n g t h a n d f r e q u e n c y o f R O H i n H o l s t e i n p o p u l a t i o n w a s c o u n t e d.T h e g e n o m e i n b r e e d i n g c o e f f i c i e n t(F R O H)w a s c a l c u-l a t e d a c c o r d i n g t o R O H,a n d t h e h i g h f r e q u e n c y R O H r e g i o n s w e r e a n n o t a t e d.R O H w a s i d e n t i-f i e d i n a l l a n i m a l s,55908R O H w e r e i d e n t i f i e d,w i t h a m e a n l e n g t h o f4.23M b.T h e e s t i m a t e d i n b r e e d i n g c o e f f i c i e n t s o f R O H i n7h e r d s r a n g e d f r o m0.082(H7)t o0.123(H2),w i t h a n a v e r-a g e F R O H o f0.106i n a l l a n i m a l s.M o r e o v e r,79g e n e s r e l a t e d t o t h e e c o n o m i c t r a i t s o f d a i r y c o w s i n t h e g e n o m i c r e g i o n w i t h h i g h f r e q u e n c y R O H w e r e i d e n t i f i e d.A m o n g t h e s e g e n e s,A K A P3, C5H12o r f4,a n d F G F6w e r e r e l a t e d t o t h e b o d y s i z e a n d h e i g h t o f c a t t l e,C A P N3w a s a s s o c i a t e d w i t h c a r c a s s a n d r e p r o d u c t i v e t r a i t s,C H S T14w a s d i r e c t l y r e l a t e d t o p r e g n a n c y m a i n t e n a n c e a n d f e t a l g r o w t h,t h e t r a i t s o f m i l k p r o t e i n c o m p o s i t i o n w e r e a f f e c t e d b y I L5R A,a n d F G F10w a s i n-v o l v e d i n r e g u l a t i n g f e t a l f o l l i c u l o g e n e s i s.N o t a b l y,a h i g h-f r e q u e n c y R O H r e g i o n w a s d e t e c t e d o n c h r o m o s o m e14(22.78-23.38M b),w h e r e m o r e t h a n80%o f i n d i v i d u a l s c a r r i e d R O H f r a g-m e n t s.T h e g e n e s T G S1,L Y N a n d C H C HD7r e l a t e d t o g r o w t h a n d f e e d c o n v e r s i o n w e r e i d e n t i-f i e d i n t h i s r e g i o n.E v a l u a t i o n o f d a i r y c a t t l e i n b r e e d i n g b a s e d o n R O H i n f o r m a t i o n c o u l d b e a u s e f u l t o o l f o r s e l e c t i o n a n d m a t i n g s t r a t e g i e s.T h e c a n d i d a t e g e n e s i d e n t i f i e d c o u l d b e u s e d f o r m a r k e r-a s s i s t e d s e l e c t i o n i n d a i r y c a t t l e b r e e d i n g.K e y w o r d s:r u n s o f h o m o z y g o s i t y(R O H);g e n o m i c i n b r e e d i n g c o e f f i c i e n t;c a n d i d a t e g e n e;C h i-n e s e H o l s t e i n c a t t l e*C o r r e s p o n d i n g a u t h o r s:HU A N G H e t i a n,E-m a i l:h u a n g h t@h e n a u.e d u.c n;Z H A N G Z h e n,E-m a i l:z z g x u@163.c o m基因组长纯合片段(r u n s o f h o m o z y g o s i t y, R OH)一般存在于二倍体生物中,它是亲代将单倍型基因中同源相同(i d e n t i t y b y d e s c e n t,I B D)的片段遗传给子代,并且在子代的基因组中形成连续性的纯合片段[1],即子代从亲代继承了同源的染色体片段,从而导致后代基因组中的纯合片段产生并上升到R O H[2]㊂连锁不平衡㊁种群瓶颈㊁遗传漂变㊁近亲交配和选择都可能是引起R O H产生的因素[1,3-4]㊂不同的群体历史会产生不同长短的R OH,长片段R O H通常由群体近几个世代近交产生,短片段R O H通常来自更远的祖先[5-7]㊂因此,通过全基因组R OH特征的检测,可以了解种群历史㊁结构㊁近交情况㊂R O H最早在人类染色体基因组发现,并被认为可能对人类健康有重要影响㊂随着R O H在人类群体遗传学中研究的深入[8-10],不同畜禽的R O H 分析研究也逐渐开展[11-13]㊂基于R O H估计基因组近交系数已成为利用全基因组信息评估近交的常用方法,即利用R O H计算基因组近交系数F R O H(i n-b r e e d i n g c a l c u l a t e d f r o m R O H),它可以准确计算个体近交系数㊂现已有多项研究证明了基于系谱信息计算的近交系数要低于真实的近交系数㊂杨湛澄等[14]利用牛54K S N P芯片数据对北京地区2107头荷斯坦牛基因组R O H分布进行了统计,并计算了基因组近交系数和系谱近交系数,发现基于R O H 计算的基因组近交系数能更准确地反映个体的真实近交情况㊂P e r i p o l l i等[15]利用770K S N P芯片数据比较了2908头吉尔牛(G y r)基于R O H(F R O H)㊁基因组关系矩阵(g e n o m i c r e l a t i o n s h i p m a t r i x, F G R M)㊁基因组纯合子百分比(h o m o z y g o s i t y, F H OM)㊁系谱信息(p e d i g r e e,F P E D)4种方法计算的近交系数,结果表明在没有系谱记录的情况下, F R O H可用作近交估计的替代方法㊂此外,通过识别群体的高频R O H片段,鉴定到了与产奶量㊁乳成分㊁热适应相关的基因㊂N a n i和P eña g a r i c a n o[16]研究发现,基因组R O H与荷斯坦公牛繁殖性状显著相关,公牛群体中高度纯合的基因组区域与公牛繁殖性状呈现负相关,并在低繁殖力公牛R O H富集区域鉴定到与精子生物学和雄性生育能力密切相关的基因㊂L i u等[17]利用简化基因组测序的方法,通过R O H与综合单倍型评分(i n t e g r a t e d h a p l o t y p e s c o r e,i H S)分析,检测到与上海荷斯坦奶牛群体健9482畜牧兽医学报54卷康㊁繁殖㊁环境适应等有关的候选基因㊂通过对全基因组R O H进行检测,可以更准确地掌握群体的近交程度,帮助研究者在育种实践中制定科学合理的选种选配方案㊂鉴定全基因组的R O H也可以更好的了解R O H在染色体上的分布规律,进而挖掘可能影响畜禽重要性状的候选基因[18-20]㊂在我国,北京[14]㊁上海[17]㊁宁夏[21]基于荷斯坦牛群体基因组R OH估算群体近交系数㊁检测与经济性状相关候选基因及选育过程中的选择信号等的研究,为中国荷斯坦奶牛育种提供了重要数据参考㊂然而,通过基因组R O H信息估计不同牧场荷斯坦奶牛群体近交水平和检测群体选择特征的研究仍然较少㊂本研究旨在利用奶牛150K S N P芯片数据对河南省7个奶牛场荷斯坦牛进行全基因组R O H检测,计算R O H的长度㊁频率㊁数目和分布以及基因组近交系数F R O H,比较不同牧场荷斯坦牛基因组近交程度,并在高频R O H区域注释与荷斯坦牛经济性状相关的候选基因㊂以期为详细了解河南省荷斯坦牛群体基因组R O H分布特征及基因组近交程度,为牧场今后选种选配提供参考㊂也可通过R OH富集区域鉴定一些与奶牛经济性状相关的基因,为奶牛标记辅助选择提供候选基因信息,为奶牛场科学选种选配提供指导㊂1材料与方法1.1试验动物根据系谱㊁生产数据记录的完整性,筛选出7个存栏量在150~5000头的规模化牧场,按存栏量10%的比例抽取牧场核心群个体进行血液样本采集,最终共采集了900头荷斯坦牛㊂具体样本分布情况详见表1㊂1.2S N P芯片分型及数据质量控制采集尾椎静脉血,提取D N A,利用G G P B o v i n e 150K芯片进行基因分型㊂用P L I N K(v1.90)[22]对原始数据进行质控,设定条件:1)S N P检出率大于95%;2)个体检出率大于99%;3)最小等位基因频率大于0.01;4)哈迪-温伯格平衡P值大于10-6;5)保留常染色体数据㊂1.3群体结构及连锁不平衡分析基于S N P信息,使用G C T A(v1.93)软件[23]对900头荷斯坦牛群体进行主成分分析(p r i n c i p a l c o m p o n e n t a n a l y s i s,P C A)㊂采用P o p L D d e c a y (v3.42)软件[24]计算每个牧场的连锁不平衡(l i n k-a g e d i s e q u i l i b r i u m,L D)程度,并使用软件自带的P l o t_M u l t i P o p.p l脚本绘制L D衰减曲线图㊂1.4R O H检测及基因组近交系数的计算R O H检测使用P L I N K软件[22],使用滑动窗口的方法对常染色体进行检测,具体检测参数如下: 1)滑动窗口阈值使用0.05;2)滑动窗口设置50个S N P s位点;3)每一个滑动窗口中允许丢失的基因型为5个;4)每一个滑动窗口中允许的杂合子数目为1个;5)组成R O H的S N P的最大间隔为1M b;6)组成R O H的S N P的最低密度为每50k b1个S N P;7)R O H片段的最小长度设为500k b;8)每个R O H至少由50个S N P s组成㊂利用R OH计算近交系数(F R O H),公式如下:F R O H=ðL R O HL g e n o m e其中,ðL R O H为常染色体上R OH片段长度之和,L g e n o m e为常染色体基因组物理长度之和(2.49G b)㊂1.5高频R O H区域候选基因鉴定使用R语言统计每个S N P在奶牛群体中参与组成R O H的次数占样本数的比例,并将前1%的S N P s区域作为高频的R O H区域㊂基于高频R O H 区段的物理位置,并通过生物数据库E n s e m b l[25]中的B i o M a r t模块与牛参考基因组(B o s_t a u r u s.A R S-U C D1.2)进行比对,检索基因,然后依据N CB I (h t t p s://w w w.n c b i.n l m.n i h.g o v/)㊁G e n eC a r d s (h t t p s://w w w.g e n e c a r d s.o r g/)网站及文献查询基因功能㊂运用K O B A S(h t t p://b i o i n f o.o r g/k o-b a s/)[26]在线数据库对注释到的基因进行K E G G 通路富集分析,当P<0.05时,则表示显著富集㊂2结果2.1S N P质控结果及群体遗传结构和连锁不平衡分析在质控后每个个体保留了96789个S N P s位点,相邻S N P s之间的平均距离为25.72k b,以供后续分析㊂图1A显示了7个牧场荷斯坦牛群体的P C A分析结果㊂从图1可以看出,7个牛场主要分为了5个亚群㊂采用P o p L D d e c a y分别计算各牧场群体的成对r2值,用于比较不同荷斯坦牛群体的L D 水平(图1B)㊂L D分析显示,7个牧场奶牛群体L D 衰减的顺序为:H7>H4&H5>H2&H3&H6>H1㊂05827期王振宇等:基于S N P芯片数据分析不同奶牛场基因组近交系数及筛选功能性基因A.主成分分析图;B .L D 衰减图㊂H 1~H 7代表牧场编号A.P r i n c i p a l c o m p o n e n t a n a l y s i s o f H o l s t e i n c a t t l e p o p u l a t i o n ;B .L D d e c a y o f H o l s t e i n c a t t l e p o p u l a t i o n .H 1-H 7r e pr e s e n t s pa s t u r e n u mb e r 图1 群体遗传结构及连锁不平衡F i g .1 P o p u l a t i o n g e n e t ic s t r u c t u r e a nd l i n k a ge d i s e qu i l i b r i u m 2.2 R O H 数目㊁长度及分布的统计由表1可以看出,在7个牧场荷斯坦牛群体中共检测出55908个R O H ,R O H 的平均长度为4.23M b ,范围在1.90~14.07M b ㊂其中H 6号牛场R O H 平均长度最小,为3.27M b ;H 2号牛场R OH 平均长度最大为4.49M b ㊂在0~5M b 长度上,R O H 总体比例占76.21%,其中H 1㊁H 6牧场R OH 比例较大(83.70%㊁84.30%),其余牧场R O H 比例范围为73.33%~76.52%;在5~10M b长度上,R O H 总体比例占15.14%,其中H 1㊁H 6牧场R O H 比例较小(10.26%㊁10.67%),其余牧场R O H 比例范围为14.89%~17.06%;在>10M b长度上,R O H 总体比例占8.64%,其中H 1㊁H 6牧场R O H 比例较小(6.03%,5.04%),其余牧场R O H 比例范围为7.61%~9.61%㊂图2展示了常染色体上不同长度R O H 的数目㊂表1 不同奶牛场荷斯坦牛R O H 长度和数量T a b l e 1 T h e m e a n l e n g t h a n d n u m b e r o f r u n s o f h o m o z y g o s i t y (R O H )i n H o l s t e i n o f d i f f e r e n t d a i r y f a r m s 牛场编号F a r m n u m b e r 牛群数量N u m b e ro f c a t t l e 成母牛数量N u m b e ro f c o w s样本数S a m pl e s i z e 总R OH 数量T o t a l n u m b e ro f R OHR OH 平均长度/M bT h e m e a n l e n gt h o f R OH 均值M e a n标准差S D最小值M i n最大值M a xH 1152721411163.470.442.754.26H 23631983624634.490.523.575.71H 3185991912234.380.653.525.91H 451522600530325494.361.262.1014.07H 513106*********4.400.872.676.50H 610055019371663.270.622.185.41H 711265109747404.211.061.908.52平均A v e r a ge 132866212979874.080.782.677.20合计T o t a l92934632900559084.231.161.9014.072.3 基因组近交系数评估不同牧场荷斯坦牛群体基于R O H 的近交系数及变化范围见表2㊂全群中基于R O H 的基因组F R O H 范围为0.021~0.447,近交系数平均值为0.106,标准差为0.040㊂其中H 2号牧场平均F R O H最高(0.123),H 7号牧场平均F R O H 最低(0.082),其他牧场分别为0.112㊁0.114㊁0.109㊁0.108㊁0.103㊂在个体层面中,F R O H 最低的个体出现在H 71582畜 牧 兽 医 学 报54卷图2 染色体上不同长度R O H 的数目F i g .2 N u m b e r o f R O H w i t h d i f f e r e n t l e n gt h o n c h r o m o s o m e 号牛场中(0.021),F R O H 最高的个体出现在H 4号牛场中(0.447)㊂2.4 高频R O H 区域及候选基因鉴定与注释㊁富集图3展示了在1~29号染色体上组成R O H 的S N P s 占群体的百分率㊂通过选择组成R O H 中前1%S N P s ,以确定统计阈值,本研究选取频率大于29.78%作为高频率的R O H 区域阈值㊂共检测到8个高频区域,并通过E n s e m b l 数据库对R O H 中的高频区域进行基因注释,共注释到79个基因,见表3㊂其中,14号染色体上22.78~23.38M b 位置的区域,80%的个体都在该区域内发生R O H 片段,并注释到3个基因㊂利用K O B A S 对注释到的基因进行K E G G 通路富集分析,结果见表4㊂分析得出表2 基于R O H 的不同奶牛场的近交系数(F R O H )T a b l e 2 I n b r e e d i n g c o e f f i c i e n t (F R O H )o f d i f f e r e n t d a i r yf a r m s b a s e d o n R O H 牛场编号F a r m n u m b e r 样本数S a m pl e s i z e 近交系数(F R O H )I n b r e e d i n g co e f f i c i e n t 均值M e a n标准差S D最小值M i n最大值M a xH 1140.1120.0260.0620.156H 2360.1230.0190.0840.163H 3190.1140.0680.0680.173H 45300.1090.0430.0290.447H 51110.1080.0360.0280.213H 6930.1030.0310.0410.196H 7970.0820.0350.0210.226平均A v e r a ge 1290.1070.0370.0470.225合计T o t a l9000.1060.0400.0210.447图3 R O H s 中S N P s 百分比曼哈顿图F i g .3 M a n h a t t a n p l o t o f S N P s p e r c e n t a ge s i n R O H s 25827期王振宇等:基于S N P 芯片数据分析不同奶牛场基因组近交系数及筛选功能性基因79个基因显著富集于酮体的合成与降解(s yn t h e s i s a n d d e gr a d a t i o n o f k e t o n e b o d i e s )㊁缬氨酸㊁亮氨酸和异亮氨酸降解(v a l i n e ,l e u c i n e a n d i s o l e u c i n ed e gr a d a t i o n )㊁丁酸代谢(b u t a n o a t e m e t a b o l i s m )㊁R a s 信号通路(r a s s i g n a l i n g p a t h w a y)等11个信号通路㊂表3 荷斯坦牛高频R O H 区域及候选基因T a b l e 3 H i g h -f r e q u e n c y R O H r e gi o n s a n d c a n d i d a t e g e n e s i n H o l s t e i n c a t t l e 染色体C h r o m o s o m e物理位置/M b P h ys i c a l d i s t a n c e S N P s 数目N u m b e r o f S N P s 基因G e n e5105.514~105.77639A K A P 3㊁C 5H 12o r f 4㊁F G F 23㊁F G F 61035.989~38.53083B A H D 1㊁C 10H 15o r f 62㊁C A P N 3㊁C C ND B P 1㊁C H A C 1㊁C H P 1㊁C H S T 14㊁D L L 4㊁G A N C ㊁G C H F R ㊁H A U S 2㊁I T P K A ㊁I V D ㊁J M J D 7㊁K N L 1㊁K N S T R N ㊁M A P K B P 1㊁M G A ㊁P L A 2G 4B ㊁R A D 51㊁R P U S D 2㊁R T F 1㊁S N A P 23㊁S P I N T 1㊁T M E M 62㊁T Y R O 3㊁Z F Y V E 19㊁V P S 181421.726~25.698323R G S 20㊁M R P L 15㊁S O X 17㊁R P 1㊁X K R 4㊁T G S 1㊁L Y N ㊁C H C HD 7㊁F AM 110B ㊁U B XN 2B ㊁S D C B P1710.153~10.55516P R M T 9205.444~6.070134C P E B 4㊁C 20H 5o r f 47㊁N S G 224.070~33.323299E S M 1㊁C S P G 4B ㊁A R L 15㊁M O C S 2㊁E M B ㊁H C N 1㊁F G F 10㊁P A I P 1㊁C 20H 5o r f 34㊁C C L 28㊁T M E M 267㊁HM G C S 1㊁S E L E N O P ㊁O X C T 1㊁P L C X D 3㊁C 62222.914~23.31715C R B N ㊁I L 5R A2937.108~39.90862M S 4A 15㊁M S 4A 10㊁C C D C 86㊁T M E M 109㊁T M E M 132A ㊁C D 6㊁C D 5㊁P A G 10㊁P A G 12㊁P A G 8㊁P G A 5㊁T K F C ㊁T M E M 138㊁T M E M 216表4 高频R O H 区域基因的K E G G 通路富集分析(P <0.05)T a b l e 4 K E G G p a t h w a y e n r i c h m e n t a n a l y s i s o f g e n e s i n h i g h -f r e q u e n c y R O H r e gi o n s (P <0.05)通路P a t h w a y注释D e s c r i pt i o n 基因数NP 值P v a l u e基因G e n eb t a 04974:P r o t e i n d i g e s t i o n a n d a b s o r pt i o n 蛋白质消化吸收42.99ˑ10-4P A G 8㊁P A G 12㊁P A G 10㊁P G A 5b t a 00280:V a l i n e ,l e u c i n e a n di s o l e u c i n e d e gr a d a t i o n 缬氨酸㊁亮氨酸和异亮氨酸降解34.36ˑ10-4I V D ㊁HM G C S 1㊁O X C T 1b t a 00072:S y n t h e s i s a n d d e gr a d a t i o n o f k e t o n e b o d i e s酮体的合成与降解25.60ˑ10-4HM G C S 1㊁O X C T 1b t a 05224:B r e a s t c a n c e r乳腺癌48.42ˑ10-4F G F 6㊁F G F 10㊁D L L 4㊁F G F 23b t a 05218:M e l a n o m a黑色素瘤31.18ˑ10-3F G F 6㊁F G F 10㊁F G F 23b t a 00650:B u t a n o a t e m e t a b o l i s m 丁酸代谢23.03ˑ10-3HM G C S 1㊁O X C T 1b t a 05200:P a t h w a ys i n c a n c e r 癌症的通路63.73ˑ10-3I L 5R A ㊁D L L 4㊁R A D 51㊁F G F 6㊁F G F 10㊁F G F 23b t a 04014:R a s s i g n a l i n g p a t h w a y R a s 信号通路44.61ˑ10-3P L A 2G 4B ㊁F G F 10㊁F G F 23㊁F G F 6b t a 04611:P l a t e l e t ac t i v a t i o n血小板活化34.76ˑ10-3P L A 2G 4B ㊁L Y N ㊁S N A P 23b t a 04010:MA P K s i g n a l i n g p a t h w a y MA P K 信号通路48.76ˑ10-3P L A 2G 4B ㊁F G F 10㊁F G F 23㊁F G F 6b t a 05226:G a s t r ic c a n c e r胃癌38.95ˑ10-3F G F 6㊁F G F 10㊁F G F 233 讨 论3.1 荷斯坦牛群体基因组R O H 基本统计分析不同育种目标及选择强度会引起不同荷斯坦牛群体中R O H 数目㊁长度及分布情况的差异[5-6,27]㊂K i m 等[7]通过比较3个北美荷斯坦牛群体在产奶性状不同选择强度下基因组R O H 的变化,揭示了总体R O H 频率和分布方面的显著差异,结果显示3582畜牧兽医学报54卷群体内R OH平均长度约为6M b,小于5M b的R OH片段数目占总片段数目的53%㊂而与K i m 等[7]的研究结果相比,本研究中荷斯坦牛群体R OH平均长度为4.23M b,小于5M b的R O H片段的数目占总片段数目的76.21%㊂另外对比不同牧场群体,小于5M b的R O H片段数目所占比例也有差异㊂在基因组R O H长度上,M a r r a s等[28]利用50K S N P芯片对5个意大利公牛品种进行R O H分析,结果表明相较于其他品种,乳用品种荷斯坦牛和意大利布朗牛的平均R O H长度更大(3.6㊁3.9M b),其中荷斯坦牛群体的R OH平均长度与本研究的结果相近㊂在牧场群体方面,H1和H6号牧场群体在小于5M b的R O H片段数目占总片段数目最高(83.70%㊁84.30%),而大于10M b的R O H片段数目占总片段数目比例最低(6.03%㊁5.04%)㊂研究显示,较近世代的共同祖先会造成长R O H片段的形成,短的R OH来源于关系较远的共同祖先[7,29]㊂此外,各个牧场奶牛群体R O H平均长度㊁变化范围也有差异,这与不同牧场奶牛群体来源以及选配过程中使用不同国别的冷冻精液有关㊂因此,本研究基于对不同牧场群体基因组R O H的数目㊁长度及分布的研究,评估群体近交情况,为牧场今后的选种选配提供参考㊂3.2基于R O H的基因组近交系数目前,R OH常用来计算个体近交系数,且具有较高的准确性[15,30-33]㊂本研究中,河南荷斯坦牛群体总平均F R O H(0.106)与宁夏[21](0.101)㊁北京[14] (0.007~0.312)荷斯坦牛群体F R O H相近,与上海[17]荷斯坦牛群体(0.363)相差较大㊂上海与北京作为我国的南㊁北奶牛养殖业的代表地区,由于选育目标㊁强度㊁气候等因素的影响,群体近交程度出现差异,河南地理位置上属于中原地区,在奶牛育种策略和群体近交情况上与北方更相近㊂近交水平在一定程度上也可以反映牧场选种选配管理状况㊂在牧场选配管理上,由表2可以看到,H1㊁H2㊁H3号牧场平均F R O H较高(0.112㊁0.123㊁0.114),H7号牧场平均F R O H较低(0.082),不同牧场之间的差异侧面反映出这些牧场在选配过程中对群体近交问题的管理程度;在牧场规模上,H1㊁H2㊁H3号牧场规模较小,群体数量较少,平均F R O H较高(0.112㊁0.123㊁0.114),H4号牧场规模较大,群体数量多,平均F R O H较低(0.109)㊂此外,在H4号牧场中有些个体的F R O H明显较高(>0.285),最大F R O H达到0.458,反映出该牧场在个体选种选配过程中未充分考虑近交问题㊂因此,通过对近交系数的计算可以了解不同牧场群体近交状况,从而在实际选种选配工作中能更有效的避免近交,减少经济损失㊂3.3基因组高频R O H区域的候选基因分析本研究在高频R O H区域中共鉴定到了79个基因,其中包含与奶牛经济性状有关的基因,如A K A P3㊁C5H12o r f4㊁C A P N3㊁A R L15㊁X K R4㊁C R B N㊁I L5R A等㊂5号染色体上A K A P3㊁C5H12o r f4㊁F G F6基因与体型㊁体高有关[34-36]㊂10号染色体上C A P N3基因与胴体㊁繁殖性状相关[37-38]㊂C H S T14基因与妊娠维持和胎儿生长直接相关[39]㊂22号染色体上I L5R A基因影响牛奶蛋白质组成[40]㊂此外还有一些基因与繁殖㊁生长等性状有关,如F G F10基因参与调节胎儿卵泡生成[41]㊂值得注意的是,14号染色体上22.78~ 23.38M b区域是R O H频率最高的区域,80%的个体都在该区域内发生R O H片段(图3)㊂发现该区域与宁夏[21]荷斯坦牛群体高频区域(21.61~ 24.99M b)高度重合,这可能与不同地区育种目标及选择强度有关,并随着选育的推进,在基因组中出现相近的长纯合区域㊂这个高频区域注释到T G S1㊁L Y N㊁C H C HD7基因,这些基因与生长㊁胴体相关性状[42-43]和饲料效率有关[35,44-45]㊂因此,本研究在R O H富集区域鉴定的基因可以为荷斯坦奶牛分子育种提供候选基因信息㊂4结论本研究对河南省荷斯坦牛群体进行全基因组R O H检测与分析,发现R OH在不同牧场群体中的数目㊁长度及频率存在差异,基于R OH计算的近交系数范围在0.082~0.123,反映出不同牧场近交水平存在差异,这有助于了解河南省荷斯坦牛群体近交程度,为牧场选育过程中避免近交提供指导㊂在全基因组范围内检测到8个高频R O H富集区域,共筛选出79个与奶牛经济性状相关的基因,如A K A P3㊁C5H12o r f4㊁C A P N3㊁A R L15㊁X K R4㊁C R B N㊁I L5R A等,可作为奶牛分子育种中进行标记辅助选择的候选基因㊂参考文献(R e f e r e n c e s):[1] C E B A L L O S F C,J O S H I P K,C L A R K D W,e t a l.R u n s o f h o m o z y g o s i t y:w i n d o w s i n t o p o p u l a t i o n45827期王振宇等:基于S N P芯片数据分析不同奶牛场基因组近交系数及筛选功能性基因h i s t o r y a n d t r a i t a r c h i t e c t u r e[J].N a t R e v G e n e t,2018,19(4):220-234.[2] B R OMA N K W,W E B E R J L.L o n g h o m o z y g o u sc h r o m o s o m a l s e g m e n t s i n r e f e r e n c e f a m i l i e s f r o m t h eC e n t r e d E t u d e d u P o l y m o r p h i s m e H u m a i n[J].A m JH u m G e n e t,1999,65(6):1493-1500.[3] C U R I K I,F E R E N㊅C A K O V I C'M,SÖL K N E R J.I n b r e e d i n g a n d r u n s o f h o m o z y g o s i t y:a p o s s i b l es o l u t i o n t o a n o l d p r o b l e m[J].L i v e s t S c i,2014,166:26-34.[4] MU L I M H A,B R I T O L F,P I N T O L F B,e t a l.C h a r a c t e r i z a t i o n o f r u n s o f h o m o z y g o s i t y,h e t e r o z y g o s i t y-e n r i c h e d r e g i o n s,a n d p o p u l a t i o ns t r u c t u r e i n c a t t l e p o p u l a t i o n s s e l e c t e d f o r d i f f e r e n tb r e e d i n g g o a l s[J].B M C G e n o m ic s,2022,23(1):209.[5] Z HA N G Q Q,G U L D B R A N D T S E N B,B O S S E M,e ta l.R u n s o f h o m o z y g o s i t y a n d d i s t r ib u t i o n o ff u n c t i o n a l v a r i a n t s i n t h e c a t t l eg e n o m e[J].B M CG e n o m i c s,2015,16(1):542.[6] P U R F I E L D D C,B E R R Y D P,M C P A R L A N D S,e ta l.R u n s o f h o m o z y g o s i t y a n d p o p u l a t i o n h i s t o r y i nc a t t l e[J].B M C G e n e t,2012,13:70.[7] K I M E S,C O L E J B,HU S O N H,e t a l.E f f e c t o fa r t i f i c i a l s e l e c t i o n o n r u n s o f h o m o z y g o s i t y i n U.S.H o l s t e i n c a t t l e[J].P L o S O n e,2013,8(11):e80813.[8] L E N C Z T,L AM B E R T C,D E R O S S E P,e t a l.R u n s o fh o m o z y g o s i t y r e v e a l h i g h l y p e n e t r a n t r e c e s s i v e l o c i i ns c h i z o p h r e n i a[J].P r o c N a t l A c a d S c i U S A,2007,104(50):19942-19947.[9] C O R R E I A-C O S T A G R,S G A R D I O L I I C,S A N T O SA P D,e t a l.I n c r e a s e d r u n s o f h o m o z y g o s i t y i n t h ea u t o s o m a l g e n o m e o f B r a z i l i a n i n d i v i d u a l s w i t hn e u r o d e v e l o p m e n t a l d e l a y/i n t e l l e c t u a l d i s a b i l i t y a n d/o r m u l t i p l e c o n g e n i t a l a n o m a l i e s i n v e s t i g a t e d b yc h r o m o s o m a l m i c r o a r r a y a n a l y s i s[J].G e n e t M o lB i o l,2022,45(1):e20200480.[10] D A C R U Z P R S,A N A N I N A G,S E C O L I N R,e t a l.D e m o g r a p h i c h i s t o r y d i f f e r e n c e s b e t w e e n H i s p a n i c sa n d B r a z i l i a n s i m p r i n t h a p l o t y p e f e a t u r e s[J].G3(B e t h e s d a),2022,12(7):j k a c111.[11]刘家鑫,魏霞,邓天宇,等.绵羊全基因组R OH检测及候选基因鉴定[J].畜牧兽医学报,2019,50(8):1554-1566.L I U J X,W E I X,D E N G T Y,e t a l.G e n o m e-w i d e s c a nf o r r u n o f h o m o z yg o s i t y a n d i d e n t i f i c a t i o n o fc o r r e s p o nd i n g c a n d i d a te g e n e s i n s h e e p p o p u l a t i o n s[J].A c t a V e t e r i n a r i a e t Z o o t e c h n i c a S i n i c a,2019,50(8):1554-1566.(i n C h i n e s e)[12] G O R S S E N W,M E Y E R MA N S R,J A N S S E N S S,e ta l.A p ub l ic l y a v a i l a b l e r e p o s i t o r y o f R OH i s l a nd sr e v e a l s s i g n a t u r e s o f s e l e c t i o n i n d i f f e r e n t l i v e s t o c ka n d p e t s p e c i e s[J].G e n e t S e l E v o l,2021,53(1):2.[13]赵国耀.基于肉牛基因组纯合片段的性状关联与预测[D].北京;中国农业科学院,2021.Z HA O G Y.A s s o c i a t i o n a n d p r e d i c t i o n o f t r a i t s b a s e do n g e n o m i c h o m o z y g o u s s e g m e n t s i n b e e f c a t t l e[D].B e i j i n g:C h i n e s e A c a d e m y o f A g r i c u l t u r a l S c i e n c e s,2021.(i n C h i n e s e)[14]杨湛澄,黄河天,闫青霞,等.利用高密度S N P标记分析中国荷斯坦牛基因组近交[J].遗传,2017,39(1):41-47.Y A N G Z C,HU A N G H T,Y A N Q X,e t a l.E s t i m a t i o n o f g e n o m i c i n b r e e d i n g c o e f f i c i e n t s b a s e do n h i g h-d e n s i t y S N P m a r k e r s i n C h i n e s e H o l s t e i nc a t t l e[J].H e r ed i t a s,2017,39(1):41-47.(i n C h i ne s e)[15] P E R I P O L L I E,S T A F U Z Z A N B,MU N A R I D P,e ta l.A s s e s s m e n t o f r u n s o f h o m o z y g o s i t y i s l a n d s a n de s t i m a t e s ofg e n o m i c i n b r e e d i n g i n G y r(B o s i n d i c u s)d a i r y c a t t l e[J].B M C Ge n o m i c s,2018,19(1):34.[16] N A N I J P,P EÑA G A R I C A N O F.W h o l e-g e n o m eh o m o z y g o s i t y m a p p i n g r e v e a l s c a n d i d a t e r e g i o n sa f f e c t i n gb u l l f e r t i l i t y i n U S H o l s t e i nc a t t l e[J].B M CG e n o m i c s,2020,21(1):338.[17] L I U D Y,C H E N Z L,Z HA O W,e t a l.G e n o m e-w i d es e l e c t i o n s i g n a t u r e s d e t e c t i o n i n S h a n g h a i H o l s t e i nc a t t l e p o p u l a t i o n ide n t if i e dg e n e s r e l a t e d t o a d a p t i o n,h e a l t h a n d r e p r o d u c t i o n t r a i t s[J].B M C G e n o m i c s,2021,22(1):747.[18] MA K A N J U O L A B O,MA L T E C C A C,M I G L I O R F,e t a l.I d e n t if i c a t i o n o f u n i q u e R OH r eg i o n s w i t hu n f a v o r a b l e e f f e c t s o n p r o d u c t i o n a n d f e r t i l i t y t r a i t s i nC a n a d i a n H o l s t e i n s[J].G e n e t S e l E v o l,2021,53(1):68.[19] L I U J X,S H I L Y,L I Y,e t a l.E s t i m a t e s o f g e n o m i ci n b r e e d i n g a n d i d e n t i f i c a t i o n o f c a n d i d a t e r e g i o n s t h a td i f fe r b e t w e e n C h i n e s e i n d i g e n o u s s h e e p b r e e d s[J].JA n i m S c iB i o t e c h n o l,2021,12(1):95.[20]史良玉,王立刚,张鹏飞,等.不同来源大白猪总产仔数近交衰退评估[J].畜牧兽医学报,2021,52(10):2772-2782.S H I L Y,WA N G L G,Z HA N G P F,e t a l.E v a l u a t i o no f i n b r e e d i n g d e p r e s s i o n o n t h e t o t a l n u m b e r s o fp i g l e t s b o r n i n d i f f e r e n t g r o u p s o f l a r g e w h i t e p i g s[J].A c t a V e t e r i n a r i a e t Z o o t e c h n i c a S i n i c a,2021,525582畜牧兽医学报54卷(10):2772-2782.(i n C h i n e s e)[21]刘丽元.GWA S㊁C N V及R OH挖掘宁夏地区荷斯坦奶牛重要性状候选基因的研究[D].银川:宁夏大学,2021.L I U L Y.I n t e g r a t i n g GWA S,C N V a n d R OH a n a l y s i sr e v e a l s c a n d i d a t e g e n e s o f i m p o r t a n t t r a i t s i n N i n g x i ah o l s t e i n c o w[D].Y i n c h u a n:N i n g x i a U n i v e r s i t y,2021.(i n C h i n e s e)[22] C HA N G C C,C HOW C C,T E L L I E R L C A M,e ta l.S e c o n d-g e n e r a t i o n P L I N K:r i s i n g t o t h e c h a l l e n g eo f l a r g e r a n d r i c h e r d a t a s e t s[J].G i g a s c i e n c e,2015,4(1):7.[23] Y A N G J A,L E E S H,G O D D A R D M E,e t a l.G C T A:a t o o l f o r g e n o m e-w i d e c o m p l e x t r a i t a n a l y s i s[J].A m J H u m G e n e t,2011,88(1):76-82.[24] Z HA N G C,D O N G S S,X U J Y,e t a l.P o p L D d e c a y:af a s t a n d e f f e c t i v e t o o l f o r l i n k ag e d i s e q u i l i b r i u m d e c a ya n a l y s i sb a s e d o n v a r i a n tc a l l f o r m a t f i l e s[J].B i o i n f o r m a t i c s,2019,35(10):1786-1788.[25] C U N N I N G HAM F,A L L E N J E,A L L E N J,e t a l.E n s e m b l2022[J].N u c l e i c A c i d s R e s,2022,50(D1):D988-D995.[26] B U D C,L U O H T,HU O P P,e t a l.K O B A S-i:i n t e l l i g e n t p r i o r i t i z a t i o n a n d e x p l o r a t o r y v i s u a l i z a t i o no f b i o l o g i c a l f u n c t i o n s f o r g e n e e n r i c h m e n t a n a l y s i s[J].N u c l e i c A c i d s R e s,2021,49(W1):W317-W325.[27] HOWA R D J T,MA L T E C C A C,HA I L E-MA R I AMM,e t a l.C h a r a c t e r i z i n g h o m o z y g o s i t y a c r o s s U n i t e dS t a t e s,N e w Z e a l a n d a n d A u s t r a l i a n J e r s e y c o w a n db u l l p o p u l a t i o n s[J].B M C G e n o m ic s,2015,16(1):187.[28] MA R R A S G,G A S P A G,S O R B O L I N I S,e t a l.A n a l y s i s o f r u n s o f h o m o z y g o s i t y a n d t h e i rr e l a t i o n s h i p w i t h i n b r e e d i n g i n f i v e c a t t l e b r e e d sf a r m e d i n I t a l y[J].A n i m G e n e t,2015,46(2):110-121.[29] K E L L E R M C,V I S S C H E R P M,G O D D A R D M E.Q u a n t i f i c a t i o n o f i n b r e e d i n g d u e t o d i s t a n t a n c e s t o r sa n d i t s d e t e c t i o n u s i n g d e n s e s i n g l e n u c l e o t i d ep o l y m o r p h i s m d a t a[J].G e n e t i c s,2012,189(1):237-249.[30] F E R E N C A K O V I C M,HAM Z I C E,G R E D L E R B,e ta l.R u n s o f h o m o z y g o s i t y r e v e a l g e n o m e-w i d ea u t o z y g o s i t y i n t h e A u s t r i a n F l e c k v i e h c a t t l e[J].A g r i c C o n s p e c S c i,2011,76(4):325-329.[31] Z HA N G Q Q,C A L U S M P L,G U L D B R A N D T S E NB,e t a l.E s t i m a t i o n o f i n b r e e d i n g u s i n g p e d i g r e e,50kS N P c h i p g e n o t y p e s a n d f u l l s e q u e n c e d a t a i n t h r e ec a t t l e b r e ed s[J].B M C Ge n e t,2015,16:88.[32] F O R U T A N M,MA H Y A R I S A,B A E S C,e t a l.I n b r e e d i n g a n d r u n s o f h o m o z y g o s i t y b e f o r e a n d a f t e rg e n o m i c s e l e c t i o n i n N o r t h A m e r i c a n H o l s t e i n c a t t l e[J].B M C G e n o m i c s,2018,19(1):98.[33] L O Z A D A-S O T O E A,T I E Z Z I F,J I A N G J C,e t a l.G e n o m i c c h a r a c t e r i z a t i o n o f a u t o z y g o s i t y a n d r e c e n ti n b r e e d i n g t r e n d s i n a l l m a j o r b r e e d s o f U S d a i r yc a t t l e[J].J D a i r y S c i,2022,105(11):8956-8971.[34]J I A N G J C,C O L E J B,F R E E B E R N E,e t a l.F u n c t i o n a l a n n o t a t i o n a n d B a y e s i a n f i n e-m a p p i n gr e v e a l s c a n d i d a t e g e n e s f o r i m p o r t a n t a g r o n o m i c t r a i t si n H o l s t e i n b u l l s[J].C o m m u n B i o l,2019,2(1):212.[35] G HO R E I S H I F A R S M,E R I K S S O N S,J OHA N S S O N A M,e t a l.S i g n a t u r e s o f s e l e c t i o nr e v e a l c a n d i d a t e g e n e s i n v o l v e d i n e c o n o m i c t r a i t s a n dc o ld a c c l i m a t i o n i n f i ve S w e d i s h c a t t l e b r e e d s[J].G e n e t S e l E v o l,2020,52(1):52.[36] F A N G L Z,C A I W T,L I U S L,e t a l.C o m p r e h e n s i v ea n a l y s e s o f723t r a n s c r i p t o m e s e n h a n c e g e n e t i c a n db i o l o g ic a l i n t e r p r e t a t i o n s f o r c o m p l e x t r a i t s i n c a t t l e[J].G e n o m e R e s,2020,30(5):790-801.[37] Z HA N G Y Y,X U E X L,L I U Y,e t a l.G e n o m e-w i d ec o m p a r a t i v e a n a l y s e s r e v e a l s e l e c t i o n s i g n a t u r e su n d e r l y i n g a d a p t a t i o n a n d p r o d u c t i o n i n T i b e t a n a n dP o l l D o r s e t s h e e p[J].S c i R e p,2021,11(1):2466.[38] WA N G J F,L I B Z,Y A N G X R,e t a l.I n t e g r a t i o n o fR N A-s e q a n d A T A C-s e q i d e n t i f i e s m u s c l e-r e g u l a t e dh u b g e n e s i n c a t t l e[J].F r o n t V e t S c i,2022,9:925590.[39] S I G D E L A,B I S I N O T T O R S,P EÑA G A R I C A N O F.G e n e s a n d p a t h w a y s a s s o c i a t e d w i t h p r e g n a n c y l o s s i nd a i r y c a t t l e[J].S c i Re p,2021,11(1):13329.[40] Z HO U C H,L I C,C A I W T,e t a l.G e n o m e-w i d ea s s o c i a t i o n s t u d y f o r m i l k p r o t e i n c o m p o s i t i o n t r a i t si n a c h i n e s e h o l s t e i n p o p u l a t i o n u s i n g a s i n g l e-s t e pa p p r o a c h[J].F r o n t G e n e t,2019,10:72.[41] F R E I T A S P H F,O L I V E I R A H R,S I L V A F F,e ta l.S h o r t c o mm u n i c a t i o n:t i m e-d e p e n d e n t g e n e t i cp a r a m e t e r s a n d s i n g l e-s t e p g e n o m e-w i d e a s s o c i a t i o na n a l y s e s f o r p r e d i c t e d m i l k f a t t y a c i d c o m p o s i t i o n i nA y r s h i r e a n d J e r s e y d a i r y c a t t l e[J].J D a i r y S c i,2020,103(6):5263-5269.[42] C H E R U I Y O T E K,B E T T R C,AM I MO J O,e t a l.S i g n a t u r e s o f s e l e c t i o n i n a d m i x e d d a i r y c a t t l e i nt a n z a n i a[J].F r o n t G e n e t,2018,9:607.6582。
Affymetrix 全基因组 SNP 芯片检测

A f f y m e t r i x全基因组S N P芯片检测单核苷酸多态性(single nucleotide polymorphism, SNP) 指基因组单个核苷酸的变异,它是最微小的变异单元,是由单个核苷酸对置换、颠换、插入或缺失所形成的变异形式。
单核苷酸多态性是基因组上高密度的遗传标志,在人类基因组中已发现的SNP数量超过3000万。
作为第三代遗传标记,SNP数量众多、分布密集、易于检测,因而是理想的基因分型目标。
SNP分型检测在疾病基因组(如疾病易感性),药物基因组(药效、药物代谢差异和不良反应)和群体进化等研究中具有重大意义。
在人研究方面,Affymetrix 公司有分别基于GeneChip和GeneTitan平台的SNP 芯片和针对中国人群设计的CHB1&2 Array,既可用于全基因组SNP分析,又可用于CNV分析,极大地方便了中国人类疾病GWAS研究。
Affymetrix公司针对多个农业物种也开发了多款商品化的基因分型芯片,如鸡、牛、水牛、鲑鱼、水稻、小麦、辣椒、草莓等,为农业育种研究、遗传图谱构建、群体基因组学研究提供研究手段。
此外,Affymetrix公司还支持定制芯片,最低起订量为480个样品。
检测原理|技术优势|产品列表|定制芯片|数据分析|基于GeneChip平台的人SNP 芯片实验流程:基于GeneTitan平台的Axiom基因分型芯片检测流程:从SNP原理谈SNP分析技术之SNP芯片日期:2012-05-21 来源:网络标签:摘要:SNP是近年来基因突变的热点研究之一。
它是指在单个的核苷酸上发生了变异,有四种不同的变异形式,而实际上只发生转换和颠换这两种。
当科学家弄清了SNP的突变原理以后,他们就着手对SNP进行分析,以求找到疾病相对应的突变位点或者是进行个性化药物治疗研究。
其中应用到的技术多达上百余种,其中包括有测序技术、质谱分析技术、HRM技术、Taqman技术以及SNP芯片技术。
SNP分析原理方法及其应用

SNP分析原理方法及其应用SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是指在基因组中的一些位置上,不同个体之间存在的碱基差异,是常见的遗传变异形式之一、SNP分析是研究SNP在基因与表型之间关联性的方法,用于揭示SNP与遗传疾病、药物反应性等的关系。
本文将介绍SNP分析的原理、方法以及其应用。
一、SNP分析原理1.SNP检测技术:SNP检测技术包括基于DNA芯片的方法、测序技术、实时荧光PCR等。
其中,高通量测序技术是最常用的SNP检测方法,可以同时检测数千个SNP位点。
2.数据分析与统计学方法:通过SNP检测技术获得的数据可以分为基因型数据(AA、AB、BB等)和等位基因频率数据(A频率、B频率等)。
统计学方法常用的有卡方检验、线性回归、逻辑回归等,用于研究SNP与表型之间的关联性。
二、SNP分析方法1.关联分析:关联分析是研究SNP与表型之间关联性的基本方法。
常用的关联分析方法包括单基因型分析、单SNP分析、基因组关联分析(GWAS)等。
单基因型分析主要是比较单个SNP的基因型在表型不同组之间的差异;单SNP分析是研究单个SNP是否与表型相关;GWAS是通过分析数万个SNP与表型之间的关系来找到与表型相关的SNP。
2. 基因型预测:基因型预测是根据已有的SNP数据,通过统计模型来预测个体的基因型。
常用的基因型预测方法有HapMap、PLINK等。
3. 功能注释:功能注释是研究SNP位点的生物学功能,揭示SNP与基因功能、表达水平之间的关系。
常用的功能注释工具有Ensembl、RegulomeDB等。
三、SNP分析应用1.遗传疾病研究:SNP与遗传疾病之间存在着密切的关系。
通过SNP分析可以发现与遗传疾病相关的SNP位点,进一步揭示疾病发生的机制,为疾病的诊断、治疗提供依据。
2.药物反应性研究:个体对药物的反应性往往存在较大差异,这与个体的遗传背景密切相关。
生命科学数据分析的方法与应用

生命科学数据分析的方法与应用近年来,生命科学领域的迅速发展,使得大量数据积累在了科研工作者的手中。
而数据的分析与应用,已经成为生命科学研究的必然趋势。
而针对如此多的数据,如何进行高效的挖掘,成为了生命科学研究领域中重要的问题之一。
本文将结合实际案例,探讨生命科学数据分析的方法与应用。
一、SNP数据分析与应用SNP(sing nucleotide polymorphisms)是指单个核苷酸的多态性,以其多变性为基础进行基因千变万化地研究。
SNP数据如何进行分析和应用呢?首先,进行SNP芯片的数据分析。
通过芯片中的探针将样本DNA与芯片固定的单核苷酸进行匹配并输出,然后对输出结果进行标准化和归一化处理。
接着进行基因型的分析,根据样本中每个SNP位点的基因型分析, 确定每个个体所携带的所有SNP位点的基因型。
其次,进行SNP数据的应用。
SNP 基因组分型技术的应用包括疾病风险预测和药物治疗反应性预测。
例如,针对遗传性疾病,可以通过SNP与疾病之间的关联,估计个体发病风险。
针对癌症治疗,可以根据药物代谢 SNP 和靶标 SNP 的情况,预测药物在患者体内的代谢效率和治疗效果。
二、生物成像数据分析与应用生物成像技术已经成为了生命科学研究中不可或缺的工具,如何进行生物成像数据的分析与应用呢?首先,进行生物成像数据的处理。
对于不同的生物成像技术,数据处理方法和软件有所不同,例如,对于荧光显微镜下的图像,需要进行图像处理和去噪,以得到清晰的细胞图像。
其次,进行生物成像数据的分析。
对生物成像样品的图像进行分析,可以获得样品中各种特征如面积、位置、大小、形态等,并可以对生命过程进行动态跟踪。
例如,在活体成像中,可以利用时间序列数据分析细胞轨迹并找出细胞的运动及各个时间点的强度。
最后,进行生物成像数据的应用。
生物成像技术的应用广泛,例如,在肿瘤病理学中,可以通过活体成像技术来实时观察肿瘤细胞的生长、扩散和药物疗效;在生物学研究中,可以通过高分辨率显微镜图像,在细胞水平上探究细胞器的组成和功能。
使用生物大数据技术进行SNP关联分析的方法与工具推荐

使用生物大数据技术进行SNP关联分析的方法与工具推荐随着生物学研究的不断发展,基因组学数据的积累和可用性不断增加。
其中,单核苷酸多态性(SNP)是一类广泛存在于基因组中的遗传变异,是研究复杂性疾病和个体差异的重要标记。
SNP关联分析是一种常用的研究方法,可以帮助我们识别与疾病发展或生物表型相关的SNP。
本文将介绍使用生物大数据技术进行SNP关联分析的方法和一些推荐的工具。
这些工具可以加快分析过程并提供丰富的数据可视化和解释。
一、SNP数据预处理进行SNP关联分析之前,首要任务是预处理SNP数据。
这包括数据清洗、格式转换、去除无关变异和处理缺失数据等步骤。
常用的SNP数据预处理工具包括PLINK、VCFtools和GATK等。
1. PLINK(Purcell et al., 2007)是一个功能强大的工具集,用于进行基因组关联分析。
它可以处理各种格式的SNP数据,包括PED/MAP、BED等,并提供了丰富的数据处理和统计分析功能。
2. VCFtools是一个专门用于VCF格式(Variant Call Format,常用于常见SNP格式)的SNP数据处理工具。
它可以用来过滤、格式转换、计算遗传群体统计信息等。
3. GATK(Genome Analysis Toolkit)是一个广泛使用的工具包,用于分析高通量测序数据。
它可以进行SNP/Indel检测、变异质量评估、基于家系或群体的SNP筛选等。
二、SNP关联分析SNP关联分析是通过比较个体的基因型和表型来寻找与表型相关的SNP。
这一步骤通常涉及人群结构分析、关联测试和多重比较校正等。
1. 人群结构分析可以帮助去除由于人群混合导致的伪关联。
常用的人群结构分析工具包括ADMIXTURE和STRUCTURE等。
这些工具可以将样本划分为亚群,并提供每个样本在亚群中的成分比例。
2. 关联测试是判断SNP与表型之间是否存在相关性的关键步骤。
一种常见的关联测试方法是单SNP关联分析,可以使用PLINK、SNPTEST或GEMMA等工具进行。
基因组学研究中SNP标记方法与数据分析

基因组学研究中SNP标记方法与数据分析SNP标记方法与数据分析在基因组学研究中起着重要的作用。
SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是基因组中最常见的变异形式,是导致个体间遗传差异的主要原因之一。
因此,对SNP标记方法和数据分析的研究对于揭示基因与表型之间的关联、为功能基因组学研究提供有效工具具有重要意义。
SNP标记方法主要分为两种:基于技术平台的方法和计算预测的方法。
技术平台包括传统的基因测序、SNP芯片和下一代测序。
传统的基因测序方法通过测序反应来确定SNP位点上的碱基,虽然准确性高,但费时费力。
SNP芯片是一种高通量的方法,可以同时检测多个SNP位点,准确性相对较低。
下一代测序则是目前最常用的方法,具有高通量、高分辨率、低成本的特点。
在SNP标记方法的选择上,需要根据研究对象、目标和预算来权衡不同方法的优缺点。
在SNP标记数据的分析中,主要涉及到数据的预处理、基因型分型和遗传关联分析。
首先,数据的预处理包括对原始数据进行质量控制、过滤掉低质量的SNP位点和个体,以及进行数据标准化和归一化。
这一步骤对后续的分析至关重要,能够减少误报率和漏报率,提高结果的可靠性。
其次,基因型分型是确定每个个体在每个SNP位点上的基因型。
由于SNP位点的碱基组合较多,需要运用一系列的算法和统计模型来进行基因型分型,其中包括Bayes算法、混合模型和机器学习方法等。
最后,遗传关联分析是研究SNP位点与表型之间关联的主要方法,可以通过构建模型、计算单个SNP的关联程度,或者进行基因组广义关联分析(GWAS),来揭示SNP位点与表型之间的关系。
在进行SNP标记方法和数据分析时,还需注意一些常见的挑战和问题。
首先,SNP标记的质量控制和过滤是一个关键的步骤,需要选择合适的阈值来确保数据的准确性。
同时,样本大小也是一个重要的考虑因素,在样本量较小时,可能会出现较大的偏差。
另外,SNP位点之间的连锁不平衡(Linkage Disequilibrium,LD)也需要在分析中进行考虑,以减少虚假关联的可能性。
snp芯片

snp芯片SNP芯片(Single Nucleotide Polymorphism chips)是一种高通量基因分型技术,目前广泛应用于基因组学研究和个体遗传变异的检测。
SNP芯片可以同时检测数千至数百万个单核苷酸多态性位点,从而快速、精准地分析个体间的遗传差异。
SNP(Single Nucleotide Polymorphism)是指基因组中单个核苷酸发生变异的位置。
它们是人类遗传变异中最常见的形式,每个人的基因组中大约有数百万个SNP。
通过对SNP的识别和分析,可以研究个体之间的遗传差异,并且了解这些差异与健康、疾病以及药物反应等方面的关系。
SNP芯片是通过DNA芯片技术实现SNP的检测和分析。
其基本原理是将经过特定设计的探针固定在芯片上,探针可以与特定的SNP序列互相匹配。
然后,样本DNA经过扩增和标记处理,与芯片上的探针进行杂交,并通过荧光等方式进行信号检测。
根据信号的强度,可以确定每个位点上的SNP类型。
SNP芯片的优势在于高通量、高准确性和高效率。
相比传统的基因分型方法,SNP芯片可以同时分析大量SNP位点,大大提高了研究效率。
同时,SNP芯片的准确性也非常高,可以达到99%以上。
此外,SNP芯片还具有良好的可重复性和可比性,可以在不同实验室之间进行数据交流和共享。
SNP芯片广泛应用于基因组学研究、人类遗传学和药物研发等领域。
在基因组学研究中,SNP芯片可以用于遗传关联研究,寻找与疾病或性状相关的SNP位点。
在人类遗传学中,SNP芯片可以用于研究不同人群之间的遗传差异,了解人类种群演化和迁移的历史。
在药物研发中,SNP芯片可以用于个体化药物治疗的研究,根据个体的遗传信息优化药物方案,提高疗效和减少副作用。
然而,SNP芯片也存在一些局限性。
首先,SNP芯片只能检测已知的SNP位点,对于未知或罕见的变异无法检测。
其次,SNP芯片无法检测复杂的遗传变异,如插入缺失、倒位等结构变异。
此外,SNP芯片的解读和分析需要复杂的生物信息学算法和数据库支持,对研究人员的专业水平提出了要求。
SNP数据结果说明文档

SNP数据结果说明文档一、数据结果说明数据结果均在Data results文件夹中Raw data为原始数据1.结尾为samplesheet.xls文件为样本说明文件2.结尾为样本编号_FinalReport.txt的文件为每个样本的分型结果数据每列的含义:SNP_ID: SNP名称其余列为每个样本的分型数据3.结尾为Samples T able.txt的文件为每个样本的call Rate值情况数据每列的含义:Index: 数据编号Sample ID: 样本编号Call Rate: 样本的call Rate值Gender: 样本的性别p05 Grn: 百分之五分位数时A等位基因的强度p50Grn: 百分之五十分位数时A等位基因的强度p95 Grn: 百分之九十五分位数时A等位基因的强度p05 Red: 百分之五分位数时B等位基因的强度p50Red: 百分之五十分位数时B等位基因的强度p95 Red: 百分之九十五分位数时B等位基因的强度p10 GC: 此样本所有SNP 百分之十分位数的Gen Call scoreP50 GC: 此样本所有SNP百分之五十分位数的Gen Call score Rep Error Rate,PC Error Rate,PPC Error Rate,Subset结果并不给出所以略过.Aux: 用户自己设置的SNP辅助值Genotype for exm-IND11-102094357: exm-IND11-102094357的基因型Array Info.Sentrix ID: 芯片条形码IDArray Info.Sentrix Position: 样本在芯片上的位置4.DNAReport.csv 包含AA,AB 等位基因频率等5.Plink文件夹中是转化为plink格式的数据。
/doc/585616010.html,V 分析说明:此分析为收费项目用penncnv软件做CNV分析包括/doc/585616010.html,V,CNVR的区域、CN 值、所含SNP位点数、基因注释等。
SNP位点数据分析和人类遗传学研究

SNP位点数据分析和人类遗传学研究SNP (Single Nucleotide Polymorphism) 位点数据分析和人类遗传学研究随着现代技术的快速发展,生物信息学领域的研究变得越来越重要。
其中,单核苷酸多态性(SNP)位点数据分析在人类遗传学研究中起着关键作用。
本文将讨论SNP位点的概念、分析方法以及其在人类遗传学研究中的应用。
首先,SNP位点是人类基因组中最常见的突变形式。
它是DNA序列中的单个核苷酸发生变异的地方,包括碱基的替换、插入和删除。
SNP位点通常在基因和表达调控区域中,对个体间的遗传差异和基因功能起着重要作用。
因此,研究SNP位点对于理解人类遗传学和疾病的发生机制至关重要。
在SNP位点数据的分析中,最常见的方法是基因型和等位基因频率分析。
基因型分析涉及确定每个个体的等位基因组合,包括纯合子(两个等位基因相同)和杂合子(两个等位基因不同)。
等位基因频率分析则是研究一个等位基因在某个群体中的频率。
通过这些分析方法,我们可以了解SNP位点的遗传多样性及其在人群间的分布情况。
此外,SNP位点数据还可以通过关联分析来研究基因与特定性状或疾病之间的联系。
关联分析(Association Analysis)是将SNP位点与某个性状或疾病之间的关联关系联系起来。
这种方法被广泛应用于复杂性疾病的研究,如肿瘤、心血管疾病和神经退行性疾病等。
通过关联分析,我们可以发现与某个特定性状或疾病相关的SNP位点,进一步了解其遗传机制,发现相关基因以及相关通路,为疾病的预测、诊断和治疗提供重要的线索。
SNP位点数据的分析离不开高通量测序技术的支持,如基因芯片和下一代测序。
这些技术的发展使得大规模SNP位点分析成为可能,相对应的数据处理和分析方法也在不断更新和改进。
然而,SNP位点数据分析中也存在一些挑战和限制,如缺乏样本数量和SNP位点的不均匀分布,这些问题需要继续研究和解决。
总结起来,SNP位点数据分析在人类遗传学研究中具有重要作用。
人类基因组研究中的SNP分析

人类基因组研究中的SNP分析SNP(Single nucleotide polymorphism,单核苷酸多态性),是指基因组中出现的一种常见的遗传变异形式,其在人类进化、疾病易感性等方面的研究具有重要意义。
SNP分析作为人类基因组研究的主要手段之一,已经在人类进化、疾病与药物研究等领域得到了广泛应用。
SNP分析通过测定与细胞相关的基因座上的多态性位点,确定细胞中的SNP型,并用以评估个体间的遗传差异。
SNP是通过与疾病发病相关的基因关联研究中的反复测定,鉴定和确认的。
SNP分析常常使用高通量测序技术,如全基因组测序或SNP芯片来获取大规模和全面的数据。
SNP分析在人类的进化研究中起到了重要作用。
通过比较不同人群之间的基因差异,科学家可以了解人类进化历程和人类族群之间的遗传关系。
例如,研究人员可以通过SNP分析来揭示人类不同地理区域人群的迁徙历史、近亲交配、适应性进化等信息。
此外,SNP分析还可以用于确定古人类的基因组信息,揭示与现存人类的共同祖先和近亲种群的关系,帮助我们更好地理解人类的进化过程。
在疾病研究中,SNP分析可以用于揭示疾病发病的遗传基础。
通过比较疾病患者和正常人群之间的SNP型分布差异,科学家可以识别与特定疾病发病相关的基因。
这为疾病的早期诊断、个体化治疗以及疾病风险评估等提供了重要依据。
例如,许多研究已经鉴定并确认了与肿瘤、心血管疾病、自身免疫性疾病等多种疾病发病相关的SNP。
这些研究有助于我们了解疾病的发病机制,并为相关疾病的预防和治疗提供了新的指导。
此外,SNP分析还在临床药物疗效和安全性评估中起到重要作用。
通过比较受试者的一些关键基因的SNP型,科学家可以预测一些药物的疗效和不良反应风险,从而实现个体化的药物治疗。
例如,一些药物代谢酶的SNP型可以影响对该药物的代谢速度,从而影响疗效和安全性。
SNP分析可以帮助医生更好地选择适合患者的药物和剂量,提高治疗效果,减少药物不良反应。
关于SNP检测服务报告的解读

关于SNP检测服务报告的解读SNP全称Single Nucleotide Polymorphisms,是指在基因组上单个核苷酸的变异,包括转换、颠换、缺失和插入,形成的遗传标记,其数量很多,多态性丰富。
我公司提供的SNP检测报告一般包括以下部分:在“峰图”文件夹中即为测序峰图文件。
Word文件“SNP分型实验报告-snapshot”提供实验操作的步骤、以及PCR参数等。
其中snapshot为检测SNP的方法。
此外我们还有LDR法等。
Excel表格“引物序列”提供实验用引物的序列。
Excel表格“SNP检测结果”为最终结果展示。
以下表为例(部分):“序号”“客户编号”“姓名”等项由客户提供。
每个SNP都有唯一的编号,一般都是以rs为字头。
[C/T]表示在这个位点上的碱基种类。
以表中rs429358为例,此例中即代表在这个位点上的碱基可能是C,也可能是T,只能是其中之一。
因为人的染色体都是成对出现,所以在成对的染色体上对应的位置会同时存在两个相同基因(称之为等位基因)。
我们的检测会同时测到两个等位基因,这两个的结果可能相同也可能不同,所以上表中的结果有“C C”“T T”以及“C T”。
“C C”或“T T”即为纯合型,“C T”即为杂合型。
本结果只提供SNP的检测结果,不包含对SNP信息的后续处理,如与临床数据的关联性分析等。
附:SNP与突变表面上来看,SNP与突变确实有着共同之处。
比如二者都是在某特定位置上的碱基变为了另一个。
但这两个概念还是有本质区别的。
1、多态性是一个群体概念,多态性指这个差异占群体的1%以上。
否则就叫突变(小于1%)。
2、SNP是多态性中的一种,只是进一步限定了差异只是单碱基。
3、SNP一般来说,是全部体细胞一样的基因型(除开嵌合体)。
4、突变一般不是一个个体全部细胞的变化。
5、如果突变发生在生殖细胞,则可以遗传,但是只要这个突变群没有达到总群体的1%,它就只是一个突变株/系。
SNP分析及其在遗传学中应用情况

SNP分析及其在遗传学中应用情况简介单核苷酸多态性(Single Nucleotide Polymorphism,SNP)是人类基因组中最常见的遗传变异形式之一。
SNP分析是研究个体之间以及不同种群之间遗传差异的有力工具。
随着高通量测序技术和生物信息学的发展,SNP分析已经成为遗传学研究中的一个重要领域,为我们理解基因变异与疾病风险、药物反应以及个体差异等提供了深入的了解。
SNP分析技术SNP分析的主要技术包括SNP芯片和基于测序的方法。
SNP芯片利用微阵列技术在一块芯片上同时检测大量的SNP位点。
而基于测序的方法则通过对个体基因组的全面测序来获取SNP信息。
两种方法各有优劣势,选择合适的方法应根据研究目的和预算来决定。
SNP在人类遗传学中的应用1. 疾病风险预测SNP与疾病之间存在密切的关联。
通过大规模SNP关联研究(Genome-wide Association Study,GWAS),研究人员已经发现了大量与疾病相关的SNP位点。
这些位点可以用来预测个体患病的风险,对疾病的早期筛查以及制定个性化的治疗方案具有重要意义。
2. 遗传进化研究SNP分析可以帮助我们了解人类和其他物种的遗传演化历程。
通过比较不同种群之间的SNP差异,研究人员可以揭示人类迁徙历史、种群形成以及适应性进化等重要信息。
此外,SNP还能用于研究个体之间的近交程度以及人类的远亲关系。
3. 药物反应预测个体对药物的反应存在很大的差异,这主要受遗传变异的影响。
SNP分析可以帮助我们预测个体对特定药物的反应情况,从而指导临床用药。
例如,根据某些特定的SNP位点,可以预测患者是否对某种药物具有耐药性,以及药物代谢速度的快慢。
4. 父权鉴定和犯罪侦查SNP分析可以利用个体之间的基因型差异来进行父权鉴定和犯罪侦查。
通过比较孩子和母亲、孩子和潜在父亲之间的SNP位点,可以确定孩子的生物学父亲。
此外,对犯罪现场的DNA样本与嫌疑人DNA样本进行SNP分析,还可以帮助警方追踪犯罪嫌疑人。
人类基因组研究中的SNP分析

人类基因组研究中的SNP分析随着现代科技的快速发展,人类已经进入了基因组时代。
在这个时代里,基因组研究是关键的一环,因此,人类基因组研究已成为当前热门科学研究领域。
SNP是人类基因组研究中非常重要的一种基因类型,其全称为“单核苷酸多态性”(Single nucleotide polymorphisms),是指基因组DNA序列上的单个核苷酸发生突变的现象。
这些突变可能会对个体的遗传特征、代谢和疾病易感性产生影响,因此,SNP分析被广泛应用于人类基因组的研究。
SNP分析的意义SNP分析作为一种高效而有效的基因分析方法,其应用范围非常广泛。
除了帮助人们更好地了解人类基因组的不同特征外,SNP分析也可以被应用于以下领域:1. 遗传病研究基因突变是遗传病发生的原因之一,而SNP的变异也可能引起明显的遗传病症状。
SNP分析可以帮助科学家更好地了解这些突变与遗传病之间的关系,从而提供更有效的治疗方法。
2. 药物研究SNP分析在药物研究过程中也可以发挥重要作用。
因为不同人群人体内的代谢和反应机制是不一样的,因此,在开发新药物的过程中,SNP分析可以提供更全面的信息,从而提高药物的效率和安全性。
3. 个性化医疗随着SNP分析的应用越来越广泛,越来越多的医疗机构开始使用它来提供更精准的治疗方案。
根据患者的基因信息,医生可以制定更适合个人的治疗方法,从而提高治疗效果和疗效持续时间。
SNP分析的方法SNP分析的方法有很多,其中最常见的两种方法是Sanger测序和芯片技术。
1. Sanger测序Sanger测序是SNP分析的传统方法,之所以广泛应用,是因为它是一种基于荧光技术的自动测序方法。
Sanger测序的具体原理如下:首先,将DNA样本与引物一起反应,通过PCR技术扩增目标基因区域。
然后,将PCR产物分离并富集,通过荧光标记的引物在ABI 3730 DNA自动测序仪上进行自动测序。
最后,通过电脑软件将测序结果转化为DNA碱基序列。
snp 芯片

snp 芯片SNP芯片,也称为单核苷酸多态性芯片(Single Nucleotide Polymorphism,SNP),是一种用于检测个体基因变异的技术。
SNP指的是DNA序列中存在的单个核苷酸变异,它是基因组中最常见的变异类型,据估计,人类基因组中大约有数百万个SNP。
SNP芯片包含了大量的DNA探针,每个探针用于检测一个SNP位点的基因型。
这些位点通常位于基因组中的编码区域或者非编码区域,与个体的基因型密切相关。
SNP芯片具有高通量、高灵敏度、高特异性和高吞吐量的特点,因此在基因组学研究、疾病诊断、药物研发等领域得到了广泛应用。
SNP芯片的工作原理是基于杂交技术。
首先,将待检测样品中的DNA片段进行扩增,然后用特定的探针与待检测样品中的DNA片段进行杂交反应。
探针上的标记物可以帮助识别杂交反应的结果。
根据标记物的信号强度,可以确定待检测样品中SNP位点的基因型。
SNP芯片可用于多种应用,包括基因组关联研究、遗传性疾病诊断、个体化药物治疗等。
在基因组关联研究中,可以比较不同个体之间的基因型差异,以探索基因与特定性状之间的关联。
在遗传性疾病诊断中,可以检测个体是否携带与特定疾病相关的致病基因。
在个体化药物治疗中,可以确定个体是否对某种药物敏感,从而指导药物的选择和剂量的调整。
SNP芯片的应用还面临一些挑战和限制。
首先,SNP芯片只能检测已知的SNP位点,不能发现新的变异。
其次,由于人群的基因座群存在较大的差异,SNP芯片的设计需要针对不同人群进行优化。
此外,SNP芯片的数据处理和解读也是一个复杂的任务,需要结合临床信息和其他遗传变异数据进行综合分析。
总的来说,SNP芯片是一种重要的基因检测技术,可以用于研究人类基因组的变异、诊断遗传性疾病和指导个体化药物治疗。
随着技术的不断发展和成本的降低,SNP芯片将在医学和生命科学领域发挥更大的作用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Affymetrix SNP芯片数据分析方案
项目一、基本分析
包括:
芯片原始数据的处理和基因分型,我们给出有统计意义的SNP列表。
描述性统计,如minor allele frequency,Hardy-Weinberg equilibrium等。
显著性检验,实验组与对照组的差异,假阳性率(FDR)的计算等。
SNP的关联分析,建立线性模型或logistic回归模型等。
(所有的统计可以选择由SAS,SPSS,或S-Plus/R给出)
项目二、Copy Number Variation(CNV)的计算。
CNV是目前的一个热点研究内容。
SNP芯片数据可以用于精确地计算CNV。
我们提供针对SNP芯片的基于CNAG(Copy Number Analyser for GeneChip), dChip(DNA-Chip Analyzer)和CNAT(Chromosome Copy Number Analysis Tool)等算法的CNV计算结果。
项目三、SNP注释
通过SNP在染色体上的位置,利用寻找SNP可能影响的基因( or EST)。
我们也可以对相应基因进行功能的注释(gene ontology ,pathway和转录因子分析等),进而解释SNP可能的作用机理。
该部分可以参考常规表达谱芯片的分析。
项目四:基于模式识别的SNP挖掘
传统的SNP挖掘使用统计学的方法来进行,往往在敏感性与特异性上有一定的限制。
利用一些模式识别/机器学习的方法可以更好解决SNP筛选问题。
我们提供基于决策树等SNP挖掘算法。
Hsiang-Yu Yuan et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Research 2006 34(Web Server issue):W635-W641
项目五:诊断模型建立
利用筛选到的SNP建立人工神经网络(ANN)、SVM、PAML等诊断模型,在临床上具有重要意义。
下图是我们使用ANN方法来构建诊断模型设计的策略:Array项目六、不同SNP平台整合分析
目前SNP public的数据越来越多,主要使用的是Illumina和affymetrix两个平台。
我们提供
公共数据的整合分析的解决方案,包括不同平台数据之间的整合。
项目七、SNP芯片与表达谱芯片,aCGH等其他高通量数据整合
SNP与表达谱芯片,aCGH在技术上各有所长。
我们提供数据整合方案实现综合所有可能的高通量方法,解决相应的生物学问题:如癌症药靶的筛选,复杂遗传疾病marker的挖掘等。
Stein Aerts et al. Gene prioritization through genomic data fusion. NA TURE BIOTECHNOLOGY VOLUME 24 NUMBER 5 MAY 2006
项目八、实验验证部分
包括数据验证实验设计,以及实验服务。
我们提供采用PCR方法,或taq-man的real-time PCR基因分型方法等对SNP芯片内容进行验证的服务。
另外,我们也提供对SNP功能的后续实验服务,这包括:
1.对于位于基因启动子区域的SNP, 我们推荐对基因蛋白表达量进行检测(western blot)。
同时利用转录因子分析点突变对转录因子结合自由能的影响。
2.对于intron中的SNP,我们推荐进行可变剪切的验证(northern blot)。
3.对于CDS区域的SNP我们建议是否为同义突变,非同义突变可以利用3D你比较建模
分析蛋白结构的变化。
4.对于3’-UTR我们推荐进行microRNA结合位点预测和常见3’-UTR元件预测(如ARE
等)。
突变会造成结合位点的丢失。